BARHEMSYS- amisulpride injection, solution
Barhemsys by
Drug Labeling and Warnings
Barhemsys by is a Prescription medication manufactured, distributed, or labeled by Acacia Pharma Ltd. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BARHEMSYS safely and effectively. See full prescribing information for BARHEMSYS.
BARHEMSYS® (amisulpride) injection, for intravenous use
Initial U.S. Approval: 2020INDICATIONS AND USAGE
BARHEMSYS is a dopamine-2 (D2) antagonist indicated in adults for:
DOSAGE AND ADMINISTRATION
The recommended dosage of BARHEMSYS:
- Prevention of PONV, either alone or in combination with another antiemetic: 5 mg as a single intravenous dose infused over 1 to 2 minutes at the time of induction of anesthesia. (2.1)
- Treatment of PONV: 10 mg as a single intravenous dose infused over 1 to 2 minutes in the event of nausea and/or vomiting after a surgical procedure. (2.1)
- See full prescribing information for preparation and administration instructions. (2.2)
DOSAGE FORMS AND STRENGTHS
Injection: 5 mg/2 mL (2.5 mg/mL) or 10 mg/4 mL (2.5 mg/mL) in a single-dose vial. (3)
CONTRAINDICATIONS
Known hypersensitivity to amisulpride. (4)
WARNINGS AND PRECAUTIONS
QT Prolongation: Occurs in a dose- and concentration-dependent manner. Avoid use in patients with congenital long QT syndrome and in patients taking droperidol. ECG monitoring is recommended in patients with pre-existing arrhythmias/cardiac conduction disorders; electrolyte abnormalities (e.g., hypokalemia or hypomagnesemia); congestive heart failure; and in patients taking other medicinal products (e.g., ondansetron) or with other medical conditions known to prolong the QT interval. (5.1, 7.2)
ADVERSE REACTIONS
Most common adverse reactions (≥ 2%) are:
- Prevention of PONV: increased blood prolactin concentrations, chills, hypokalemia, procedural hypotension, and abdominal distension. (6.1)
- Treatment of PONV: infusion site pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Acacia Pharma at 1-877-357-9237 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Lactation: A lactating woman may pump and discard breast milk for 48 hours after BARHEMSYS administration. (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 9/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 QT Prolongation
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Dopamine Agonists
7.2 Drugs Prolonging the QT Interval
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Prevention of Postoperative Nausea and Vomiting
14.2 Treatment of Postoperative Nausea and Vomiting
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
BARHEMSYS® is indicated in adults for:
- prevention of postoperative nausea and vomiting (PONV), either alone or in combination with an antiemetic of a different class.
- treatment of PONV in patients who have received antiemetic prophylaxis with an agent of a different class or have not received prophylaxis.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended adult dosage of BARHEMSYS and infusion rate by indication is shown in the table below:
Indication Adult Dosage Regimen Prevention of PONV 5 mg as a single intravenous injection infused over 1 to 2 minutes at the time of induction of anesthesia [see Dosage and Administration (2.2)]. Treatment of PONV 10 mg as a single intravenous injection infused over 1 to 2 minutes in the event of nausea and/or vomiting after a surgical procedure [see Dosage and Administration (2.2)]. 2.2 Preparation and Administration
- Dilution of BARHEMSYS is not required before administration. BARHEMSYS is chemically and physically compatible with Water for Injection, 5% Dextrose Injection, 0.9% Sodium Chloride Injection, and Lactated Ringer's Solution (also known as Ringer's Lactate Solution, Compound Sodium Lactate Solution, and Hartmann's Solution), any of which may be used to flush an intravenous line before or after administration of BARHEMSYS.
- Protect from light. BARHEMSYS is subject to photodegradation. Administer BARHEMSYS within 12 hours of removal of the vial from the protective carton.
- Prior to administration, inspect the BARHEMSYS solution visually for particulate matter and discoloration. Discard if particulate matter or discoloration is observed.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
BARHEMSYS is contraindicated in patients with known hypersensitivity to amisulpride [see Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 QT Prolongation
BARHEMSYS causes dose- and concentration-dependent prolongation of the QT interval [see Clinical Pharmacology (12.2)]. The recommended dosage is 5 or 10 mg as a single intravenous dose infused over 1 to 2 minutes [see Dosage and Administration (2.1)].
Avoid use in patients with congenital long QT syndrome and in patients taking droperidol.
