TYENNE- tocilizumab-aazg injection, solution
TYENNE by
Drug Labeling and Warnings
TYENNE by is a Prescription medication manufactured, distributed, or labeled by Fresenius Kabi USA, LLC , Fresenius Kabi Austria GmbH, Merck Serono S.p.A . Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TYENNE safely and effectively. See full prescribing information for TYENNE.
TYENNE® (tocilizumab-aazg) injection, for intravenous or subcutaneous use
Initial U.S. Approval: 2024
TYENNE® (tocilizumab-aazg) is biosimilar* to ACTEMRA® (tocilizumab).WARNING: RISK OF SERIOUS INFECTIONS
See full prescribing information for complete boxed warning.
- Serious infections leading to hospitalization or death including tuberculosis (TB), bacterial, invasive fungal, viral, and other opportunistic infections have occurred in patients receiving tocilizumab products. (5.1)
- If a serious infection develops, interrupt TYENNE until the infection is controlled. (5.1)
- Perform test for latent TB (except patients with COVID-19); if positive, start treatment for TB prior to starting TYENNE. (5.1)
- Monitor all patients for active TB during treatment, even if initial latent TB test is negative. (5.1)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
TYENNE® (tocilizumab-aazg) is an interleukin-6 (IL-6) receptor antagonist indicated for treatment of:
Rheumatoid Arthritis (RA) (1.1)
- Adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more Disease-Modifying Anti-Rheumatic Drugs (DMARDs).
Giant Cell Arteritis (GCA) (1.2)
- Adult patients with giant cell arteritis.
Polyarticular Juvenile Idiopathic Arthritis (PJIA) (1.3)
- Patients 2 years of age and older with active polyarticular juvenile idiopathic arthritis.
Systemic Juvenile Idiopathic Arthritis (SJIA) (1.4)
- Patients 2 years of age and older with active systemic juvenile idiopathic arthritis.
Cytokine Release Syndrome (CRS) (1.5)
- Adults and pediatric patients 2 years of age and older with chimeric antigen receptor (CAR) T cell-induced severe or life-threatening cytokine release syndrome.
Coronavirus Disease 2019 (COVID-19) (1.6)
- Hospitalized adult patients with coronavirus disease 2019 (COVID-19) who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO).
DOSAGE AND ADMINISTRATION
For RA, pJIA and sJIA, TYENNE may be used alone or in combination with methotrexate; and in RA, other non-biologic DMARDs may be used. (2)
General Administration and Dosing Information (2.1)
- RA, GCA, PJIA and SJIA – It is recommended that TYENNE not be initiated in patients with an absolute neutrophil count (ANC) below 2,000 per mm3, platelet count below 100,000 per mm3, or ALT or AST above 1.5 times the upper limit of normal (ULN) (5.3, 5.4).
- COVID-19 – It is recommended that TYENNE not be initiated in patients with an absolute neutrophil count (ANC) below 1,000 per mm3, platelet count below 50,000 mm3, or ALT or AST above 10 times ULN (5.3, 5.4).
- In RA, CRS or COVID-19 patients, TYENNE doses exceeding 800 mg per infusion are not recommended. (2.2, 2.9, 12.3)
- In GCA patients, TYENNE doses exceeding 600 mg per infusion are not recommended. (2.3, 12.3)
Rheumatoid Arthritis (2.2)
Recommended Adult Intravenous Dosage:
When used in combination with non-biologic DMARDs or as monotherapy the recommended starting dose is 4 mg per kg every 4 weeks followed by an increase to 8 mg per kg every 4 weeks based on clinical response.
Recommended Adult Subcutaneous Dosage:
Patients less than 100 kg weight
162 mg administered subcutaneously every other week, followed by an increase to every week based on clinical response
Patients at or above 100 kg weight
162 mg administered subcutaneously every week
Giant Cell Arteritis (2.3)
Recommended Adult Intravenous Dosage: The recommended dose is 6 mg per kg every 4 weeks in combination with a tapering course of glucocorticoids. TYENNE can be used alone following discontinuation of glucocorticoids.
Recommended Adult Subcutaneous Dosage: The recommended dose is 162 mg given once every week as a subcutaneous injection, in combination with a tapering course of glucocorticoids.
A dose of 162 mg given once every other week as a subcutaneous injection, in combination with a tapering course of glucocorticoids, may be prescribed based on clinical considerations.
TYENNE can be used alone following discontinuation of glucocorticoids.
Polyarticular Juvenile Idiopathic Arthritis (2.4)
Recommended Intravenous PJIA Dosage Every 4 Weeks
Patients less than 30 kg weight
10 mg per kg
Patients at or above 30 kg weight
8 mg per kg
Recommended Subcutaneous PJIA Dosage
Patients less than 30 kg weight
162 mg once every three weeks
Patients at or above 30 kg weight
162 mg once every two weeks
Systemic Juvenile Idiopathic Arthritis (2.5)
Recommended Intravenous SJIA Dosage Every 2 Weeks
Patients less than 30 kg weight
12 mg per kg
Patients at or above 30 kg weight
8 mg per kg
Recommended Subcutaneous SJIA Dosage
Patients less than 30 kg weight
162 mg every two weeks
Patients at or above 30 kg weight
162 mg every week
Cytokine Release Syndrome (2.6)
Recommended Intravenous CRS Dosage
Patients less than 30 kg weight
12 mg per kg
Patients at or above 30 kg weight
8 mg per kg
Alone or in combination with corticosteroids.
Coronavirus Disease 2019 (2.7)
The recommended dosage of TYENNE for adult patients with COVID-19 is 8 mg per kg administered by a 60-minute intravenous infusion.
Administration of Intravenous formulation (2.8)
- For patients with RA, GCA, COVID-19, CRS, PJIA, SJIA patients at or above 30 kg, dilute to 100 mL in 0.9% or 0.45% Sodium Chloride Injection, USP for intravenous infusion using aseptic technique.
- For PJIA, SJIA and CRS patients less than 30 kg, dilute to 50 mL in 0.9% or 0.45% Sodium Chloride Injection, USP for intravenous infusion using aseptic technique.
- Administer as a single intravenous drip infusion over 1 hour; do not administer as bolus or push.
Administration of Subcutaneous formulation (2.9)
- Follow the Instructions for Use for prefilled syringe and prefilled autoinjector
Dose Modifications (2.10)
Recommended for management of certain dose-related laboratory changes including elevated liver enzymes, neutropenia, and thrombocytopenia.
DOSAGE FORMS AND STRENGTHS
Intravenous Infusion
Injection: 80 mg/4 mL (20 mg/mL), 200 mg/10 mL (20 mg/mL), 400 mg/20 mL (20 mg/mL) in single-dose vials for further dilution prior to intravenous infusion (3)
Subcutaneous Injection
Injection: 162 mg/0.9 mL in a single-dose prefilled syringe or single-dose prefilled autoinjector (3)
CONTRAINDICATIONS
Known hypersensitivity to tocilizumab products. (4)
WARNINGS AND PRECAUTIONS
- Serious Infections – do not administer TYENNE during an active infection, including localized infections. If a serious infection develops, interrupt TYENNE until the infection is controlled. (5.1)
- Gastrointestinal (GI) perforation-use with caution in patients who may be at increased risk. (5.2)
- Hepatotoxicity- Monitor patients for signs and symptoms of hepatic injury. Modify or discontinue TYENNE if abnormal liver tests persist or worsen or if clinical signs and symptoms of liver disease develop. (2.10, 5.3)
- Laboratory monitoring-recommended due to potential consequences of treatment-related changes in neutrophils, platelets, lipids, and liver function tests. (2.10, 5.4)
- Hypersensitivity reactions, including anaphylaxis and death and serious cutaneous reactions including Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) - discontinue TYENNE, treat promptly, and monitor until reaction resolves (5.6)
- Live vaccines-Avoid use with TYENNE (5.9, 7.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence of at least 5%): upper respiratory tract infections, nasopharyngitis, headache, hypertension, increased ALT, injection site reactions. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Fresenius Kabi USA, LLC at 1-800-551-7176 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
- * Biosimilar means that the biological product is approved based on data demonstrating that it is highly similar to an FDA-approved biological product, known as a reference product, and that there are no clinically meaningful differences between the biosimilar product and the reference product. Biosimilarity of TYENNE has been demonstrated for the condition(s) of use (e.g., indication(s), dosing regimen(s), strength(s), dosage form(s), and route(s) of administration) described in its Full Prescribing Information.
Revised: 2/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: RISK OF SERIOUS INFECTIONS
1 INDICATIONS AND USAGE
1.1 Rheumatoid Arthritis (RA)
1.2 Giant Cell Arteritis (GCA)
1.3 Polyarticular Juvenile Idiopathic Arthritis (PJIA)
1.4 Systemic Juvenile Idiopathic Arthritis (SJIA)
1.5 Cytokine Release Syndrome (CRS)
1.6 Coronavirus Disease 2019 (COVID-19)
2 DOSAGE AND ADMINISTRATION
2.1 General Considerations for Administration
2.2 Recommended Dosage for Rheumatoid Arthritis
2.3 Recommended Dosage for Giant Cell Arteritis
2.4 Recommended Dosage for Polyarticular Juvenile Idiopathic Arthritis
2.5 Recommended Dosage for Systemic Juvenile Idiopathic Arthritis
2.6 Recommended Dosage for Cytokine Release Syndrome (CRS)
2.7 Coronavirus Disease 2019 (COVID-19)
2.8 Preparation and Administration Instructions for Intravenous Infusion
2.9 Preparation and Administration Instructions for Subcutaneous Injection
2.10 Dosage Modifications due to Serious Infections or Laboratory Abnormalities
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
5.2 Gastrointestinal Perforations
5.3 Hepatotoxicity
5.4 Changes in Laboratory Parameters
5.5 Immunosuppression
5.6 Hypersensitivity Reactions, Including Anaphylaxis
5.7 Demyelinating Disorders
5.8 Active Hepatic Disease and Hepatic Impairment
5.9 Vaccinations
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience in Rheumatoid Arthritis Patients Treated with Intravenous Tocilizumab (tocilizumab-IV)
6.2 Clinical Trials Experience in Rheumatoid Arthritis Patients Treated with Subcutaneous Tocilizumab (tocilizumab-SC)
6.3 Clinical Trials Experience in Giant Cell Arteritis Patients Treated with Subcutaneous Tocilizumab (tocilizumab-SC)
6.4 Clinical Trials Experience in Giant Cell Arteritis Patients Treated With Intravenous Tocilizumab (tocilizumab-IV)
6.5 Clinical Trials Experience in Polyarticular Juvenile Idiopathic Arthritis Patients Treated With Intravenous Tocilizumab (tocilizumab-IV)
6.6 Clinical Trials Experience in Polyarticular Juvenile Idiopathic Arthritis Patients Treated With Subcutaneous Tocilizumab (tocilizumab-SC)
6.7 Clinical Trials Experience in Systemic Juvenile Idiopathic Arthritis Patients Treated with Intravenous Tocilizumab (tocilizumab-IV)
6.8 Clinical Trials Experience in Systemic Juvenile Idiopathic Arthritis Patients Treated with Subcutaneous Tocilizumab (tocilizumab-SC)
6.9 Clinical Trials Experience in Patients with Cytokine Release Syndrome Treated with Intravenous Tocilizumab (tocilizumab-IV)
6.10 Clinical Trials Experience in COVID-19 Patients Treated with Intravenous Tocilizumab (tocilizumab-IV)
6.11 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Concomitant Drugs for Treatment of Adult Indications
7.2 Interactions with CYP450 Substrates
7.3 Live Vaccines
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
9 DRUG ABUSE AND DEPENDENCE
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Rheumatoid Arthritis – Intravenous Administration
14.2 Rheumatoid Arthritis – Subcutaneous Administration
14.3 Giant Cell Arteritis – Subcutaneous Administration
14.4 Giant Cell Arteritis – Intravenous Administration
14.5 Polyarticular Juvenile Idiopathic Arthritis – Intravenous Administration
14.6 Polyarticular Juvenile Idiopathic Arthritis – Subcutaneous Administration
14.7 Systemic Juvenile Idiopathic Arthritis - Intravenous Administration
14.8 Systemic Juvenile Idiopathic Arthritis - Subcutaneous Administration
14.9 Cytokine Release Syndrome – Intravenous Administration
14.10 COVID-19 – Intravenous Administration
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: RISK OF SERIOUS INFECTIONS
Patients treated with tocilizumab products including TYENNE are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1), Adverse Reactions (6.1)]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
If a serious infection develops, interrupt TYENNE until the infection is controlled.
Reported infections include:
- Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients, except those with COVID-19, should be tested for latent tuberculosis before TYENNE use and during therapy. Treatment for latent infection should be initiated prior to TYENNE use.
- Invasive fungal infections, including candidiasis, aspergillosis, and pneumocystis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
- Bacterial, viral and other infections due to opportunistic pathogens.
The risks and benefits of treatment with TYENNE should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with TYENNE including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
1.1 Rheumatoid Arthritis (RA)
TYENNE® (tocilizumab-aazg) is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more Disease-Modifying Anti-Rheumatic Drugs (DMARDs).
1.2 Giant Cell Arteritis (GCA)
TYENNE® (tocilizumab-aazg) is indicated for the treatment of giant cell arteritis (GCA) in adult patients.
1.3 Polyarticular Juvenile Idiopathic Arthritis (PJIA)
TYENNE® (tocilizumab-aazg) is indicated for the treatment of active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older.
1.4 Systemic Juvenile Idiopathic Arthritis (SJIA)
TYENNE® (tocilizumab-aazg) is indicated for the treatment of active systemic juvenile idiopathic arthritis in patients 2 years of age and older.
1.5 Cytokine Release Syndrome (CRS)
TYENNE® (tocilizumab-aazg) is indicated for the treatment of chimeric antigen receptor (CAR) T cell-induced severe or life-threatening cytokine release syndrome in adults and pediatric patients 2 years of age and older.
1.6 Coronavirus Disease 2019 (COVID-19)
TYENNE® (tocilizumab-aazg) is indicated for the treatment of coronavirus disease 2019 (COVID-19) in hospitalized adult patients who are receiving systemic corticosteroids and require supplemental oxygen, noninvasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO).
-
2 DOSAGE AND ADMINISTRATION
2.1 General Considerations for Administration
Not Recommended for Concomitant Use with Biological DMARDs
Tocilizumab products have not been studied in combination with biological DMARDs such as TNF antagonists, IL-1R antagonists, anti-CD20 monoclonal antibodies and selective co-stimulation modulators because of the possibility of increased immunosuppression and increased risk of infection. Avoid using TYENNE with biological DMARDs.
Baseline Laboratory Evaluation Prior to Treatment
Obtain and assess baseline complete blood count (CBC) and liver function tests prior to treatment.
RA, GCA, PJIA and SJIA – It is recommended that TYENNE not be initiated in patients with an absolute neutrophil count (ANC) below 2,000 per mm3, platelet count below 100,000 per mm3, or ALT or AST above 1.5 times the upper limit of normal (ULN) [see Warnings and Precautions (5.3, 5.4)].
CRS – Patients with severe or life-threatening CRS frequently have cytopenias or elevated ALT or AST due to the lymphodepleting chemotherapy or the CRS. The decision to administer TYENNE should take into account the potential benefit of treating the CRS versus the risks of short-term treatment with TYENNE.
COVID-19 – It is recommended that TYENNE not be initiated in patients with an absolute neutrophil count (ANC) below 1,000 per mm3, platelet count below 50,000 mm3, or ALT or AST above 10 times ULN [see Warnings and Precautions (5.3, 5.4)].
