ITVISMA- onasemnogene abeparvovec-brve injection, suspension
ITVISMA by
Drug Labeling and Warnings
ITVISMA by is a Prescription medication manufactured, distributed, or labeled by Novartis Gene Therapies, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ITVISMA safely and effectively. See full prescribing information for ITVISMA.
ITVISMA® (onasemnogene abeparvovec-brve) suspension, for intrathecal injection
Initial U.S. Approval: 2025WARNING: SERIOUS LIVER INJURY
See full prescribing information for complete boxed warning.
- Acute serious liver injury and elevated aminotransferases can occur with ITVISMA. (5.1)
- Patients with preexisting liver impairment may be at higher risk. (5.1)
- Prior to intrathecal injection, assess liver function by clinical examination and laboratory testing. Administer systemic corticosteroid before and after ITVISMA injection. Continue to monitor liver function for at least 3 months after injection, and at other times as clinically indicated. (2.1, 2.4)
INDICATIONS AND USAGE
ITVISMA is an adeno-associated virus (AAV) vector-based gene therapy indicated for the treatment of spinal muscular atrophy (SMA) in adult and pediatric patients 2 years of age and older with confirmed mutation in SMN1 gene. (1)
DOSAGE AND ADMINISTRATION
For single-dose intrathecal injection only. (2)
- The recommended dose of ITVISMA is 1.2 × 1014 vector genomes (vg). (2.2)
- Administer ITVISMA as an intrathecal bolus injection over approximately 1 to 2 minutes. (2.4)
- Postpone ITVISMA in patients with infections until the infection has resolved and the patient is clinically stable. (2.1)
- Starting one day prior to ITVISMA injection, administer systemic corticosteroids equivalent to oral prednisolone at 1 mg/kg of body weight per day for a total of 30 days. At the end of the 30-day period, check liver function by clinical examination and by laboratory testing. For patients with unremarkable findings, taper the corticosteroid dose gradually over the next 28 days. If liver function abnormalities persist, continue systemic corticosteroids (equivalent to oral prednisolone at 1 mg/kg/day) until findings become unremarkable, and then taper the corticosteroid dose gradually over the next 28 days or longer if needed. Do not stop systemic corticosteroids abruptly. (2.2)
- If at any time patients do not respond adequately to the equivalent of 1 mg/kg/day oral prednisolone, based on the patient’s clinical course, prompt consultation with a gastroenterologist or hepatologist and adjustment to the recommended corticosteroid regimen may be considered. (2.2)
DOSAGE FORMS AND STRENGTHS
Each single-dose vial contains 1.2 × 1014 vg of onasemnogene abeparvovec in 3 mL of suspension. ITVISMA has a nominal concentration of 4 × 1013 vg/mL, and each vial contains an extractable volume of not less than 3 mL. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Hepatotoxicity: Prior to ITVISMA injection, assess liver function of patients by clinical examination and laboratory testing. Continue to monitor liver function for at least 3 months after injection, and at other times as clinically indicated. (2.1, 2.4, 5.1)
- Thrombocytopenia: Monitor platelet counts before ITVISMA injection, and at least weekly for the first month and as clinically indicated until platelet counts return to baseline. (2.1, 2.4, 5.2)
- Peripheral Sensory Neuropathy: Consider complete neurologic evaluation and other testing and/or symptom management based on the patient's clinical presentation. (5.3)
- Thrombotic Microangiopathy (TMA): Prompt attention to signs and symptoms of TMA is advised, as TMA can result in life-threatening or fatal outcomes. If clinical signs, symptoms and/or laboratory findings occur, consult a hematologist and/or nephrologist immediately to manage as clinically indicated. (5.4)
- Elevated Cardiac Troponin I: Increases in cardiac troponin I have occurred following ITVISMA injection. Consider cardiac evaluation after ITVISMA administration and consult a cardiologist as needed. (5.5)
- AAV Vector Integration and Risk of Tumorigenicity: There is a theoretical risk of tumorigenicity due to integration of AAV vector DNA into the genome. Report cases of tumors in patients who received ITVISMA, to Novartis Gene Therapies, Inc. (5.6)
ADVERSE REACTIONS
The most common adverse reactions that occurred in at least 10% of patients were upper respiratory tract infection, upper gastrointestinal symptoms, pyrexia, and headache. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Gene Therapies at 1-833-828-3947 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Adjust patient’s vaccination schedule to accommodate concomitant corticosteroid administration prior to and following ITVISMA injection. (7)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 11/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SERIOUS LIVER INJURY
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Critical Dosing Information
2.2 Dose
2.3 Preparation
2.4 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
5.2 Thrombocytopenia
5.3 Peripheral Sensory Neuropathy
5.4 Thrombotic Microangiopathy
5.5 Elevated Cardiac Troponin I
5.6 AAV Vector Integration and Risk of Tumorigenicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.6 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SERIOUS LIVER INJURY
- Acute serious liver injury and elevated aminotransferases can occur with ITVISMA. [see Warnings and Precautions (5.1)]
- Patients with preexisting liver impairment may be at higher risk. [see Warnings and Precautions (5.1)]
- Prior to intrathecal injection, assess liver function by clinical examination and laboratory testing. Administer systemic corticosteroid before and after ITVISMA injection. Continue to monitor liver function for at least 3 months after injection, and at other times as clinically indicated. [see Dosage and Administration (2.1, 2.4)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Critical Dosing Information
For single-dose intrathecal injection only.