Electrocardiogram (ECG) monitoring is recommended in patients with pre-existing arrhythmias/cardiac conduction disorders; electrolyte abnormalities (e.g., hypokalemia or hypomagnesemia); congestive heart failure; and in patients taking other medicinal products (e.g., ondansetron) or with other medical conditions known to prolong the QT interval [see Drug Interactions (7.2)].
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to BARHEMSYS in 1,166 patients treated in placebo-controlled trials. 748 of these patients received a dose of 5 mg for prevention of PONV (of whom 572 received another antiemetic concomitantly) and 418 patients received 10 mg for treatment of PONV [see Clinical Studies (14.1, 14.2)]. The mean age of the population was 49 years (range 18 to 91 years), 87% female, 80% White/Caucasian, 9% Black, and 1% Asian.
Prevention of PONV
Common adverse reactions reported in at least 2% of adult patients who received BARHEMSYS 5 mg and at a higher rate than placebo in Studies 1 and 2 for the prevention of PONV are shown in Table 1.
Table 1. Common Adverse Reactions* in Adult Patients in Studies 1 and 2 of BARHEMSYS for Prevention of PONV BARHEMSYS 5 mg Placebo N=748 N=741 - * Reported in at least 2% of patients treated with BARHEMSYS and at a higher rate than placebo
Chills 4% 3% Hypokalemia 4% 2% Procedural hypotension 3% 2% Abdominal distension 2% 1% Serum prolactin concentrations were measured in Study 1 where 5% (9/176) of BARHEMSYS-treated patients vs 1% (1/166) of placebo-treated patients had increased blood prolactin reported as an adverse reaction. Serum prolactin concentrations increased from a mean of 10 ng/mL at baseline to 32 ng/mL after BARHEMSYS treatment in 112 females (upper limit of normal 29 ng/mL) and from 10 ng/mL to 19 ng/mL in 61 males (upper limit of normal 18 ng/mL). No clinical consequences due to elevated prolactin levels were reported.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval chronic oral use of amisulpride outside of the United States (BARHEMSYS is not approved for oral dosing or chronic use). Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Blood and lymphatic system disorders: agranulocytosis
- Cardiac disorders: bradycardia, torsades de pointes, ventricular tachycardia, prolonged QT by electrocardiogram
- General disorders: neuroleptic malignant syndrome
- Immune system disorders: angioedema, hypersensitivity, urticaria
- Hepatic disorders: increased hepatic enzymes
- Nervous system disorders: agitation, anxiety, dystonia, extrapyramidal disorder, seizure
- Psychiatric disorders: confusional state, insomnia, somnolence
- Vascular disorders: hypotension
-
7 DRUG INTERACTIONS
7.1 Dopamine Agonists
Reciprocal antagonism of effects occurs between dopamine agonists (e.g., levodopa) and BARHEMSYS. Avoid using levodopa with BARHEMSYS.
7.2 Drugs Prolonging the QT Interval
BARHEMSYS causes dose- and concentration-dependent QT prolongation [see Clinical Pharmacology (12.2)]. To avoid potential additive effects, avoid use of BARHEMSYS in patients taking droperidol. ECG monitoring is recommended in patients taking other drugs known to prolong the QT interval (e.g., ondansetron) [see Warnings and Precautions (5.1)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data with amisulpride use in pregnant women are insufficient to establish a drug associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In animal reproduction studies, there were no adverse developmental effects observed with oral administration of amisulpride in rats and rabbits during the period of organogenesis at exposures about 43 and 645 times, respectively, the exposure delivered by the highest recommended human dose (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Reproduction studies of amisulpride were conducted in pregnant rats administered oral doses up to 160 mg/kg/day (43 times the exposure based on area under the curve (AUC) at the highest recommended dose of 10 mg) throughout the period of organogenesis. No adverse embryo-fetal developmental effects were observed at any dose level. Maternal animals exhibited a dose-related decrease in overall mean body weight gain. In rabbits administered amisulpride throughout the period of organogenesis, oral doses up to 210 mg/kg/day (645 times the exposure based on AUC at the highest recommended dose of 10 mg) had no adverse developmental effects on the fetus. Maternal animals exhibited reduced mean body weight gain at doses of 100 and 210 mg/kg/day and reduced food intake was observed at 210 mg/kg/day.