2.2 Recommended Dosage for Rheumatoid Arthritis
TYENNE may be used as monotherapy or concomitantly with methotrexate or other non-biologic DMARDs as an intravenous infusion or as a subcutaneous injection.
Recommended Intravenous Dosage Regimen:
The recommended dosage of TYENNE for adult patients given as a 60-minute single intravenous drip infusion is 4 mg per kg every 4 weeks followed by an increase to 8 mg per kg every 4 weeks based on clinical response.
- Reduction of dose from 8 mg per kg to 4 mg per kg is recommended for management of certain dose-related laboratory changes including elevated liver enzymes, neutropenia, and thrombocytopenia [see Dosage and Administration (2.10), Warnings and Precautions (5.3, 5.4), and Adverse Reactions (6.1)].
- Doses exceeding 800 mg per infusion are not recommended in RA patients [see Clinical Pharmacology (12.3)].
Recommended Subcutaneous Dosage Regimen:
Patients less than 100 kg weight
162 mg administered subcutaneously every other week, followed by an increase to every week based on clinical response
Patients at or above 100 kg weight
162 mg administered subcutaneously every week
When transitioning from TYENNE intravenous therapy to subcutaneous administration administer the first subcutaneous dose instead of the next scheduled intravenous dose.
Interruption of dose or reduction in frequency of administration of subcutaneous dose from every week to every other week dosing is recommended for management of certain dose-related laboratory changes including elevated liver enzymes, neutropenia, and thrombocytopenia [see Dosage and Administration (2.10), Warnings and Precautions (5.3, 5.4), and Adverse Reactions (6.2)].
2.3 Recommended Dosage for Giant Cell Arteritis
Recommended Intravenous Dosage Regimen:
The recommended dosage of TYENNE for adult patients given as a 60-minute single intravenous drip infusion is 6 mg per kg every 4 weeks in combination with a tapering course of glucocorticoids.
TYENNE can be used alone following discontinuation of glucocorticoids.
- Interruption of dosing may be needed for management of dose-related laboratory abnormalities including elevated liver enzymes, neutropenia, and thrombocytopenia [see Dosage and Administration (2.10)].
- Doses exceeding 600 mg per infusion are not recommended in GCA patients [see Clinical Pharmacology (12.3)].
Recommended Subcutaneous Dosage Regimen:
The recommended dose of TYENNE for adult patients with GCA is 162 mg given once every week as a subcutaneous injection in combination with a tapering course of glucocorticoids.
A dose of 162 mg given once every other week as a subcutaneous injection in combination with a tapering course of glucocorticoids may be prescribed based on clinical considerations.
TYENNE can be used alone following discontinuation of glucocorticoids.
When transitioning from TYENNE intravenous therapy to subcutaneous administration, administer the first subcutaneous dose instead of the next scheduled intravenous dose.
Interruption of dose or reduction in frequency of administration of subcutaneous dose from every week to every other week dosing may be needed for management of dose-related laboratory abnormalities including elevated liver enzymes, neutropenia, and thrombocytopenia [see Dosage and Administration (2.10)].
2.4 Recommended Dosage for Polyarticular Juvenile Idiopathic Arthritis
TYENNE may be used as an intravenous infusion or as a subcutaneous injection alone or in combination with methotrexate. Do not change dose based solely on a single visit body weight measurement, as weight may fluctuate.
Recommended Intravenous Dosage Regimen:
The recommended dosage of TYENNE for PJIA patients given once every 4 weeks as a 60-minute single intravenous drip infusion is:
Recommended Intravenous PJIA Dosage Every 4 Weeks
Patients less than 30 kg weight
10 mg per kg
Patients at or above 30 kg weight
8 mg per kg
Recommended Subcutaneous Dosage Regimen:
Recommended Subcutaneous PJIA Dosage
Patients less than 30 kg weight
162 mg once every 3 weeks
Patients at or above 30 kg weight
162 mg once every 2 weeks
When transitioning from TYENNE intravenous therapy to subcutaneous administration, administer the first subcutaneous dose instead of the next scheduled intravenous dose.
Interruption of dosing may be needed for management of dose-related laboratory abnormalities including elevated liver enzymes, neutropenia, and thrombocytopenia [see Dosage and Administration (2.10)].
2.5 Recommended Dosage for Systemic Juvenile Idiopathic Arthritis
TYENNE may be used as an intravenous infusion or as a subcutaneous injection alone or in combination with methotrexate. Do not change a dose based solely on a single visit body weight measurement, as weight may fluctuate.
Recommended Intravenous Dosage Regimen:
The recommended dose of TYENNE for SJIA patients given once every 2 weeks as a 60-minute single intravenous drip infusion is:
Recommended Intravenous SJIA Dosage Every 2 Weeks
Patients less than 30 kg weight
12 mg per kg
Patients at or above 30 kg weight
8 mg per kg
Recommended Subcutaneous Dosage Regimen:
Recommended Subcutaneous SJIA Dosage
Patients less than 30 kg weight
162 mg once every two weeks
Patients at or above 30 kg weight
162 mg once every week
When transitioning from TYENNE intravenous therapy to subcutaneous administration, administer the first subcutaneous dose when the next scheduled intravenous dose is due.
Interruption of dosing may be needed for management of dose-related laboratory abnormalities including elevated liver enzymes, neutropenia, and thrombocytopenia [see Dosage and Administration (2.10)].
2.6 Recommended Dosage for Cytokine Release Syndrome (CRS)
Use only the intravenous route for treatment of CRS. The recommended dose of TYENNE for treatment of CRS given as a 60-minute intravenous infusion is:
Recommended Intravenous CRS Dosage
Patients less than 30 kg weight
12 mg per kg
Patients at or above 30 kg weight
8 mg per kg
Alone or in combination with corticosteroids
- If no clinical improvement in the signs and symptoms of CRS occurs after the first dose, up to 3 additional doses of TYENNE may be administered. The interval between consecutive doses should be at least 8 hours.
- Doses exceeding 800 mg per infusion are not recommended in CRS patients.
- Subcutaneous administration is not approved for CRS.
2.7 Coronavirus Disease 2019 (COVID-19)
Administer TYENNE by intravenous infusion only.
The recommended dosage of TYENNE for treatment of adult patients with COVID-19 is 8 mg per kg administered as a single 60-minute intravenous infusion. If clinical signs or symptoms worsen or do not improve after the first dose, one additional infusion of TYENNE may be administered at least 8 hours after the initial infusion.
- Doses exceeding 800 mg per infusion are not recommended in patients with COVID-19.
- Subcutaneous administration is not approved for COVID-19.
2.8 Preparation and Administration Instructions for Intravenous Infusion
TYENNE for intravenous infusion should be diluted by a healthcare professional using aseptic technique as follows:
- Use a sterile needle and syringe to prepare TYENNE.
- Patients less than 30 kg: use a 50 mL infusion bag or bottle of 0.9% or 0.45% Sodium Chloride Injection, USP, and then follow steps 1 and 2 below.
- Patients at or above 30 kg weight: use a 100 mL infusion bag or bottle, and then follow steps 1 and 2 below.
- - Step 1. Withdraw a volume of 0.9% or 0.45% Sodium Chloride Injection, USP, equal to the volume of the TYENNE injection required for the patient's dose from the infusion bag or bottle [see Dosage and Administration (2.2, 2.4, 2.5, 2.6)].
For Intravenous Use: Volume of TYENNE Injection per kg of Body Weight
Dosage
Indication
Volume of TYENNE injection per kg of body weight
4 mg/kg
Adult RA
0.2 mL/kg
6 mg/kg
Adult GCA
0.3 mL/kg
8 mg/kg
Adult RA
Adult COVID-19
SJIA, PJIA and CRS (greater than or equal to 30 kg of body weight)
0.4 mL/kg
10 mg/kg
PJIA (less than 30 kg of body weight)
0.5 mL/kg
12 mg/kg
SJIA and CRS (less than 30 kg of body weight)
0.6 mL/kg
- - Step 2. Withdraw the amount of TYENNE for intravenous infusion from the vial(s) and add slowly into the 0.9% or 0.45% Sodium Chloride Injection, USP infusion bag or bottle. To mix the solution, gently invert the bag to avoid foaming.
- The prepared solution for infusion should be used immediately. If not used immediately, the diluted tocilizumab solutions may be refrigerated at 36°F to 46°F (2°C to 8°C) up to 24 hours, or stored at room temperature at or below 77°F (25°C) for up to 4 hours and should be protected from light. Administration of diluted TYENNE solution must be completed within this period of time.
- TYENNE solutions do not contain preservatives; therefore, unused product remaining in the vials should not be used.
- Allow the fully diluted TYENNE solution to reach room temperature prior to infusion.
- The infusion should be administered over 60 minutes and must be administered with an infusion set. Do not administer as an intravenous push or bolus.
- TYENNE should not be infused concomitantly in the same intravenous line with other drugs. No physical or biochemical compatibility studies have been conducted to evaluate the co-administration of TYENNE with other drugs.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. If particulates and discolorations are noted, the product should not be used.
- Fully diluted TYENNE solutions are compatible with polypropylene, polyethylene and polyvinyl chloride infusion bags and bottles, and glass infusion bottles.
2.9 Preparation and Administration Instructions for Subcutaneous Injection
- TYENNE for subcutaneous injection is not intended for intravenous drip infusion.
-
Assess suitability of patient for subcutaneous home use and instruct patients to inform a healthcare professional before administering the next dose if they experience any symptoms of allergic reaction. Patients should seek immediate medical attention if they develop symptoms of serious allergic reactions. TYENNE subcutaneous injection is intended for use under the guidance of a healthcare practitioner. After proper training in subcutaneous injection technique, a patient may self-inject TYENNE or the patient's caregiver may administer TYENNE if a healthcare practitioner determines that it is appropriate. PJIA and SJIA patients may self-inject with the TYENNE prefilled syringe or autoinjector, or the patient's caregiver may administer TYENNE if both the healthcare practitioner and the parent/legal guardian determine it is appropriate [see Use in Specific Populations (8.4)].
Patients, or patient caregivers, should be instructed to follow the directions provided in the Instructions for Use (IFU) for additional details on medication administration. - Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use TYENNE prefilled syringes (PFS) or prefilled autoinjectors exhibiting particulate matter, cloudiness, or discoloration. TYENNE for subcutaneous administration should be clear and colorless to pale yellow. Do not use if any part of the PFS or autoinjector appears to be damaged.
- Patients using TYENNE for subcutaneous administration should be instructed to inject the full amount in the syringe (0.9 mL) or full amount in the autoinjector (0.9 mL), which provides 162 mg of TYENNE, according to the directions provided in the IFU.
- Injection sites should be rotated with each injection and should never be given into moles, scars, or areas where the skin is tender, bruised, red, hard, or not intact.
2.10 Dosage Modifications due to Serious Infections or Laboratory Abnormalities
Serious Infections
Hold TYENNE treatment if a patient develops a serious infection until the infection is controlled.
Laboratory Abnormalities
Rheumatoid Arthritis and Giant Cell Arteritis
Liver Enzyme Abnormalities [see Warnings and Precautions (5.3, 5.4)]
Lab Value
Recommendation for RA
Recommendation for GCA
Greater than 1 to 3x ULN
Dose modify concomitant DMARDs if appropriate
For persistent increases in this range:
- For patients receiving intravenous TYENNE, reduce dose to 4 mg per kg or hold TYENNE until ALT or AST have normalized
- For patients receiving subcutaneous TYENNE, reduce injection frequency to every other week or hold dosing until ALT or AST have normalized. Resume TYENNE at every other week and increase frequency to every week as clinically appropriate
Dose modify immunomodulatory agents if appropriate
For persistent increases in this range:
- For patients receiving Intravenous TYENNE, hold TYENNE until ALT or AST have normalized
- For patients receiving subcutaneous TYENNE, reduce injection frequency to every other week or hold dosing until ALT or AST have normalized. Resume TYENNE at every other week and increase frequency to every week as clinically appropriate
Greater than 3 to 5x ULN (confirmed by repeat testing)
Hold TYENNE dosing until less than 3x ULN and follow recommendations above for greater than 1 to 3x ULN
For persistent increases greater than 3x ULN, discontinue TYENNEHold TYENNE dosing until less than 3x ULN and follow recommendations above for greater than 1 to 3x ULN
For persistent increases greater than 3x ULN, discontinue TYENNEGreater than
5x ULNDiscontinue TYENNE
Discontinue TYENNE
Low Absolute Neutrophil Count (ANC) [see Warnings and Precautions (5.4)]
Lab Value
(cells per mm3)Recommendation for RA
Recommendation for GCA
ANC greater than 1,000
Maintain dose
Maintain dose
ANC 500 to 1,000
Hold TYENNE dosing
When ANC greater than 1,000 cells per mm3:
- For patients receiving intravenous TYENNE, resume TYENNE at 4 mg per kg and increase to 8 mg per kg as clinically appropriate
- For patients receiving subcutaneous TYENNE, resume TYENNE at every other week and increase frequency to every week as clinically appropriate
Hold TYENNE dosing
When ANC greater than 1,000 cells per mm3:
- For patients receiving intravenous TYENNE, resume TYENNE at 6 mg per kg
- For patients receiving subcutaneous TYENNE, resume TYENNE at every other week and increase frequency to every week as clinically appropriate
ANC less than 500
Discontinue TYENNE
Discontinue TYENNE
Low Platelet Count [see Warnings and Precautions (5.4)]
Lab Value
(cells per mm3)Recommendation for RA
Recommendation for GCA
50,000 to 100,000
Hold TYENNE dosing
When platelet count is greater than 100,000 cells per mm3:
- For patients receiving intravenous TYENNE, resume TYENNE at 4 mg per kg and increase to 8 mg per kg as clinically appropriate
- For patients receiving subcutaneous TYENNE, resume TYENNE at every other week and increase frequency to every week as clinically appropriate
Hold TYENNE dosing
When platelet count is greater than 100,000 cells per mm3:
- For patients receiving intravenous TYENNE, resume TYENNE at 6 mg per kg
- For patients receiving subcutaneous TYENNE, resume TYENNE at every other week and increase frequency to every week as clinically appropriate
Less than 50,000
Discontinue TYENNE
Discontinue TYENNE
Polyarticular and Systemic Juvenile Idiopathic Arthritis
Dose reduction of tocilizumab products has not been studied in the PJIA and SJIA populations. Dose interruptions of TYENNE are recommended for liver enzyme abnormalities, low neutrophil counts, and low platelet counts in patients with PJIA and SJIA at levels similar to what is outlined above for patients with RA and GCA. If appropriate, dose modify or stop concomitant methotrexate and/or other medications and hold TYENNE dosing until the clinical situation has been evaluated. In PJIA and SJIA the decision to discontinue TYENNE for a laboratory abnormality should be based upon the medical assessment of the individual patient.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
TYENNE is contraindicated in patients with known hypersensitivity to tocilizumab products [see Warnings and Precautions (5.6)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
Serious and sometimes fatal infections due to bacterial, mycobacterial, invasive fungal, viral, protozoal, or other opportunistic pathogens have been reported in patients receiving immunosuppressive agents including tocilizumab products. The most common serious infections included pneumonia, urinary tract infection, cellulitis, herpes zoster, gastroenteritis, diverticulitis, sepsis and bacterial arthritis [see Adverse Reactions (6.1)]. Among opportunistic infections, tuberculosis, cryptococcus, aspergillosis, candidiasis, and pneumocystosis were reported with tocilizumab products. Other serious infections, not reported in clinical studies, may also occur (e.g., histoplasmosis, coccidioidomycosis, listeriosis). Patients have presented with disseminated rather than localized disease, and were often taking concomitant immunosuppressants such as methotrexate or corticosteroids which in addition to rheumatoid arthritis may predispose them to infections.