- Patients previously treated with ZOLGENSMA (onasemnogene abeparvovec-xioi) should not be treated with ITVISMA [see Clinical Pharmacology (12.1)].
- ITVISMA should only be administered intrathecally using a lumbar puncture by healthcare professionals (e.g., interventional radiologist or neurologist) experienced in performing lumbar punctures.
-
Prior to ITVISMA injection:
- Due to the increased risk of serious systemic immune response, administer ITVISMA to patients who are clinically stable in their overall baseline health status (e.g., hydration and nutritional status, absence of infection, respiratory status) prior to administration. Postpone ITVISMA in patients with active or recent infections, until the infection has resolved, and the patient is clinically stable. Clinical signs or symptoms of infection should not be evident at the time of ITVISMA injection.
- Assess vaccination status. Vaccination status should be up-to-date prior to ITVISMA administration. Recommend seasonal prophylaxis against respiratory syncytial virus (RSV).
- Assess liver function (clinical examination and laboratory testing including aspartate aminotransferase (AST), alanine aminotransferase (ALT), albumin, prothrombin time, partial thromboplastin time (PTT), international normalized ratio (INR), and total bilirubin) [see Warnings and Precautions (5.1), Use in Specific Populations (8.6)].
- Obtain creatinine and complete blood count (including hemoglobin and platelet count) [see Warnings and Precautions (5.2, 5.4)].
- Perform baseline testing for the presence of anti-AAV9 antibodies.
- One day prior to ITVISMA injection, begin administration of systemic corticosteroids equivalent to oral prednisolone at 1 mg per kg of body weight per day (mg/kg/day) for a total of 30 days. Do not stop systemic corticosteroids abruptly. After the 30-day period, taper prednisolone (or equivalent) as needed according to the clinical status and liver function testing [see Warnings and Precautions (5.1, 5.2)]. See Table 1 for the recommended corticosteroid regimen.
- Do not re-administer ITVISMA.
2.2 Dose
The recommended dose of ITVISMA is 1.2 × 1014 vector genomes (vg).
Table 1 includes the recommended corticosteroid regimen prior to and following ITVISMA injection.
If at any time patients do not respond adequately to the equivalent of 1 mg/kg/day oral prednisolone, based on the patient’s clinical course, obtain prompt consultation with a gastroenterologist or hepatologist and consider adjustment to the recommended corticosteroid regimen, including increased dose, longer duration or prolongation of corticosteroid taper [see Warnings and Precautions (5.1)]. If oral corticosteroid therapy is not tolerated or not effective, consider intravenous corticosteroids, as clinically indicated.
Table 1: Recommended Corticosteroid Regimen Pre- and Post- ITVISMA Injection Pre-Injection - 24 hours prior to ITVISMA injection Oral prednisolone 1 mg/kg/day (or equivalent) Post-Injection - 30 days (including the day of ITVISMA administration) Oral prednisolone 1 mg/kg/day (or equivalent) Followed by 28 days:
For patients with unremarkable findings (normal clinical exam, total bilirubin, and ALT and AST levels below 2 × ULN)
or
For patients with liver function abnormalities at the end of the 30 day period: continue until the AST and ALT values are both below 2 × ULN and all other assessments return to normal range, and then taper the corticosteroid dose over the next 28 days or longer if needed.Systemic corticosteroids should be tapered gradually
Taper prednisolone (or equivalent)
Systemic corticosteroids (equivalent to oral prednisolone 1 mg/kg/day)
Systemic corticosteroids should be tapered gradually2.3 Preparation
Required supplies and materials (not supplied)
- Needle for withdrawal
- Syringe
- Syringe cap
- Spinal needle
The supplies and materials compatible with ITVISMA are listed in Table 2. Device components must be indicated for intrathecal or neuraxial use. Ensure all device components use the same connector type. Incompatible device connections may result in dose loss during administration.
