ROTATEQ- rotavirus vaccine, live, oral, pentavalent solution
RotaTeq by
Drug Labeling and Warnings
RotaTeq by is a Other medication manufactured, distributed, or labeled by Merck Sharp & Dohme LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use RotaTeq safely and effectively. See full prescribing information for RotaTeq.
RotaTeq® (Rotavirus Vaccine, Live, Oral, Pentavalent) Oral Solution
Initial U.S. Approval: 2006INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- FOR ORAL USE ONLY. NOT FOR INJECTION. (2)
- The vaccination series consists of three ready-to-use liquid doses of RotaTeq administered orally starting at 6 to 12 weeks of age, with the subsequent doses administered at 4- to 10-week intervals. The third dose should not be given after 32 weeks of age. (2)
DOSAGE FORMS AND STRENGTHS
2 mL solution for oral administration of 5 live human-bovine reassortant rotaviruses which contains a minimum of 2.0 – 2.8 x 106 infectious units (IU) per reassortant dose, depending on the reassortant, and not greater than 116 x 106 IU per aggregate dose. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- No safety or efficacy data are available from clinical trials regarding the administration of RotaTeq to infants who are potentially immunocompromised (e.g., HIV/AIDS). (5.2)
- In a post-marketing study, cases of intussusception were observed in temporal association within 21 days following the first dose of RotaTeq, with a clustering of cases in the first 7 days. (5.3, 6.2)
- No safety or efficacy data are available for the administration of RotaTeq to infants with a history of gastrointestinal disorders (e.g., active acute gastrointestinal illness, chronic diarrhea, failure to thrive, history of congenital abdominal disorders, and abdominal surgery). (5.4)
- Vaccine virus transmission from vaccine recipient to non-vaccinated contacts has been reported. Caution is advised when considering whether to administer RotaTeq to individuals with immunodeficient contacts. (5.5)
ADVERSE REACTIONS
Most common adverse events included diarrhea, vomiting, irritability, otitis media, nasopharyngitis, and bronchospasm. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., at 1-877-888-4231 or VAERS at 1-800-822-7967 or www.vaers.hhs.gov{4}.USE IN SPECIFIC POPULATIONS
Pediatric Use: Safety and efficacy have not been established in infants less than 6 weeks of age or greater than 32 weeks of age. Data are available from clinical studies to support the use of RotaTeq in:
- Pre-term infants according to their age in weeks since birth
- Infants with controlled gastroesophageal reflux disease. (8.4)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Use with Other Vaccines
2.2 Instructions for Use
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Hypersensitivity
4.2 Severe Combined Immunodeficiency Disease
4.3 History of Intussusception
5 WARNINGS AND PRECAUTIONS
5.1 Managing Allergic Reactions
5.2 Immunocompromised Populations
5.3 Intussusception
5.4 Gastrointestinal Illness
5.5 Shedding and Transmission
5.6 Febrile Illness
5.7 Incomplete Regimen
5.8 Limitations of Vaccine Effectiveness
5.9 Post-Exposure Prophylaxis
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
6.2 Post-Marketing Experience
7 DRUG INTERACTIONS
7.1 Concomitant Vaccine Administration
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Rotavirus Efficacy and Safety Trial (Study 006)
14.2 Study 007
14.3 Multiple Rotavirus Seasons
14.4 Rotavirus Gastroenteritis Regardless of Type
14.5 Rotavirus Gastroenteritis by Type
14.6 Immunogenicity
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
RotaTeq® is indicated for the prevention of rotavirus gastroenteritis in infants and children caused by types G1, G2, G3, G4, and G9 when administered as a 3-dose series to infants between the ages of 6 to 32 weeks. The first dose of RotaTeq should be administered between 6 and 12 weeks of age [see Dosage and Administration (2)].
-
2 DOSAGE AND ADMINISTRATION
FOR ORAL USE ONLY. NOT FOR INJECTION.
The vaccination series consists of three ready-to-use liquid doses of RotaTeq administered orally starting at 6 to 12 weeks of age, with the subsequent doses administered at 4- to 10-week intervals. The third dose should not be given after 32 weeks of age [see Clinical Studies (14)].
There are no restrictions on the infant's consumption of food or liquid, including breast milk, either before or after vaccination with RotaTeq.
Do not mix the RotaTeq vaccine with any other vaccines or solutions. Do not reconstitute or dilute [see Dosage and Administration (2.2)].
For storage instructions [see How Supplied/Storage and Handling (16.1)].
Each dose is supplied in a container consisting of a squeezable plastic dosing tube with a twist-off cap, allowing for direct oral administration. The dosing tube is contained in a pouch [see Dosage and Administration (2.2)].
2.1 Use with Other Vaccines
In clinical trials, RotaTeq was administered concomitantly with other licensed pediatric vaccines [see Adverse Reactions (6.1), Drug Interactions (7.1), and Clinical Studies (14)].
2.2 Instructions for Use
To administer the vaccine: 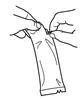
Tear open the pouch and remove the dosing tube. 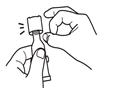
Clear the fluid from the dispensing tip by holding tube vertically and tapping cap. Open the dosing tube in 2 easy motions: 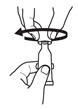
1. Puncture the dispensing tip by screwing cap clockwise
until it becomes tight.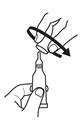
2. Remove cap by turning it counterclockwise. 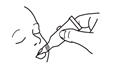
Administer dose by gently squeezing liquid into infant's mouth toward the inner cheek until dosing tube is empty. (A residual drop may remain in the tip of the tube.) If for any reason an incomplete dose is administered (e.g., infant spits or regurgitates the vaccine), a replacement dose is not recommended, since such dosing was not studied in the clinical trials. The infant should continue to receive any remaining doses in the recommended series.
Discard the empty tube and cap in approved biological waste containers according to local regulations. -
3 DOSAGE FORMS AND STRENGTHS
RotaTeq, 2 mL for oral use, is a ready-to-use solution of live reassortant rotaviruses, containing G1, G2, G3, G4 and P1A[8] which contains a minimum of 2.0 – 2.8 x 106 infectious units (IU) per individual reassortant dose, depending on the reassortant and not greater than 116 x 106 IU per aggregate dose.
Each dose is supplied in a container consisting of a squeezable plastic dosing tube with a twist-off cap, allowing for direct oral administration. The dosing tube is contained in a pouch.