Do not administer TYENNE in patients with an active infection, including localized infections. The risks and benefits of treatment should be considered prior to initiating TYENNE in patients:
- with chronic or recurrent infection;
- who have been exposed to tuberculosis;
- with a history of serious or an opportunistic infection;
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses; or
- with underlying conditions that may predispose them to infection.
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with TYENNE, as signs and symptoms of acute inflammation may be lessened due to suppression of the acute phase reactants [see Dosage and Administration (2.1), Adverse Reactions (6.1), and Patient Counseling Information (17)].
Hold TYENNE if a patient develops a serious infection, an opportunistic infection, or sepsis. A patient who develops a new infection during treatment with TYENNE should undergo a prompt and complete diagnostic workup appropriate for an immunocompromised patient, initiate appropriate antimicrobial therapy, and closely monitor the patient.
COVID-19
In patients with COVID-19, monitor for signs and symptoms of new infections during and after treatment with TYENNE. There is limited information regarding the use of tocilizumab products in patients with COVID-19 and concomitant active serious infections. The risks and benefits of treatment with TYENNE in COVID-19 patients with other concurrent infections should be considered.
Tuberculosis
Evaluate patients for tuberculosis risk factors and test for latent infection prior to initiating TYENNE. In patients with COVID-19, testing for latent infection is not necessary prior to initiating treatment with TYENNE.
Consider anti-tuberculosis therapy prior to initiation of TYENNE in patients with a past history of latent or active tuberculosis in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent tuberculosis but having risk factors for tuberculosis infection. Consultation with a physician with expertise in the treatment of tuberculosis is recommended to aid in the decision whether initiating anti-tuberculosis therapy is appropriate for an individual patient.
Closely monitor patients for the development of signs and symptoms of tuberculosis including patients who tested negative for latent tuberculosis infection prior to initiating therapy.
The incidence of tuberculosis in worldwide clinical development programs is 0.1%. Patients with latent tuberculosis should be treated with standard antimycobacterial therapy before initiating TYENNE.
Viral Reactivation
Viral reactivation has been reported with immunosuppressive biologic therapies and cases of herpes zoster exacerbation were observed in clinical studies with tocilizumab. No cases of Hepatitis B reactivation were observed in the trials; however patients who screened positive for hepatitis were excluded.
5.2 Gastrointestinal Perforations
Events of gastrointestinal perforation have been reported in clinical trials, primarily as complications of diverticulitis in patients treated with tocilizumab. Use TYENNE with caution in patients who may be at increased risk for gastrointestinal perforation. Promptly evaluate patients presenting with fever, new onset abdominal symptoms, and a change in bowel habits for early identification of gastrointestinal perforation [see Adverse Reactions (6.1)].
5.3 Hepatotoxicity
Serious cases of hepatic injury have been observed in patients taking intravenous or subcutaneous tocilizumab products. Some of these cases have resulted in liver transplant or death. Time to onset for cases ranged from months to years after treatment initiation with tocilizumab products. While most cases presented with marked elevations of transaminases (> 5 times ULN), some cases presented with signs or symptoms of liver dysfunction and only mildly elevated transaminases.
During randomized controlled studies, treatment with tocilizumab was associated with a higher incidence of transaminase elevations [see Adverse Reactions (6.1, 6.2, 6.5, 6.7)]. Increased frequency and magnitude of these elevations was observed when potentially hepatotoxic drugs (e.g., MTX) were used in combination with tocilizumab.
For RA and GCA patients, obtain a liver test panel (serum alanine aminotransferase [ALT], aspartate aminotransferase [AST], alkaline phosphatase, and total bilirubin) before initiating TYENNE, every 4 to 8 weeks after start of therapy for the first 6 months of treatment and every 3 months thereafter. It is not recommended to initiate TYENNE treatment in RA or GCA patients with elevated transaminases ALT or AST greater than 1.5x ULN. In patients who develop elevated ALT or AST greater than 5x ULN, discontinue TYENNE. For recommended modifications based upon increase in transaminases [see Dosage and Administration (2.10)].
Patients hospitalized with COVID-19 may have elevated ALT or AST levels. Multi-organ failure with involvement of the liver is recognized as a complication of severe COVID-19. The decision to administer TYENNE should balance the potential benefit of treating COVID-19 against the potential risks of acute treatment with TYENNE. It is not recommended to initiate TYENNE treatment in COVID-19 patients with elevated ALT or AST above 10 x ULN. Monitor ALT and AST during treatment.
Measure liver tests promptly in patients who report symptoms that may indicate liver injury, such as fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice. In this clinical context, if the patient is found to have abnormal liver tests (e.g., ALT greater than three times the upper limit of the reference range, serum total bilirubin greater than two times the upper limit of the reference range), TYENNE treatment should be interrupted and investigation done to establish the probable cause. TYENNE should only be restarted in patients with another explanation for the liver test abnormalities after normalization of the liver tests.
A similar pattern of liver enzyme elevation is noted with tocilizumab products treatment in the PJIA and SJIA populations. Monitor liver test panel at the time of the second administration and thereafter every 4 to 8 weeks for PJIA and every 2 to 4 weeks for SJIA.
5.4 Changes in Laboratory Parameters
Patients with Rheumatoid Arthritis, Giant Cell Arteritis and Coronavirus Disease 2019
Neutropenia
Treatment with tocilizumab products was associated with a higher incidence of neutropenia. Infections have been uncommonly reported in association with treatment-related neutropenia in long-term extension studies and postmarketing clinical experience.
- - It is not recommended to initiate TYENNE treatment in RA and GCA patients with a low neutrophil count, i.e., absolute neutrophil count (ANC) less than 2000 per mm3. In patients who develop an absolute neutrophil count less than 500 per mm3 treatment is not recommended.
- - Monitor neutrophils 4 to 8 weeks after start of therapy and every 3 months thereafter [see Clinical Pharmacology (12.2)]. For recommended modifications based on ANC results [ see Dosage and Administration (2.10)].
- - It is not recommended to initiate TYENNE treatment in COVID-19 patients with an ANC less than 1000 per mm3. Neutrophils should be monitored.
Thrombocytopenia
Treatment with tocilizumab products was associated with a reduction in platelet counts. Treatment-related reduction in platelets was not associated with serious bleeding events in clinical trials [see Adverse Reactions (6.1, 6.2)].
- - It is not recommended to initiate TYENNE treatment in RA and GCA patients with a platelet count below 100,000 per mm3. In patients who develop a platelet count less than 50,000 per mm3 treatment is not recommended.
- - Monitor platelets 4 to 8 weeks after start of therapy and every 3 months thereafter. For recommended modifications based on platelet counts [see Dosage and Administration (2.10)].
- - In COVID-19 patients with a platelet count less than 50,000 per mm3, treatment is not recommended. Platelets should be monitored.
Elevated Liver Enzymes
Refer to 5.3 Hepatotoxicity. For recommended modifications [see Dosage and Administration (2.10)].
Lipid Abnormalities
Treatment with tocilizumab products was associated with increases in lipid parameters such as total cholesterol, triglycerides, LDL cholesterol, and/or HDL cholesterol [see Adverse Reactions (6.1, 6.2)].
- - Assess lipid parameters approximately 4 to 8 weeks following initiation of TYENNE therapy.
- - Subsequently, manage patients according to clinical guidelines [e.g., National Cholesterol Educational Program (NCEP)] for the management of hyperlipidemia.
Patients with Polyarticular and Systemic Juvenile Idiopathic Arthritis
A similar pattern of liver enzyme elevation, low neutrophil count, low platelet count and lipid elevations is noted with tocilizumab products treatment in the PJIA and SJIA populations. Monitor neutrophils, platelets, ALT and AST at the time of the second administration and thereafter every 4 to 8 weeks for PJIA and every 2 to 4 weeks for SJIA. Monitor lipids as above for approved adult indications [see Dosage and Administration (2.10)].
5.5 Immunosuppression
The impact of treatment with tocilizumab products on the development of malignancies is not known but malignancies were observed in clinical studies [see Adverse Reactions (6.1)]. TYENNE is an immunosuppressant, and treatment with immunosuppressants may result in an increased risk of malignancies.
5.6 Hypersensitivity Reactions, Including Anaphylaxis
Hypersensitivity reactions, including anaphylaxis, have been reported in association with tocilizumab products and anaphylactic events with a fatal outcome have been reported with intravenous infusion of tocilizumab products. Anaphylaxis and other hypersensitivity reactions that required treatment discontinuation were reported in 0.1% (3 out of 2644) of patients in the 6-month controlled trials of intravenous tocilizumab, 0.2% (8 out of 4009) of patients in the intravenous all-exposure RA population, 0.7% (8 out of 1068) in the subcutaneous 6-month controlled RA trials, and in 0.7% (10 out of 1465) of patients in the subcutaneous all-exposure population. In the SJIA controlled trial with intravenous tocilizumab, 1 out of 112 patients (0.9%) experienced hypersensitivity reactions that required treatment discontinuation. In the PJIA controlled trial with intravenous tocilizumab, 0 out of 188 patients (0%) in the tocilizumab all-exposure population experienced hypersensitivity reactions that required treatment discontinuation. Reactions that required treatment discontinuation included generalized erythema, rash, and urticaria. Injection site reactions were categorized separately [see Adverse Reactions (6)].
In the postmarketing setting, events of hypersensitivity reactions, including anaphylaxis and death have occurred in patients treated with a range of doses of intravenous tocilizumab products, with or without concomitant therapies. Events have occurred in patients who received premedication. Hypersensitivity, including anaphylaxis events, have occurred both with and without previous hypersensitivity reactions and as early as the first infusion of tocilizumab products [see Adverse Reactions (6.11)]. In addition, serious cutaneous reactions, including Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), have been reported in patients with autoinflammatory conditions treated with tocilizumab products.
TYENNE for intravenous use should only be infused by a healthcare professional with appropriate medical support to manage anaphylaxis. For TYENNE subcutaneous injection, advise patients to seek immediate medical attention if they experience any symptoms of a hypersensitivity reaction. If a hypersensitivity reaction occurs, immediately discontinue TYENNE, treat promptly and monitor until signs and symptoms resolve.
5.7 Demyelinating Disorders
The impact of treatment with tocilizumab products on demyelinating disorders is not known, but multiple sclerosis and chronic inflammatory demyelinating polyneuropathy were reported rarely in RA clinical studies. Monitor patients for signs and symptoms potentially indicative of demyelinating disorders. Prescribers should exercise caution in considering the use of TYENNE in patients with preexisting or recent onset demyelinating disorders.
5.8 Active Hepatic Disease and Hepatic Impairment
Treatment with TYENNE is not recommended in patients with active hepatic disease or hepatic impairment [see Adverse Reactions (6.1), Use in Specific Populations (8.6)].
5.9 Vaccinations
Avoid use of live vaccines concurrently with TYENNE as clinical safety has not been established. No data are available on the secondary transmission of infection from persons receiving live vaccines to patients receiving tocilizumab products.
No data are available on the effectiveness of vaccination in patients receiving tocilizumab products. Because IL-6 inhibition may interfere with the normal immune response to new antigens, it is recommended that all patients, particularly pediatric or elderly patients, if possible, be brought up to date with all immunizations in agreement with current immunization guidelines prior to initiating TYENNE therapy. The interval between live vaccinations and initiation of TYENNE therapy should be in accordance with current vaccination guidelines regarding immunosuppressive agents.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in labeling:
- Serious Infections [see Warnings and Precautions (5.1)]
- Gastrointestinal Perforations [see Warnings and Precautions (5.2)]
- Laboratory Parameters [see Warnings and Precautions (5.4)]
- Immunosuppression [see Warnings and Precautions (5.5)]
- Hypersensitivity Reactions, Including Anaphylaxis [see Warnings and Precautions (5.6)]
- Demyelinating Disorders [see Warnings and Precautions (5.7)]
- Active Hepatic Disease and Hepatic Impairment [see Warnings and Precautions (5.8)]
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not predict the rates observed in a broader patient population in clinical practice.
6.1 Clinical Trials Experience in Rheumatoid Arthritis Patients Treated with Intravenous Tocilizumab (tocilizumab-IV)
The tocilizumab-IV data in rheumatoid arthritis (RA) includes 5 double-blind, controlled, multicenter studies. In these studies, patients received doses of tocilizumab-IV 8 mg per kg monotherapy (288 patients), tocilizumab-IV 8 mg per kg in combination with DMARDs (including methotrexate) (1582 patients), or tocilizumab-IV 4 mg per kg in combination with methotrexate (774 patients).
The all exposure population includes all patients in registration studies who received at least one dose of tocilizumab-IV. Of the 4009 patients in this population, 3577 received treatment for at least 6 months, 3309 for at least one year; 2954 received treatment for at least 2 years and 2189 for 3 years.
All patients in these studies had moderately to severely active rheumatoid arthritis. The study population had a mean age of 52 years, 82% were female and 74% were Caucasian.
The most common serious adverse reactions were serious infections [see Warnings and Precautions (5.1)]. The most commonly reported adverse reactions in controlled studies up to 24 weeks (occurring in at least 5% of patients treated with tocilizumab-IV monotherapy or in combination with DMARDs) were upper respiratory tract infections, nasopharyngitis, headache, hypertension and increased ALT.
The proportion of patients who discontinued treatment due to any adverse reactions during the double-blind, placebo-controlled studies was 5% for patients taking tocilizumab-IV and 3% for placebo-treated patients. The most common adverse reactions that required discontinuation of tocilizumab-IV were increased hepatic transaminase values (per protocol requirement) and serious infections.
Overall Infections
In the 24 week, controlled clinical studies, the rate of infections in the tocilizumab-IV monotherapy group was 119 events per 100 patient-years and was similar in the methotrexate monotherapy group. The rate of infections in the 4 mg per kg and 8 mg per kg tocilizumab-IV plus DMARD group was 133 and 127 events per 100 patient-years, respectively, compared to 112 events per 100 patient-years in the placebo plus DMARD group. The most commonly reported infections (5% to 8% of patients) were upper respiratory tract infections and nasopharyngitis.
The overall rate of infections with tocilizumab-IV in the all exposure population remained consistent with rates in the controlled periods of the studies.
Serious Infections
In the 24 week, controlled clinical studies, the rate of serious infections in the tocilizumab-IV monotherapy group was 3.6 per 100 patient-years compared to 1.5 per 100 patient-years in the methotrexate group. The rate of serious infections in the 4 mg per kg and 8 mg per kg tocilizumab-IV plus DMARD group was 4.4 and 5.3 events per 100 patient-years, respectively, compared to 3.9 events per 100 patient-years in the placebo plus DMARD group.
In the all-exposure population, the overall rate of serious infections remained consistent with rates in the controlled periods of the studies. The most common serious infections included pneumonia, urinary tract infection, cellulitis, herpes zoster, gastroenteritis, diverticulitis, sepsis and bacterial arthritis. Cases of opportunistic infections have been reported [see Warnings and Precautions (5.1)].
In the cardiovascular outcomes Study WA25204, the rate of serious infections in the tocilizumab 8 mg/kg IV every 4 weeks group, with or without DMARD, was 4.5 per 100 patient-years, and the rate in the etanercept 50 mg weekly SC group, with or without DMARD, was 3.2 per 100 patient-years [see Clinical Studies (14.1)].