Table 2: Component Materials Compatible With ITVISMA aNot to be manufactured with Polyvinylchloride (PVC), Bisphenol-A (BPA), Bis(2- ethylhexyl) phthalate (DEHP) or Latex Component Material of Construction 18G to 19G Needle for withdrawal, maximum 1.5” long Stainless steel 5mL to 10mL Syringea Polypropylene Syringe capa Polypropylene or Polyethylene or Methacrylate-Acrylonitrile-Butadiene-Styrene 22G to 27G Spinal needle, maximum 150mm long Stainless steel Vial Preparation:
- ITVISMA should be prepared aseptically.
- Thaw ITVISMA in the refrigerator for approximately 4 hours, or at room temperature for approximately 1 hour. If thawed in the refrigerator, remove ITVISMA from refrigerator on day of dosing.
- Do not use ITVISMA unless thawed.
- Prior to intrathecal injection, ITVISMA should be brought to room temperature.
- When thawed, ITVISMA is a clear to slightly opaque, colorless to faint white liquid, free of particles. After withdrawal of ITVISMA from the vial, a visual inspection is required. DO NOT use if particulates, cloudiness, or discoloration are visible.
- DO NOT SHAKE.
- Immediately prior to dosing, draw the content from the vial into the syringe, remove air from syringe, confirm the dose volume of 3 mL in the syringe, cap syringe and deliver to patient injection location.
- Once dose is drawn into the syringe, it may be held in the refrigerator at 2°C to 8°C (36°F to 46°F) for up to 24 hours, including a 5-hour maximum time out-of-refrigeration allowance within the 24-hour period. Discard the vector-containing syringe if not injected within this time period.
- DO NOT REFREEZE.
Procedural Preparation Instructions:
- Consider sedation if indicated by the patient’s clinical status.
- Consider imaging techniques to guide intrathecal injection of ITVISMA.
- Evaluate patient prior to and after intrathecal injection for conditions that may contraindicate lumbar puncture or increase procedural risk to prevent serious complications.
2.4 Administration
Intrathecal Injection Instructions:
- Prior to administration, remove 3 mL of cerebrospinal fluid (CSF) using a lumbar puncture needle to create space for injection volume.
- Administer ITVISMA as an intrathecal bolus injection over approximately 1 to 2 minutes through the lumbar puncture needle.
- Place patient in Trendelenburg position (head down at 30 degrees for 15 minutes). Adjust patient positioning and duration based on the patient’s clinical status to enhance distribution.
- Follow standard post-lumbar puncture care protocols.
Monitoring Following ITVISMA Injection:
- Liver function (AST, ALT, total bilirubin) weekly for the month after ITVISMA injection and during the corticosteroid taper period (over the next 28 days or longer if needed). If the patient is clinically stable with unremarkable findings (normal clinical exam, total bilirubin, and ALT and AST levels below 2 × ULN) at the end of the corticosteroid taper period, continue to monitor liver function every other week for another month [see Warnings and Precautions (5.1)].
- Platelet counts weekly for the first month and as clinically indicated until platelet counts return to baseline [see Warnings and Precautions (5.2)].
-
3 DOSAGE FORMS AND STRENGTHS
ITVISMA is a clear to slightly opaque, colorless to faint white suspension for intrathecal injection.
Each single-dose vial contains 1.2 × 1014 vg of onasemnogene abeparvovec in 3 mL of suspension.
ITVISMA has a nominal concentration of 4 × 1013 vg/mL, and each vial contains an extractable volume of not less than 3 mL.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
Hepatotoxicity, with elevated ALT and/or AST levels, has occurred with ITVISMA [see Adverse Reactions (6.1)]. Patients with preexisting hepatic impairment or acute hepatic viral infection may be at higher risk of liver injury. In order to mitigate potential aminotransferase elevations, administer systemic corticosteroid before and after ITVISMA injection. Immune-mediated hepatotoxicity may require adjustment of the corticosteroid treatment regimen, including longer duration, increased dose, or prolongation of the corticosteroid taper [see Dosage and Administration (2.2)].
Prior to ITVISMA injection, assess liver function by clinical examination and laboratory testing. Continue to monitor liver function for at least 3 months after ITVISMA administration, and at other times as clinically indicated. Monitor AST, ALT and total bilirubin weekly for the month after ITVISMA administration and during the corticosteroid taper period. If the patient is clinically stable with unremarkable findings at the end of the corticosteroid taper period, continue to monitor liver function every other week for another month. Tapering of systemic corticosteroids should not be considered until AST/ALT levels are less than 2 × ULN [see Dosage and Administration (2.1, 2.4)].
Monitor patients with worsening liver function test results and/or signs or symptoms of acute illness (e.g., vomiting, deterioration in health). In case hepatic injury is suspected, further testing is recommended (e.g., albumin, prothrombin time, partial thromboplastin time (PTT) and international normalized ratio (INR)). Promptly consult with a gastroenterologist or hepatologist, as necessary.