The pre- and post-natal developmental effects of amisulpride were assessed in rats administered oral doses of 60, 100 or 160 mg/kg/day during the periods of organogenesis and lactation. At 160 mg/kg/day (43 times the exposure based on AUC at the highest recommended dose of 10 mg), maternal animals exhibited a reduction in mean body weight gain and decrease in food intake during lactation. Amisulpride had no effect on maternal pregnancy parameters, litter survival or pup growth, development or maturation at any dose tested.
8.2 Lactation
Risk Summary
Based on case reports in published literature, amisulpride is present in human milk at concentrations that are 11- to 20-fold higher than human plasma in patients taking multiple oral doses of amisulpride (200 to 400 mg/day). The estimated infant daily dose ranged from 5% to 11% of the maternal dose. There are ways to minimize drug exposure to a breastfed infant (see Clinical Considerations). There are no reports of adverse effects on the breastfed child and no information on the effects of amisulpride on milk production. The pharmacological action of amisulpride, a dopamine-2 (D2) receptor antagonist, may result in an increase in serum prolactin levels, which may lead to a reversible increase in maternal milk production [see Adverse Reactions (6.1)]. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for BARHEMSYS and any potential adverse effects on the breastfed child from BARHEMSYS or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Infertility
In animal fertility studies, administration of repeated doses of amisulpride over a 10-day period to female rats resulted in infertility that was reversible [see Nonclinical Toxicology (13.1)].
8.5 Geriatric Use
Of the total number of patients enrolled in controlled clinical trials who received BARHEMSYS 5 mg for prevention of PONV or 10 mg for treatment of PONV, 235 (17%) were 65 years of age and older, while 59 (4%) were 75 years of age and older. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
-
10 OVERDOSAGE
Doses of oral amisulpride (BARHEMSYS is not approved for oral dosing) above 1200 mg/day have been associated with adverse reactions related to dopamine-2 (D2) antagonism, in particular:
- cardiovascular adverse reactions (e.g., prolongation of the QT interval, torsades de pointes, bradycardia and hypotension) [see Warnings and Precautions (5.1)].
- neuropsychiatric adverse reactions (e.g., sedation, coma, seizures, and dystonic and extrapyramidal reactions).
There is no specific antidote for amisulpride overdose. Management includes cardiac monitoring and treatment of severe extrapyramidal symptoms.
Since amisulpride is weakly dialyzed, hemodialysis should not be used to eliminate the drug.
-
11 DESCRIPTION
The active ingredient of BARHEMSYS is amisulpride, a dopamine-2 (D2) receptor antagonist. Its chemical name is 4-Amino-N-[(1-ethyl-2-pyrrolidinyl)methyl]-5-(ethylsulfonyl)-o-anisamide. It has the following chemical structure:
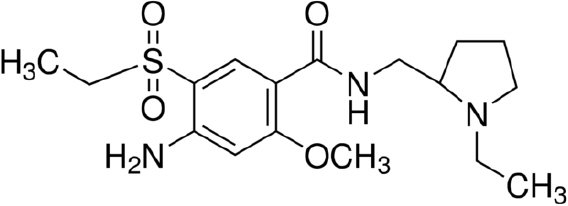
(racemic) The empirical formula is C17H27N3O4S representing a molecular weight of 369.48.
Amisulpride is a white or almost white crystalline powder. It is practically insoluble in water, sparingly soluble in ethanol and freely soluble in methylene chloride, and has a melting point of around 126°C. The compound is racemic and shows no optical rotation and is not hygroscopic. No other polymorphs of amisulpride have been reported.
BARHEMSYS (amisulpride) injection is a clear, colorless, nonpyrogenic, sterile solution formulation of amisulpride 5 mg/2 mL (2.5 mg/mL) or 10 mg/4 mL (2.5 mg/mL) for intravenous infusion presented in a single-dose vial. It has a pH of approximately 5.0 and the osmolality of the product is between 250 and 330 mOsmol/kg.
Each 2 mL vial of BARHEMSYS contains 5 mg of amisulpride; 18.7 mg of citric acid monohydrate USP; 3.6 mg of sodium chloride USP; 32.64 mg of trisodium citrate dihydrate; hydrochloric acid NF and sodium hydroxide NF as needed to adjust the pH (4.75 to 5.25); and Water for Injection USP to make up to volume.
