ATOVAQUONE AND PROGUANIL HYDROCHLORIDE tablet, film coated
Atovaquone and Proguanil Hydrochloride by
Drug Labeling and Warnings
Atovaquone and Proguanil Hydrochloride by is a Prescription medication manufactured, distributed, or labeled by BluePoint Laboratories, Glenmark Generics Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ATOVAQUONE AND PROGUANIL HYDROCHLORIDE TABLETS safely and effectively. See full prescribing information for ATOVAQUONE AND PROGUANIL HYDROCHLORIDE TABLETS.
ATOVAQUONE AND PROGUANIL HYDROCHLORIDE TABLETS, for oral administration
ATOVAQUONE AND PROGUANIL HYDROCHLORIDE TABLETS pediatric tablets, for oral administration
Initial U.S. Approval: 2000INDICATIONS AND USAGE
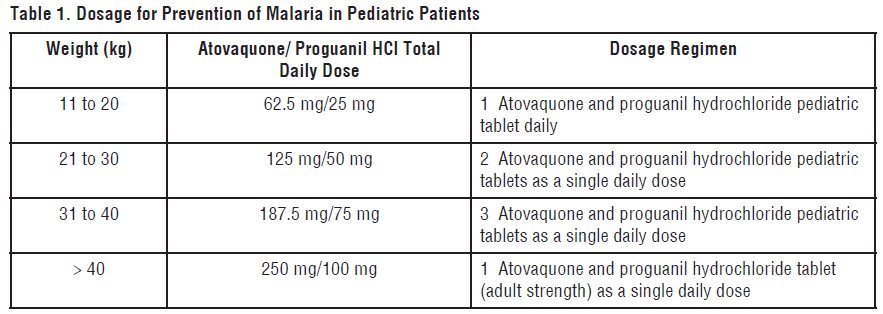
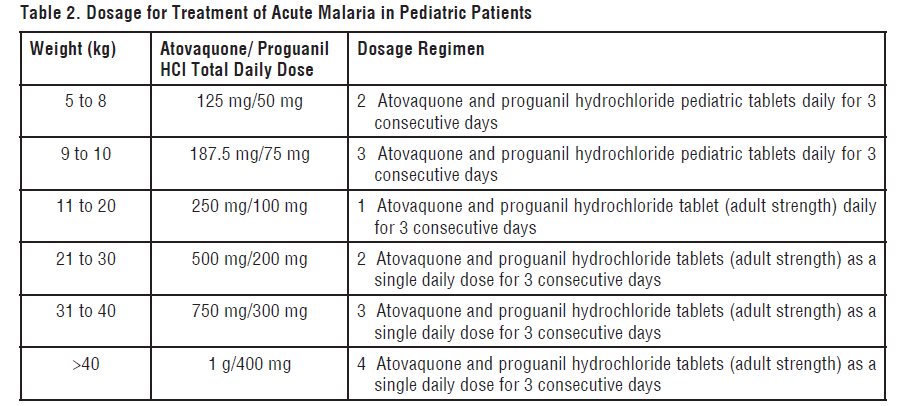
DOSAGE AND ADMINISTRATION
- Atovaquone and proguanil hydrochloride tablets should be taken with food or a milky drink.
Prophylaxis (2.1):
- Start prophylaxis 1 or 2 days before entering a malaria-endemic area and continue daily during the stay and for 7 days after return.
- Adults: One adult strength tablet per day.
- Pediatric Patients: Dosage based on body weight (see Table 1)
Treatment (2.2):
- Adults: Four adult strength tablets as a single daily dose for 3 days.
- Pediatric Patients: Dosage based on body weight (see Table 2)
Renal Impairment (2.3):
- Do not use for prophylaxis of malaria in patients with severe renal impairment.
- Use with caution for treatment of malaria in patients with severe renal impairment.
DOSAGE FORMS AND STRENGTHS
- Tablets (adult strength): 250 mg atovaquone and 100 mg proguanil hydrochloride. (3)
- Pediatric tablets: 62.5 mg atovaquone and 25 mg proguanil hydrochloride. (3)
CONTRAINDICATIONS
- Known serious hypersensitivity reactions to atovaquone or proguanil hydrochloride or any component of the formulation. (4)
- Prophylaxis of P. falciparum malaria in patients with severe renal impairment (creatinine clearance <30 mL/min). (4)
WARNINGS AND PRECAUTIONS
- Atovaquone absorption may be reduced in patients with diarrhea or vomiting. If used in patients who are vomiting, parasitemia should be closely monitored and the use of an antiemetic considered. In patients with severe or persistent diarrhea or vomiting, alternative antimalarial therapy may be required. (5.1)
- In mixed P. falciparum and Plasmodium vivax infection, P. vivax relapse occurred commonly when patients were treated with atovaquone and proguanil hydrochloride alone. (5.2)
- In the event of recrudescent P. falciparum infections after treatment or prophylaxis failure, patients should be treated with a different blood schizonticide. (5.2)
- Elevated liver laboratory tests and cases of hepatitis and hepatic failure requiring liver transplantation have been reported with prophylactic use. (5.3)
- Atovaquone and proguanil hydrochloride has not been evaluated for the treatment of cerebral malaria or other severe manifestations of complicated malaria. Patients with severe malaria are not candidates for oral therapy. (5.4)
ADVERSE REACTIONS
- Prophylaxis: Common adverse reactions (≥4%) in adults were diarrhea, dreams, oral ulcers, and headache; these events occurred in a similar or lower proportion of subjects receiving atovaquone and proguanil hydrochloride than an active comparator. Common adverse reactions (≥5%) in pediatric patients included abdominal pain, headache, cough, and vomiting. (6.1)
- Treatment: Common adverse reactions (≥5%) in adolescents and adults were abdominal pain, nausea, vomiting, headache, diarrhea, asthenia, anorexia, and dizziness. Common adverse reactions (≥6%) in pediatric patients included vomiting, pruritus, and diarrhea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Glenmark Pharmaceuticals Inc., USA at 1 (888)721-7115 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Administration with rifampin or rifabutin is known to reduce atovaquone concentrations; concomitant use with atovaquone and proguanil hydrochloride is not recommended. (7.1)
- Proguanil may potentiate anticoagulant effect of warfarin and other coumarin-based anticoagulants. Caution advised when initiating or withdrawing atovaquone and proguanil hydrochloride in patients on anticoagulants; coagulation tests should be closely monitored. (7.2)
- Tetracycline may reduce atovaquone concentrations; parasitemia should be closely monitored. (7.3)
USE IN SPECIFIC POPULATIONS
- Renal impairment: contraindicated for prophylaxis of P. falciparum malaria in patients with severe renal impairment. (8.6)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Prevention of Malaria
1.2 Treatment of Malaria
2 DOSAGE AND ADMINISTRATION
2.1 Prevention of Malaria
2.2 Treatment of Acute Malaria
2.3 Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Vomiting and Diarrhea
5.2 Relapse of Infection
5.3 Hepatotoxicity
5.4 Severe or Complicated Malaria
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Rifampin/Rifabutin
7.2 Anticoagulants
7.3 Tetracycline
7.4 Metoclopramide
7.5 Indinavir
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Prevention of P. falciparum Malaria
14.2 Treatment of Acute, Uncomplicated P. falciparum Malaria Infections
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Prevention of Malaria
Atovaquone and proguanil hydrochloride tablets are indicated for the prophylaxis of Plasmodium falciparum malaria, including in areas where chloroquine resistance has been reported.
1.2 Treatment of Malaria
Atovaquone and proguanil hydrochloride tablets are indicated for the treatment of acute, uncomplicated P. falciparum malaria. Atovaquone and proguanil hydrochloride tablets have been shown to be effective in regions where the drugs chloroquine, halofantrine, mefloquine, and amodiaquine may have unacceptable failure rates, presumably due to drug resistance.