Gastrointestinal Perforations
During the 24 week, controlled clinical trials, the overall rate of gastrointestinal perforation was 0.26 events per 100 patient-years with tocilizumab-IV therapy.
In the all-exposure population, the overall rate of gastrointestinal perforation remained consistent with rates in the controlled periods of the studies. Reports of gastrointestinal perforation were primarily reported as complications of diverticulitis including generalized purulent peritonitis, lower GI perforation, fistula and abscess. Most patients who developed gastrointestinal perforations were taking concomitant nonsteroidal anti-inflammatory medications (NSAIDs), corticosteroids, or methotrexate [see Warnings and Precautions (5.2)]. The relative contribution of these concomitant medications versus tocilizumab-IV to the development of GI perforations is not known.
Infusion Reactions
In the 24 week, controlled clinical studies, adverse events associated with the infusion (occurring during or within 24 hours of the start of infusion) were reported in 8% and 7% of patients in the 4 mg per kg and 8 mg per kg tocilizumab-IV plus DMARD group, respectively, compared to 5% of patients in the placebo plus DMARD group. The most frequently reported event on the 4 mg per kg and 8 mg per kg dose during the infusion was hypertension (1% for both doses), while the most frequently reported event occurring within 24 hours of finishing an infusion were headache (1% for both doses) and skin reactions (1% for both doses), including rash, pruritus and urticaria. These events were not treatment limiting.
Anaphylaxis
Hypersensitivity reactions requiring treatment discontinuation, including anaphylaxis, associated with tocilizumab-IV were reported in 0.1% (3 out of 2644) in the 24 week, controlled trials and in 0.2% (8 out of 4009) in the all-exposure population. These reactions were generally observed during the second to fourth infusion of tocilizumab-IV. Appropriate medical treatment should be available for immediate use in the event of a serious hypersensitivity reaction [see Warnings and Precautions 5.6)].
Laboratory Abnormalities
Neutropenia
In the 24 week, controlled clinical studies, decreases in neutrophil counts below 1000 per mm3 occurred in 1.8% and 3.4% of patients in the 4 mg per kg and 8 mg per kg tocilizumab-IV plus DMARD group, respectively, compared to 0.1% of patients in the placebo plus DMARD group.
Approximately half of the instances of ANC below 1000 per mm3 occurred within 8 weeks of starting therapy. Decreases in neutrophil counts below 500 per mm3 occurred in 0.4% and 0.3% of patients in the 4 mg per kg and 8 mg per kg tocilizumab-IV plus DMARD, respectively, compared to 0.1% of patients in the placebo plus DMARD group. There was no clear relationship between decreases in neutrophils below 1000 per mm3 and the occurrence of serious infections.
In the all-exposure population, the pattern and incidence of decreases in neutrophil counts remained consistent with what was seen in the 24 week controlled clinical studies [see Warnings and Precautions (5.4)].
Thrombocytopenia
In the 24 week, controlled clinical studies, decreases in platelet counts below 100,000 per mm3 occurred in 1.3% and 1.7% of patients on 4 mg per kg and 8 mg per kg tocilizumab-IV plus DMARD, respectively, compared to 0.5% of patients on placebo plus DMARD, without associated bleeding events.
In the all-exposure population, the pattern and incidence of decreases in platelet counts remained consistent with what was seen in the 24 week controlled clinical studies [see Warnings and Precautions 5.4)].
Elevated Liver Enzymes
Liver enzyme abnormalities are summarized in Table 1. In patients experiencing liver enzyme elevation, modification of treatment regimen, such as reduction in the dose of concomitant DMARD, interruption of tocilizumab-IV, or reduction in tocilizumab-IV dose, resulted in decrease or normalization of liver enzymes [see Dosage and Administration (2.1)]. These elevations were not associated with clinically relevant increases in direct bilirubin, nor were they associated with clinical evidence of hepatitis or hepatic insufficiency [see Warnings and Precautions (5.3, 5.4)].
Table 1 Incidence of Liver Enzyme Abnormalities in the 24 Week Controlled Period of Studies I to V* ULN = Upper Limit of Normal
*For a description of these studies, see Section 14, Clinical Studies.Tocilizumab
8 mg per kg MONOTHERAPYMethotrexate
Tocilizumab 4 mg per kg + DMARDs
Tocilizumab 8 mg per kg + DMARDs
Placebo + DMARDs
N = 288
N = 284
N = 774
N = 1582
N = 1170
(%)
(%)
(%)
(%)
(%)
AST (U/L)
> ULN to 3x ULN
22
26
34
41
17
> 3x ULN to 5x ULN
0.3
2
1
2
0.3
> 5x ULN
0.7
0.4
0.1
0.2
< 0.1
ALT (U/L)
> ULN to 3x ULN
36
33
45
48
23
> 3x ULN to 5x ULN
1
4
5
5
1
> 5x ULN
0.7
1
1.3
1.5
0.3
In the all-exposure population, the elevations in ALT and AST remained consistent with what was seen in the 24 week, controlled clinical trials.
In Study WA25204, of the 1538 patients with moderate to severe RA [see Clinical Studies (14.1)] and treated with tocilizumab, elevations in ALT or AST >3 x ULN occurred in 5.3% and 2.2% patients, respectively. One serious event of drug induced hepatitis with hyperbilirubinemia was reported in association with tocilizumab.
Lipids
Elevations in lipid parameters (total cholesterol, LDL, HDL, triglycerides) were first assessed at 6 weeks following initiation of tocilizumab-IV in the controlled 24 week clinical trials.
Increases were observed at this time point and remained stable thereafter. Increases in triglycerides to levels above 500 mg per dL were rarely observed. Changes in other lipid parameters from baseline to week 24 were evaluated and are summarized below:
- - Mean LDL increased by 13 mg per dL in the tocilizumab 4 mg per kg+DMARD arm, 20 mg per dL in the tocilizumab 8 mg per kg+DMARD, and 25 mg per dL in tocilizumab 8 mg per kg monotherapy.
- - Mean HDL increased by 3 mg per dL in the tocilizumab 4 mg per kg+DMARD arm, 5 mg per dL in the tocilizumab 8 mg per kg+DMARD, and 4 mg per dL in tocilizumab 8 mg per kg monotherapy.
- - Mean LDL/HDL ratio increased by an average of 0.14 in the tocilizumab 4 mg per kg+DMARD arm, 0.15 in the tocilizumab 8 mg per kg+DMARD, and 0.26 in tocilizumab 8 mg per kg monotherapy.
- - ApoB/ApoA1 ratios were essentially unchanged in tocilizumab-treated patients.
- - Elevated lipids responded to lipid lowering agents.
In the all-exposure population, the elevations in lipid parameters remained consistent with what was seen in the 24 week, controlled clinical trials.
Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of tocilizumab or of other tocilizumab products.
In the 24 week, controlled clinical studies, a total of 2876 patients have been tested for anti-tocilizumab antibodies. Forty-six patients (2%) developed positive anti-tocilizumab antibodies, of whom 5 had an associated, medically significant, hypersensitivity reaction leading to withdrawal. Thirty patients (1%) developed neutralizing antibodies.
Malignancies
During the 24 week, controlled period of the studies, 15 malignancies were diagnosed in patients receiving tocilizumab-IV, compared to 8 malignancies in patients in the control groups. Exposure-adjusted incidence was similar in the tocilizumab-IV groups (1.32 events per 100 patient-years) and in the placebo plus DMARD group (1.37 events per 100 patient-years).
In the all-exposure population, the rate of malignancies remained consistent with the rate observed in the 24 week, controlled period [see Warnings and Precautions (5.5)].
Other Adverse Reactions
Adverse reactions occurring in 2% or more of patients on 4 or 8 mg per kg tocilizumab-IV plus DMARD and at least 1% greater than that observed in patients on placebo plus DMARD are summarized inTable 2.
Table 2 Adverse Reactions Occurring in at Least 2% or More of Patients on 4 or 8 mg per kg Tocilizumab plus DMARD and at Least 1% Greater Than That Observed in Patients on Placebo plus DMARD 24 Week Phase 3 Controlled Study Population Tocilizumab
8 mg per kg MONOTHERAPYMethotrexate Tocilizumab 4 mg per kg + DMARDs Tocilizumab 8 mg per kg + DMARDs Placebo + DMARDs Preferred Term N = 288 N = 284 N = 774 N = 1582 N = 1170 (%) (%) (%) (%) (%) Upper Respiratory Tract Infection
7
5
6
8
6
Nasopharyngitis
7
6
4
6
4
Headache
7
2
6
5
3
Hypertension
6
2
4
4
3
ALT increased
6
4
3
3
1
Dizziness
3
1
2
3
2
Bronchitis
3
2
4
3
3
Rash
2
1
4
3
1
Mouth Ulceration
2
2
1
2
1
Abdominal Pain Upper
2
2
3
3
2
Gastritis
1
2
1
2
1
Transaminase increased
1
5
2
2
1
Other infrequent and medically relevant adverse reactions occurring at an incidence less than 2% in rheumatoid arthritis patients treated with tocilizumab-IV in controlled trials were:
Infections and Infestations: oral herpes simplex
Gastrointestinal disorders: stomatitis, gastric ulcer
Investigations: weight increased, total bilirubin increased
Blood and lymphatic system disorders: leukopenia
General disorders and administration site conditions: edema peripheral
Respiratory, thoracic, and mediastinal disorders: dyspnea, cough
Eye disorders: conjunctivitis
Renal disorders: nephrolithiasis
Endocrine disorders: hypothyroidism
6.2 Clinical Trials Experience in Rheumatoid Arthritis Patients Treated with Subcutaneous Tocilizumab (tocilizumab-SC)
The tocilizumab-SC data in rheumatoid arthritis (RA) includes 2 double-blind, controlled, multicenter studies. Study SC-I was a non-inferiority study that compared the efficacy and safety of tocilizumab 162 mg administered every week subcutaneously and 8 mg/kg intravenously every four weeks in 1262 adult subjects with rheumatoid arthritis. Study SC-II was a placebo-controlled superiority study that evaluated the safety and efficacy of tocilizumab 162 mg administered every other week subcutaneously or placebo in 656 patients. All patients in both studies received background non-biologic DMARDs.
The safety observed for tocilizumab-SC administered subcutaneously was consistent with the known safety profile of intravenous tocilizumab, with the exception of injection site reactions (ISRs), which were more common with tocilizumab-SC compared with placebo SC injections (IV arm).
Injection Site Reactions
In the 6-month control period, in SC-I, the frequency of ISRs was 10.1% (64/631) and 2.4% (15/631) for the weekly tocilizumab-SC and placebo SC (IV-arm) groups, respectively. In SC-II, the frequency of ISRs was 7.1% (31/437) and 4.1% (9/218) for the every other week tocilizumab-SC and placebo groups, respectively. These ISRs (including erythema, pruritus, pain and hematoma) were mild to moderate in severity. The majority resolved without any treatment and none necessitated drug discontinuation.
Immunogenicity
In the 6-month control period in SC-I, 0.8% (5/625) in the tocilizumab-SC arm and 0.8% (5/627) in the IV arm developed anti-tocilizumab antibodies; of these, all developed neutralizing antibodies. In SC-II, 1.6% (7/434) in the tocilizumab-SC arm compared with 1.4 % (3/217) in the placebo arm developed anti-tocilizumab antibodies; of these, 1.4% (6/434) in the tocilizumab-SC arm and 0.5% (1/217) in the placebo arm also developed neutralizing antibodies.
A total of 1454 (>99%) patients who received tocilizumab-SC in the all exposure group have been tested for anti- tocilizumab antibodies. Thirteen patients (0.9%) developed anti-tocilizumab antibodies, and, of these, 12 patients (0.8%) developed neutralizing antibodies.
The rate is consistent with previous intravenous experience. No correlation of antibody development to adverse events or loss of clinical response was observed.
Laboratory Abnormalities
Neutropenia
During routine laboratory monitoring in the 6-month controlled clinical trials, a decrease in neutrophil count below 1 × 109/L occurred in 2.9% and 3.7% of patients receiving tocilizumab-SC weekly and every other week, respectively.
There was no clear relationship between decreases in neutrophils below 1 x 109/L and the occurrence of serious infections.
Thrombocytopenia
During routine laboratory monitoring in the tocilizumab-SC 6-month controlled clinical trials, none of the patients had a decrease in platelet count to ≤ 50,000/mm3.
Elevated Liver Enzymes
During routine laboratory monitoring in the 6-month controlled clinical trials, elevation in ALT or AST ≥3 x ULN occurred in 6.5% and 1.4% of patients, respectively, receiving tocilizumab-SC weekly and 3.4% and 0.7% receiving tocilizumab-SC every other week.
Lipid Parameters Elevations
During routine laboratory monitoring in the tocilizumab-SC 6-month clinical trials, 19% of patients dosed weekly and 19.6% of patients dosed every other week and 10.2% of patients on placebo experienced sustained elevations in total cholesterol > 6.2 mmol/l (240 mg/dL), with 9%, 10.4% and 5.1% experiencing a sustained increase in LDL to 4.1 mmol/l (160 mg/dL) receiving tocilizumab-SC weekly, every other week and placebo, respectively.
6.3 Clinical Trials Experience in Giant Cell Arteritis Patients Treated with Subcutaneous Tocilizumab (tocilizumab-SC)
The safety of subcutaneous tocilizumab has been studied in one Phase III study (WA28119) with 251 GCA patients. The total patient years duration in the tocilizumab-SC GCA all exposure population was 138.5 patient years during the 12-month double blind, placebo-controlled phase of the study. The overall safety profile observed in the tocilizumab-SC treatment groups was generally consistent with the known safety profile of tocilizumab. There was an overall higher incidence of infections in GCA patients relative to RA patients.
The rate of infection/serious infection events was 200.2/9.7 events per 100 patient years in the tocilizumab-SC weekly group and 160.2/4.4 events per 100 patient years in the tocilizumab-SC every other week group as compared to 156.0/4.2 events per 100 patient years in the placebo + 26 week prednisone taper and 210.2/12.5 events per 100 patient years in the placebo + 52 week taper groups.
6.4 Clinical Trials Experience in Giant Cell Arteritis Patients Treated With Intravenous Tocilizumab (tocilizumab-IV)
The safety of tocilizumab-IV was studied in an open label PK-PD and safety study in 24 patients with GCA who were in remission on tocilizumab-IV at time of enrollment. Patients received tocilizumab 7 mg/kg every 4 weeks for 20 weeks, followed by 6 mg/kg every 4 weeks for 20 weeks. The total patient years exposure to treatment was 17.5 years. The overall safety profile observed for tocilizumab administered intravenously in GCA patients was consistent with the known safety profile of tocilizumab.
6.5 Clinical Trials Experience in Polyarticular Juvenile Idiopathic Arthritis Patients Treated With Intravenous Tocilizumab (tocilizumab-IV)
The safety of tocilizumab-IV was studied in 188 pediatric patients 2 to 17 years of age with PJIA who had an inadequate clinical response or were intolerant to methotrexate. The total patient exposure in the tocilizumab-IV all exposure population (defined as patients who received at least one dose of tocilizumab-IV) was 184.4 patient years. At baseline, approximately half of the patients were taking oral corticosteroids and almost 80% were taking methotrexate. In general, the types of adverse drug reactions in patients with PJIA were consistent with those seen in RA and SJIA patients [see Adverse Reactions (6.1 and 6.7)].
Infections
The rate of infections in the tocilizumab-IV all exposure population was 163.7 per 100 patient years. The most common events observed were nasopharyngitis and upper respiratory tract infections. The rate of serious infections was numerically higher in patients weighing less than 30 kg treated with 10 mg/kg tocilizumab (12.2 per 100 patient years) compared to patients weighing at or above 30 kg, treated with 8 mg/kg tocilizumab (4.0 per 100 patient years).