-
4 CONTRAINDICATIONS
4.1 Hypersensitivity
A demonstrated history of hypersensitivity to any component of the vaccine.
Infants who develop symptoms suggestive of hypersensitivity after receiving a dose of RotaTeq should not receive further doses of RotaTeq.
4.2 Severe Combined Immunodeficiency Disease
Infants with Severe Combined Immunodeficiency Disease (SCID) should not receive RotaTeq. Post-marketing reports of gastroenteritis, including severe diarrhea and prolonged shedding of vaccine virus, have been reported in infants who were administered RotaTeq and later identified as having SCID [see Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Managing Allergic Reactions
Appropriate medical treatment and supervision must be available to manage possible anaphylactic reactions following administration of the vaccine.
5.2 Immunocompromised Populations
No safety or efficacy data are available from clinical trials regarding the administration of RotaTeq to infants who are potentially immunocompromised including:
- Infants with blood dyscrasias, leukemia, lymphomas of any type, or other malignant neoplasms affecting the bone marrow or lymphatic system.
- Infants on immunosuppressive therapy (including high-dose systemic corticosteroids). RotaTeq may be administered to infants who are being treated with topical corticosteroids or inhaled steroids.
- Infants with primary and acquired immunodeficiency states, including HIV/AIDS or other clinical manifestations of infection with human immunodeficiency viruses; cellular immune deficiencies; and hypogammaglobulinemic and dysgammaglobulinemic states. There are insufficient data from the clinical trials to support administration of RotaTeq to infants with indeterminate HIV status who are born to mothers with HIV/AIDS.
- Infants who have received a blood transfusion or blood products, including immunoglobulins within 42 days.
Vaccine virus transmission from vaccine recipient to non-vaccinated contacts has been reported [see Warnings and Precautions (5.5)].
5.3 Intussusception
Following administration of a previously licensed live rhesus rotavirus reassortant vaccine, an increased risk of intussusception was observed.{1}
In a post-marketing observational study in the US cases of intussusception were observed in temporal association within 21 days following the first dose of RotaTeq, with a clustering of cases in the first 7 days. [See Adverse Reactions (6.2).]
In worldwide passive post-marketing surveillance, cases of intussusception have been reported in temporal association with RotaTeq. [See Adverse Reactions (6.2).]
5.4 Gastrointestinal Illness
No safety or efficacy data are available for administration of RotaTeq to infants with a history of gastrointestinal disorders including infants with active acute gastrointestinal illness, infants with chronic diarrhea and failure to thrive, and infants with a history of congenital abdominal disorders, and abdominal surgery. Caution is advised when considering administration of RotaTeq to these infants.
5.5 Shedding and Transmission
Shedding of vaccine virus was evaluated among a subset of subjects in the Rotavirus Efficacy and Safety Trial (Study 006, also known as REST) 4 to 6 days after each dose and among all subjects who submitted a stool antigen rotavirus positive sample at any time. RotaTeq was shed in the stools of 32 of 360 [8.9%, 95% CI (6.2%, 12.3%)] vaccine recipients tested after dose 1; 0 of 249 [0.0%, 95% CI (0.0%, 1.5%)] vaccine recipients tested after dose 2; and in 1 of 385 [0.3%, 95% CI (<0.1%, 1.4%)] vaccine recipients after dose 3. In phase 3 studies, shedding was observed as early as 1 day and as late as 15 days after a dose. Transmission of vaccine virus was not evaluated in phase 3 studies.
Transmission of vaccine virus strains from vaccinees to non-vaccinated contacts has been observed post-marketing.
The potential risk of transmission of vaccine virus should be weighed against the risk of acquiring and transmitting natural rotavirus.
Caution is advised when considering whether to administer RotaTeq to individuals with immunodeficient close contacts such as:
- Individuals with malignancies or who are otherwise immunocompromised;
- Individuals with primary immunodeficiency; or
- Individuals receiving immunosuppressive therapy.
5.6 Febrile Illness
Febrile illness may be reason for delaying use of RotaTeq except when, in the opinion of the physician, withholding the vaccine entails a greater risk. Low-grade fever (<100.5°F [38.1°C]) itself and mild upper respiratory infection do not preclude vaccination with RotaTeq.
5.7 Incomplete Regimen
The clinical studies were not designed to assess the level of protection provided by only one or two doses of RotaTeq.
-
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
71,725 infants were evaluated in 3 placebo-controlled clinical trials including 36,165 infants in the group that received RotaTeq and 35,560 infants in the group that received placebo. Parents/guardians were contacted on days 7, 14, and 42 after each dose regarding intussusception and any other serious adverse events. The racial distribution was as follows: White (69% in both groups); Hispanic-American (14% in both groups); Black (8% in both groups); Multiracial (5% in both groups); Asian (2% in both groups); Native American (RotaTeq 2%, placebo 1%); and Other (<1% in both groups). The gender distribution was 51% male and 49% female in both vaccination groups.
Because clinical trials are conducted under conditions that may not be typical of those observed in clinical practice, the adverse reaction rates presented below may not be reflective of those observed in clinical practice.
Serious Adverse Events
Serious adverse events occurred in 2.4% of recipients of RotaTeq when compared to 2.6% of placebo recipients within the 42-day period of a dose in the phase 3 clinical studies of RotaTeq. The most frequently reported serious adverse events for RotaTeq compared to placebo were:
bronchiolitis (0.6% RotaTeq vs. 0.7% Placebo),
gastroenteritis (0.2% RotaTeq vs. 0.3% Placebo),
pneumonia (0.2% RotaTeq vs. 0.2% Placebo),
fever (0.1% RotaTeq vs. 0.1% Placebo), and
urinary tract infection (0.1% RotaTeq vs. 0.1% Placebo).
Deaths
Across the clinical studies, 52 deaths were reported. There were 25 deaths in the RotaTeq recipients compared to 27 deaths in the placebo recipients. The most commonly reported cause of death was sudden infant death syndrome, which was observed in 8 recipients of RotaTeq and 9 placebo recipients.
Intussusception
In Study 006, 34,837 vaccine recipients and 34,788 placebo recipients were monitored by active surveillance to identify potential cases of intussusception at 7, 14, and 42 days after each dose, and every 6 weeks thereafter for 1 year after the first dose.
For the primary safety outcome, cases of intussusception occurring within 42 days of any dose, there were 6 cases among RotaTeq recipients and 5 cases among placebo recipients (see Table 1). The data did not suggest an increased risk of intussusception relative to placebo.