-
2 DOSAGE AND ADMINISTRATION
The daily dose should be taken at the same time each day with food or a milky drink. In the event of vomiting within 1 hour after dosing, a repeat dose should be taken.
Atovaquone and proguanil hydrochloride tablets may be crushed and mixed with condensed milk just prior to administration to patients who may have difficulty swallowing tablets.
2.1 Prevention of Malaria
Start prophylactic treatment with atovaquone and proguanil hydrochloride tablets 1 or 2 days before entering a malaria-endemic area and continue daily during the stay and for 7 days after return.
Adults:
One atovaquone and proguanil hydrochloride tablet (adult strength = 250 mg atovaquone/100 mg proguanil hydrochloride) per day.
Pediatric Patients
The dosage for prevention of malaria in pediatric patients is based upon body weight (Table 1)
2.2 Treatment of Acute Malaria
Adults:
Four atovaquone and proguanil hydrochloride tablets (adult strength; total daily dose 1 g atovaquone/400 mg proguanil hydrochloride) as a single daily dose for 3 consecutive day.
Pediatric Patients
The dosage for treatment of acute malaria in pediatric patients is based upon body weight (Table 2)
2.3 Renal Impairment
Do not use atovaquone and proguanil hydrochloride tablets for malaria prophylaxis in patients with severe renal impairment (creatinine clearance <30 mL/min) [See Contraindications (4)]. Use with caution for the treatment of malaria in patients with severe renal impairment, only if the benefits of the 3 day treatment regimen outweigh the potential risks associated with increased drug exposure. No dosage adjustments are needed in patients with mild (creatinine clearance 50 to 80 mL/min) or moderate (creatinine clearance 30 to 50 mL/min) renal impairment. [See Clinical Pharmacology (12.3).
-
3 DOSAGE FORMS AND STRENGTHS
Each atovaquone and proguanil hydrochloride tablets (adult strength) contains 250 mg atovaquone USP and 100 mg proguanil hydrochloride USP. Atovaquone and proguanil hydrochloride tablets are pinkish brown to brown colored, circular, biconvex beveled edge, film-coated tablets with ‘404’ debossed on one side and ‘G’ debossed on the other side.
Atovaquone and proguanil hydrochloride pediatric tablets contain 62.5 mg atovaquone USP and 25 mg proguanil hydrochloride USP. Atovaquone and proguanil hydrochloride pediatric tablets are pinkish brown to brown colored, circular, biconvex beveled edged, film coated tablets with ‘70’ debossed on one side and ‘G’ debossed on the other side.
-
4 CONTRAINDICATIONS
- Atovaquone and proguanil hydrochloride is contraindicated in individuals with known hypersensitivity reactions (e.g., anaphylaxis, erythema multiforme or Stevens-Johnson syndrome, angioedema, vasculitis) to atovaquone or proguanil hydrochloride or any component of the formulation
- Atovaquone and proguanil hydrochloride is contraindicated for prophylaxis of P. falciparum malaria in patients with severe renal impairment (creatinine clearance <30 mL/min) because of pancytopenia in patients with severe renal impairment treated with proguanil [see Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Vomiting and Diarrhea
Absorption of atovaquone may be reduced in patients with diarrhea or vomiting. If atovaquone and proguanil hydrochloride is used in patients who are vomiting, parasitemia should be closely monitored and the use of an antiemetic considered. [See Dosage and Administration (2).] Vomiting occurred in up to 19% of pediatric patients given treatment doses of atovaquone and proguanil hydrochloride. In the controlled clinical trials, 15.3% of adults received an antiemetic when they received atovaquone/proguanil and 98.3% of these patients were successfully treated. In patients with severe or persistent diarrhea or vomiting, alternative antimalarial therapy may be required.
5.2 Relapse of Infection
In mixed P. falciparum and Plasmodium vivax infections, P. vivax parasite relapse occurred commonly when patients were treated with atovaquone and proguanil hydrochloride alone.
In the event of recrudescent P. falciparum infections after treatment with atovaquone and proguanil hydrochloride or failure of chemoprophylaxis with atovaquone and proguanil hydrochloride, patients should be treated with a different blood schizonticide.
5.3 Hepatotoxicity
Elevated liver laboratory tests and cases of hepatitis and hepatic failure requiring liver transplantation have been reported with prophylactic use of atovaquone and proguanil hydrochloride.
5.4 Severe or Complicated Malaria
Atovaquone and proguanil hydrochloride has not been evaluated for the treatment of cerebral malaria or other severe manifestations of complicated malaria, including hyperparasitemia, pulmonary edema, or renal failure. Patients with severe malaria are not candidates for oral therapy
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Because atovaquone and proguanil hydrochloride tablets contain atovaquone and proguanil hydrochloride, the type and severity of adverse reactions associated with each of the compounds may be expected. The lower prophylactic doses of atovaquone and proguanil hydrochloride were better tolerated than the higher treatment doses.
Prophylaxis of P. falciparum Malaria:
In 3 clinical trials (2 of which were placebo-controlled) 381 adults (mean age 31 years) received atovaquone and proguanil hydrochloride for the prophylaxis of malaria; the majority of adults were black (90%) and 79% were male. In a clinical trial for the prophylaxis of malaria, 125 pediatric patients (mean age 9 years) received atovaquone and proguanil hydrochloride; all subjects were black and 52% were male. Adverse experiences reported in adults and pediatric patients considered attributable to therapy occurred in similar proportions of subjects receiving atovaquone and proguanil hydrochloride or placebo in all studies. Prophylaxis with atovaquone and proguanil hydrochloride was discontinued prematurely due to a treatment-related adverse experience in 3 of 381 (0.8%) adults and 0 of 125 pediatric patients.
In a placebo-controlled study of malaria prophylaxis with atovaquone and proguanil hydrochloride involving 330 pediatric patients (aged 4 to 14 years) in Gabon, a malaria-endemic area, the safety profile of atovaquone and proguanil hydrochloride was consistent with that observed in the earlier prophylactic studies in adults and pediatric patients. The most common treatment-emergent adverse events with atovaquone and proguanil hydrochloride were abdominal pain (13%), headache (13%), and cough (10%). Abdominal pain (13% vs. 8%) and vomiting (5% vs. 3%) were reported more often with atovaquone and proguanil hydrochloride than with placebo. No patient withdrew from the study due to an adverse experience with atovaquone and proguanil hydrochloride. No routine laboratory data were obtained during this study.
Non-immune travelers visiting a malaria-endemic area received MALARONE (n = 1,004) for prophylaxis of malaria in 2 active-controlled clinical trials. In one study (n = 493), the mean age of subjects was 33 years and 53% were male; 90% of subjects were white, 6% of subjects were black and the remaining were of other racial/ethnic groups. In the other study (n = 511), the mean age of subjects was 36 years and 51% were female; the majority of subjects (97%) were white. Adverse experiences occurred in a similar or lower proportion of subjects receiving atovaquone and proguanil hydrochloride tablets than an active comparator (Table 3). Fewer neuropsychiatric adverse experiences occurred in subjects who received atovaquone and proguanil hydrochloride tablets than mefloquine. Fewer gastrointestinal adverse experiences occurred in subjects receiving atovaquone and proguanil hydrochloride tablets than chloroquine/proguanil. Compared with active comparator drugs, subjects receiving atovaquone and proguanil hydrochloride tablets had fewer adverse experiences overall that were attributed to prophylactic therapy (Table 3). Prophylaxis with atovaquone and proguanil hydrochloride tablets was discontinued prematurely due to a treatment-related adverse experience in 7 of 1,004 travelers.