5.2 Thrombocytopenia
Transient decreases in platelet counts were observed within the first week after ITVISMA administration [see Adverse Reactions (6.1)]. The platelets counts are expected to return to baseline two weeks following ITVISMA injection.
Monitor platelet counts before ITVISMA injection and on a regular basis afterwards (at least weekly for the first month and as clinically indicated until platelet counts return to baseline) [see Dosage and Administration (2.1, 2.4)].
5.3 Peripheral Sensory Neuropathy
Peripheral sensory neuropathy has occurred with ITVISMA administration [see Adverse Reactions (6.1)]. Signs and symptoms may include numbness, tingling, prickling, or pain in the arms, hands, legs and/or feet, with onset seen at approximately three weeks post-injection in clinical studies.
Consider complete neurologic evaluation and other testing and/or symptom management based on the patient's clinical presentation. Inform patients and caregivers about the signs and symptoms of peripheral sensory neuropathy, and advise patients and caregivers to notify their physician promptly if such symptoms occur.
5.4 Thrombotic Microangiopathy
Thrombotic microangiopathy (TMA) may occur with ITVISMA administration. TMA is characterized by thrombocytopenia, microangiopathic hemolytic anemia, and acute kidney injury. Concurrent immune system activation (e.g., infections, vaccinations) may be a contributing factor.
Prompt attention to signs and symptoms of TMA is advised, as TMA can result in life-threatening or fatal outcomes.
Monitor platelet counts on a regular basis following ITVISMA injection [see Warnings and Precautions (5.2)], as well as signs and symptoms of TMA, such as hypertension, bruising easily, seizures, or decreased urine output. In case these signs and symptoms occur in the presence of thrombocytopenia, further diagnostic evaluation for hemolytic anemia and renal dysfunction should be promptly undertaken. If clinical signs, symptoms and/or laboratory findings consistent with TMA occur, consult a hematologist and/or nephrologist immediately to manage TMA as clinically indicated.
5.5 Elevated Cardiac Troponin I
Increases in cardiac troponin I levels have occurred following ITVISMA administration without clinical sequelae [see Adverse Reactions (6.1)]. Cardiac toxicity was observed in animal studies [see Nonclinical Toxicology (13.2)]. Consider cardiac evaluation after ITVISMA administration and consult a cardiologist as needed.
5.6 AAV Vector Integration and Risk of Tumorigenicity
There is a theoretical risk of tumorigenicity due to integration of AAV vector DNA into the genome.
ITVISMA is composed of a recombinant, non-replicating AAV9 vector whose DNA persists largely in episomal form. Random integration of recombinant AAV-vector DNA into human DNA has been reported with AAV gene therapies. The clinical relevance of individual integration events is unknown, but it is acknowledged that individual integration events could potentially contribute to a risk of tumorigenicity. If a tumor develops in a patient receiving ITVISMA, health care providers should contact and report the tumor to Novartis Gene Therapies, Inc. at 1-833-828-3947.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another product and may not reflect the rates observed in practice.
The safety data described in this section reflects exposure of ITVISMA in two clinical studies, Study 1, a randomized, sham-controlled study which evaluated the safety of ITVISMA in 126 patients with spinal muscular atrophy (SMA) and Study 2, an open-label-single arm study which evaluated safety of ITVISMA in 27 patients with SMA who were previously treated with nusinersen (at least 4 months washout) or risdiplam (at least 15 days washout). In Study 1, a total of 75 patients received a single intrathecal injection of ITVISMA at a fixed dose of 1.2 x 1014 vg and 51 patients underwent a sham-procedure [see Clinical Studies (14)]. In Study 2, a total of 27 patients received a single intrathecal injection of ITVISMA at a fixed dose of 1.2 x 1014 vg. The patients were followed for a duration of 52 weeks for both studies.
In Study 1, serious adverse reactions were reported in four patients (5%) including elevated liver enzymes (n=1), sensory disturbance (n=2), and vomiting (n=1).
The most frequent adverse reactions occurring in ≥ 2% of patients in Study 1 are summarized in Table 3 below.
Table 3: Adverse Reactions Occurring in ≥2% of Patients or with higher frequency in ITVISMA-treated Patients compared to Sham group in Study 1 Adverse reactions ITVISMA Sham (N = 75), n (%) (N = 51), n (%) *Is a composite that includes multiple related terms
a)Two patients had ALT elevations of 20 times the upper limit of normal (ULN)
b)Signs and symptoms that may be suggestive of dorsal root ganglion (DRG) toxicity occurred within 3 weeks of ITVISMA injection and stabilized but remained unresolved at the end of study period.