Table 1: Confirmed cases of intussusception in recipients of RotaTeq as compared with placebo recipients during Study 006 - * Relative risk and 95% confidence interval based upon group sequential design stopping criteria employed in Study 006.
RotaTeq (n=34,837) Placebo (n=34,788) Confirmed intussusception cases within 42 days of any dose 6 5 Relative risk (95% CI) * 1.6 (0.4, 6.4) Confirmed intussusception cases within 365 days of dose 1 13 15 Relative risk (95% CI) 0.9 (0.4, 1.9) Among vaccine recipients, there were no confirmed cases of intussusception within the 42-day period after the first dose, which was the period of highest risk for the rhesus rotavirus-based product (see Table 2).
Table 2: Intussusception cases by day range in relation to dose in Study 006 Dose 1 Dose 2 Dose 3 Any Dose Day Range RotaTeq Placebo RotaTeq Placebo RotaTeq Placebo RotaTeq Placebo 1-7 0 0 1 0 0 0 1 0 1-14 0 0 1 0 0 1 1 1 1-21 0 0 3 0 0 1 3 1 1-42 0 1 4 1 2 3 6 5 All of the children who developed intussusception recovered without sequelae with the exception of a 9-month-old male who developed intussusception 98 days after dose 3 and died of post-operative sepsis. There was a single case of intussusception among 2,470 recipients of RotaTeq in a 7-month-old male in the phase 1 and 2 studies (716 placebo recipients).
Hematochezia
Hematochezia reported as an adverse experience occurred in 0.6% (39/6,130) of vaccine and 0.6% (34/5,560) of placebo recipients within 42 days of any dose. Hematochezia reported as a serious adverse experience occurred in <0.1% (4/36,150) of vaccine and <0.1% (7/35,536) of placebo recipients within 42 days of any dose.
Seizures
All seizures reported in the phase 3 trials of RotaTeq (by vaccination group and interval after dose) are shown in Table 3.
Table 3: Seizures reported by day range in relation to any dose in the phase 3 trials of RotaTeq Day range 1-7 1-14 1-42 RotaTeq 10 15 33 Placebo 5 8 24 Seizures reported as serious adverse experiences occurred in <0.1% (27/36,150) of vaccine and <0.1% (18/35,536) of placebo recipients (not significant). Ten febrile seizures were reported as serious adverse experiences, 5 were observed in vaccine recipients and 5 in placebo recipients.
Kawasaki Disease
In the phase 3 clinical trials, infants were followed for up to 42 days of vaccine dose. Kawasaki disease was reported in 5 of 36,150 vaccine recipients and in 1 of 35,536 placebo recipients with unadjusted relative risk 4.9 (95% CI 0.6, 239.1).
Most Common Adverse Events
Solicited Adverse Events
Detailed safety information was collected from 11,711 infants (6,138 recipients of RotaTeq) which included a subset of subjects in Study 006 and all subjects from Studies 007 and 009 (Detailed Safety Cohort). A Vaccination Report Card was used by parents/guardians to record the child's temperature and any episodes of diarrhea and vomiting on a daily basis during the first week following each vaccination. Table 4 summarizes the frequencies of these adverse events and irritability.
Table 4: Solicited adverse experiences within the first week after doses 1, 2, and 3 (Detailed Safety Cohort) - * Temperature ≥100.5°F [38.1°C] rectal equivalent obtained by adding 1 degree F to otic and oral temperatures and 2 degrees F to axillary temperatures
Adverse experience Dose 1 Dose 2 Dose 3 RotaTeq Placebo RotaTeq Placebo RotaTeq Placebo Elevated temperature* n=5,616
17.1%n=5,077
16.2%n=5,215
20.0%n=4,725
19.4%n=4,865
18.2%n=4,382
17.6%n=6,130 n=5,560 n=5,703 n=5,173 n=5,496 n=4,989 Vomiting 6.7% 5.4% 5.0% 4.4% 3.6% 3.2% Diarrhea 10.4% 9.1% 8.6% 6.4% 6.1% 5.4% Irritability 7.1% 7.1% 6.0% 6.5% 4.3% 4.5% Other Adverse Events
Parents/guardians of the 11,711 infants were also asked to report the presence of other events on the Vaccination Report Card for 42 days after each dose.
Fever was observed at similar rates in vaccine (N=6,138) and placebo (N=5,573) recipients (42.6% vs. 42.8%). Adverse events that occurred at a statistically higher incidence (i.e., 2-sided p-value <0.05) within the 42 days of any dose among recipients of RotaTeq as compared with placebo recipients are shown in Table 5.
Table 5: Adverse events that occurred at a statistically higher incidence within 42 days of any dose among recipients of RotaTeq as compared with placebo recipients Adverse event RotaTeq
N=6,138Placebo
N=5,573n (%) n (%) Diarrhea 1,479 (24.1%) 1,186 (21.3%) Vomiting 929 (15.2%) 758 (13.6%) Otitis media 887 (14.5%) 724 (13.0%) Nasopharyngitis 422 (6.9%) 325 (5.8%) Bronchospasm 66 (1.1%) 40 (0.7%) Safety in Pre-Term Infants
RotaTeq or placebo was administered to 2,070 pre-term infants (25 to 36 weeks gestational age, median 34 weeks) according to their age in weeks since birth in Study 006. All pre-term infants were followed for serious adverse experiences; a subset of 308 infants was monitored for all adverse experiences. There were 4 deaths throughout the study, 2 among vaccine recipients (1 SIDS and 1 motor vehicle accident) and 2 among placebo recipients (1 SIDS and 1 unknown cause). No cases of intussusception were reported. Serious adverse experiences occurred in 5.5% of vaccine and 5.8% of placebo recipients. The most common serious adverse experience was bronchiolitis, which occurred in 1.4% of vaccine and 2.0% of placebo recipients. Parents/guardians were asked to record the child's temperature and any episodes of vomiting and diarrhea daily for the first week following vaccination. The frequencies of these adverse experiences and irritability within the week after dose 1 are summarized in Table 6.