Table 3. Adverse Experiences in Active-Controlled Clinical Trials of atovaquone and proguanil hydrochloride for Prophylaxis of P. falciparum Malaria
Percent of Subjects With Adverse Experiencesa (Percent of Subjects With Adverse Experiences Attributable to Therapy)
Study 1
Study 2
Atovaquone and proguanil hydrochloride n = 493 (28 days)b
Mefloquine n = 483 (53 days)b
Atovaquone and proguanil hydrochloride
n = 511 (26 days)b
Chloroquine plus Proguanil n = 511 (49 days)b
Diarrhea
38 (8)
36 (7)
34 (5)
39 (7)
Nausea
14 (3)
20 (8)
11 (2)
18 (7)
Abdominal pain
17 (5)
16 (5)
14 (3)
22 (6)
Headache
12 (4)
17 (7)
12 (4)
14 (4)
Dreams
7 (7)
16 (14)
6 (4)
7 (3)
Insomnia
5 (3)
16 (13)
4 (2)
5 (2)
Fever
9 (<1)
11 (1)
8 (<1)
8 (<1)
Dizziness
5 (2)
14 (9)
7 (3)
8 (4)
Vomiting
8 (1)
10 (2)
8 (0)
14 (2)
Oral ulcers
9 (6)
6 (4)
5 (4)
7 (5)
Pruritus
4 (2)
5 (2)
3 (1)
2 (<1)
Visual difficulties
2 (2)
5 (3)
3 (2)
3 (2)
Depression
<1 (<1)
5 (4)
<1 (<1)
1 (<1)
Anxiety
1 (<1)
5 (4)
<1 (<1)
1 (<1)
Any adverse experience
64 (30)
69 (42)
58 (22)
66 (28)
Any neuropsychiatric event
20 (14)
37 (29)
16 (10)
20 (10)
Any GI event
49 (16)
50 (19)
43 (12)
54 (20)
a Adverse experiences that started while receiving active study drug.
b Mean duration of dosing based on recommended dosing regimens.
In a third active-controlled study, atovaquone and proguanil hydrochloride (n = 110) was compared with chloroquine/proguanil (n = 111) for the prophylaxis of malaria in 221 non-immune pediatric patients (2 to 17 years). The mean duration of exposure was 23 days for atovaquone and proguanil hydrochloride, 46 days for chloroquine, and 43 days for proguanil, reflecting the different recommended dosage regimens for these products. Fewer patients treated with atovaquone and proguanil hydrochloride reported abdominal pain (2% vs. 7%) or nausea (<1% vs. 7%) than children who received chloroquine/proguanil. Oral ulceration (2% vs. 2%), vivid dreams (2% vs. <1%), and blurred vision (0% vs. 2%) occurred in similar proportions of patients receiving either atovaquone and proguanil hydrochloride or chloroquine/proguanil, respectively. Two patients discontinued prophylaxis with chloroquine/proguanil due to adverse events, while none of those receiving atovaquone and proguanil hydrochloride discontinued due to adverse events.
Treatment of Acute, Uncomplicated P. falciparum Malaria:
In 7 controlled trials, 436 adolescents and adults received atovaquone and proguanil hydrochloride for treatment of acute, uncomplicated P. falciparum malaria. The range of mean ages of subjects was 26 to 29 years; 79% of subjects were male. In these studies, 48% of subjects were classified as other racial/ethnic groups, primarily Asian; 42% of subjects were black and the remaining subjects were white. Attributable adverse experiences that occurred in ≥5% of patients were abdominal pain (17%), nausea (12%), vomiting (12%), headache (10%), diarrhea (8%), asthenia (8%), anorexia (5%), and dizziness (5%). Treatment was discontinued prematurely due to an adverse experience in 4 of 436 (0.9%) adolescents and adults treated with atovaquone and proguanil hydrochloride .
In 2 controlled trials, 116 pediatric patients (weighing 11 to 40 kg) (mean age 7 years) received atovaquone and proguanil hydrochloride for the treatment of malaria. The majority of subjects were black (72%); 28% were of other racial/ethnic groups, primarily Asian. Attributable adverse experiences that occurred in ≥5% of patients were vomiting (10%) and pruritus (6%). Vomiting occurred in 43 of 319 (13%) pediatric patients who did not have symptomatic malaria but were given treatment doses of atovaquone and proguanil hydrochloride for 3 days in a clinical trial. The design of this clinical trial required that any patient who vomited be withdrawn from the trial. Among pediatric patients with symptomatic malaria treated with atovaquone and proguanil hydrochloride, treatment was discontinued prematurely due to an adverse experience in 1 of 116 (0.9%).
In a study of 100 pediatric patients (5 to <11 kg body weight) who received atovaquone and proguanil hydrochloride for the treatment of uncomplicated P. falciparum malaria, only diarrhea (6%) occurred in ≥5% of patients as an adverse experience attributable to atovaquone and proguanil hydrochloride. In 3 patients (3%), treatment was discontinued prematurely due to an adverse experience.
Abnormalities in laboratory tests reported in clinical trials were limited to elevations of transaminases in patients with malaria being treated with atovaquone and proguanil hydrochloride . The frequency of these abnormalities varied substantially across trials of treatment and were not observed in the randomized portions of the prophylaxis trials.
One active-controlled trial evaluated the treatment of malaria in Thai adults (n = 182); the mean age of subjects was 26 years (range: 15 to 63 years); 80% of subjects were male. Early elevations of ALT and AST occurred more frequently in patients treated with atovaquone and proguanil hydrochloride (n = 91) compared to patients treated with an active control, mefloquine (n = 91). On Day 7, rates of elevated ALT and AST with atovaquone and proguanil hydrochloride and mefloquine (for patients who had normal baseline levels of these clinical laboratory parameters) were ALT 26.7% vs. 15.6%; AST 16.9% vs. 8.6%, respectively. By Day 14 of this 28-day study, the frequency of transaminase elevations equalized across the 2 groups.
6.2 Postmarketing Experience
In addition to adverse events reported from clinical trials, the following events have been identified during postmarketing use of atovaquone and proguanil hydrochloride. Because they are reported voluntarily from a population of unknown size, estimates of frequency cannot be made. These events have been chosen for inclusion due to a combination of their seriousness, frequency of reporting, or potential causal connection to atovaquone and proguanil hydrochloride.
Blood and Lymphatic System Disorders:
Neutropenia and anemia. Pancytopenia in patients with severe renal impairment treated with proguanil [see Contraindications (4)].
Immune System Disorders:
Allergic reactions including anaphylaxis, angioedema, and urticaria, and vasculitis.
Nervous System Disorders:
Seizures and psychotic events (such as hallucinations); however, a causal relationship has not been established.
Gastrointestinal Disorders:
Stomatitis.
Hepatobiliary Disorders:
Elevated liver laboratory tests, hepatitis, cholestasis; hepatic failure requiring transplant has been reported.
Skin and Subcutaneous Tissue Disorders:
Photosensitivity, rash, erythema multiforme, and Stevens-Johnson syndrome
-
7 DRUG INTERACTIONS
7.1 Rifampin/Rifabutin
Concomitant administration of rifampin or rifabutin is known to reduce atovaquone concentrations [see Clinical Pharmacology (12.3)]. The concomitant administration of atovaquone and proguanil hydrochloride and rifampin or rifabutin is not recommended.
7.2 Anticoagulants
Proguanil may potentiate the anticoagulant effect of warfarin and other coumarin-based anticoagulants. The mechanism of this potential drug interaction has not been established. Caution is advised when initiating or withdrawing malaria prophylaxis or treatment with atovaquone and proguanil hydrochloride in patients on continuous treatment with coumarin-based anticoagulants. When these products are administered concomitantly, coagulation tests should be closely monitored.
7.3 Tetracycline
Concomitant treatment with tetracycline has been associated with a reduction in plasma concentrations of atovaquone [see Clinical Pharmacology (12.3)]. Parasitemia should be closely monitored in patients receiving tetracycline.
7.4 Metoclopramide
While antiemetics may be indicated for patients receiving atovaquone and proguanil hydrochloride, metoclopramide may reduce the bioavailability of atovaquone and should be used only if other antiemetics are not available [see Clinical Pharmacology (12.3)].