The incidence of infections leading to dose interruptions was also numerically higher in patients weighing less than 30 kg treated with 10 mg/kg tocilizumab (21%) compared to patients weighing at or above 30 kg, treated with 8 mg/kg tocilizumab (8%).
Infusion Reactions
In PJIA patients, infusion-related reactions are defined as all events occurring during or within 24 hours of an infusion. In the tocilizumab-IV all exposure population, 11 patients (6%) experienced an event during the infusion, and 38 patients (20.2%) experienced an event within 24 hours of an infusion. The most common events occurring during infusion were headache, nausea and hypotension, and occurring within 24 hours of infusion were dizziness and hypotension. In general, the adverse drug reactions observed during or within 24 hours of an infusion were similar in nature to those seen in RA and SJIA patients [see Adverse Reactions (6.1 and 6.7)].
No clinically significant hypersensitivity reactions associated with tocilizumab and requiring treatment discontinuation were reported.
Immunogenicity
One patient, in the 10 mg/kg less than 30 kg group, developed positive anti-tocilizumab antibodies without developing a hypersensitivity reaction and subsequently withdrew from the study.
Neutropenia
During routine laboratory monitoring in the tocilizumab-IV all exposure population, a decrease in neutrophil counts below 1 × 109 per L occurred in 3.7% of patients.
There was no clear relationship between decreases in neutrophils below 1 x 109 per L and the occurrence of serious infections.
Thrombocytopenia
During routine laboratory monitoring in the tocilizumab-IV all exposure population, 1% of patients had a decrease in platelet count at or less than 50,000 per mm3 without associated bleeding events.
Elevated Liver Enzymes
During routine laboratory monitoring in the tocilizumab-IV all exposure population, elevation in ALT or AST at or greater than 3 x ULN occurred in 4% and less than 1% of patients, respectively.
Lipids
During routine laboratory monitoring in the tocilizumab all exposure population, elevation in total cholesterol greater than 1.5-2 x ULN occurred in one patient (0.5%) and elevation in LDL greater than 1.5-2 x ULN occurred in one patient (0.5%).
6.6 Clinical Trials Experience in Polyarticular Juvenile Idiopathic Arthritis Patients Treated With Subcutaneous Tocilizumab (tocilizumab-SC)
The safety of tocilizumab-SC was studied in 52 pediatric patients 1 to 17 years of age with PJIA who had an inadequate clinical response or were intolerant to methotrexate. The total patient exposure in the PJIA tocilizumab-SC population (defined as patients who received at least one dose of tocilizumab-SC and accounting for treatment discontinuation) was 49.5 patient years.
In general, the safety observed for tocilizumab administered subcutaneously was consistent with the known safety profile of intravenous tocilizumab, with the exception of injection site reactions (ISRs), and neutropenia.
Injection Site Reactions
During the 1-year study, a frequency of 28.8% (15/52) ISRs was observed in tocilizumab-SC treated PJIA patients. These ISRs occurred in a greater proportion of patients at or above 30 kg (44.0%) compared with patients below 30 kg (14.8%). All ISRs were mild in severity and none of the ISRs required patient withdrawal from treatment or dose interruption. A higher frequency of ISRs was observed in tocilizumab-SC treated PJIA patients compared to what was seen in adult RA or GCA patients [see Adverse Reactions (6.2 and 6.3)].
Immunogenicity
Three patients, 1 patient below 30 kg and 2 patients at or above 30 kg, developed positive anti-tocilizumab antibodies with neutralizing potential without developing a serious or clinically significant hypersensitivity reaction. One patient subsequently withdrew from the study.
Neutropenia
During routine laboratory monitoring in the tocilizumab-SC all exposure population, a decrease in neutrophil counts below 1 × 109 per L occurred in 15.4% of patients, and was more frequently observed in the patients less than 30 kg (25.9%) compared to patients at or above 30 kg (4.0%). There was no clear relationship between decreases in neutrophils below 1 x 109 per L and the occurrence of serious infections.
6.7 Clinical Trials Experience in Systemic Juvenile Idiopathic Arthritis Patients Treated with Intravenous Tocilizumab (tocilizumab-IV)
The data described below reflect exposure to tocilizumab-IV in one randomized, double-blind, placebo-controlled trial of 112 pediatric patients with SJIA 2 to 17 years of age who had an inadequate clinical response to nonsteroidal anti-inflammatory drugs (NSAIDs) or corticosteroids due to toxicity or lack of efficacy. At baseline, approximately half of the patients were taking 0.3 mg/kg/day corticosteroids or more, and almost 70% were taking methotrexate. The trial included a 12 week controlled phase followed by an open-label extension. In the 12 week double-blind, controlled portion of the clinical study 75 patients received treatment with tocilizumab-IV (8 or 12 mg per kg based upon body weight). After 12 weeks or at the time of escape, due to disease worsening, patients were treated with tocilizumab-IV in the open-label extension phase.
The most common adverse events (at least 5%) seen in tocilizumab-IV treated patients in the 12 week controlled portion of the study were: upper respiratory tract infection, headache, nasopharyngitis and diarrhea.
Infections
In the 12 week controlled phase, the rate of all infections in the tocilizumab-IV group was 345 per 100 patient-years and 287 per 100 patient-years in the placebo group. In the open label extension over an average duration of 73 weeks of treatment, the overall rate of infections was 304 per 100 patient-years.
In the 12 week controlled phase, the rate of serious infections in the tocilizumab-IV group was 11.5 per 100 patient years. In the open label extension over an average duration of 73 weeks of treatment, the overall rate of serious infections was 11.4 per 100 patient years. The most commonly reported serious infections included pneumonia, gastroenteritis, varicella, and otitis media.
Macrophage Activation Syndrome
In the 12 week controlled study, no patient in any treatment group experienced macrophage activation syndrome (MAS) while on assigned treatment; 3 per 112 (3%) developed MAS during open-label treatment with tocilizumab-IV. One patient in the placebo group escaped to tocilizumab-IV 12 mg per kg at Week 2 due to severe disease activity, and ultimately developed MAS at Day 70. Two additional patients developed MAS during the long-term extension. All 3 patients had tocilizumab-IV dose interrupted (2 patients) or discontinued (1 patient) for the MAS event, received treatment, and the MAS resolved without sequelae. Based on a limited number of cases, the incidence of MAS does not appear to be elevated in the tocilizumab-IV SJIA clinical development experience; however no definitive conclusions can be made.
Infusion Reactions
Patients were not premedicated, however most patients were on concomitant corticosteroids as part of their background treatment for SJIA. Infusion related reactions were defined as all events occurring during or within 24 hours after an infusion. In the 12 week controlled phase, 4% of tocilizumab-IV and 0% of placebo treated patients experienced events occurring during infusion. One event (angioedema) was considered serious and life-threatening, and the patient was discontinued from study treatment.
Within 24 hours after infusion, 16% of patients in the tocilizumab-IV treatment group and 5% of patients in the placebo group experienced an event. In the tocilizumab-IV group the events included rash, urticaria, diarrhea, epigastric discomfort, arthralgia and headache. One of these events, urticaria, was considered serious.
Anaphylaxis
Anaphylaxis was reported in 1 out of 112 patients (less than 1%) treated with tocilizumab-IV during the controlled and open label extension study [see Warnings and Precautions (5.6)].
Immunogenicity
All 112 patients were tested for anti-tocilizumab antibodies at baseline. Two patients developed positive anti- tocilizumab antibodies: one of these patients experienced serious adverse events of urticaria and angioedema consistent with an anaphylactic reaction which led to withdrawal; the other patient developed macrophage activation syndrome while on escape therapy and was discontinued from the study.
Neutropenia
During routine monitoring in the 12 week controlled phase, a decrease in neutrophil below 1 × 109 per L occurred in 7% of patients in the tocilizumab-IV group, and in no patients in the placebo group. In the open label extension over an average duration of 73 weeks of treatment, a decreased neutrophil count occurred in 17% of the tocilizumab-IV group. There was no clear relationship between decrease in neutrophils below 1 x 109 per L and the occurrence of serious infections.
Thrombocytopenia
During routine monitoring in the 12 week controlled phase, 1% of patients in the tocilizumab-IV group and 3% in the placebo group had a decrease in platelet count to no more than 100,000 per mm3.
In the open label extension over an average duration of 73 weeks of treatment, decreased platelet count occurred in 4% of patients in the tocilizumab-IV group, with no associated bleeding.
Elevated Liver Enzymes
During routine laboratory monitoring in the 12 week controlled phase, elevation in ALT or AST at or above 3x ULN occurred in 5% and 3% of patients, respectively in the tocilizumab-IV group and in 0% of placebo patients.
In the open label extension over an average duration of 73 weeks of treatment, the elevation in ALT or AST at or above 3x ULN occurred in 13% and 5% of tocilizumab-IV treated patients, respectively.
Lipids
During routine laboratory monitoring in the 12 week controlled phase, elevation in total cholesterol greater than 1.5x ULN – 2x ULN occurred in 1.5% of the tocilizumab-IV group and in 0% of placebo patients. Elevation in LDL greater than 1.5x ULN – 2x ULN occurred in 1.9% of patients in the tocilizumab-IV group and 0% of the placebo group.
In the open label extension study over an average duration of 73 weeks of treatment, the pattern and incidence of elevations in lipid parameters remained consistent with the 12 week controlled study data.
6.8 Clinical Trials Experience in Systemic Juvenile Idiopathic Arthritis Patients Treated with Subcutaneous Tocilizumab (tocilizumab-SC)
The safety profile of tocilizumab-SC was studied in 51 pediatric patients 1 to 17 years of age with SJIA who had an inadequate clinical response to NSAIDs and corticosteroids. In general, the safety observed for tocilizumab administered subcutaneously was consistent with the known safety profile of intravenous tocilizumab, with the exception of ISRs where a higher frequency was observed in tocilizumab-SC treated SJIA patients compared to PJIA patients and adult RA or GCA patients [see Adverse Reactions (6.2, 6.3 and 6.6)].
Injection Site Reactions (ISRs)
A total of 41.2% (21/51) SJIA patients experienced ISRs to tocilizumab-SC. The most common ISRs were erythema, pruritus, pain, and swelling at the injection site. The majority of ISRs reported were Grade 1 events and all ISRs reported were non-serious and none required patient withdrawal from treatment or dose interruption.
6.9 Clinical Trials Experience in Patients with Cytokine Release Syndrome Treated with Intravenous Tocilizumab (tocilizumab-IV)
In a retrospective analysis of pooled outcome data from multiple clinical trials 45 patients were treated with tocilizumab 8 mg/kg (12 mg/kg for patients less than 30 kg) with or without additional high-dose corticosteroids for severe or life-threatening CAR T-cell-induced CRS. A median of 1 dose of tocilizumab (range, 1-4 doses) was administered. No adverse reactions related to tocilizumab were reported [see Clinical Studies (14.10)].
6.10 Clinical Trials Experience in COVID-19 Patients Treated with Intravenous Tocilizumab (tocilizumab-IV)
The safety of tocilizumab in hospitalized COVID-19 patients was evaluated in a pooled safety population that includes patients enrolled in EMPACTA, COVACTA, AND REMDACTA. The analysis of adverse reactions included a total of 974 patients exposed to tocilizumab. Patients received a single, 60-minute infusion of intravenous tocilizumab 8 mg/kg (maximum dose of 800 mg). If clinical signs or symptoms worsened or did not improve, one additional dose of tocilizumab 8 mg/kg could be administered between 8- 24 hours after the initial dose.
Adverse reactions summarized in Table 3 occurred in at least 3% of tocilizumab-treated patients and more commonly than in patients on placebo in the pooled safety population.
Table 3 Adverse Reactions1 Identified From the Pooled COVID-19 Safety Population 1Patients are counted once for each category regardless of the number of reactions Adverse Reaction
Tocilizumab 8 mg per kg
N = 974 (%)Placebo
N = 483 (%)Hepatic Transaminases increased
10%
8%
Constipation
9%
8%
Urinary tract infection
5%
4%
Hypertension
4%
1%
Hypokalaemia
4%
3%
Anxiety
4%
2%
Diarrhea
4%
2%
Insomnia
4%
3%
Nausea
3%
2%
In the pooled safety population, the rates of infection/serious infection events were 30%/19% in patients receiving tocilizumab versus 32%/23% receiving placebo.
Laboratory Abnormalities
In the pooled safety population of EMPACTA, COVACTA, and REMDACTA, neutrophil counts <1000 cells/mcl occurred in 3.4% of patients who received tocilizumab and 0.5% of patients who received placebo. Platelet counts <50,000 cells/mcl occurred in 3.2% of patients who received tocilizumab and 1.5% of patients who received placebo. ALT or AST at or above 5x ULN occurred in 11.7% of patients who received tocilizumab and 9.9% of patients who received placebo.
6.11 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of tocilizumab products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
-
7 DRUG INTERACTIONS
7.1 Concomitant Drugs for Treatment of Adult Indications
In RA patients, population pharmacokinetic analyses did not detect any effect of methotrexate (MTX), non-steroidal anti-inflammatory drugs or corticosteroids on tocilizumab clearance. Concomitant administration of a single intravenous dose of 10 mg/kg tocilizumab with 10-25 mg MTX once weekly had no clinically significant effect on MTX exposure. Tocilizumab products have not been studied in combination with biological DMARDs such as TNF antagonists [see Dosage and Administration (2.2)].
In GCA patients, no effect of concomitant corticosteroid on tocilizumab exposure was observed.
7.2 Interactions with CYP450 Substrates
Cytochrome P450s in the liver are down-regulated by infection and inflammation stimuli including cytokines such as IL-6. Inhibition of IL-6 signaling in RA patients treated with tocilizumab products may restore CYP450 activities to higher levels than those in the absence of tocilizumab products leading to increased metabolism of drugs that are CYP450 substrates. In vitro studies showed that tocilizumab has the potential to affect expression of multiple CYP enzymes including CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. Its effect on CYP2C8 or transporters is unknown. In vivo studies with omeprazole, metabolized by CYP2C19 and CYP3A4, and simvastatin, metabolized by CYP3A4, showed up to a 28% and 57% decrease in exposure one week following a single dose of tocilizumab, respectively. The effect of tocilizumab products on CYP enzymes may be clinically relevant for CYP450 substrates with narrow therapeutic index, where the dose is individually adjusted.
Upon initiation or discontinuation of TYENNE, in patients being treated with these types of medicinal products, perform therapeutic monitoring of effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) and the individual dose of the medicinal product adjusted as needed. Exercise caution when coadministering TYENNE with CYP3A4 substrate drugs where decrease in effectiveness is undesirable, e.g., oral contraceptives, lovastatin, atorvastatin, etc. The effect of tocilizumab products on CYP450 enzyme activity may persist for several weeks after stopping therapy [see Clinical Pharmacology (12.3)].