Table 6: Solicited adverse experiences within the first week of doses 1, 2, and 3 among pre-term infants - * Temperature ≥100.5°F [38.1°C] rectal equivalent obtained by adding 1 degree F to otic and oral temperatures and 2 degrees F to axillary temperatures
Dose 1 Dose 2 Dose 3 Adverse event RotaTeq Placebo RotaTeq Placebo RotaTeq Placebo N=127 N=133 N=124 N=121 N=115 N=108 Elevated temperature* 18.1% 17.3% 25.0% 28.1% 14.8% 20.4% N=154 N=154 N=137 N=137 N=135 N=129 Vomiting 5.8% 7.8% 2.9% 2.2% 4.4% 4.7% Diarrhea 6.5% 5.8% 7.3% 7.3% 3.7% 3.9% Irritability 3.9% 5.2% 2.9% 4.4% 8.1% 5.4% 6.2 Post-Marketing Experience
The following adverse events have been identified during post-approval use of RotaTeq from reports to the Vaccine Adverse Event Reporting System (VAERS).
Reporting of adverse events following immunization to VAERS is voluntary, and the number of doses of vaccine administered is not known; therefore, it is not always possible to reliably estimate the adverse event frequency or establish a causal relationship to vaccine exposure using VAERS data.
In post-marketing experience, the following adverse events have been reported following the use of RotaTeq:
Immune system disorders:
Anaphylactic reaction
Gastrointestinal disorders:
Intussusception (including death)
Hematochezia
Gastroenteritis with vaccine viral shedding in infants with Severe Combined Immunodeficiency Disease (SCID)
Skin and subcutaneous tissue disorders:
Urticaria
Angioedema
Infections and infestations:
Kawasaki disease
Transmission of vaccine virus strains from vaccine recipient to non-vaccinated contacts.
Post-Marketing Observational Safety Surveillance Studies
The temporal association between vaccination with RotaTeq and intussusception was evaluated in the Post-licensure Rapid Immunization Safety Monitoring (PRISM) program {2}, an electronic active surveillance program comprised of 3 US health insurance plans.
More than 1.2 million RotaTeq vaccinations (507,000 of which were first doses) administered to infants 5 through 36 weeks of age were evaluated. From 2004 through 2011, potential cases of intussusception in either the inpatient or emergency department setting and vaccine exposures were identified through electronic procedure and diagnosis codes. Medical records were reviewed to confirm intussusception and rotavirus vaccination status.
The risk of intussusception was assessed using self-controlled risk interval and cohort designs, with adjustment for age. Risk windows of 1-7 and 1-21 days were evaluated. Cases of intussusception were observed in temporal association within 21 days following the first dose of RotaTeq, with a clustering of cases in the first 7 days. Based on the results, approximately 1 to 1.5 excess cases of intussusception occur per 100,000 vaccinated US infants within 21 days following the first dose of RotaTeq. In the first year of life, the background rate of intussusception hospitalizations in the US has been estimated to be approximately 34 per 100,000 infants.{3}
In an earlier prospective post-marketing observational cohort study conducted using a large US medical claims database, the risks of intussusception or Kawasaki disease resulting in emergency department visits or hospitalizations during the 30 days following any dose of vaccine were analyzed among 85,150 infants receiving one or more doses of RotaTeq from February 2006 through March 2009. Medical charts were reviewed to confirm these diagnoses. Evaluation included concurrent (n = 62,617) and historical (n=100,000 from 2001-2005) control groups of infants who received diphtheria, tetanus and acellular pertussis vaccine (DTaP) but not RotaTeq.
Confirmed intussusception cases in the RotaTeq group were compared with those in the concurrent DTaP control group and in the historical control group. The data were analyzed post-dose 1 and post any dose, in both 7 day and 30 day risk windows. A statistically significant increased risk of intussusception after RotaTeq vaccination was not observed.
One confirmed case of Kawasaki disease (23 days post-dose 3) was identified among infants vaccinated with RotaTeq and one confirmed case of Kawasaki disease (22 days post-dose 2) was identified among concurrent DTaP controls (relative risk = 0.7; 95% CI: 0.01-55.56).
In addition, general safety was monitored by electronic search of the automated records database for all emergency department visits and hospitalizations in the 30-day period after each dose of RotaTeq compared with: 1) days 31-60 after each dose of RotaTeq (self-matched controls) and 2) the 30-day period after each dose of DTaP vaccine (historical control subset from 2004-2005, n=40,000). In safety analyses which evaluated multiple follow-up windows after vaccination (days: 0-7, 1-7, 8-14 and 0-30), no safety concerns were identified for infants vaccinated with RotaTeq when compared with self-matched controls and the historical control subset.
Reporting Adverse Events
Parents or guardians should be instructed to report any adverse reactions to their health care provider.
Health care providers should report all adverse events to the U.S. Department of Health and Human Services' Vaccine Adverse Events Reporting System (VAERS).
VAERS accepts all reports of suspected adverse events after the administration of any vaccine, including but not limited to the reporting of events required by the National Childhood Vaccine Injury Act of 1986. For information or a copy of the vaccine reporting form, call the VAERS toll-free number at 1-800-822-7967 or report on line to www.vaers.hhs.gov.{4}
-
7 DRUG INTERACTIONS
Immunosuppressive therapies including irradiation, antimetabolites, alkylating agents, cytotoxic drugs and corticosteroids (used in greater than physiologic doses), may reduce the immune response to vaccines.
7.1 Concomitant Vaccine Administration
In clinical trials, RotaTeq was administered concomitantly with diphtheria and tetanus toxoids and acellular pertussis (DTaP), inactivated poliovirus vaccine (IPV), H. influenzae type b conjugate (Hib), hepatitis B vaccine, and pneumococcal conjugate vaccine [see Clinical Studies (14)]. The safety data available are in the ADVERSE REACTIONS section [see Adverse Reactions (6.1)]. There was no evidence for reduced antibody responses to the vaccines that were concomitantly administered with RotaTeq.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
RotaTeq is not approved for individuals 32 weeks of age and older. No human or animal data are available to assess vaccine-associated risks in pregnancy.
8.2 Lactation
No human or animal data are available to assess the impact of RotaTeq on milk production, its presence in breast milk, or its effect on the breastfed infant.
8.4 Pediatric Use
Safety and effectiveness of RotaTeq have not been established in infants less than 6 weeks of age or greater than 32 weeks of age.
Data are available from clinical studies to support the use of RotaTeq in pre-term infants according to their age in weeks since birth [see Adverse Reactions (6.1)].
Data are available from clinical studies to support the use of RotaTeq in infants with controlled gastroesophageal reflux disease.
-
10 OVERDOSAGE
There have been post-marketing reports of infants who received more than one dose or a replacement dose of RotaTeq after regurgitation [see Dosage and Administration (2.2)]. In limited post-marketing experience of reported overdosage, the adverse events reported after incorrect administration of higher than recommended doses of RotaTeq were similar to adverse events observed with the approved dosage and schedule.