Each 4 mL vial of BARHEMSYS contains 10 mg of amisulpride; 37.4 mg of citric acid monohydrate USP; 7.2 mg of sodium chloride USP; 65.3 mg of trisodium citrate dihydrate; hydrochloric acid NF and sodium hydroxide NF as needed to adjust the pH (4.75 to 5.25); and Water for Injection USP to make up to volume.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Amisulpride is a selective dopamine-2 (D2) and dopamine-3 (D3) receptor antagonist. D2 receptors are located in the chemoreceptor trigger zone (CTZ) and respond to the dopamine released from the nerve endings. Activation of CTZ relays stimuli to the vomiting center which is involved in emesis. Studies in multiple species indicate that D3 receptors in the area postrema also play a role in emesis. Studies conducted in ferrets have shown that amisulpride inhibits emesis caused by apomorphine, with an estimated ED50 of less than 1 mcg/kg, subcutaneously; and inhibits cisplatin-induced emesis at 2 mg/kg and morphine-induced emesis at 3 to 6 mg/kg, when given intravenously.
Amisulpride has no appreciable affinity for any other receptor types apart from low affinities for 5-HT2B and 5-HT7 receptors.
12.2 Pharmacodynamics
Cardiac Electrophysiology
A significant exposure-response relationship was identified between amisulpride concentration and ΔΔQTcF. In 40 healthy Caucasian and Japanese subjects, the maximum mean difference (95% upper confidence bound) in QTcF from placebo after baseline-correction (ΔΔQTcF) was 5.0 (7.1) milliseconds after a 2-minute intravenous infusion of 5 mg BARHEMSYS and 23.4 (25.5) milliseconds after an 8-minute intravenous infusion of 40 mg BARHEMSYS [see Warnings and Precautions (5.1)].
The recommended infusion rate is 1 to 2 minutes for 5 mg or 10 mg of BARHEMSYS [see Dosage and Administration (2.1)].
12.3 Pharmacokinetics
After an intravenous infusion, the peak plasma concentration of amisulpride is achieved at the end of the infusion period and the plasma concentration decreases to about 50% of the peak value within approximately 15 minutes. The AUC(0-∞) increases dose-proportionally in the dose range from 5 mg to 40 mg (4-times the maximum recommended dose).
The following mean pharmacokinetic parameters of amisulpride were observed following a single 5 or 10 mg intravenous dose in adult healthy subjects and surgical patients.
Table 2. Summary of Key Pharmacokinetic Parameters of Amisulpride from Clinical Studies Single Dose Infusion Duration (minutes) Number of Subjects Mean (SD) Peak Plasma Concentration
(ng/mL)AUC(0-∞)
(ng∙h/mL)- * AUC(0-2 hr)
- † Patients in a clinical trial for prophylaxis of PONV
- ‡ Patients in clinical trials for treatment of PONV
Healthy Subjects 5 mg 2 39 200 (139) 154 (30) Healthy Subjects 10 mg 1 29 451 (230) 136 (28)* Patients 5 mg 1 to 2 26† 161 (58) 260 (65) 27‡ 127 (64) 204 (94) Patients 10 mg 1 to 2 31‡ 285 (446) 401 (149) Distribution
Following intravenous infusion, the mean volume of distribution of amisulpride is estimated to be 127 to 144 L in surgical patients and 171 L in healthy subjects.
Amisulpride distributes into erythrocytes. Plasma protein binding is 25% to 30% in the concentration range from 37 to 1850 ng/mL.
Elimination
The mean elimination half-life is approximately 4 to 5 hours and similar between healthy subjects and surgical patients. Population pharmacokinetic analysis estimated that the plasma clearance of amisulpride is 20.6 L/h in surgical patients and 24.1 L/h in healthy subjects.
Metabolism
In a mass balance study, no metabolites were detectable in plasma while four metabolites were identified in urine and feces. Each metabolite accounts for less than 7% of the dose. In vitro amisulpride is not metabolized by major cytochrome P450 enzymes.
Excretion
After intravenous administration of amisulpride, 74% and 23% of the administered dose was recovered in urine and feces, respectively. 58% and 20% of the dose was excreted as unchanged amisulpride in urine and feces, respectively.
Renal clearance was estimated to be 20.5 L/hr (342 mL/min) in healthy subjects suggesting that amisulpride undergoes active renal secretion.
Specific Populations
Age, Sex, and Racial Groups
No clinically significant effect on the pharmacokinetics of amisulpride was observed based on age (18 to 90 years), sex, or race.