c)Occurred 154 days after the sham procedure and resolved after 15 days without intervention.Upper respiratory tract infection* 31 (41) 15 (29) Pyrexia 19 (25) 12 (24) Upper gastrointestinal symptoms* 20 (27) 8 (16) Hepatic enzyme increased* 6 (8)a 5 (10) Headache 8 (11) 2 (4) Dizziness 4 (5) 1 (2) Pain in extremity 3 (4) 1 (2) Thrombocytopenia* 3 (4) 0 Sensory disturbance* 2 (3)b 1 (2)c The safety evaluated in Study 2 did not identify any additional safety events with ITVISMA administration.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ZOLGENSMA, a similar product containing the same active ingredient (onasemnogene abeparvovec) administered intravenously. Because these reactions are reported voluntarily, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: thrombotic microangiopathy
Hepatobiliary Disorders: acute liver failure (fatal and non-fatal), acute liver injury
General Disorders and Administration Site Conditions: pyrexia, infusion-related reactions
Investigations: troponin increased
-
7 DRUG INTERACTIONS
Adjust patient’s vaccination schedule to accommodate concomitant corticosteroid administration prior to and following ITVISMA injection [see Dosage and Administration (2.1)]. Certain vaccines, such as measles, mumps, and rubella (MMR) and varicella, are contraindicated for patients on a substantially immunosuppressive steroid dose (i.e., ≥ 2 weeks of daily receipt of 20 mg or 2 mg/kg body weight of prednisone or equivalent).
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no clinical studies in pregnant women to inform a product-associated risk. There are no available data in animal embryo-fetal development studies with ITVISMA. It is not known whether ITVISMA has the potential to be transferred to the fetus. Therefore, women who are pregnant or desire to become pregnant should only be treated with ITVISMA after a thorough benefit-risk evaluation.
In the United States general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
8.2 Lactation
Risk Summary
There is no information available on the presence of ITVISMA in human milk, the effects on the breastfed infant or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ITVISMA and any potential adverse effects on the breastfed child from ITVISMA or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
The pregnancy status of females of reproductive potential should be verified prior to treatment with ITVISMA [see Use in Specific Populations (8.1)].
Contraception
Females of reproductive potential should use effective contraception (methods that result in less than 1% pregnancy rates) and should refrain from egg donation for 6 months following ITVISMA administration. Men capable of fathering a child should use a barrier method of contraception and should refrain from sperm donation for 3 months following ITVISMA administration.
Infertility
There is no data on the effect of ITVISMA on human fertility. In animal fertility studies, onasemnogene abeparvovec did not impact fertility in male and female mice at a dose of 1.1 × 1014 vg/kg administered intravenously [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and efficacy of ITVISMA have been established in pediatric patients 2 years of age and older. The use of ITVISMA was supported by data from one clinical study (Study 1) which enrolled 126 pediatric patients 2 years of age and older [see Adverse Reaction (6) and Clinical Studies (14)].
The safety and effectiveness of ITVISMA have not been established in pediatric patients younger than 2 years of age.
-
11 DESCRIPTION
ITVISMA (onasemnogene abeparvovec-brve) is a suspension of an adeno-associated viral vector-based gene therapy for intrathecal injection. It is a recombinant self-complementary AAV9 containing a transgene encoding the human survival motor neuron (SMN) protein, under the control of a cytomegalovirus enhancer/chickenβactin hybrid promoter.
ITVISMA has a nominal concentration of 4 × 1013 vg/mL. Each vial contains an extractable volume of not less than 3 mL and the excipients 20 mM Tris (pH 8.0), 1 mM magnesium chloride (MgCl2), 200 mM sodium chloride (NaCl) and 0.005% poloxamer 188. ITVISMA is packaged as a sterile suspension and contains no preservative.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
ITVISMA is a non-replicating recombinant AAV vector that utilizes AAV9 capsid to deliver a functional copy of human survival motor neuron 1 gene (SMN1). The transgene DNA persists largely in episomal form in the nucleus of transduced cells. Expression of the transgene is driven by a constitutive promoter (cytomegalovirus enhanced chicken β actin hybrid), resulting in continuous and sustained SMN expression. SMA is caused by a bi-allelic mutation in the SMN1 gene, which results in insufficient SMN protein expression. By providing an alternative source of SMN protein expression in motor neurons, it is expected to promote the survival and function of transduced motor neurons.
12.3 Pharmacokinetics
Nonclinical Biodistribution
Intrathecal (IT) administration of onasemnogene abeparvovec to non-human primates (NHPs) at dose levels of 1.2 × 1013 (equivalent to the clinical therapeutic dose), 3.0 × 1013, or 6.0 × 1013 vector genomes (vg)/animal, resulted in biodistribution of the vector to all the CNS and peripheral tissues assessed. Vector DNA concentrations were highest in the liver, followed by the dorsal root ganglia (DRG) and spinal cord, with the lowest concentrations detected in the gonads. Vector DNA concentrations in the spinal cord tended to remain stable between 6-weeks and 12-months post-administration at all dose levels assessed.