-
11 DESCRIPTION
RotaTeq is a live, oral pentavalent vaccine that contains 5 live reassortant rotaviruses. The rotavirus parent strains of the reassortants were isolated from human and bovine hosts. Four reassortant rotaviruses express one of the outer capsid proteins (G1, G2, G3, or G4) from the human rotavirus parent strain and the attachment protein (type P7) from the bovine rotavirus parent strain. The fifth reassortant virus expresses the attachment protein, P1A (genotype P[8]), herein referred to as type P1A[8], from the human rotavirus parent strain and the outer capsid protein of type G6 from the bovine rotavirus parent strain (see Table 7).
Table 7 Name of Reassortant Human Rotavirus Parent Strains and Outer Surface Protein Compositions Bovine Rotavirus Parent Strain and Outer Surface Protein Composition Reassortant Outer Surface Protein Composition (Human Rotavirus Component in Bold) Minimum Dose Levels (106 infectious units) G1 WI79 – G1P1A[8] WC3 - G6, P7[5] G1P7[5] 2.2 G2 SC2 – G2P2[6] G2P7[5] 2.8 G3 WI78 – G3P1A[8] G3P7[5] 2.2 G4 BrB – G4P2[6] G4P7[5] 2.0 P1A[8] WI79 – G1P1A[8] G6P1A[8] 2.3 The reassortants are propagated in Vero cells using standard cell culture techniques in the absence of antifungal agents.
The reassortants are suspended in a buffered stabilizer solution. Each vaccine dose contains sucrose, sodium citrate, sodium phosphate monobasic monohydrate, sodium hydroxide, polysorbate 80, cell culture media, and trace amounts of fetal bovine serum. RotaTeq contains no preservatives.
In the manufacturing process for RotaTeq, a porcine-derived material is used. DNA from porcine circoviruses (PCV) 1 and 2 has been detected in RotaTeq. PCV-1 and PCV-2 are not known to cause disease in humans.
RotaTeq is a pale yellow clear liquid that may have a pink tint.
The plastic dosing tube and cap do not contain latex.
-
12 CLINICAL PHARMACOLOGY
Rotavirus is a leading cause of severe acute gastroenteritis in infants and young children, with over 95% of these children infected by the time they are 5 years old.{5} The most severe cases occur among infants and young children between 6 months and 24 months of age.{6}
12.1 Mechanism of Action
The exact immunologic mechanism by which RotaTeq protects against rotavirus gastroenteritis is unknown [see Clinical Studies (14.6)]. RotaTeq is a live viral vaccine that replicates in the small intestine and induces immunity.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
Overall, 73,086 infants were randomized in 4 placebo-controlled, phase 3 studies conducted in 12 countries on 4 continents. The data demonstrating the efficacy of RotaTeq in preventing rotavirus gastroenteritis come from 7,744 of these infants from the US (including Navajo and White Mountain Apache Nations), Finland, and Japan who were enrolled in 3 of these studies: Study 006, Study 007, and Study 029. A fourth trial, Study 009, provided clinical evidence supporting the consistency of manufacture and contributed data to the overall safety evaluation.
The racial distribution of the efficacy subset was as follows: White (RotaTeq 61%, placebo 62%); Hispanic-American (RotaTeq 9%, placebo 8%); Black (2% in both groups); Multiracial (4% in both groups); Asian (10% in both groups); Native American (13% in both groups); and Other (<1% in both groups). The gender distribution was 52% male and 48% female in both vaccination groups.
The efficacy evaluations in these studies included: 1) Prevention of any grade of severity of rotavirus gastroenteritis; 2) Prevention of severe rotavirus gastroenteritis, as defined by a clinical scoring system; and 3) Reduction in hospitalizations due to rotavirus gastroenteritis.
The vaccine was given as a three-dose series to healthy infants with the first dose administered between 6 and 12 weeks of age and followed by two additional doses administered at 4- to 10-week intervals. The age of infants receiving the third dose was 32 weeks of age or less. Oral polio vaccine administration was not permitted; however, other childhood vaccines could be concomitantly administered. Breast-feeding was permitted in all studies.
The case definition for rotavirus gastroenteritis used to determine vaccine efficacy required that a subject meet both of the following clinical and laboratory criteria: (1) greater than or equal to 3 watery or looser-than-normal stools within a 24-hour period and/or forceful vomiting; and (2) rotavirus antigen detection by enzyme immunoassay (EIA) in a stool specimen taken within 14 days of onset of symptoms. The severity of rotavirus acute gastroenteritis was determined by a clinical scoring system that took into account the intensity and duration of symptoms of fever, vomiting, diarrhea, and behavioral changes.
The primary efficacy analyses included cases of rotavirus gastroenteritis caused by types G1, G2, G3, G4 (and G types containing P1A8 (in Study 029 only)) that occurred at least 14 days after the third dose through the first rotavirus season post vaccination.
Analyses were also done to evaluate the efficacy of RotaTeq against rotavirus gastroenteritis caused by any of types G1, G2, G3, and G4 (and G types containing P1A8 (in Study 029 only)) at any time following the first dose through the first rotavirus season postvaccination among infants who received at least one vaccination (Intent-to-treat, ITT).
14.1 Rotavirus Efficacy and Safety Trial (Study 006)
Primary efficacy against any grade of severity of rotavirus gastroenteritis caused by naturally occurring types G1, G2, G3, or G4 through the first rotavirus season after vaccination was 74.0% (95% CI: 66.8, 79.9) and the ITT efficacy was 60.0% (95% CI: 51.5, 67.1). Primary efficacy against severe rotavirus gastroenteritis caused by naturally occurring types G1, G2, G3, or G4 through the first rotavirus season after vaccination was 98.0% (95% CI: 88.3, 100.0), and ITT efficacy was 96.4% (95% CI: 86.2, 99.6). See Table 8.
Table 8: Efficacy of RotaTeq against any grade of severity of and severe* G1-4 rotavirus gastroenteritis through the first rotavirus season postvaccination in Study 006 - * Severe gastroenteritis defined by a clinical scoring system based on the intensity and duration of symptoms of fever, vomiting, diarrhea, and behavioral changes
- † ITT analysis includes all subjects in the efficacy cohort who received at least one dose of vaccine.