7.3 Live Vaccines
Avoid use of live vaccines concurrently with TYENNE [see Warnings and Precautions (5.9)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The available data with tocilizumab products from a pregnancy exposure registry, retrospective cohort study, pharmacovigilance, and published literature are insufficient to draw conclusions about a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. These studies had methodological limitations, including small sample size of tocilizumab exposed groups, missing exposure and outcomes information, and lack of adjustment for cofounders. Monoclonal antibodies, such as tocilizumab products, are actively transported across the placenta during the third trimester of pregnancy and may affect immune response in the in utero exposed infant [see Clinical Considerations]. In animal reproduction studies, intravenous administration of tocilizumab to Cynomolgus monkeys during organogenesis caused abortion/embryo-fetal death at doses 1.25 times and higher than the maximum recommended human dose by the intravenous route of 8 mg per kg every 2 to 4 weeks. The literature in animals suggests that inhibition of IL-6 signaling may interfere with cervical ripening and dilatation and myometrial contractile activity leading to potential delays of parturition [see Data]. Based on the animal data, there may be a potential risk to the fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Fetal/Neonatal adverse reactions
Monoclonal antibodies are increasingly transported across the placenta as pregnancy progresses, with the largest amount transferred during the third trimester. Risks and benefits should be considered prior to administering live or live-attenuated vaccines to infants exposed to TYENNE in utero [see Warnings and Precautions 5.9)].
Disease-associated Maternal Risk
Published data suggest that the risk of adverse pregnancy outcomes in women with rheumatoid arthritis is associated with increased disease activity. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Data
Animal Data
An embryo-fetal developmental toxicity study was performed in which pregnant Cynomolgus monkeys were treated intravenously with tocilizumab at daily doses of 2, 10, or 50 mg/ kg during organogenesis from gestation day (GD) 20-50. Although there was no evidence for a teratogenic/dysmorphogenic effect at any dose, tocilizumab produced an increase in the incidence of abortion/embryo-fetal death at doses 1.25 times and higher the MRHD by the intravenous route at maternal intravenous doses of 10 and 50 mg/ kg. Testing of a murine analogue of tocilizumab in mice did not yield any evidence of harm to offspring during the pre- and postnatal development phase when dosed at 50 mg/kg intravenously with treatment every three days from implantation (GD 6) until post-partum day 21 (weaning). There was no evidence for any functional impairment of the development and behavior, learning ability, immune competence and fertility of the offspring.
Parturition is associated with significant increases of IL-6 in the cervix and myometrium. The literature suggests that inhibition of IL-6 signaling may interfere with cervical ripening and dilatation and myometrial contractile activity leading to potential delays of parturition. For mice deficient in IL-6 (ll6-/- null mice), parturition was delayed relative to wild-type (ll6+/+) mice.
Administration of recombinant IL-6 to ll6-/- null mice restored the normal timing of delivery.
8.2 Lactation
Risk Summary
No information is available on the presence of tocilizumab products in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. Maternal immunoglobulin G (IgG) is present in human milk. If tocilizumab products are transferred into human milk, the effects of local exposure in the gastrointestinal tract and potential limited systemic exposure in the infant to tocilizumab products are unknown.
The lack of clinical data during lactation precludes clear determination of the risk of tocilizumab products to an infant during lactation; therefore the developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TYENNE and the potential adverse effects on the breastfed child from TYENNE or from the underlying maternal condition.
8.4 Pediatric Use
TYENNE by intravenous use is indicated for the treatment of pediatric patients with:
- Active systemic juvenile idiopathic arthritis in patients 2 years of age and older
- Active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older
- Severe or life-threatening CAR T cell-induced cytokine release syndrome (CRS) in patients 2 years of age and older.
TYENNE by subcutaneous use is indicated for the treatment of pediatric patients with:
- Active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older
- Active systemic juvenile idiopathic arthritis in patients 2 years of age and older
The safety and effectiveness of TYENNE in pediatric patients with conditions other than PJIA, SJIA or CRS have not been established. The safety and effectiveness in pediatric patients below the age of 2 have not been established in PJIA, SJIA or CRS.
Systemic Juvenile Idiopathic Arthritis – Intravenous Use
A multicenter, open-label, single arm study to evaluate the PK, safety and exploratory PD and efficacy of tocilizumab over 12-weeks in SJIA patients (N=11) under 2 years of age was conducted. Patients received intravenous tocilizumab 12 mg/kg every two weeks. Concurrent use of stable background treatment with corticosteroids, MTX, and/or non-steroidal anti-inflammatory drugs was permitted.
Patients who completed the 12-week period could continue to the optional extension period (a total of 52-weeks or until the age of 2 years, whichever was longer).
The primary PK endpoints (Cmax, Ctrough and AUC2weeks) of tocilizumab at steady-state in this study were within the ranges of these parameters observed in patients with SJIA aged 2 to 17 years.
The safety and immunogenicity of tocilizumab for patien ts with SJIA under 2 years of age was assessed descriptively. SAEs, AEs leading to discontinuation, and infectious AEs were reported by 27.3%, 36.4%, and 81.8% of patients. Six patients (54.5%) experienced hypersensitivity reactions, defined as all adverse events occurring during or within 24 hours after an infusion considered related to tocilizumab. Three of these patients experienced serious hypersensitivity reactions and were withdrawn from the study. Three patients with hypersensitivity reactions (two with serious hypersensitivity reactions) developed treatment induced anti- tocilizumab antibodies after the event. There were no cases of MAS based on the protocol-specified criteria, but 2 cases of suspected MAS based on Ravelli criteria1.
Cytokine Release Syndrome – Intravenous Use
In the retrospective analysis of pooled outcome data for patients treated with tocilizumab for CAR T cell-induced CRS, 25 patients were children (2 years up to 12 years of age), and 17 patients were adolescents (12 years up to 18 years of age). There were no differences between the pediatric patients and the adults for safety or efficacy.
- 1 Ravelli A, Minoia F, Davì S on behalf of the Paediatric Rheumatology International Trials Organisation, the Childhood Arthritis and Rheumatology Research Alliance, the Pediatric Rheumatology Collaborative Study Group, and the Histiocyte Society, et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis. Annals of the Rheumatic Diseases 2016;75:481-489.
8.5 Geriatric Use
Of the 2644 patients who received tocilizumab in Studies I to V [see Clinical Studies (14)], a total of 435 rheumatoid arthritis patients were 65 years of age and older, including 50 patients 75 years and older. Of the 1069 patients who received tocilizumab-SC in studies SC-I and SC-II there were 295 patients 65 years of age and older, including 41 patients 75 years and older. The frequency of serious infection among tocilizumab treated subjects 65 years of age and older was higher than those under the age of 65. As there is a higher incidence of infections in the elderly population in general, caution should be used when treating the elderly.
Clinical studies that included tocilizumab for CRS did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients.
In the EMPACTA, COVACTA, and REMDACTA studies, of the 974 COVID-19 patients in the tocilizumab arm, 375 (39%) were 65 years of age or older. No overall differences in safety or effectiveness of tocilizumab were observed between patients 65 years of age and older and those under the age of 65 years of age in these studies [see Adverse Reactions (6.1) and Clinical Studies (14.10)].In the RECOVERY study, of the 2022 COVID-19 patients in the tocilizumab arm, 930 (46%) were 65 years of age or older. No overall differences in effectiveness of tocilizumab were observed between patients 65 years of age and older and those under the age 65 years of age in this study [see Clinical Studies (14.10)].
8.6 Hepatic Impairment
The safety and efficacy of tocilizumab products have not been studied in patients with hepatic impairment, including patients with positive HBV and HCV serology [see Warnings and Precautions (5.8)].
8.7 Renal Impairment
No dose adjustment is required in patients with mild or moderate renal impairment. Tocilizumab products have not been studied in patients with severe renal impairment [see Clinical Pharmacology (12.3)].
- 9 DRUG ABUSE AND DEPENDENCE
-
10 OVERDOSAGE
There are limited data available on overdoses with tocilizumab products. One case of accidental overdose was reported with intravenous tocilizumab in which a patient with multiple myeloma received a dose of 40 mg per kg. No adverse drug reactions were observed. No serious adverse drug reactions were observed in healthy volunteers who received single doses of up to 28 mg per kg, although all 5 patients at the highest dose of 28 mg per kg developed dose-limiting neutropenia.
In case of an overdose, it is recommended that the patient be monitored for signs and symptoms of adverse reactions. Patients who develop adverse reactions should receive appropriate symptomatic treatment.
-
11 DESCRIPTION
Tocilizumab-aazg is a recombinant humanized anti-human interleukin 6 (IL-6) receptor monoclonal antibody of the immunoglobulin IgG1κ (gamma 1, kappa) subclass with a typical H2L2 polypeptide structure. Each light chain and heavy chain consists of 214 and 448 amino acids (excluding the C-terminal lysine), respectively.
The four polypeptide chains are linked intra- and inter-molecularly by disulfide bonds. Tocilizumab-aazg has a molecular weight of approximately 148 kDa. The antibody is produced in mammalian (Chinese hamster ovary) cells.
Intravenous Infusion
TYENNE (tocilizumab-aazg) injection is a sterile, clear and colorless to pale yellow, histidine buffered preservative-free solution with a pH of approximately 6 for further dilution prior to intravenous infusion. Each single-dose vial is available at a concentration of 20 mg/mL containing 80 mg/4 mL, 200 mg/10 mL, or 400 mg/20 mL of TYENNE. Each mL of solution contains arginine (17.4 mg), histidine (3.1 mg), lactic acid (0.9 mg), polysorbate 80 (0.2 mg), sodium chloride (0.6 mg), and Water for Injection, USP. Hydrochloric acid and sodium hydroxide are added to adjust the pH.
Subcutaneous Injection
TYENNE (tocilizumab-aazg) injection is a sterile, clear, colorless to pale yellow, preservative-free, histidine buffered solution with a pH of approximately 6 for subcutaneous use. It is supplied in a ready-to-use, single-dose 0.9 mL prefilled syringe (PFS) with a needle safety device or in a ready-to-use, single-dose 0.9 mL autoinjector that delivers 162 mg tocilizumab-aazg, arginine (16.7 mg), histidine (2.0 mg), lactic acid (0.9 mg), polysorbate 80 (0.2 mg), sodium chloride (0.6 mg), and Water for Injection, USP. Hydrochloric acid and sodium hydroxide are added to adjust the pH.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tocilizumab products bind to both soluble and membrane-bound IL-6 receptors (sIL-6R and mIL-6R), and have been shown to inhibit IL-6-mediated signaling through these receptors. IL-6 is a pleiotropic pro-inflammatory cytokine produced by a variety of cell types including T- and B-cells, lymphocytes, monocytes and fibroblasts. IL-6 has been shown to be involved in diverse physiological processes such as T-cell activation, induction of immunoglobulin secretion, initiation of hepatic acute phase protein synthesis, and stimulation of hematopoietic precursor cell proliferation and differentiation. IL-6 is also produced by synovial and endothelial cells leading to local production of IL-6 in joints affected by inflammatory processes such as rheumatoid arthritis.
12.2 Pharmacodynamics
In clinical studies in RA patients with the 4 mg per kg and 8 mg per kg intravenous doses or the 162 mg weekly and every other weekly subcutaneous doses of tocilizumab, decreases in levels of C-reactive protein (CRP) to within normal ranges were seen as early as week 2. Changes in pharmacodynamic parameters were observed (i.e., decreases in rheumatoid factor, erythrocyte sedimentation rate (ESR), serum amyloid A, fibrinogen and increases in hemoglobin) with doses, however the greatest improvements were observed with 8 mg per kg tocilizumab. Pharmacodynamic changes were also observed to occur after tocilizumab administration in GCA, PJIA, and SJIA patients (decreases in CRP, ESR, and increases in hemoglobin). The relationship between these pharmacodynamic findings and clinical efficacy is not known.
In healthy subjects administered tocilizumab in doses from 2 to 28 mg per kg intravenously and 81 to 162 mg subcutaneously, absolute neutrophil counts decreased to the nadir 3 to 5 days following tocilizumab administration. Thereafter, neutrophils recovered towards baseline in a dose dependent manner. Rheumatoid arthritis and GCA patients demonstrated a similar pattern of absolute neutrophil counts following tocilizumab administration [see Warnings and Precautions (5.4)].
12.3 Pharmacokinetics
PK of tocilizumab is characterized by nonlinear elimination which is a combination of linear clearance and Michaelis-Menten elimination. The nonlinear part of tocilizumab elimination leads to an increase in exposure that is more than dose-proportional. The pharmacokinetic parameters of tocilizumab do not change with time. Due to the dependence of total clearance on tocilizumab serum concentrations, the half-life of tocilizumab is also concentration-dependent and varies depending on the serum concentration level. Population pharmacokinetic analyses in any patient population tested so far indicate no relationship between apparent clearance and the presence of anti-drug antibodies.
Rheumatoid Arthritis – Intravenous and Subcutaneous Administration
The pharmacokinetics in healthy subjects and RA patients suggest that PK is similar between the two populations.
The population PK model was developed from an analysis dataset composed of an IV dataset of 1793 patients from Study I, Study III, Study IV, and Study V, and from an IV and SC dataset of 1759 patients from Studies SC-I and SC-II. Cmean is included in place of AUCtau, since for dosing regimens with different inter-dose intervals, the mean concentration over the dosing period characterizes the comparative exposure better than AUCtau.
At high serum concentrations, when total clearance of tocilizumab is dominated by linear clearance, a terminal half-life of approximately 21.5 days was derived from the population parameter estimates.
For doses of 4 mg/kg tocilizumab given every 4 weeks intravenously, the estimated median (range) Cmax, Ctrough, and Cmean of tocilizumab at steady state were 86.1 (44.8–202) mcg/mL, 0.1 (0.0–14.6) mcg/mL, and 18.0 (8.9– 50.7) mcg/mL, respectively. For doses of 8 mg/kg tocilizumab given every 4 weeks intravenously, the estimated median (range) Cmax, Ctrough, and Cmean of tocilizumab were 176 (75.4–557) mcg/mL, 13.4 (0.1–154) mcg/mL, and 54.0 (17–260) mcg/mL, respectively. Cmax increased dose-proportionally between doses of 4 and 8 mg/kg IV every 4 weeks, while a greater than dose-proportional increase was observed in Cmean and Ctrough. At steady-state, Cmean and Ctrough were 3.0 and 134 fold higher at 8 mg/kg as compared to 4 mg/kg, respectively.
The accumulation ratios for AUC and Cmax after multiple doses of 4 and 8 mg/kg IV Q4W are low, while the accumulation ratios for Ctrough are higher (2.62 and 2.47, respectively). For Cmax, greater than 90% of the steady-state value was reached after the 1st IV infusion. For AUCtau and Cmean, 90% of the steady-state value was reached after the 1st and 3rd infusion for 4 mg/kg and 8 mg/kg IV, while for Ctrough, approximately 90% of the steady-state value was reached after the 4th IV infusion after both doses.
For doses of 162 mg given every other week subcutaneously, the estimated median (range) steady-state Cmax, Ctrough, and Cmean of tocilizumab were 12.1 (0.4–49.3) mcg/mL, 4.1 (0.0–34.2) mcg/mL, and 9.2 (0.2– 43.6) mcg/mL, respectively.
For doses of 162 mg given every week subcutaneously, the estimated median (range) steady-state Cmax, Ctrough, and Cmean of tocilizumab were 49.8 (3–150) mcg/mL, 42.9 (1.3–144) mcg/mL, and 47.3 (2.4–147) mcg/mL, respectively. Exposures after the 162 mg SC QW regimen were greater by 5.1 (Cmean) to 10.5 fold (Ctrough) compared to the 162 mg SC Q2W regimen.
Accumulation ratios after multiple doses of either SC regimen were higher than after IV regimen with the highest ratios for Ctrough (6.02 and 6.30, for 162 mg SC Q2W and 162 mg SC QW, respectively). The higher accumulation for Ctrough was expected based on the nonlinear clearance contribution at lower concentrations. For Cmax, greater than 90% of the steady-state value was reached after the 5th SC and the 12th SC injection with the Q2W and QW regimens, respectively. For AUCtau and Cmean, 90% of the steady-state value was reached after the 6th and 12th injections for the 162 mg SC Q2W and QW regimens, respectively. For Ctrough, approximately 90% of the steady-state value was reached after the 6th and 12th injections for the 162 mg SC Q2W and QW regimens, respectively.