Intrathecal or intra-cisterna magna administration of a tool scAAV9CB-GFP vector to adult NHPs resulted in the detection of vector DNA in the oocytes and ovarian stromal cells of females administered 1.0 × 1013 and 3.0 × 1013 vg/animal at 28-days post-administration of the product. In mice, IV, or intracerebroventricular (ICV) administration of onasemnogene abeparvovec resulted in no detection of vector DNA in the germline cells of males and females at 24 weeks post-administration.
In non-human primates, high pre-existing serum anti-AAV9 antibody titers (corresponding to human titer values of up to approximately 1:25000) were not shown to affect scAAV9 vector (utilized in onasemnogene abeparvovec) DNA distribution in the spinal cord following intrathecal administration.
Clinical Vector Shedding
ITVISMA vector shedding studies, which assess the amount of vector DNA eliminated from the body through saliva, urine, feces and nasal secretions were performed following intrathecal administration in 134 patients.
Vector DNA was detectable in shedding samples in 134 patients following intrathecal injection of ITVISMA. Shedding of onasemnogene abeparvovec DNA was primarily via feces. Peak shedding in participants was observed within 10-, 3-, 2-, and 8 days post-dose for stool, urine, saliva and nasal secretion, respectively. The majority of the vector DNA (> 90%) is excreted within 2 weeks after dose administration.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of ITVISMA or of other AAV gene therapy products.
In Study 1 and Study 2, anti-AAV9 antibody titers were evaluated in 102 patients following a single intrathecal injection of ITVISMA. In these studies, patients were required to have baseline anti-AAV9 antibody titers ≤ 1:50.
Increases from baseline in anti-AAV9 antibody titers were reported in all patients with a median anti-AAV9 antibody titers of ≥ 1:819,200 at 12 months following ITVISMA injection in both studies.
During the 12-month period following ITVISMA injection in Study 1 and Study 2, positive anti-SMN antibodies were observed in 5/75 (6.7%) and 2/27 (7.4%) of ITVISMA-treated patients, respectively. There was no identified clinical effect of anti-SMN and anti-AAV9 antibodies on safety or efficacy of ITVISMA over the follow-up period of 12 months post-dose.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No animal studies have been performed to evaluate the effects of onasemnogene abeparvovec on carcinogenesis or mutagenesis.
In fertility and early embryonic development studies conducted in male and female mice, intravenous administration of onasemnogene abeparvovec at dose levels up to 1.1 × 1014 vector genomes (vg)/kg body weight had no effect on fertility, fecundity, or mating indices. There was no evidence of germ cell transduction or germline transmission.
13.2 Animal Toxicology and/or Pharmacology
A single intrathecal (IT) dose of onasemnogene abeparvovec was administered at three dose levels: 1.20 × 1013 vg/animal (equivalent to the clinical therapeutic dose), 3.0 × 1013 vg/animal, and 6.0 × 1013 vg/animal. At 6 weeks post-administration, all dose levels resulted in acute, minimal to moderate mononuclear cell inflammation and neuronal degeneration across multiple anatomical sites, including the cerebrum, cerebellar white matter, brain stem, dorsal root ganglia (DRG), and various nerve structures (trigeminal, dorsal spinal, spinal, and peripheral nerves). Additionally, axonal degeneration and/or gliosis was observed in the spinal cord. These findings partially or fully resolved by 12 months post-administration and had no correlative clinical observations. Elevated liver enzyme levels (aspartate and alanine amino transferase) and single cell necrosis of hepatocytes were observed in select NHPs at 6 weeks post-administration but resolved by 12 months post-administration.
-
14 CLINICAL STUDIES
The efficacy of ITVISMA was evaluated in a randomized, double-blind, sham-controlled study (Study 1; NCT05089656). The study enrolled patients with spinal muscular atrophy (SMA) who were treatment-naive, and able to sit but never able to walk independently. Patients with elevated (reference to > 1:50) baseline serum anti-AAV9 antibody titer were excluded.
A total of 136 patients were randomized in 3:2 ratio to receive either ITVISMA at a dose of 1.2 x 1014 vg by single lumbar intrathecal injection or sham procedure. Randomization was stratified by age and pre-treatment Hammersmith Functional Motor Scale – Expanded (HFMSE) score at screening. A total of 126 patients received the assigned treatment and were included in the efficacy evaluation.