Per Protocol Intent-to-Treat† RotaTeq Placebo RotaTeq Placebo Subjects vaccinated 2,834 2,839 2,834 2,839 Gastroenteritis cases Any grade of severity 82 315 150 371 Severe* 1 51 2 55 Efficacy estimate % and (95% confidence interval) Any grade of severity 74.0
(66.8, 79.9)60.0
(51.5, 67.1)Severe* 98.0
(88.3, 100.0)96.4
(86.2, 99.6)The efficacy of RotaTeq against severe disease was also demonstrated by a reduction in hospitalizations for rotavirus gastroenteritis among all subjects enrolled in Study 006. RotaTeq reduced hospitalizations for rotavirus gastroenteritis caused by types G1, G2, G3, and G4 through the first two years after the third dose by 95.8% (95% CI: 90.5, 98.2). The ITT efficacy in reducing hospitalizations was 94.7% (95% CI: 89.3, 97.3) as shown in Table 9.
Table 9: Efficacy of RotaTeq in reducing G1-4 rotavirus-related hospitalizations in Study 006 - * ITT analysis includes all subjects who received at least one dose of vaccine.
Per Protocol Intent-to-Treat* RotaTeq Placebo RotaTeq Placebo Subjects vaccinated 34,035 34,003 34,035 34,003 Number of hospitalizations 6 144 10 187 Efficacy estimate % and
(95% confidence interval)95.8
(90.5, 98.2)94.7
(89.3, 97.3)14.2 Study 007
Primary efficacy against any grade of severity of rotavirus gastroenteritis caused by naturally occurring types G1, G2, G3, or G4 through the first rotavirus season after vaccination was 72.5% (95% CI: 50.6, 85.6) and the ITT efficacy was 58.4% (95% CI: 33.8, 74.5). Primary efficacy against severe rotavirus gastroenteritis caused by naturally occurring types G1, G2, G3, or G4 through the first rotavirus season after vaccination was 100% (95% CI: 13.0, 100.0) and ITT efficacy against severe rotavirus disease was 100% (95% CI: 30.2, 100.0) as shown in Table 10.
Table 10: Efficacy of RotaTeq against any grade of severity of and severe* G1-4 rotavirus gastroenteritis through the first rotavirus season postvaccination in Study 007 - * Severe gastroenteritis defined by a clinical scoring system based on the intensity and duration of symptoms of fever, vomiting, diarrhea, and behavioral change
- † ITT analysis includes all subjects in the efficacy cohort who received at least one dose of vaccine.
Per Protocol Intent-to-Treat† RotaTeq Placebo RotaTeq Placebo Subjects vaccinated 650 660 650 660 Gastroenteritis cases Any grade of severity 15 54 27 64 Severe* 0 6 0 7 Efficacy estimate % and (95% confidence interval) Any grade of severity 72.5
(50.6, 85.6)58.4
(33.8, 74.5)Severe* 100.0
(13.0, 100.0)100.0
(30.2, 100.0)14.3 Multiple Rotavirus Seasons
The efficacy of RotaTeq through a second rotavirus season was evaluated in a single study (Study 006). Efficacy against any grade of severity of rotavirus gastroenteritis caused by rotavirus types G1, G2, G3, and G4 through the two rotavirus seasons after vaccination was 71.3% (95% CI: 64.7, 76.9). The efficacy of RotaTeq in preventing cases occurring only during the second rotavirus season postvaccination was 62.6% (95% CI: 44.3, 75.4). The efficacy of RotaTeq beyond the second season postvaccination was not evaluated.
14.4 Rotavirus Gastroenteritis Regardless of Type
The rotavirus types identified in the efficacy subset of Study 006 and Study 007 were G1P1A[8]; G2P1[4]; G3P1A[8]; G4P1A[8]; and G9P1A[8].
In Study 006, the efficacy of RotaTeq against any grade of severity of naturally occurring rotavirus gastroenteritis regardless of type was 71.8% (95% CI: 64.5, 77.8) and efficacy against severe rotavirus disease was 98.0% (95% CI: 88.3, 99.9). The ITT efficacy starting at dose 1 was 50.9% (95% CI: 41.6, 58.9) for any grade of severity of rotavirus disease and was 96.4% (95% CI: 86.3, 99.6) for severe rotavirus disease.
In Study 007, the primary efficacy of RotaTeq against any grade of severity of rotavirus gastroenteritis regardless of type was 72.7% (95% CI: 51.9, 85.4) and efficacy against severe rotavirus disease was 100% (95% CI: 12.7, 100). The ITT efficacy starting at dose 1 was 48.0% (95% CI: 21.6, 66.1) for any grade of severity of rotavirus disease and was 100% (95% CI: 30.4, 100.0) for severe rotavirus disease.
14.5 Rotavirus Gastroenteritis by Type
The efficacy against any grade of severity of rotavirus gastroenteritis by type was evaluated in Study 006 and Study 029. The efficacy cohort analysis from Study 006 is shown in Table 11.
Table 11: Type-specific efficacy of RotaTeq against any grade of severity of rotavirus gastroenteritis among infants in Study 006 efficacy cohort through the first rotavirus season postvaccination (Per Protocol) - * Includes rotavirus antigen-positive samples in which the specific type could not be identified by PCR
Type identified by PCR Number of cases % Efficacy
(95% Confidence Interval)RotaTeq
(N=2,834)Placebo
(N=2,839)G1P1A[8] 72 286 74.9 (67.3, 80.9) G2P1[4] 6 17 63.4 (2.6, 88.2) G3P1A[8] 1 6 NS G4P1A[8] 3 6 NS G9P1A[8] 1 3 NS Unidentified* 11 15 NS N=number vaccinated
NS=not significantAdditional analyses were conducted to evaluate efficacy in the prevention of rotavirus gastroenteritis due to G9P1A[8].
- In Study 029 (a Phase 3 randomized, blinded, placebo-controlled study conducted in Japan), efficacy on the pre-specified primary endpoint (rotavirus gastroenteritis caused by G1, G2, G3, G4, and G-serotypes associated with serotype P1A[8] (e.g., G9)) was 74.5% (95% CI: 39.9, 90.6). G9P1A[8]-associated gastroenteritis was observed in 0/356 and 5/354 subjects in the RotaTeq and placebo groups, respectively (100% (95% CI: -9.0, 100)).
- In a post hoc analysis of health care utilization data from 68,038 infants (RotaTeq 34,035 and placebo 34,003) in Study 006, using a case definition that included culture confirmation, hospitalization and emergency departments visits for rotavirus gastroenteritis, cases due to G9P1A[8] were reduced (RotaTeq 0 cases: placebo 14 cases) by 100% (95% CI: 69.6, 100.0).