Patients with Renal Impairment
In subjects with severe renal impairment (eGFR < 30 mL/min/1.73 m2) and no requirement for dialysis, the Cmax of amisulpride after a single 10 mg intravenous dose was not greater than that in healthy subjects with normal renal function, and the AUC(0-∞) was approximately twice as high.
In surgical patients with mild or moderate renal impairment (eGFR 30 to 89 mL/min/1.73 m2), the Cmax of amisulpride following a single 5 mg or 10 mg intravenous dose was not significantly different and the AUC(0-∞) of amisulpride increased about 1.3-fold compared to patients with normal renal function.
The changes in the pharmacokinetics of amisulpride following a single 5 mg or 10 mg intravenous dose in subjects or patients with mild, moderate, or severe renal impairment compared to subjects or patients with normal renal function are not considered to be clinically meaningful.
Drug Interaction Studies
No clinical drug interaction trials have been conducted with BARHEMSYS.
In Vitro Studies
Cytochrome P450-Related Metabolism
In vitro, amisulpride did not inhibit CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, or CYP3A4, or induce CYP1A2, CYP2C9, CYP2C19, or CYP3A4.
In vitro, amisulpride was not a substrate of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4.
Transporters
Amisulpride inhibits MATE1 and MATE2-K transporters.
Amisulpride does not inhibit P-gp, BCRP, OCT1, OCT2, OAT1, OAT3, OATP1B1, OATP1B3 at therapeutic concentrations.
Amisulpride is a substrate for P-gp, BCRP, OCT1, MATE1 and MATE2-K, but not a substrate for OATP1B1, OATP1B3, OAT1, OAT3 and OCT2.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies to assess the carcinogenic potential of amisulpride have not been conducted. Amisulpride was not genotoxic in the bacterial reverse mutation assay, the in vitro human peripheral blood lymphocytes assay, and the in vivo rat bone marrow micronucleus assay.
The effect of amisulpride on fertility was studied in rats at oral doses up to 160 mg/kg/day (43 times the exposure based on AUC at the highest recommended dose of 10 mg). Most female animals (90% to 95%) at each dose level remained in diestrus and failed to mate. However, this effect on mating reversed following cessation of treatment. No treatment-related effects were observed on uterine/implantation parameters or sperm counts, sperm motility or sperm morphology.
-
14 CLINICAL STUDIES
14.1 Prevention of Postoperative Nausea and Vomiting
The efficacy of BARHEMSYS for the prevention of PONV was evaluated in two randomized, double-blind, placebo-controlled, multi-center trials in patients undergoing general anesthesia and elective surgery (Study 1 and Study 2). In Study 1 (NCT01991860), patients received monotherapy with BARHEMSYS; while in Study 2 (NCT02337062), patients received BARHEMSYS in combination with one other intravenously administered, non-dopaminergic antiemetic (ondansetron, dexamethasone or betamethasone). In both trials, patients were administered BARHEMSYS at the induction of anesthesia.
Study 1 was conducted in the United States in 342 patients. The mean age was 54 years (range 21 to 88 years); 65% female; 87% White/Caucasian, 12% Black, and 1% Asian race. The treatment groups were similarly matched in terms of risk for PONV with 30% of patients having two risk factors, 47% of patients having three risk factors and 23% of patients having four risk factors.
Study 2 was conducted in the United States and Europe in 1,147 patients. The mean age was 49 years (range 18 to 91 years); 97% female; 75% White/Caucasian, 9% Black, 1% Asian, and 14% of unreported race. The treatment groups were similarly matched in terms of risk for PONV with 56% of patients having three risk factors and 43% of patients having four risk factors.
The primary efficacy endpoint in both trials was Complete Response, defined as absence of any episode of emesis or use of rescue medication within the first 24 hours postoperatively. Results for both trials are shown in Table 3.