7.5 Indinavir
Concomitant administration of atovaquone and indinavir did not result in any change in the steady-state AUC and Cmax of indinavir but resulted in a decrease in the Ctrough of indinavir [see Clinical Pharmacology (12.3)]. Caution should be exercised when prescribing atovaquone with indinavir due to the decrease in trough concentrations of indinavir
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data for published literature and postmarketing experience with the use of atovaquone and proguanil hydrochloride in pregnant women are insufficient to identify a drug-associated risk for major birth defects, miscarriage, or adverse maternal or fetal outcomes. The proguanil component of atovaquone and proguanil hydrochloride tablets acts to inhibit parasitic dihydrofolate reductase; however, pregnant women and females of reproductive potential should continue folate supplementation to prevent neural tube defects [see Clinical Pharmacology (12.4)]. Pregnant women with malaria are at increased risk for adverse pregnancy outcomes (see Clinical Considerations).
Atovaquone administered by oral gavage to pregnant rats and rabbits during the period of organogenesis was not associated with fetal malformations at plasma exposures approximately 7 times and equal to, respectively, the estimated human exposure for the treatment of malaria based on AUC. Proguanil administered to pregnant rats and rabbits during the period of organogenesis was not associated with embryo-fetal toxicity at maternally toxic plasma exposures approximately 0.07 and 0.8 times, respectively, the estimated human exposure for treatment of malaria based on AUC (see Data).
The combination of atovaquone and proguanil hydrochloride given orally by gavage during the period of organogenesis was not associated with embryo-fetal developmental effects in pregnant rats or rabbits at atovaquone: proguanil hydrochloride doses of 50:20 mg/kg/day and 100:40 mg/kg/day, respectively (1.7 and 0.1 times and 0.3 and 0.5 times, respectively, the estimated human exposure for treatment of malaria). In a pre-and post-natal study with atovaquone and another pre-and post-natal study with proguanil, neither compound impaired the growth, development, or reproductive ability of first generation offspring at maternal AUC exposures of approximately 7.3 and 0.04 times, respectively, the estimated human AUC exposure for treatment of malaria (see Data).
The estimated background risk of major birth defects and miscarriage for the indicates population in unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S general population, the background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk: Malaria during pregnancy increases the risk for adverse pregnancy outcomes, including maternal anemia, prematurity, spontaneous abortion, and stillbirth.
Data
Animal Data: Atovaquone: Atovaquone administered in oral doses of 250, 500, and 1,000 mg/kg/day during organogenesis (Gestation Day [GD] 6 to GD15) in pregnant rats did not cause maternal or embryo-fetal toxicity at doses up to 1,000 mg/kg/day corresponding to maternal plasma exposures up to 7.3 times the estimated human exposure for the treatment of malaria based on AUC. In pregnant rabbits, atovaquone administered in oral doses of 300, 600, and 1,200 mg/kg/day by gavage during organogenesis (GD6 to GD18) was associated with decreased fetal body length at a maternally toxic dose of 1,200 mg/kg/day corresponding to plasmas exposures that were approximately 1.3 times the estimated human exposure during treatment of malaria based on AUC. In a pre- and pot-natal study in rats, atovaquone administered in oral doses of 250, 500 and 1,000 mg/day/day form GD15 until Lactation Day (LD) 20 did not impair the growth or development effects in the first generation offspring at dosed up to 1,000 mg/kg/day corresponding to AUC exposures of approximately 7.3 times the estimated human exposure during treatment of malaria. Atovaquone crossed the placenta and was present in fetal rat and rabbit tissue.
Proguanil: Proguanil administered orally to pregnant rats during organogenesis (GD6 to GD17) was not associated with fetal malformations, but increased ureter variations at a maternally toxic dose of 20 mg/kg/day corresponding to a plasma concentration approximately equal to 0.07 times the estimated human exposure for the treatment of malaria based on AUC. Proguanil given orally by gavage at a maternally toxic dose of 40 mg/kg/day to pregnant rabbits during organogenesis (GD6 to GD20) did not produce adverse embryo-fetal effects at a plasma concentration up to 0.8 times the estimated human exposure for the treatment of malaria based on AUC. In a pre- and post-natal study in female rats, proguanil hydrochloride administered in oral doses of 4, 8, of 16 mg/kg/day from GD6 until LD20 did not impair the growth, development, or reproductive ability of first generation offspring or the survivability of second generation offspring at doses up to 16 mg/kg/day (0.04 times the average human exposure based on AUC). Pre- and post-natal studies of proguanil in animals at exposures similar to or greater than those observed in humans have not been conducted.
Atovaquone and Proguanil: The combination of atovaquone and proguanil hydrochloride administered orally to pregnant rats in atovaquone; proguanil hydrochloride doses of 12.5:5, 25:10, and 50:20 mg/kg/day during organogenesis (GD6 to GD17) did not produce maternal toxicity or adverse embryo-fetal developmental effects with doses up to 50:20 mg/kg/day corresponding to plasma concentrations up to 1.7 and 0.1 times, respectively, the estimated human exposure during treatment of malaria based on AUC. In pregnant rabbits, the combination of atovaquone and proguanil hydrochloride administered orally in atovaquone; proguanil hydrochloride doses of 25:10, 50:20, or 100:40 mg/kg/day during organogenesis (GD6 to GD20) did not produce adverse embryo-fetal developmental effects at a maternally toxic dose of 100:40 mg/kg/day corresponding to plasma concentration of approximately 0.3 and 0.5 times, respectively, the estimated human exposure during treatment of malaria based on AUC.
8.2 Lactation
Risk Summary
There are no data on the presence of atovaquone in human milk; however, proguanil is present in human milk. Atovaquone is present in rat milk (see Data). When a drug is present in animal milk, it is likely the drug will be present in human milk. There are no data on the effect of atovaquone and proguanil on the breastfed child or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for atovaquone and proguanil hydrochloride and any potential adverse effect on the breastfed child from atovaquone and proguanil hydrochloride or from the underlying maternal condition.
Data
In a rat study with doses of 10 and 250 mg/kg, given orally by gavage on postpartum Day 11, atovaquone concentrations in the milk were 30% of the concurrent atovaquone concentrations in the maternal plasma at both doses. The concentration of drug in animal milk does not necessarily predict the concentration of drug in human milk.
8.4 Pediatric Use
Prophylaxis of Malaria:
Safety and effectiveness have not been established in pediatric patients who weigh less than 11 kg. The efficacy and safety of atovaquone and proguanil hydrochloride have been established for the prophylaxis of malaria in controlled trials involving weighing pediatric patients 11 kg or more [see Clinical Studies (14.1)].
Treatment of Malaria:
Safety and effectiveness have not been established in pediatric patients who weigh less than 5 kg. The efficacy and safety of atovaquone and proguanil hydrochloride for the treatment of malaria have been established in controlled trials involving pediatric patients weighing 5 kg or more [see Clinical Studies (14.2)].
8.5 Geriatric Use
Clinical trials of atovaquone and proguanil hydrochloride did not include sufficient numbers of subjects aged 65 years and older to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, the higher systemic exposure to cycloguanil, and the greater frequency of concomitant disease or other drug therapy. [See Clinical Pharmacology (12.3).]
8.6 Renal Impairment
Do not use atovaquone and proguanil hydrochloride for malaria prophylaxis in patients with severe renal impairment (creatinine clearance <30 mL/min). Use with caution for the treatment of malaria in patients with severe renal impairment, only if the benefits of the 3-day treatment regimen outweigh the potential risks associated with increased drug exposure. No dosage adjustments are needed in patients with mild (creatinine clearance 50 to 80 mL/min) or moderate (creatinine clearance 30 to 50 mL/min) renal impairment. [See Clinical Pharmacology (12.3).]