The demographic characteristics of the population were as follows: the mean age was 6 years (range 2 to 17 years), 62 patients (49%) were male, 74 patients (59%) were Asian, 14 patients (11%) were White, 9 patients (7%) were Black or African American, and 7 patients (6%) were American Indian or Alaska Native, 22 patients (18%) were of “unknown” race. One hundred and twenty-two patients (97%) had confirmed biallelic deletion (0 copies) of the SMN1 gene, while 4 patients (3%) had 1 copy. Patients' highest motor function ever achieved were as follows: 66 patients (52%) sitting without support, 33 patients (26%) standing with assistance, 24 patients (19%) walking with assistance, and 3 patients (2%) standing independently. At baseline, the mean HFMSE total score was 17.97 (range 1.0 - 41.0) and 18.17 (range 2.0 - 42.5) in the ITVISMA-treated group and sham group, respectively.
The primary endpoint was the change from baseline in HFMSE total score at the end of follow-up, defined as the average of the Week 48 and Week 52 assessment, in ITVISMA compared to sham. The HFMSE evaluates motor function in patients with SMA who have limited ambulation, comprising of 33 graded items that assess various motor skills ranging from sitting to using the stairs. Each item is scored from 0-2, with a maximum total score of 66. Higher scores indicate better motor function.
The efficacy results from Study 1 are summarized in Table 4 below.
Table 4: Efficacy Results from the Study 1 (n=126) Endpoint ITVISMA
(N = 75)Sham
(N = 51)Treatment Difference ITVISMA-Sham
(95% CI)p-value CI = confidence interval
1Assessed using the Full Analysis Set (FAS) population, which included all participants who were dosed with ITVISMA (n=75) or who underwent sham procedure (n=51). The follow-up period was defined as the average of the Week 48 and Week 52 assessment.
2Least squares (LS) mean.
3Estimated using a linear mixed model repeated measures (MMRM) model with the observed change from Baseline in HFMSE total score at all post-baseline visits as the dependent variable. The fixed effects included treatment, visit, treatment by visit interaction, the strata, and the baseline HFMSE total score as covariate. An unstructured covariance matrix was used.
4Standard error of the mean (SEM)Mean change from baseline in HFMSE total score at the end of follow-up1, 2, 3 2.39 (0.439)4 0.51 (0.532)4 1.88
(0.51 – 3.25)0.0074 -
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
ITVISMA is supplied as a sterile, preservative-free, clear to slightly opaque, colorless to faint white suspension for intrathecal injection. ITVISMA contains 4 × 1013 vg per mL.
Each carton of ITVISMA contains a single-dose vial with an extractable volume of not less than 3 mL, containing 1.2 × 1014 vg.
Carton containing one 1.2 × 1014 vg/3 mL (4 × 1013 vg/mL) single-dose vial.
Container (vial): NDC Number 71894-200-01
Package (carton): NDC Number 71894-200-02
16.2 Storage and Handling
- Product is shipped and delivered frozen (≤ -60°C [-76°F]) in a single-dose clear vial.
- Upon receipt, immediately place the carton in a refrigerator at 2°C to 8°C (36°F to 46°F).
- ITVISMA is stable for 14 days from receipt when stored at 2°C to 8°C (36°F to 46°F).
- DO NOT REFREEZE.
- Must use within 14 days of receipt.
-
17 PATIENT COUNSELING INFORMATION
Hepatotoxicity
Inform patients and caregivers that ITVISMA could increase liver enzyme levels. Inform patients and caregivers that patients will receive an oral corticosteroid medication before and after ITVISMA injection, and will undergo regular blood tests to monitor liver function. Advise patients and caregivers to contact their healthcare provider immediately if the patient’s skin and/or whites of the eyes appear yellowish, if the patient misses a dose of corticosteroid or vomits it up, or if the patient experiences a decrease in alertness [see Warnings and Precautions (5.1)].
Vaccination Before and After ITVISMA Injection
Advise patients and caregivers to consult with their healthcare provider to determine if adjustments to the patient’s vaccination schedule are necessary during corticosteroid use. Inform patients and caregivers that where feasible, the vaccination schedule should be adjusted appropriately to accommodate treatment with corticosteroid. Prophylaxis against influenza and RSV is recommended and vaccination status should be up-to-date prior to ITVISMA administration. Please consult your health care provider [see Dosage and Administration (2.1), Drug Interactions (7)].
Concurrent Infections
Patients and caregivers should be aware that an infection (e.g., cold, flu, gastroenteritis, otitis media, bronchiolitis, etc.) before or after ITVISMA injection could lead to more serious complications. Patients, caregivers and close contacts of patients should follow infection prevention practices (e.g., hand hygiene, coughing/sneezing etiquette, limiting potential contacts). Advise patients and caregivers of the signs of a possible infection, such as coughing, wheezing, sneezing, runny nose, sore throat, or fever. Patients and caregivers should contact their healthcare provider immediately if the patient experiences any symptoms suggestive of infection before or after ITVISMA injection [see Dosage and Administration (2.1)].