Table 3. Complete Response Rates in Adult Patients for the Prevention of PONV Within 24 Hours After End of Surgery in Studies 1 and 2 Study 1 Study 2 BARHEMSYS 5 mg
(n=176)Placebo
(n=166)BARHEMSYS 5 mg with Another Antiemetic
(n=572)Placebo with Another Antiemetic
(n=575)- * Unadjusted, nominal 95% confidence interval
Complete Response 78 (44%) 54 (33%) 330 (58%) 268 (47%) Difference
(95% CI)*12%
(2%, 22%)11%
(5%, 17%)14.2 Treatment of Postoperative Nausea and Vomiting
The efficacy of BARHEMSYS 10 mg as a single dose was evaluated in two randomized, double-blind, placebo-controlled, multi-center trials in patients experiencing PONV after general anesthesia and elective surgery (Study 3 and Study 4). Study 3 (NCT02449291) enrolled patients who had not received prior PONV prophylaxis, whereas Study 4 (NCT02646566) included patients who had received and failed PONV prophylaxis with an antiemetic of another class. Patients were excluded if they had received a D2 receptor antagonist antiemetic.
Study 3 was conducted in 369 patients (mean age 47 years, range 19 to 82 years; 76% female; 82% White/Caucasian, 8% Black, 2% Asian, and 8% of unreported race). Most of the patients had either two risk factors (36%) or three risk factors (53%) for PONV and these percentages were similar between treatment groups.
Study 4 was conducted in 465 patients (mean age 46 years, range 18 to 85 years; 90% female; 82% White/Caucasian, 9% Black, 3% Asian, and 6% of unreported race). Patients had received prior PONV prophylaxis with one or more non-dopaminergic antiemetics: a 5-HT3-antagonist in 77%, dexamethasone in 65% and another antiemetic class in 10%. Most of the patients had either three risk factors (43%) or four risk factors (51%) for PONV and these percentages were similar between treatment groups.
For both trials, the primary efficacy endpoint was Complete Response defined as absence of any episode of emesis or use of rescue medication within the first 24 hours after treatment (excluding emesis within the first 30 minutes).
BARHEMSYS Complete Response in both studies is shown in Table 4.
Table 4. Complete Response Rates in Adult Patients for the Treatment of PONV Within 24 Hours After Treatment* in Studies 3 and 4 Study 3
(no prophylaxis)Study 4
(prior prophylaxis)†BARHEMSYS 10 mg
(n=188)Placebo
(n=181)BARHEMSYS 10 mg
(n=230)Placebo
(n=235)- * Excluding emesis within the first 30 minutes
- † Received prior PONV prophylaxis with one or more non-dopaminergic antiemetics: a 5-HT3-antagonist in 77%, dexamethasone in 65% and another antiemetic class in 10%
- ‡ Unadjusted, nominal 95% confidence interval
Complete Response 59 (31%) 39 (22%) 96 (42%) 67 (29%) Difference (95% CI)‡ 10% (1%, 19%) 13% (5%, 22%) -
16 HOW SUPPLIED/STORAGE AND HANDLING
BARHEMSYS (amisulpride) injection is supplied as follows:
NDC: 71390-125-20: Package of 10 cartons. Each carton (NDC: 71390-125-21) contains one single-dose vial of clear, colorless, sterile solution of BARHEMSYS (amisulpride) injection, 5 mg in 2 mL (2.5 mg/mL).
NDC: 71390-125-50: Package of 10 cartons. Each carton (NDC: 71390-125-51) contains one single-dose vial of clear, colorless, sterile solution of BARHEMSYS (amisulpride) injection, 10 mg in 4 mL (2.5 mg/mL).
Store vials at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature].
Protect from light. Administer BARHEMSYS within 12 hours after the vial is removed from the protective carton [see Dosage and Administration (2.2)].
-
17 PATIENT COUNSELING INFORMATION
QT Prolongation
Instruct patients to contact their healthcare provider immediately if they perceive a change in their heart rate, if they feel lightheaded, or if they have a syncopal episode [see Warnings and Precautions (5.1)].
Drug Interactions
Advise patients to report to their healthcare provider if they are taking drugs which prolong the QT interval [see Warnings and Precautions (5.1)].
Lactation
Women may consider reducing infant exposure through pumping and discarding breastmilk for 48 hours after BARHEMSYS administration [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 2 mL Vial Label
NDC: 71390-125-21
Rx ONLYBarhemsys®
(amisulpride) injection5 mg | 2 mL
(2.5 mg/mL)Single-dose sterile vial,
discard unused portion.For Intravenous Infusion Only.
-
PRINCIPAL DISPLAY PANEL - 2 mL Vial Carton
NDC: 71390-125-21
Barhemsys®
(amisulpride) injection5 mg | 2 mL
(2.5 mg/mL)Single-dose sterile vial.
For Intravenous
Infusion Only.Discard Unused Portion.