8.7 Hepatic Impairment
No dosage adjustments are needed in patients with mild or moderate hepatic impairment [see Clinical Pharmacology (12.3)]. No trials have been conducted in patients with severe hepatic impairment
-
10 OVERDOSAGE
There is no information on overdoses of atovaquone and proguanil hydrochloride substantially higher than the doses recommended for treatment.
There is no known antidote for atovaquone, and it is currently unknown if atovaquone is dialyzable. Overdoses up to 31,500 mg of atovaquone have been reported. In one such patient who also took an unspecified dose of dapsone, methemoglobinemia occurred. Rash has also been reported after overdose.
Overdoses of proguanil hydrochloride as large as 1,500 mg have been followed by complete recovery, and doses as high as 700 mg twice daily have been taken for over 2 weeks without serious toxicity. Adverse experiences occasionally associated with proguanil hydrochloride doses of 100 to 200 mg/day, such as epigastric discomfort and vomiting would be likely to occur with overdose. There are also reports of reversible hair loss and scaling of the skin on the palms and/or soles, reversible aphthous ulceration, and hematologic side effects.
-
11 DESCRIPTION
Atovaquone and proguanil hydrochloride tablets (adult strength) and Atovaquone and proguanil hydrochloride pediatric tablets, for oral administration, contain a fixed-dose combination of the antimalarial agents atovaquone USP and proguanil hydrochloride USP.
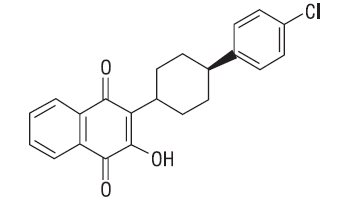
The chemical name of atovaquone USP is trans-2-[4-(4-chlorophenyl)cyclohexyl]-3-hydroxy-1,4-naphthalenedione. Atovaquone USP is a yellow crystalline solid that is freely soluble in N-methyl-2-pyrrolidone and in tetrahydrofuran; soluble in chloroform; sparingly soluble in acetone and dimethyl sulfoxide; slightly soluble in octanol, ethyl acetate, polyethylene glycol 200; very slightly soluble in 0.1N sodium hydroxide; insoluble in water. It has a molecular weight of 366.84 and the molecular formula C22H19ClO3. The compound has the following structural formula:
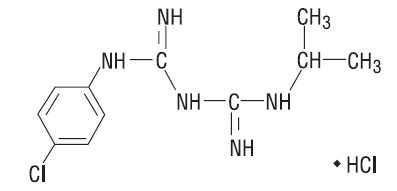
The chemical name of proguanil hydrochloride USP is 1-(4-chlorophenyl)-5-isopropyl-biguanide hydrochloride. Proguanil hydrochloride USP is a white crystalline powder slightly soluble in water, sparingly soluble in alcohol, practically insoluble in methylene chloride. It has a molecular weight of 290.22 g/mol and the molecular formula C11H16ClN5HCl. The compound has the following structural formula:
Each atovaquone and proguanil hydrochloride tablet contains 250 mg of atovaquone USP and 100 mg of proguanil hydrochloride USP and each atovaquone and proguanil hydrochloride pediatric tablets contains 62.5 mg of atovaquone USP and 25 mg of proguanil hydrochloride USP. The inactive ingredients in the tablet are colloidal silicon dioxide, ferric oxide red, hypromellose 2910, low substituted hydroxypropyl cellulose, magnesium stearate, microcrystalline cellulose, poloxamer 188, polyethylene glycol 400, polyethylene glycol 8000, povidone K30, sodium starch glycolate and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Atovaquone and proguanil hydrochloride tablets, fixed-dose combination of atovaquone and proguanil hydrochloride, is an antimalarial agent [see Microbiology (12.4)].
12.2 Pharmacodynamics
Cardiac Effects
The effect of atovaquone and proguanil hydrochloride on the QT interval is unknown in humans.
12.3 Pharmacokinetics
Absorption:
Atovaquone is a highly lipophilic compound with low aqueous solubility. The bioavailability of atovaquone shows considerable inter-individual variability.
Effect of Food: Atovaquone and proguanil hydrochloride should be taken with food or a milky drink. Dietary fat taken with atovaquone increases the rate and extend of absorption, increasing AUC 2 to 3 times and Cmax 5 times over fasting. The absolute bioavailability of the tablet formulation of atovaquone when taken with food is 23%.
Distribution:
Atovaquone is highly protein bound (> 99%) over the concentration range of 1 to 90 mcg/mL. A population pharmacokinetic analysis demonstrated that the apparent volume of distribution of atovaquone (V/F) in adult and pediatric patients after oral administration is approximately 8.8 L/kg.
Proguanil is 75% protein bound. A population pharmacokinetic analysis demonstrated that the apparent V/F of proguanil in adult and pediatric patients older than 15 years of age with body weights from 31 to 110 kg ranged from 1,617 to 2,502 L. In pediatric patients 15 years and younger with body weights from 11 to 56 kg, the V/F of proguanil ranged from 462 to 966 L.
In human plasma, the binding of atovaquone and proguanil was unaffected by the presence of the other.
Elimination:
The elimination half-life of atovaquone is about 2 to 3 days in adult patients.
The elimination half-life of proguanil is 12 to 21 hours in both adult patients and pediatric patients, but may be longer in individuals who are slow metabolizers.
The main routes of elimination are hepatic biotransformation and renal excretion.
Metabolism: In a study where 14C-labeled atovaquone was administered to healthy volunteers, greater than 94% of the dose was recovered as unchanged atovaquone in the feces over 21 days. There was little or no excretion of atovaquone in the urine (less than 0.6%). There is indirect evidence that atovaquone may undergo limited metabolism; however, a specific metabolite has not been identified. Between 40% to 60% of proguanil is excreted by the kidneys. Proguanil is metabolized to cycloguanil (primarily via cytochrome [CYP2C19]) and 4-chlorophenylbiguanide.
Excretion: A population pharmacokinetic analysis in adult and pediatric patients showed that the apparent clearance (CL/F) of both atovaquone and proguanil are related to the body weight. The values CL/F for both atovaquone and proguanil in subjects with body weight ≥11 kg are shown in Table 4.
Table 4. Apparent Clearance for Atovaquone and Proguanil in Patients as a Function of Body Weight
Atovaquone
Proguanil
Body Weight
N
CL/F (L/hr) Mean ± SDa (range)
n
CL/F (L/hr) Mean ± SDa (range)
11 to 20 kg
159
1.34 ± 0.63 (0.52 to 4.26)
146
29.5 ± 6.5 (10.3 to 48.3)
21 to 30 kg
117
1.87 ± 0.81 (0.52 to 5.38)
113
40.0 ± 7.5 (15.9 to 62.7)
31 to 40 kg
95
2.76 ± 2.07 (0.97 to 12.5)
91
49.5 ± 8.30 (25.8 to 71.5)
>40 kg
368
6.61 ± 3.92 (1.32 to 20.3)
282
67.9 ± 19.9 (14.0 to 145)
a SD = standard deviation.
The pharmacokinetics of atovaquone and proguanil in patients with body weight below 11 kg have not been adequately characterized.
Specific Populations
Pediatrics Patients: The pharmacokinetics of proguanil and cycloguanil are similar in adult patients and pediatric patients. However, the elimination half-life of atovaquone is shorter in pediatric patients (1 to 2 days) than in adult patients (2 to 3 days). In clinical trials, plasma trough concentrations of atovaquone and proguanil in pediatric patients weighing 5 to 40 kg were within the range observed in adults after dosing by body weight.
Geriatrics Patients: In a single-dose study, the pharmacokinetics of atovaquone, proguanil, and cycloguanil were compared in 13 elderly subjects (age 65 to 79 years) with those of 13 younger subjects (age 30 to 45 years). In the elderly subjects, the extent of systemic exposure (AUC) of cycloguanil was increased (point estimate: 2.36, 90% CI: 1.70, 3.28). Tmax was longer in elderly subjects (median 8 hours) compared with younger subjects (median 4 hours) and average elimination half-life was longer in elderly subjects (mean: 14.9 hours) compared with younger subjects (mean: 8.3 hours).