Thrombocytopenia
Inform patients and caregivers that ITVISMA could decrease blood platelet count and increase the risk of bruising or bleeding. Inform patients and caregivers that decreases in platelet counts were observed within the first week after ITVISMA injection. Advise patients and caregivers to seek medical attention if the patient experiences unexpected bruising or bleeding [see Warnings and Precautions (5.2)].
Peripheral Sensory Neuropathy
Inform patients and caregivers that peripheral sensory neuropathy has occurred with ITVISMA administration. Advise patients and caregivers to contact their healthcare provider promptly if the patient experiences numbness, tingling, prickling, or pain in the arms, hands, legs and/or feet [see Warnings and Precautions (5.3)].
Thrombotic Microangiopathy
Inform patients and/or caregivers that decreased blood platelet and red blood cell counts, acute kidney injury, and increased bruising or bleeding, which may be indicative of TMA, can occur. Advise patients and/or caregivers to seek immediate medical attention if the patient experiences unexpected bruising or bleeding, seizures, or decreased urine output [see Warnings and Precautions (5.4)].
AAV Vector Integration and Risk of Tumorigenicity
Inform patients and/or caregivers that there is a theoretical risk of tumorigenicity with AAV therapies such as ITVISMA. Advise patients and/or caregivers to contact their healthcare provider and Novartis Gene Therapies, Inc. (1-833-828-3947) if the patient who received ITVISMA develops a tumor [see Warnings and Precautions (5.6)].
Contraception and Egg/Sperm Donation
Advise women of childbearing potential to use an effective method of contraception and to refrain from egg donation for 6 months following ITVISMA injection. Advise men capable of fathering a child to use a barrier method of contraception and to refrain from sperm donation for 3 months following ITVISMA injection [see Use in Specific Populations (8.3)].
Manufactured by, Packed by, Distributed by:
Novartis Gene Therapies, Inc.
2275 Half Day Road
Bannockburn, IL 60015 USA
U.S. License Number 2250
©2025 Novartis Gene Therapies, Inc.
T2025-67
-
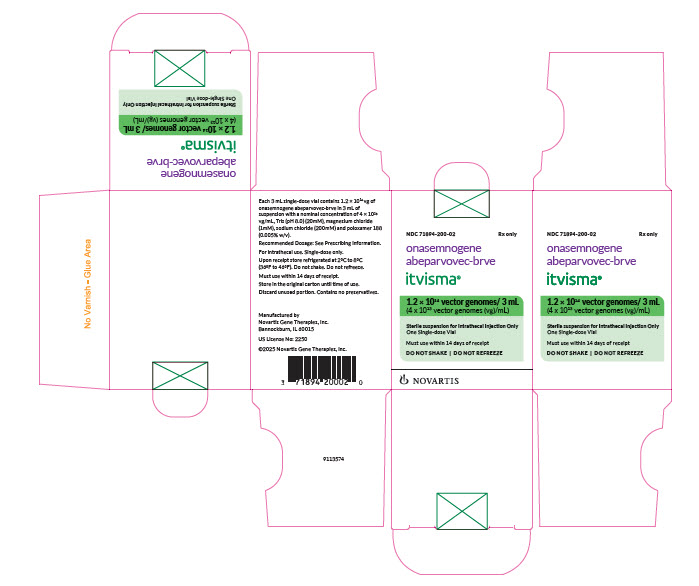
PRINCIPAL DISPLAY PANEL
NDC: 71894-200-02
Rx only
onasemnogene
abeparvovec-brveitvisma®
1.2 × 1014 vector genomes/ 3 mL
(4 x 1013 vector genomes (vg)/mL)
Sterile solution for Intrathecal Injection Only
One Single-dose Vial
Must use within 14 days of receipt
DO NOT SHAKE | DO NOT REFREEZE
NOVARTIS
-
INGREDIENTS AND APPEARANCE
ITVISMA
onasemnogene abeparvovec-brve injection, suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71894-200 Route of Administration INTRATHECAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ONASEMNOGENE ABEPARVOVEC (UNII: MLU3LU3EVV) (ONASEMNOGENE ABEPARVOVEC - UNII:MLU3LU3EVV) ONASEMNOGENE ABEPARVOVEC 40000000000000 [arb'U] in 1 mL Inactive Ingredients Ingredient Name Strength TROMETHAMINE (UNII: 023C2WHX2V) MAGNESIUM CHLORIDE (UNII: 02F3473H9O) SODIUM CHLORIDE (UNII: 451W47IQ8X) POLOXAMER 188 (UNII: LQA7B6G8JG) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71894-200-02 1 in 1 CARTON 11/24/2025 1 NDC: 71894-200-01 3 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125856 11/24/2025 Labeler - Novartis Gene Therapies, Inc. (080136787)
Trademark Results [ITVISMA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ITVISMA 79379551 not registered Live/Pending |
NOVARTIS AG 2023-07-26 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.