Rx ONLY
-
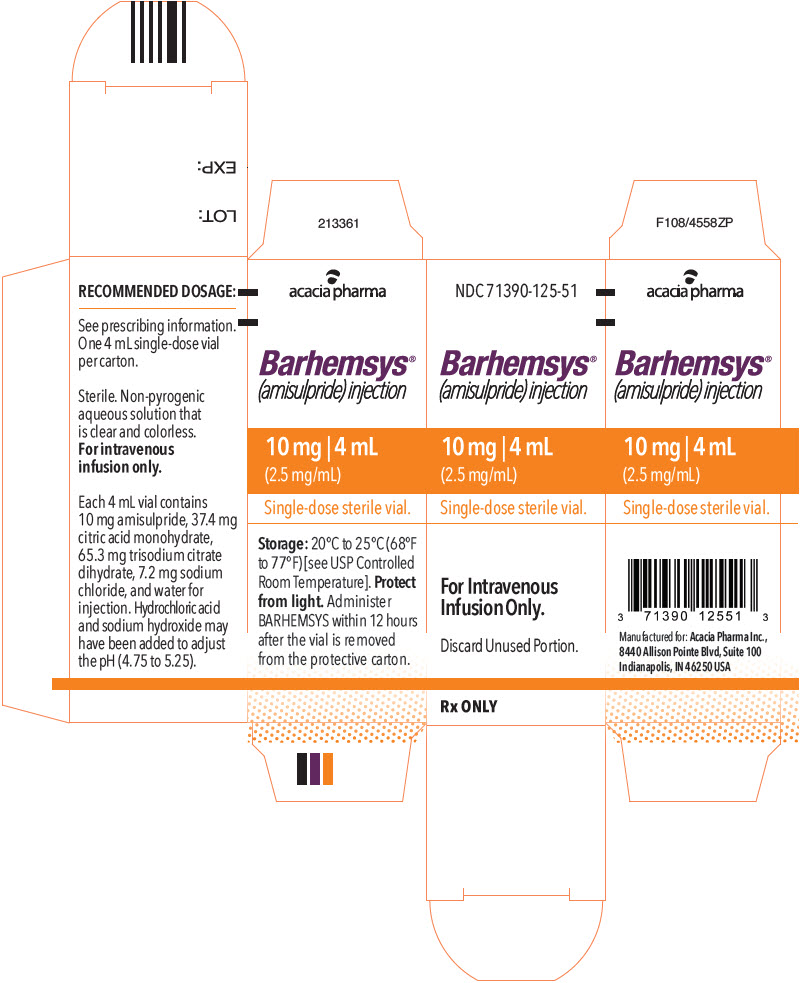
PRINCIPAL DISPLAY PANEL - 4 mL Vial Label
NDC: 71390-125-51
Rx ONLYBarhemsys®
(amisulpride) injection10 mg | 4 mL
(2.5 mg/mL)Single-dose sterile vial,
discard unused portion.For Intravenous Infusion Only.
-
PRINCIPAL DISPLAY PANEL - 4 mL Vial Carton
NDC: 71390-125-51
Barhemsys®
(amisulpride) injection10 mg | 4 mL
(2.5 mg/mL)Single-dose sterile vial.
For Intravenous
Infusion Only.Discard Unused Portion.
Rx ONLY
-
INGREDIENTS AND APPEARANCE
BARHEMSYS
amisulpride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71390-125 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Amisulpride (UNII: 8110R61I4U) (Amisulpride - UNII:8110R61I4U) Amisulpride 2.5 mg in 1 mL Inactive Ingredients Ingredient Name Strength Citric Acid Monohydrate (UNII: 2968PHW8QP) Sodium Chloride (UNII: 451W47IQ8X) Trisodium Citrate Dihydrate (UNII: B22547B95K) Hydrochloric Acid (UNII: QTT17582CB) Sodium Hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71390-125-20 10 in 1 PACKAGE 02/27/2020 1 NDC: 71390-125-21 1 in 1 CARTON 1 2 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 2 NDC: 71390-125-50 10 in 1 PACKAGE 02/27/2020 2 NDC: 71390-125-51 1 in 1 CARTON 2 4 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209510 02/27/2020 Labeler - Acacia Pharma Ltd (779660930)
Trademark Results [Barhemsys]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 BARHEMSYS 87808035 5620798 Live/Registered |
Acacia Pharma Limited 2018-02-22 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.