Patients with Renal Impairment:In patients with mild renal impairment (creatinine clearance 50 to 80 mL/min), oral clearance and/or AUC data for atovaquone, proguanil, and cycloguanil are within the range of values observed in patients with normal renal function (creatinine clearance >80 mL/min). In patients with moderate renal impairment (creatinine clearance 30 to 50 mL/min), mean oral clearance for proguanil was reduced by approximately 35% compared with patients with normal renal function (creatinine clearance >80 mL/min) and the oral clearance of atovaquone was comparable between patients with normal renal function and mild renal impairment. No data exist on the use of atovaquone and proguanil hydrochloride for long-term prophylaxis (over 2 months) in individuals with moderate renal failure. In patients with severe renal impairment (creatinine clearance <30 mL/min), atovaquone Cmax and AUC are reduced but the elimination half-lives for proguanil and cycloguanil are prolonged, with corresponding increases in AUC, resulting in the potential of drug accumulation and toxicity with repeated dosing [see Contraindications (4)].
Patients with Hepatic Impairment:In a single-dose study, the pharmacokinetics of atovaquone, proguanil, and cycloguanil were compared in 13 subjects with hepatic impairment (9 mild, 4 moderate, as indicated by the Child-Pugh method) with those of 13 subjects with normal hepatic function. In subjects with mild or moderate hepatic impairment as compared with healthy subjects, there were no marked differences (<50%) in the rate or extent of systemic exposure of atovaquone. However, in subjects with moderate hepatic impairment, the elimination half-life of atovaquone was increased (point estimate: 1.28, 90% CI: 1 to 1.63). Proguanil AUC, Cmax, and its elimination half-life increased in subjects with mild hepatic impairment when compared to healthy subjects (Table 5). Also, the proguanil AUC and its elimination half-life increased in subjects with moderate hepatic impairment when compared with healthy subjects. Consistent with the increase in proguanil AUC, there were marked decreases in the systemic exposure of cycloguanil (Cmax and AUC) and an increase in its elimination half-life in subjects with mild hepatic impairment when compared to healthy volunteers (Table 5). There were few measurable cycloguanil concentrations in subjects with moderate hepatic impairment. The pharmacokinetics of atovaquone, proguanil, and cycloguanil after administration of atovaquone and proguanil hydrochloride have not been studied in patients with severe hepatic impairment.
Table 5. Point Estimates (90% CI) for Proguanil and Cycloguanil Parameters in Subjects With Mild and Moderate Hepatic Impairment Compared to Healthy Volunteers
Parameter
Comparison
Proguanil
Cycloguanil
AUC(0-inf)a
mild:healthy
1.96 (1.51, 2.54)
0.32 (0.22, 0.45)
Cmaxa
mild:healthy
1.41 (1.16, 1.71)
0.35 (0.24, 0.50)
t1/2b
mild:healthy
1.21 (0.92, 1.60)
0.86 (0.49, 1.48)
AUC(0-inf)a
moderate:healthy
1.64 (1.14, 2.34)
ND
Cmaxa
moderate:healthy
0.97 (0.69, 1.36)
ND
t1/2b
moderate:healthy
1.46 (1.05, 2.05)
ND
ND = not determined due to lack of quantifiable data.
a Ratio of geometric means.
b Mean difference.
Drug Interactions Studies
There are no pharmacokinetic interactions between atovaquone and proguanil at the recommended dose.
Atovaquone is highly protein bound (>99%) but does not displace other highly protein-bound drugs in vitro.
Proguanil is metabolized primarily by CYP2C19. Potential pharmacokinetic interactions between proguanil or cycloguanil and other drugs that are CYP2C19 substrates or inhibitors are unknown.
Rifampin/Rifabutin: Concomitant administration of rifampin or rifabutin is known to reduce atovaquone concentrations by approximately 50% and 34%, respectively. The mechanisms of these interactions are unknown.
Tetracyline: Concomitant treatment with tetracycline has been associated with approximately a 40% reduction in plasma concentrations of atovaquone.
Metoclopramide: Concomitant treatment with metoclopramide has been associated with decreased bioavailability of atovaquone.
Indinavir: Concomitant administration of atovaquone (750 mg twice daily with food for 14 days) and indinavir (800 mg three times daily without food for 14 days) did not result in any change in the steady-state AUC and Cmax of indinavir but resulted in a decrease in the Ctrough of indinavir (23% decrease [90% CI: 8%, 35%]).
12.4 Microbiology
Mechanism of Action
The constituents of atovaquone and proguanil hydrochloride tablets, atovaquone and proguanil hydrochloride, interfere with 2 different pathways involved in the biosynthesis of pyrimidines required for nucleic acid replication. Atovaquone is a selective inhibitor of parasite mitochondrial electron transport. Proguanil hydrochloride primarily exerts its effect by means of the metabolite cycloguanil, a dihydrofolate reductase inhibitor. Inhibition of dihydrofolate reductase in the Plasmodium parasite disrupts deoxythymidylate synthesis.
Antimicrobial Activity
Atovaquone and cycloguanil (an active metabolite of proguanil) are active against the erythrocytic and exoerythrocytic stages of P. Falciparum. Enhaced efficacy of the combination compared with either atovaquone or proguanil hydrochloride alone was demonstrated in clinical trials in both immune and non-immune patients [see Clinical Studies (14.1, 14.2)].
Resistance
Strains of P. falciparum with decreased susceptibility to atovaquone or proguanil/cycloguanil alone can be selected in vitro of in vivo. The combination of atovaquone and proguanil hydrochloride may not be effective for treatment of recrudescent malaria that develops after prior therapy with the combination.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Genotoxicity studies have not been performed with atovaquone in combination with proguanil. Effects of atovaquone and proguanil hydrochloride on male and female reproductive performance are unknown.
Atovaquone:
A 24-month carcinogenicity study in CD rats was negative for neoplasms at doses up to 500 mg/kg/day corresponding to approximately 54 times the average steady-state plasma concentrations in humans during prophylaxis of malaria. In CD-1 mice, a 24-month study showed treatment-related increases in incidence of hepatocellular adenoma and hepatocellular carcinoma at all doses tested (50, 100, and 200 mg/kg/day) which correlated with at least 15 times the average steady-state plasma concentrations in humans during prophylaxis of malaria.
Atovaquone was negative with or without metabolic activation in the Ames Salmonella mutagenicity assay, the Mouse Lymphoma mutagenesis assay, and the cultured human lymphocyte cytogenetic assay. No evidence of genotoxicity was observed in the in vivo Mouse Micronucleus assay.
Atovaquone administered by oral gavage in doses of 100, 300, or 1,000 mg/kg/day to adult male rats from 73 days prior to mating until 20 days after mating and to adult female rats from 14 days prior to mating until LD20 did not impair male or female fertility or early embryonic development at doses up to 1,000 mg/kg/day corresponding to plasma exposures of approximately 7.3 times the estimated human exposure for the treatment of malaria based on AUC.
Proguanil:
No evidence of a carcinogenic effect was observed in 24-month studies conducted in CD-1 mice at doses up to 16 mg/kg/day corresponding to 1.5 times the average human plasma exposure during prophylaxis of malaria based on AUC, and in Wistar Hannover rats at doses up 20 mg/kg/day corresponding to 1.1 times the average human plasma exposure during prophylaxis of malaria based on AUC.
Proguanil was negative with or without metabolic activation in the Ames Salmonella mutagenicity assay and the Mouse Lymphoma mutagenesis assay. No evidence of genotoxicity was observed in the in vivo Mouse Micronucleus assay.
Cycloguanil, the active metabolite of proguanil, was also negative in the Ames test, but was positive in the Mouse Lymphoma assay and the Mouse Micronucleus assay. These positive effects with cycloguanil, a dihydrofolate reductase inhibitor, were significantly reduced or abolished with folinic acid supplementation.