14.6 Immunogenicity
A relationship between antibody responses to RotaTeq and protection against rotavirus gastroenteritis has not been established. In phase 3 studies, 92.9% to 100% of 439 recipients of RotaTeq achieved a 3-fold or more rise in serum anti-rotavirus IgA after a three-dose regimen when compared to 12.3%-20.0% of 397 placebo recipients.
-
15 REFERENCES
- Murphy TV, Gargiullo PM, Massoudi MS et al. Intussusception among infants given an oral rotavirus vaccine. N Engl J Med 2001;344:564-572.
- Yih WK, Lieu TA, Kulldorff M, et al. Intussusception risk after rotavirus vaccination in US infants. Mini-Sentinel. www.mini-sentinel.org.
- Tate JE, Simonsen L, Viboud C, et al. Trends in intussusception hospitalizations among US infants, 1993-2004: implications for monitoring the safety of the new rotavirus vaccination program. Pediatrics 2008;121(5):e1125-e1132.
- Centers for Disease Control and Prevention. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP) and the American Academy of Family Physicians (AAFP). MMWR 2002;51(RR-2):1-35.
- Parashar UD et al. Global illness and deaths caused by rotavirus disease in children. Emerg Infect Dis 2003;9(5):565-572.
- Parashar UD, Holman RC, Clarke MJ, Bresee JS, Glass RI. Hospitalizations associated with rotavirus diarrhea in the United States, 1993 through 1995: surveillance based on the new ICD-9-CM rotavirus-specific diagnostic code. J Infect Dis 1998;177:13-7.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
RotaTeq, 2 mL, a solution for oral use, is a pale yellow clear liquid that may have a pink tint. It is supplied as follows:
NDC 0006-4047-41 package of 10 individually pouched single-dose tubes.
NDC 0006-4047-20 package of 25 individually pouched single-dose tubes.
The plastic dosing tube and cap do not contain latex.
16.1 Storage and Handling
Store and transport refrigerated at 2-8°C (36-46°F). RotaTeq should be administered as soon as possible after being removed from refrigeration. For information regarding stability under conditions other than those recommended, call 1-800-MERCK-90.
Protect from light.
RotaTeq should be discarded in approved biological waste containers according to local regulations.
The product must be used before the expiration date.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Parents or guardians should be given a copy of the required vaccine information and be given the "Patient Information" appended to this insert. Parents and/or guardians should be encouraged to read the patient information that describes the benefits and risks associated with the vaccine and ask any questions they may have during the visit [see Warnings and Precautions (5) and Patient Information].
-
SPL UNCLASSIFIED SECTION
Manuf. and Dist. by: Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC., Whitehouse Station, NJ 08889, USAFor patent information: www.merck.com/product/patent/home.html
Copyright © 2006-2018 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reserved.uspi-v260-os-1803r023
-
PATIENT PACKAGE INSERT
Patient Information
RotaTeq® (pronounced "RŌ-tuh-tek")
rotavirus vaccine, live, oral, pentavalentRead this information carefully before your child receives each dose of RotaTeq® in case any information about the vaccine changes. Your child will need 3 doses of the vaccine over the course of a few months. This leaflet is a summary of certain information about RotaTeq and does not take the place of talking with your child's doctor, who can give you more complete information written for health care professionals.
What is RotaTeq?
RotaTeq is an oral vaccine used to help prevent rotavirus infection in children. Rotavirus infection can cause fever, vomiting, and diarrhea that can be severe and can lead to loss of body fluids (dehydration), hospitalization and even death in some children. RotaTeq may not fully protect all children that get the vaccine, and if your child already has the virus it will not help them.
Who should not receive RotaTeq?
Your child should not get RotaTeq if:
- He or she had an allergic reaction after getting a dose of this vaccine.
- He or she is allergic to any of the ingredients of the vaccine. A list of ingredients can be found at the end of this leaflet.
- He or she has Severe Combined Immunodeficiency Disease (SCID).
- He or she has ever had intussusception, a form of blockage of the intestines.
What should I tell the doctor before my child gets RotaTeq?
Tell your doctor if your child:
- Has illness with fever. A mild fever or cold by itself is not reason to delay taking the vaccine.
- Has diarrhea or has been vomiting.
- Has not been gaining weight or is not growing as expected.
- Has a blood disorder.
- Has any type of cancer.
- Has a weak immune system because of a disease (this includes HIV/AIDS).
- Gets treatment or takes medicines that may weaken the immune system (such as high doses of steroids) or has received a blood transfusion or blood products within the past 42 days.
- Was born with gastrointestinal problems, or has had a blockage or abdominal surgery.
- Has regular close contact with a member of family or household who has a weak immune system such as someone with cancer or someone taking medicines that weaken their immune system.
What are the possible side effects of RotaTeq?
The most common side effects reported after taking RotaTeq were diarrhea, vomiting, fever, runny nose and sore throat, wheezing or coughing, and ear infection.
Call your child's doctor or go to the emergency department right away if your child has any of the following problems after getting RotaTeq, even if it has been several weeks since the last dose because these may be signs of a serious problem called intussusception:
- bad vomiting
- bad diarrhea
- severe stomach pain
- blood in the stool.
Intussusception happens when a part of the intestine gets blocked or twisted.
Since FDA approval, reports of infants with intussusception following RotaTeq have been received by the Vaccine Adverse Event Reporting System (VAERS). Intussusception occurred days and sometimes weeks after vaccination. Some infants needed hospitalization, surgery on their intestines, or a special enema to treat this problem. Death due to intussusception has occurred.
A study conducted after approval of RotaTeq showed an increased risk of intussusception in the 21 days after the first dose of RotaTeq, but especially in the first 7 days.
Other reported side effects include:
- allergic reactions, which may be severe and may include face and mouth swelling, difficulty breathing, wheezing, hives, and/or skin rash; and
- Kawasaki disease (a serious condition that can affect the heart; symptoms may include fever, rash, red eyes, red mouth, swollen glands, swollen hands and feet and, if not treated, death can occur).
Call your doctor right away if your child has any side effects that concern you or seem to get worse.
These are NOT all the possible side effects of RotaTeq. You can ask your doctor for a more complete list.
You, as a parent or guardian, may also report any adverse reactions to your child's doctor or directly to VAERS. The VAERS toll-free number is 1-800-822-7967 or report online to www.vaers.hhs.gov.