Population PK analysis identified body weight as a significant covariate impacting the pharmacokinetics of tocilizumab. When given IV on a mg/kg basis, individuals with body weight ≥ 100 kg are predicted to have mean steady-state exposures higher than mean values for the patient population. Therefore, tocilizumab doses exceeding 800 mg per infusion are not recommended in patients with RA [see Dosage and Administration (2.2)]. Due to the flat dosing employed for SC administration of tocilizumab, no modifications are necessary by this dosing route.
Giant Cell Arteritis – Subcutaneous and Intravenous Administration
The pharmacokinetics of tocilizumab S C in GCA patients was determined using a population pharmacokinetic analysis on a dataset composed of 149 GCA patients treated with 162 mg subcutaneously every week or with 162 mg subcutaneously every other week.
For the 162 mg every week dose, the estimated median (range) steady-state Cmax, Ctrough and Cmean of tocilizumab SC were 72.1 (12.2–151) mcg/mL, 67.2 (10.7–145) mcg/mL, and 70.6 (11.7–149) mcg/mL, respectively. The accumulation ratios for Cmean or AUCtau, Ctrough, and Cmax were 10.9, 9.6, and 8.9, respectively. Steady state was reached after 17 weeks. For the 162 mg every other week dose, the estimated median (range) steady-state Cmax, Ctrough, and Cmean of tocilizumab were 17.2 (1.1–56.2) mcg/mL, 7.7 (0.1–37.3) mcg/mL, and 13.7 (0.5– 49) mcg/mL, respectively. The accumulation ratios for Cmean or AUCtau, Ctrough, and Cmax were 2.8, 5.6, and 2.3 respectively. Steady-state was reached after 14 weeks.
The pharmacokinetics of tocilizumab IV in GCA patients was characterized by a non-compartmental pharmacokinetic analysis which included 22 patients treated with 6 mg/kg intravenously every 4 weeks for 20 weeks. The median (range) Cmax, Ctrough and Cmean of tocilizumab at steady state were 178 (115-320) mcg/mL, 22.7 (3.38-54.5) mcg/mL and 57.5 (32.9-110) mcg/mL, respectively. Steady state trough concentrations were within the range observed in GCA patients treated with 162 mg TCZ SC administered every week or every other week. Based on pharmacokinetic exposure and extrapolation between RA and GCA patients, when given IV on a mg/kg basis, tocilizumab doses exceeding 600 mg per infusion are not recommended in patients with GCA [see Dosage and Administration (2.3)].
Polyarticular Juvenile Idiopathic Arthritis – Intravenous and Subcutaneous Administration
The pharmacokinetics of tocilizumab (TCZ) in PJIA patients was characterized by a population pharmacokinetic analysis which included 188 patients who were treated with TCZ IV or 52 patients treated with TCZ SC.
For doses of 8 mg/kg tocilizumab (patients with a body weight at or above 30 kg) given every 4 weeks intravenously, the estimated median (range) Cmax, Ctrough, and Cmean of tocilizumab at steady state were 181 (114–331) mcg/mL, 3.28 (0.02–35.4) mcg/mL, and 38.6 (22.2–83.8) mcg/mL, respectively. For doses of 10 mg/kg tocilizumab (patients with a body weight less than 30 kg) given every 4 weeks intravenously, the estimated median (range) Cmax, Ctrough, and Cmean of tocilizumab were 167 (125–220) mcg/mL, 0.35 (0– 11.8) mcg/mL, and 30.8 (16.0–48.0) mcg/mL, respectively.
The accumulation ratios were 1.05 and 1.16 for AUC4weeks, and 1.43 and 2.22 for Ctrough for 10 mg/kg (BW less than 30 kg) and 8 mg/kg (BW at or above 30 kg) intravenous doses, respectively. No accumulation for Cmax was observed. Following 10 mg/kg and 8 mg/kg TCZ IV every 4 weeks doses in PJIA patients (aged 2 to 17 years), steady state concentrations (trough and average) were within the range of exposures in adult RA patients following 4 mg/kg and 8 mg/kg every 4 weeks, and steady state peak concentrations in PJIA patients were comparable to those following 8 mg/kg every 4 weeks in adult RA patients.
For doses of 162 mg tocilizumab (patients with a body weight at or above 30 kg) given every 2 weeks subcutaneously, the estimated median (range) Cmax, Ctrough, and Cmean of tocilizumab were 29.7 (7.56–50.3) mcg/mL, 12.7 (0.19–23.8) mcg/mL, and 23.0 (3.86–36.9) mcg/mL, respectively. For doses of 162 mg tocilizumab (patients with a body weight less than 30 kg) given every 3 weeks subcutaneously, the estimated median (range) Cmax, Ctrough, and Cmean of tocilizumab were 62.4 (39.4–121) mcg/mL, 13.4 (0.21–52.3) mcg/mL, and 35.7 (17.4–91.8) mcg/mL, respectively.
The accumulation ratios were 1.46 and 2.04 for AUC4weeks, 2.08 and 3.58 for Ctrough, and 1.32 and 1.72 for Cmax, for 162 mg given every 3 weeks (BW less than 30 kg) and 162 mg given every 2 weeks (BW at or above 30 kg) subcutaneous doses, respectively. Following subcutaneous dosing, steady state Ctrough was comparable for patients in the two body weight groups, while steady-state Cmax and Cmean were higher for patients in the less than 30 kg group compared to the group at or above 30 kg. All patients treated with TCZ SC had steady-state Ctrough at or higher than that achieved with TCZ IV across the spectrum of body weights. The average and trough concentrations in patients after subcutaneous dosing were within the range of those achieved in adult patients with RA following the subcutaneous administration of the recommended regimens.
Systemic Juvenile Idiopathic Arthritis – Intravenous and Subcutaneous Administration
The pharmacokinetics of tocilizumab (TCZ) in SJIA patients was characterized by a population pharmacokinetic analysis which included 89 patients who were treated with TCZ IV or 51 patients treated with TCZ SC.
For doses of 8 mg/kg tocilizumab (patients with a body weight at or above 30 kg) given every 2 weeks intravenously, the estimated median (range) Cmax, Ctrough, and Cmean of tocilizumab were 253 (120–404) mcg/mL, 70.7 (5.26–127) mcg/mL, and 117 (37.6–199) mcg/mL, respectively. For doses of 12 mg/kg tocilizumab (patients with a body weight less than 30 kg) given every 2 weeks intravenously, the estimated median (range) Cmax, Ctrough, and Cmean of tocilizumab were 274 (149–444) mcg/mL, 65.9 (19.0–135) mcg/mL, and 124 (60–194) mcg/mL, respectively.
The accumulation ratios were 1.95 and 2.01 for AUC4weeks, and 3.41 and 3.20 for Ctrough for 12 mg/kg (BW less than 30 kg) and 8 mg/kg (BW at or above 30 kg) intravenous doses, respectively. Accumulation data for Cmax were 1.37 and 1.42 for 12 mg/kg (BW less than 30 kg) and 8 mg/kg (BW at or above 30 kg) intravenous doses, respectively. Following every other week dosing with tocilizumab IV, steady state was reached by 8 weeks for both body weight groups. Mean estimated tocilizumab exposure parameters were similar between the two dose groups defined by body weight.
For doses of 162 mg tocilizumab (patients with a body weight at or above 30 kg) given every week subcutaneously, the estimated median (range) Cmax, Ctrough, and Cmean of tocilizumab were 89.8 (26.4–190) mcg/mL, 72.4 (19.5–158) mcg/mL, and 82.4 (23.9–169) mcg/mL, respectively. For doses of 162 mg tocilizumab (patients with a body weight less than 30 kg) given every 2 weeks subcutaneously, the estimated median (range) Cmax, Ctrough, and Cmean of tocilizumab were 127 (51.7–266) mcg/mL, 64.2 (16.6–136) mcg/mL, and 92.7 (38.5–199) mcg/mL, respectively.
The accumulation ratios were 2.27 and 4.28 for AUC4weeks, 3.21 and 4.39 for Ctrough, and 1.88 and 3.66 for Cmax, for 162 mg given every 2 weeks (BW less than 30 kg) and 162 mg given every week (BW at or above 30 kg) subcutaneous doses, respectively. Following subcutaneous dosing, steady state was reached by 12 weeks for both body weight groups. All patients treated with tocilizumab SC had steady-state Cmax lower than that achieved with tocilizumab IV across the spectrum of body weights. Trough and mean concentrations in patients after SC dosing were similar to those achieved with tocilizumab IV across body weights.
COVID-19 -Intravenous Administration
The pharmacokinetics of tocilizumab in COVID-19 patients was characterized by a population pharmacokinetic analysis of a dataset composed of 380 adult patients treated with tocilizumab 8 mg/kg intravenously (IV) in the COVACTA study [see Clinical Studies (14.10)] and another clinical study.
For one dose of 8 mg/kg tocilizumab IV, the estimated median (range) Cmax and Cday28 of tocilizumab were 151 (77.5-319) mcg/mL and 0.229 (0.00119-19.4) mcg/mL, respectively. For two doses of 8 mg/kg tocilizumab IV separated by at least 8 hours, the estimated median (range) Cmax and Cday28 of tocilizumab was 290 (152-604) mcg/mL and 7.04 (0.00474-54.8) mcg/mL, respectively. The weight-tiered dosing used in RECOVERY study, 800 mg for patients >90 kg, 600 mg for patients >65 and ≤90 kg, 400 mg for patients >40 and ≤65 kg, and 8mg/kg for patients ≤40 kg, is comparable to 8 mg/kg dosing and is expected to have similar exposure.
Absorption
Following subcutaneous dosing, the absorption half-life was around 4 days in RA and GCA patients. The bioavailability for the subcutaneous formulation was 80%.
Following subcutaneous dosing in PJIA patients, the absorption half-life was around 2 days, and the bioavailability for the subcutaneous formulation in PJIA patients was 96%.
Following subcutaneous dosing in SJIA patients, the absorption half-life was around 2 days, and the bioavailability for the SC formulation in SJIA patients was 95%.
In RA patients the median values of Tmax were 2.8 days after the tocilizumab every week dose and 4.7 days after the tocilizumab every other week dose.
In GCA patients, the median values of Tmax were 3 days after the tocilizumab every week dose and 4.5 days after the tocilizumab every other week dose.
Distribution
Following intravenous dosing, tocilizumab undergoes biphasic elimination from the circulation. In rheumatoid arthritis patients the central volume of distribution was 3.5 L and the peripheral volume of distribution was 2.9 L, resulting in a volume of distribution at steady state of 6.4 L.
In GCA patients, the central volume of distribution was 4.09 L, the peripheral volume of distribution was 3.37 L resulting in a volume of distribution at steady state of 7.46 L.
In pediatric patients with PJIA, the central volume of distribution was 1.98 L, the peripheral volume of distribution was 2.1 L, resulting in a volume of distribution at steady state of 4.08 L.
In pediatric patients with SJIA, the central volume of distribution was 1.87 L, the peripheral volume of distribution was 2.14 L resulting in a volume of distribution at steady state of 4.01 L.
In COVID-19 patients treated with one or two infusions of tocilizumab 8 mg/kg intravenously separated by 8 hours, the estimated central volume of distribution was 4.52 L, and the estimated peripheral volume of distribution was 4.23 L, resulting in a volume of distribution of 8.75 L.
Elimination
Tocilizumab is eliminated by a combination of linear clearance and nonlinear elimination. The concentration-dependent nonlinear elimination plays a major role at low tocilizumab concentrations. Once the nonlinear pathway is saturated, at higher tocilizumab concentrations, clearance is mainly determined by the linear clearance. The saturation of the nonlinear elimination leads to an increase in exposure that is more than dose-proportional. The pharmacokinetic parameters of tocilizumab do not change with time.
Population pharmacokinetic analyses in any patient population tested so far indicate no relationship between apparent clearance and the presence of anti-drug antibodies.
The linear clearance in the population pharmacokinetic analysis was estimated to be 12.5 mL per h in RA patients, 6.7 mL per h in GCA patients, 5.8 mL per h in pediatric patients with PJIA, and 5.7 mL per h in pediatric patients with SJIA. In COVID-19 patients, serum concentrations were below the limit of quantification after 35 days on average following one infusion of tocilizumab 8 mg/kg intravenously. The average linear clearance in the population pharmacokinetic analysis was estimated to be 17.6 mL per hour in patients with baseline ordinal scale category 3 (OS 3, patients requiring supplemental oxygen), 22.5 mL per hour in patients with baseline OS 4 (patients requiring high-flow oxygen or non-invasive ventilation), 29 mL per hour in patients with baseline OS 5 (patients requiring mechanical ventilation), and 35.4 mL per hour in patients with baseline OS 6 (patients requiring extracorporeal membrane oxygenation (ECMO) or mechanical ventilation and additional organ support).
Due to the dependence of total clearance on tocilizumab serum concentrations, the half-life of tocilizumab is also concentration-dependent and varies depending on the serum concentration level.
For intravenous administration in RA patients, the concentration-dependent apparent t1/2 is up to 11 days for 4 mg per kg and up to 13 days for 8 mg per kg every 4 weeks in patients with RA at steady-state. For subcutaneous administration in RA patients, the concentration-dependent apparent t1/2 is up to 13 days for 162 mg every week and 5 days for 162 mg every other week in patients with RA at steady-state.
In GCA patients at steady state, the effective t1/2 of tocilizumab varied between 18.3 and 18.9 days for 162 mg subcutaneously every week dosing regimen and between 4.2 and 7.9 days for 162 mg subcutaneously every other week dosing regimen. For intravenous administration in GCA patients, the TCZ concentration-dependent apparent t1/2 was 13.2 days following 6 mg/kg every 4 weeks.
The t1/2 of tocilizumab in children with PJIA is up to 17 days for the two body weight categories (8 mg/kg for body weight at or above 30 kg or 10 mg/kg for body weight below 30 kg) during a dosing interval at steady state. For subcutaneous administration, the t1/2 of tocilizumab in PJIA patients is up to 10 days for the two body weight categories (every other week regimen for body weight at or above 30 kg or every 3 week regimen for body weight less than 30 kg) during a dosing interval at steady state.
The t1/2 of tocilizumab intravenous in pediatric patients with SJIA is up to 16 days for the two body weight categories (8 mg/kg for body weight at or above 30 kg and 12 mg/kg for body weight below 30 kg every other week) during a dosing interval at steady-state. Following subcutaneous administration, the effective t1/2 of tocilizumab subcutaneous in SJIA patients is up to 14 days for both the body weight categories (162 mg every week for body weight at or above 30 kg and 162 mg every two weeks for body weight below 30 kg) during a dosing interval at steady state.
Specific Populations
Population pharmacokinetic analyses in adult rheumatoid arthritis patients and GCA patients showed that age, gender and race did not affect the pharmacokinetics of tocilizumab. Linear clearance was found to increase with body size. In RA patients, the body weight-based dose (8 mg per kg) resulted in approximately 86% higher exposure in patients who are greater than 100 kg in comparison to patients who are less than 60 kg. There was an inverse relationship between tocilizumab exposure and body weight for flat dose subcutaneous regimens.