Proguanil administered orally in doses of 4, 8, and 16 mg/kg/day to male rats from 29 days prior to mating until 27 days after mating and to females from 15 days prior to mating through GD7 revealed no adverse effects on adult male or female fertility or early embryonic development at doses up to 16 mg/kg/day (up to 0.04 times the average human exposure for the treatment of malaria based on AUC). Fertility studies or proguanil in animals at exposures similar to or greater than those observed in humans have not been conducted.
13.2 Animal Toxicology and/or Pharmacology
Fibrovascular proliferation in the right atrium, pyelonephritis, bone marrow hypocellularity, lymphoid atrophy, and gastritis/enteritis were observed in dogs treated with proguanil hydrochloride for 6 months at a dose of 12 mg/kg/day (approximately 3.9 times the recommended daily human dose for malaria prophylaxis on a mg/m2 basis). Bile duct hyperplasia, gall bladder mucosal atrophy, and interstitial pneumonia were observed in dogs treated with proguanil hydrochloride for 6 months at a dose of 4 mg/kg/day (approximately 1.3 times the recommended daily human dose for malaria prophylaxis on a mg/m2 basis). Mucosal hyperplasia of the cecum and renal tubular basophilia were observed in rats treated with proguanil hydrochloride for 6 months at a dose of 20 mg/kg/day (approximately 1.6 times the recommended daily human dose for malaria prophylaxis on a mg/m2 basis). Adverse heart, lung, liver, and gall bladder effects observed in dogs and kidney effects observed in rats were not shown to be reversible.
-
14 CLINICAL STUDIES
14.1 Prevention of P. falciparum Malaria
Atovaquone and proguanil hydrochloride was evaluated for prophylaxis of P. falciparum malaria in 5 clinical trials in malaria-endemic areas and in 3 active-controlled trials in non-immune travelers to malaria-endemic areas.
Three placebo-controlled trials of 10 to 12 weeks’ duration were conducted among residents of malaria-endemic areas in Kenya, Zambia, and Gabon. The mean age of subjects was 30 (range: 17 to 55), 32 (range: 16 to 64), and 10 (range: 5 to16) years, respectively. Of a total of 669 randomized patients (including 264 pediatric patients 5 to 16 years), 103 were withdrawn for reasons other than falciparum malaria- or drug-related adverse events (55% of these were lost to follow-up and 45% were withdrawn for protocol violations). The results are listed in Table 6.
Table 6. Prevention of Parasitemiaa in Placebo-Controlled Clinical Trials of Atovaquone and Proguanil hydrochloride for Prophylaxis of P. falciparum Malaria in Residents of Malaria-Endemic Areas
Atovaquone and Proguanil hydrochloride
Placebo
Total number of patients randomized
326
343
Failed to complete study
57
46
Developed parasitemia (P. falciparum)
2
92
a Free of parasitemia during the 10- to 12-week period of prophylactic therapy.
In another study, 330 Gabonese pediatric patients (weighing 13 to 40 kg, and aged 4 to 14 years) who had received successful open-label radical cure treatment with artesunate, were randomized to receive either atovaquone and proguanil hydrochloride (dosage based on body weight) or placebo in a double-blind fashion for 12 weeks. Blood smears were obtained weekly and any time malaria was suspected. Nineteen of the 165 children given atovaquone and proguanil hydrochloride and 18 of 165 patients given placebo withdrew from the study for reasons other than parasitemia (primary reason was lost to follow-up). One out of 150 evaluable patients (<1%) who received atovaquone and proguanil hydrochloride developed P. falciparum parasitemia while receiving prophylaxis with atovaquone and proguanil hydrochloride compared with 31 (22%) of the 144 evaluable placebo recipients.
In a 10-week study in 175 South African subjects who moved into malaria-endemic areas and were given prophylaxis with 1 atovaquone and proguanil hydrochloride Tablet daily, parasitemia developed in 1 subject who missed several doses of medication. Since no placebo control was included, the incidence of malaria in this study was not known.
Two active-controlled trials were conducted in non-immune travelers who visited a malaria-endemic area. The mean duration of travel was 18 days (range: 2 to 38 days). Of a total of 1,998 randomized patients who received atovaquone and proguanil hydrochloride or controlled drug, 24 discontinued from the study before follow-up evaluation 60 days after leaving the endemic area. Nine of these were lost to follow-up, 2 withdrew because of an adverse experience, and 13 were discontinued for other reasons. These trials were not large enough to allow for statements of comparative efficacy. In addition, the true exposure rate to P. falciparum malaria in both trials is unknown. The results are listed in Table 7.
Table 7. Prevention of Parasitemiaa in Active-Controlled Clinical Trials of Atovaquone and Proguanil hydrochloride for Prophylaxis of P. falciparum Malaria in Non-Immune Travelers
Atovaquone and proguanil hydrochloride
Mefloquine
Chloroquine plus Proguanil
Total number of randomized patients who received study drug
1,004
483
511
Failed to complete study
14
6
4
Developed parasitemia (P. falciparum)
0
0
3
a Free of parasitemia during the period of prophylactic therapy.
A third randomized, open-label study was conducted which included 221 otherwise healthy pediatric patients (weighing ≥11 kg and 2 to 17 years of age) who were at risk of contracting malaria by traveling to an endemic area. The mean duration of travel was 15 days (range: 1 to 30 days). Prophylaxis with atovaquone and proguanil hydrochloride (n = 110, dosage based on body weight) began 1 or 2 days before entering the endemic area and lasted until 7 days after leaving the area. A control group (n = 111) received prophylaxis with chloroquine/proguanil dosed according to WHO guidelines. No cases of malaria occurred in either group of children. However, the study was not large enough to allow for statements of comparative efficacy. In addition, the true exposure rate to P. falciparum malaria in this study is unknown.
Causal Prophylaxis
In separate trials with small numbers of volunteers, atovaquone and proguanil hydrochloride were independently shown to have causal prophylactic activity directed against liver-stage parasites of P. falciparum. Six patients given a single dose of atovaquone 250 mg 24 hours prior to malaria challenge were protected from developing malaria, whereas all 4 placebo-treated patients developed malaria.
During the 4 weeks following cessation of prophylaxis in clinical trial participants who remained in malaria-endemic areas and were available for evaluation, malaria developed in 24 of 211 (11.4%) subjects who took placebo and 9 of 328 (2.7%) who took atovaquone and proguanil hydrochloride. While new infections could not be distinguished from recrudescent infections, all but 1 of the infections in patients treated with atovaquone and proguanil hydrochloride occurred more than 15 days after stopping therapy. The single case occurring on day 8 following cessation of therapy with atovaquone and proguanil hydrochloride probably represents a failure of prophylaxis with atovaquone and proguanil hydrochloride. The possibility that delayed cases of P. falciparum malaria may occur some time after stopping prophylaxis with atovaquone and proguanil hydrochloride cannot be ruled out. Hence, returning travelers developing febrile illnesses should be investigated for malaria.
14.2 Treatment of Acute, Uncomplicated P. falciparum Malaria Infections
In 3 phase 2 clinical trials, atovaquone alone, proguanil hydrochloride alone, and the combination of atovaquone and proguanil hydrochloride were evaluated for the treatment of acute, uncomplicated malaria caused by P. falciparum. Among 156 evaluable patients, the parasitological cure rate (elimination of parasitemia with no recurrent parasitemia during follow-up for 28 days) was 59/89 (66%) with atovaquone alone, 1/17 (6%) with proguanil hydrochloride alone, and 50/50 (100%) with the combination of atovaquone and proguanil hydrochloride.