Events that have been identified or reported as side effects following RotaTeq can happen when no vaccine has been given.
What other important information should I know?
Since FDA approval, the spread of vaccine virus to non-vaccinated contacts has been reported. Tell your doctor if you have someone in your household who has a weak immune system, cancer or is taking medications that can weaken the immune system so that your doctor can provide further advice. Hand washing is recommended after diaper changes to help prevent the spread of vaccine virus.
Can RotaTeq be given with other vaccines?
Your child may get RotaTeq at the same time as other childhood vaccines.
How is RotaTeq given?
The vaccine is given by mouth. Your child will receive 3 doses of the vaccine. The first dose is given when your child is 6 to 12 weeks of age, the second dose is given 4 to 10 weeks later and the third dose is given 4 to 10 weeks after the second dose. The last (third) dose should be given to your child by 32 weeks of age.
Your doctor will gently squeeze the vaccine into your child's mouth (see Figure 1). Your infant may spit out some or all of it. If this happens, the dose does not need to be given again during that visit.
Figure 1
What do I do if my child misses a dose of RotaTeq?
All 3 doses of the vaccine should be given to your child by 32 weeks of age. Your doctor will tell you when your child should come for the follow-up doses. It is important to keep those appointments. If you forget or are not able to go back at the planned time, ask your doctor for advice.
What else should I know about RotaTeq?
This leaflet gives a summary of certain information about the vaccine. If you have any questions or concerns about RotaTeq, talk to your doctor.
What are the ingredients in RotaTeq?
5 live rotavirus strains (G1, G2, G3, G4, and P1).
Sucrose, sodium citrate, sodium phosphate monobasic monohydrate, sodium hydroxide, polysorbate 80 and also fetal bovine serum.
Parts of porcine circovirus (a virus that infects pigs) types 1 and 2 have been found in RotaTeq. Porcine circovirus type 1 (PCV-1) and porcine circovirus type 2 (PCV-2) are not known to cause disease in humans.
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - Carton 10 Single-Dose 2 mL TubesManuf. and Dist. by:
Merck Sharp & Dohme Corp., a sub. of
MERCK & CO., INC.
Whitehouse Station, NJ 08889, USANDC: 0006-4047-41
10 Single-Dose
2 mL TubesRotavirus Vaccine, Live,
Oral, Pentavalent
RotaTeq®FOR ORAL USE ONLY. NOT FOR INJECTION.
Administer orally without mixing with any
other vaccines or solutions.Each 2 mL oral dose contains 5 rotavirus reassortants that have been propagated
in Vero cells. The minimum dose levels (infectious units, IU) are as follows:
G1 (2.2 x 106) IU; G2 (2.8 x 106) IU; G3 (2.2 x 106) IU; G4 (2.0 x 106) IU and P1 (2.3 x 106) IU.RotaTeq contains no preservatives.
Rx only
-
INGREDIENTS AND APPEARANCE
ROTATEQ
rotavirus vaccine, live, oral, pentavalent solutionProduct Information Product Type VACCINE Item Code (Source) NDC: 0006-4047 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HUMAN ROTAVIRUS A TYPE G1P7(5) STRAIN WI79 LIVE ANTIGEN (UNII: 25VC15141Q) (HUMAN ROTAVIRUS A TYPE G1P7(5) STRAIN WI79 LIVE ANTIGEN - UNII:25VC15141Q) HUMAN ROTAVIRUS A TYPE G1P7(5) STRAIN WI79 LIVE ANTIGEN 2200000 [iU] in 2 mL HUMAN ROTAVIRUS A TYPE G2P7(5) STRAIN SC2 LIVE ANTIGEN (UNII: JU499IS53H) (HUMAN ROTAVIRUS A TYPE G2P7(5) STRAIN SC2 LIVE ANTIGEN - UNII:JU499IS53H) HUMAN ROTAVIRUS A TYPE G2P7(5) STRAIN SC2 LIVE ANTIGEN 2800000 [iU] in 2 mL HUMAN ROTAVIRUS A TYPE G3P7(5) STRAIN WI78 LIVE ANTIGEN (UNII: 236YGP181O) (HUMAN ROTAVIRUS A TYPE G3P7(5) STRAIN WI78 LIVE ANTIGEN - UNII:236YGP181O) HUMAN ROTAVIRUS A TYPE G3P7(5) STRAIN WI78 LIVE ANTIGEN 2200000 [iU] in 2 mL HUMAN ROTAVIRUS A TYPE G4P7(5) STRAIN BRB LIVE ANTIGEN (UNII: 6334XMP4KC) (HUMAN ROTAVIRUS A TYPE G4P7(5) STRAIN BRB LIVE ANTIGEN - UNII:6334XMP4KC) HUMAN ROTAVIRUS A TYPE G4P7(5) STRAIN BRB LIVE ANTIGEN 2000000 [iU] in 2 mL HUMAN ROTAVIRUS A TYPE G6P1A(8) STRAIN WI79 LIVE ANTIGEN (UNII: L1977Q86S5) (HUMAN ROTAVIRUS A TYPE G6P1A(8) STRAIN WI79 LIVE ANTIGEN - UNII:L1977Q86S5) HUMAN ROTAVIRUS A TYPE G6P1A(8) STRAIN WI79 LIVE ANTIGEN 2300000 [iU] in 2 mL Inactive Ingredients Ingredient Name Strength ALBUMIN BOVINE (UNII: 27432CM55Q) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM HYDROXIDE (UNII: 55X04QC32I) SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) SUCROSE (UNII: C151H8M554) SODIUM CITRATE, UNSPECIFIED FORM (UNII: 1Q73Q2JULR) Product Characteristics Color YELLOW, PINK (pale yellow clear, may have a pink tint) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0006-4047-41 10 in 1 CARTON 1 NDC: 0006-4047-01 1 in 1 POUCH 1 2 mL in 1 TUBE; Type 0: Not a Combination Product 2 NDC: 0006-4047-20 25 in 1 CARTON 2 NDC: 0006-4047-01 1 in 1 POUCH 2 2 mL in 1 TUBE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125122 02/03/2006 Labeler - Merck Sharp & Dohme Corp. (001317601)
Trademark Results [RotaTeq]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ROTATEQ 76494554 not registered Dead/Abandoned |
Merck & Co., Inc. 2003-03-04 |
 ROTATEQ 75666657 2742325 Live/Registered |
MERCK SHARP & DOHME CORP. 1999-03-24 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.