Atovaquone and proguanil hydrochloride was evaluated for treatment of acute, uncomplicated malaria caused by P. falciparum in 8 phase III randomized, open-label, controlled clinical trials (N = 1,030 enrolled in both treatment goups). The mean age of subjects was 27 years and 16% were children 12 years and younger; 74% of subjects were male. Evaluable patients included those whose outcome at 28 days were known. Among 471 evaluable patients treated with the equivalent of 4 atovaquone and proguanil hydrochloride tablets once daily for 3 days, 464 had a sensitive response (elimination of parasitemia with no recurrent parasitemia during follow-up for 28 days) (Table 8). Seven patients had a response of RI resistance (elimination of parasitemia but with recurrent parasitemia between 7 and 28 days after starting treatment). In these trials, the response to treatment with atovaquone and proguanil hydrochloride was similar to treatment with the comparator drug in 4 trials.
Table 8. Parasitological Response in 8 Clinical Trials of Atovaquone and Proguanil hydrochloride for Treatment of P.falciparum Malaria
Study Site
Atovaquone and Proguanil hydrochloride a
Comparator
Evaluable Patients (n)
% Sensitive Responseb
Drug(s)
Evaluable Patients (n)
% Sensitive Responseb
Brazil
74
98.6%
Quinine and tetracycline
76
100.0%
Thailand
79
100.0%
Mefloquine
79
86.1%
Francec
21
100.0%
Halofantrine
18
100.0%
Kenyac,d
81
93.8%
Halofantrine
83
90.4%
Zambia
80
100.0%
Pyrimethamine/ sulfadoxine (P/S)
80
98.8%
Gabonc
63
98.4%
Amodiaquine
63
81.0%
Philippines
54
100.0%
Chloroquine (Cq) Cq and P/S
23
32
30.4%
87.5%
Peru
19
100.0%
Chloroquine
P/S
13
7
7.7%
100.0%
a
b
Atovaquone and proguanil hydrochloride tablets = 1,000 mg atovaquone and 400 mg proguanil hydrochloride (or equivalent based on body weight for patients weighing ≤40 kg) once daily for 3 days.
Elimination of parasitemia with no recurrent parasitemia during follow-up for 28 days.
c
Patients hospitalized only for acute care. Follow-up conducted in outpatients.
d
Study in pediatric patients 3 to 12 years of age.
When these 8 trials were pooled and 2 additional trials evaulating atovaquone and proguanil hydrochloride alone (without a comparator arm) were added to the analysis, the overall efficacy (elimination of parasitemia with no recurrent parasitemia during follow-up for 28 days) in 521 evaluable patients was 98.7%.
The efficacy of atovaquone and proguanil hydrochloride in the treatment of the erythrocytic phase of non-falciparum malaria was assessed in a small number of patients. Of the 23 patients in Thailand infected with P. vivax and treated with atovaquone/proguanil hydrochloride 1,000 mg/400 mg daily for 3 days, parasitemia cleared in 21 (91.3%) at 7 days. Parasite relapse occurred commonly when P. vivax malaria was treated with atovaquone and proguanil hydrochloride alone. Relapsing malarias including P. vivax and P. ovale require additional treatment to prevent relapse.
The efficacy of atovaquone and proguanil hydrochloride in treating acute uncomplicated P. falciparum malaria in children weighing ≥5 and <11 kg was examined in an open-label, randomized trial conducted in Gabon. Patients received either atovaquone or proguanil hydrochloride (2 or 3 atovaquone and proguanil hydrochloride Pediatric Tablets once daily depending upon body weight) for 3 days (n = 100) or amodiaquine (10 mg/kg/day) for 3 days (n = 100). In this study, the atovaquone and proguanil hydrochloride tablets were crushed and mixed with condensed milk just prior to administration. An adequate clinical response (elimination of parasitemia with no recurrent parasitemia during follow-up for 28 days) was obtained in 95% (87/92) of the evaluable pediatric patients who received atovaquone and proguanil hydrochloride and in 53% (41/78) of those evaluable who received amodiaquine. A response of RI resistance (elimination of parasitemia but with recurrent parasitemia between 7 and 28 days after starting treatment) was noted in 3% and 40% of the patients, respectively. Two cases of RIII resistance (rising parasite count despite therapy) were reported in the patients receiving atovaquone and proguanil hydrochloride. There were 4 cases of RIII in the amodiaquine arm.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Atovaquone and proguanil hydrochloride tablets, containing 250 mg atovaquone USP and 100 mg proguanil hydrochloride USP, are pinkish brown to brown colored, circular, biconvex beveled edge, film-coated tablets with ‘404’ debossed on one side and ‘G’ debossed on the other side.
Atovaquone and proguanil hydrochloride tablets 250 mg / 100 mg
NDC: 68001-245-00 bottles of 100
NDC: 68001-245-15 Carton containing two blister cards (NDC: 68001-245-14) each containing 12 tablets.
Storage Conditions
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) (see USP Controlled Room Temperature).
-
17 PATIENT COUNSELING INFORMATION
Patients should be instructed:
- to take atovaquone and proguanil hydrochloride tablets at the same time each day with food or a milky drink.
- to take a repeat dose of atovaquone and proguanil hydrochloride if vomiting occurs within 1 hour after dosing.
- to take a dose as soon as possible if a dose is missed, then return to their normal dosing schedule. However, if a dose is skipped, the patient should not double the next dose.
- that rare serious adverse events such as hepatitis, severe skin reactions, neurological, and hematological events have been reported when atovaquone and proguanil hydrochloride was used for the prophylaxis or treatment of malaria.
- to consult a healthcare professional regarding alternative forms of prophylaxis if prophylaxis with atovaquone and proguanil hydrochloride is prematurely discontinued for any reason.
- that protective clothing, insect repellents, and bednets are important components of malaria prophylaxis.
- that no chemoprophylactic regimen is 100% effective; therefore, patients should seek medical attention for any febrile illness that occurs during or after return from a malaria-endemic area and inform their healthcare professional that they may have been exposed to malaria.
- that falciparum malaria carries a higher risk of death and serious complications in pregnant women than in the general population. Pregnant women anticipating travel to malarious areas should discuss the risks and benefits of such travel with their physicians.
Manufactured By:
Glenmark Pharmaceuticals Ltd
Colvale-Bardez, Goa 403 513, India
For BluePoint Laboratories
Questions? 1 (888)721-7115
Rev 05/19
-
Package/Label Display Panel
Bottle Label
Atovaquone and Proguanil Hydrochloride Tablets 250 mg/100 mg
NDC: 68001-245-00, 100 Tablets
-
Package/Label Display Panel
Carton Label
Atovaquone and Proguanil Hydrochloride Tablets 250 mg/100 mg
NDC: 68001-245-15, 24 (2 x 12) Unit Dose Tablets
-
INGREDIENTS AND APPEARANCE
ATOVAQUONE AND PROGUANIL HYDROCHLORIDE
atovaquone and proguanil hydrochloride tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68001-245 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ATOVAQUONE (UNII: Y883P1Z2LT) (ATOVAQUONE - UNII:Y883P1Z2LT) ATOVAQUONE 250 mg PROGUANIL HYDROCHLORIDE (UNII: R71Y86M0WT) (PROGUANIL - UNII:S61K3P7B2V) PROGUANIL HYDROCHLORIDE 100 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) POLOXAMER 188 (UNII: LQA7B6G8JG) POVIDONE K30 (UNII: U725QWY32X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color PINK (pinkish brown to brown) Score no score Shape ROUND (circular, biconvex, beveled edge) Size 11mm Flavor Imprint Code 404;G Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68001-245-00 100 in 1 BOTTLE; Type 0: Not a Combination Product 03/13/2014 2 NDC: 68001-245-15 2 in 1 CARTON 03/13/2014 2 NDC: 68001-245-14 12 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA091211 03/13/2014 Labeler - BluePoint Laboratories (985523874) Establishment Name Address ID/FEI Business Operations Glenmark Generics Limited 677318665 ANALYSIS(68001-245) , MANUFACTURE(68001-245)


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.