In GCA patients treated with tocilizumab-SC, higher exposure was observed in patients with lower body weight. For the 162 mg every week subcutaneous dosing regimen, the steady-state Cmean was 51% higher in patients with body weight less than 60 kg compared to patients weighing between 60 to 100 kg. For the 162 mg every other week subcutaneous regimen, the steady-state Cmean was 129% higher in patients with body weight less than 60 kg compared to patients weighing between 60 to 100 kg. There is limited data for patients above 100 kg (n=7).
In COVID-19 patients, exposure following body-weight-based intravenous dosing (8 mg per kg tocilizumab up to 100 kg body weight with a maximum dose of 800 mg) was dependent on body weight and disease severity assessed by an ordinal scale (OS). Within an OS category, compared to patients with a mean body weight of 80 kg, exposure was 20% lower in patients weighing less than 60 kg. Exposure in patients weighing more than 100 kg was in the same range as exposure in patients with a mean body weight of 80 kg. For an 80 kg patient, exposure decreases as OS category increases; for each category increase, exposure decreases by 13%.
Patients with Hepatic Impairment
No formal study of the effect of hepatic impairment on the pharmacokinetics of tocilizumab was conducted.
Patients with Renal Impairment
No formal study of the effect of renal impairment on the pharmacokinetics of tocilizumab was conducted.
Most of the RA and GCA patients in the population pharmacokinetic analysis had normal renal function or mild renal impairment. Mild renal impairment (estimated creatinine clearance less than 80 mL per min and at or above 50 mL per min based on Cockcroft-Gault formula) did not impact the pharmacokinetics of tocilizumab.
Approximately one-third of the patients in the tocilizumab-SC GCA clinical trial had moderate renal impairment at baseline (estimated creatinine clearance of 30-59 mL/min). No impact on tocilizumab exposure was noted in these patients.
No dose adjustment is required in patients with mild or moderate renal impairment.
Drug Interaction Studies
In vitro data suggested that IL-6 reduced mRNA expression for several CYP450 isoenzymes including CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6 and CYP3A4, and this reduced expression was reversed by co-incubation with tocilizumab at clinically relevant concentrations. Accordingly, inhibition of IL-6 signaling in RA patients treated with tocilizumab may restore CYP450 activities to higher levels than those in the absence of tocilizumab leading to increased metabolism of drugs that are CYP450 substrates. Its effect on CYP2C8 or transporters (e.g., P-gp) is unknown. This is clinically relevant for CYP450 substrates with a narrow therapeutic index, where the dose is individually adjusted. Upon initiation of TYENNE, in patients being treated with these types of medicinal products, therapeutic monitoring of the effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) should be performed and the individual dose of the medicinal product adjusted as needed.
Caution should be exercised when TYENNE is coadministered with drugs where decrease in effectiveness is undesirable, e.g., oral contraceptives (CYP3A4 substrates) [see Drug Interactions (7.2)].
Simvastatin
Simvastatin is a CYP3A4 and OATP1B1 substrate. In 12 RA patients not treated with tocilizumab, receiving 40 mg simvastatin, exposures of simvastatin and its metabolite, simvastatin acid, was 4- to 10-fold and 2-fold higher, respectively, than the exposures observed in healthy subjects. One week following administration of a single infusion of tocilizumab (10 mg per kg), exposure of simvastatin and simvastatin acid decreased by 57% and 39%, respectively, to exposures that were similar or slightly higher than those observed in healthy subjects. Exposures of simvastatin and simvastatin acid increased upon withdrawal of tocilizumab in RA patients. Selection of a particular dose of simvastatin in RA patients should take into account the potentially lower exposures that may result after initiation of TYENNE (due to normalization of CYP3A4) or higher exposures after discontinuation of TYENNE.
Omeprazole
Omeprazole is a CYP2C19 and CYP3A4 substrate. In RA patients receiving 10 mg omeprazole, exposure to omeprazole was approximately 2 fold higher than that observed in healthy subjects. In RA patients receiving 10 mg omeprazole, before and one week after tocilizumab infusion (8 mg per kg), the omeprazole AUCinf decreased by 12% for poor (N=5) and intermediate metabolizers (N=5) and by 28% for extensive metabolizers (N=8) and were slightly higher than those observed in healthy subjects.
Dextromethorphan
Dextromethorphan is a CYP2D6 and CYP3A4 substrate. In 13 RA patients receiving 30 mg dextromethorphan, exposure to dextromethorphan was comparable to that in healthy subjects. However, exposure to its metabolite, dextrorphan (a CYP3A4 substrate), was a fraction of that observed in healthy subjects. One week following administration of a single infusion of tocilizumab (8 mg per kg), dextromethorphan exposure was decreased by approximately 5%. However, a larger decrease (29%) in dextrorphan levels was noted after tocilizumab infusion.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term animal studies have been performed to establish the carcinogenicity potential of tocilizumab products. Literature indicates that the IL-6 pathway can mediate anti-tumor responses by promoting increased immune cell surveillance of the tumor microenvironment. However, available published evidence also supports that IL-6 signaling through the IL-6 receptor may be involved in pathways that lead to tumorigenesis. The malignancy risk in humans from an antibody that disrupts signaling through the IL-6 receptor, such as tocilizumab, is presently unknown.
Fertility and reproductive performance were unaffected in male and female mice that received a murine analogue of tocilizumab administered by the intravenous route at a dose of 50 mg/kg every three days.
-
14 CLINICAL STUDIES
14.1 Rheumatoid Arthritis – Intravenous Administration
The efficacy and safety of intravenously administered tocilizumab was assessed in five randomized, double-blind, multicenter studies in patients greater than 18 years with active rheumatoid arthritis diagnosed according to American College of Rheumatology (ACR) criteria. Patients had at least 8 tender and 6 swollen joints at baseline. Tocilizumab was given intravenously every 4 weeks as monotherapy (Study I), in combination with methotrexate (MTX) (Studies II and III) or other disease-modifying anti-rheumatic drugs (DMARDs) (Study IV) in patients with an inadequate response to those drugs, or in combination with MTX in patients with an inadequate response to TNF antagonists (Study V).
Study I (NCT00109408) evaluated patients with moderate to severe active rheumatoid arthritis who had not been treated with MTX within 24 weeks prior to randomization, or who had not discontinued previous methotrexate treatment as a result of clinically important toxic effects or lack of response. In this study, 67% of patients were MTX-naïve, and over 40% of patients had rheumatoid arthritis less than 2 years. Patients received tocilizumab 8 mg per kg monotherapy or MTX alone (dose titrated over 8 weeks from 7.5 mg to a maximum of 20 mg weekly). The primary endpoint was the proportion of tocilizumab patients who achieved an ACR 20 response at Week 24.
Study II (NCT00106535) was a 104-week study with an optional 156-week extension phase that evaluated patients with moderate to severe active rheumatoid arthritis who had an inadequate clinical response to MTX. Patients received tocilizumab 8 mg per kg, tocilizumab 4 mg per kg, or placebo every four weeks, in combination with MTX (10 to 25 mg weekly). Upon completion of 52-weeks, patients received open-label treatment with tocilizumab 8 mg per kg through 104 weeks or they had the option to continue their double-blind treatment if they maintained a greater than 70% improvement in swollen/tender joint count. Two pre-specified interim analyses at week 24 and week 52 were conducted. The primary endpoint at week 24 was the proportion of patients who achieved an ACR 20 response. At weeks 52 and 104, the primary endpoints were change from baseline in modified total Sharp-Genant score and the area under the curve (AUC) of the change from baseline in HAQ-DI score.
Study III (NCT00106548) evaluated patients with moderate to severe active rheumatoid arthritis who had an inadequate clinical response to MTX. Patients received tocilizumab 8 mg per kg, tocilizumab 4 mg per kg, or placebo every four weeks, in combination with MTX (10 to 25 mg weekly). The primary endpoint was the proportion of patients who achieved an ACR 20 response at week 24.
Study IV (NCT00106574) evaluated patients who had an inadequate response to their existing therapy, including one or more DMARDs. Patients received tocilizumab 8 mg per kg or placebo every four weeks, in combination with the stable DMARDs. The primary endpoint was the proportion of patients who achieved an ACR 20 response at week 24.
Study V (NCT00106522) evaluated patients with moderate to severe active rheumatoid arthritis who had an inadequate clinical response or were intolerant to one or more TNF antagonist therapies. The TNF antagonist therapy was discontinued prior to randomization. Patients received tocilizumab 8 mg per kg, tocilizumab 4 mg per kg, or placebo every four weeks, in combination with MTX (10 to 25 mg weekly). The primary endpoint was the proportion of patients who achieved an ACR 20 response at week 24.
Clinical Response
The percentages of intravenous tocilizumab-treated patients achieving ACR 20, 50 and 70 responses are shown in Table 4. In all intravenous studies, patients treated with 8 mg per kg tocilizumab had higher ACR 20, ACR 50, and ACR 70 response rates versus MTX- or placebo-treated patients at week 24.
During the 24 week controlled portions of Studies I to V, patients treated with tocilizumab at a dose of 4 mg per kg in patients with inadequate response to DMARDs or TNF antagonist therapy had lower response rates compared to patients treated with tocilizumab 8 mg per kg.
Table 4 Clinical Response at Weeks 24 and 52 in Active and Placebo Controlled Trials of Intravenous Tocilizumab (Percent of Patients) a CI: 95% confidence interval of the weighted difference to placebo adjusted for site (and disease duration for Study I only)
b Major clinical response is defined as achieving an ACR 70 response for a continuous 24 week periodPercent of Patients
Study I
Study II
Study III
Study IV
Study V
Response Rate
MTX
Tocilizumab 8 mg per kg
Placebo + MTX
Tocilizumab 4 mg per kg + MTX
Tocilizumab 8 mg per kg + MTX
Placebo + MTX
Tocilizumab 4 mg per kg + MTX
Tocilizumab 8 mg per kg + MTX
Placebo + DMARDs
Tocilizumab 8 mg per kg + DMARDs
Placebo + MTX
Tocilizumab 4 mg per kg + MTX
Tocilizumab 8 mg per kg + MTX
N=284
N=286
N=393
N=399
N=398
N=204
N=213
N=205
N=413
N=803
N=158
N=161
N=170
(95% CI)a
(95% CI)a
(95% CI)a
(95% CI)a
(95% CI)a
(95% CI)a
(95% CI)a
(95% CI)a
ACR 20
Week 24
53%
70%
27%
51%
56%
27%
48%
59%
24%
61%
10%
30%
50%
(0.11, 0.27)
(0.17, 0.29)
(0.23, 0.35)
(0.15, 0.32)
(0.23, 0.41)
(0.30, 0.40)
(0.15, 0.36)
(0.36, 0.56)
Week 52
N/A
N/A
25%
47%
56%
N/A
N/A
N/A
N/A
N/A
N/A
N/A
N/A
(0.15, 0.28)
(0.25, 0.38)
ACR 50
Week 24
34%
44%
10%
25%
32%
11%
32%
44%
9%
38%
4%
17%
29%
(0.04, 0.20)
(0.09, 0.20)
(0.16, 0.28)
(0.13, 0.29)
(0.25, 0.41)
(0.23, 0.33)
(0.05, 0.25)
(0.21, 0.41)
Week 52
N/A
N/A
10%
29%
36%
N/A
N/A
N/A
N/A
N/A
N/A
N/A
N/A
(0.14, 0.25)
(0.21, 0.32)
ACR 70
Week 24
15%
28%
2%
11%
13%
2%
12%
22%
3%
21%
1%
5%
12%
(0.07, 0.22)
(0.03, 0.13)
(0.05, 0.15)
(0.04, 0.18)
(0.12, 0.27)
(0.13, 0.21)
(-0.06, 0.14)
(0.03, 0.22)
Week 52
N/A
N/A
4%
16%
20%
N/A
N/A
N/A
N/A
N/A
(0.08, 0.17)
(0.12, 0.21)
N/A
N/A
N/A
Major Clinical Responses b
Week 52
N/A
N/A
1%
4%
7%
N/A
N/A
N/A
N/A
N/A
N/A
N/A
N/A
(0.01, 0.06)
(0.03, 0.09)
In study II, a greater proportion of patients treated with 4 mg per kg and 8 mg per kg tocilizumab + MTX achieved a low level of disease activity as measured by a DAS 28-ESR less than 2.6 compared with placebo+ MTX treated patients at week 52. The proportion of tocilizumab-treated patients achieving DAS 28-ESR less than 2.6, and the number of residual active joints in these responders in Study II are shown in Table 5.
Table 5 Proportion of Patients with DAS28-ESR Less Than 2.6 with Number of Residual Active Joints in Trials of Intravenous Tocilizumab *n denotes numerator of all the percentage. Denominator is the intent-to-treat population. Not all patients received DAS28 assessments at Week 52. Study II
Placebo + MTX
N = 393Tocilizumab 4 mg per kg
+ MTX
N = 399Tocilizumab 8 mg per kg
+ MTX
N = 398DAS28-ESR less than 2.6
Proportion of responders at week 52 (n) 95% confidence interval
3% (12)
18% (70)
0.10, 0.1932% (127)
0.24, 0.34Of responders, proportion with 0 active joints (n)
33% (4)
27% (19)
21% (27)
Of responders, proportion with 1 active joint (n)
8% (1)
19% (13)
13% (16)
Of responders, proportion with 2 active joints (n)
25% (3)
13% (9)
20% (25)
Of responders, proportion with 3 or more active joints (n)
33% (4)
41% (29)
47% (59)
The results of the components of the ACR response criteria for Studies III and V are shown in Table 6. Similar results to Study III were observed in Studies I, II and IV.
Table 6 Components of ACR Response at Week 24 in Trials of Intravenous Tocilizumab a Data shown is mean at week 24, difference in adjusted mean change from baseline compared with placebo + MTX at week 24 and 95% confidence interval for that difference
b Visual analog scale: 0 = best, 100 = worst
c Health Assessment Questionnaire: 0 = best, 3 = worst; 20 questions; 8 categories: dressing and grooming, arising, eating, walking, hygiene, reach, grip, and activitiesStudy III
Study V
Tocilizumab 4 mg per kg + MTX
N=213Tocilizumab 8 mg per kg + MTX
N=205Placebo + MTX
N=204Tocilizumab 4 mg per kg + MTX
N=161Tocilizumab 8 mg per kg + MTX
N=170Placebo + MTX
N=158Component (mean)
Baseline
Week 24a
Baseline
Week 24 a
Baseline
Week 24
Baseline
Week 24 a
Baseline
Week 24 a
Baseline
Week 24
Number of
33
19
32
14.5
33
25
31
21
32
17
30
30
tender joints
-7.0
-9.6
-10.8
-15.1
(0-68)
(-10.0, -4.1)
(-12.6, -6.7)
(-14.6, -7.1)
(-18.8, -11.4)
Number of
20
10
19.5
8
21
15
19.5
13
19
11
19
18
swollen
-4.2
-6.2
-6.2
-7.2
joints (0-66)
(-6.1, -2.3)
(-8.1, -4.2)
(-9.0, -3.5)
(-9.9, -4.5)
Pain b
61
33
-11.060
30
-15.857
43
63.5
43
-12.465
33
-23.964
48
(-17.0, -5.0)
(-21.7, -9.9)
(-22.1, -2.1)
(-33.7, -14.1)
Patient
66
34
65
31
64
45
70
46
70
36
71
51
global
-10.9
-14.9
-10.0
-17.4
Assessment b
(-17.1, -4.8)
(-20.9, -8.9)
(-20.3, 0.3)
(-27.8, -7.0)
Physician
64
26
64
23
64
32
66.5
39
66
28
67.5
43
global
-5.6
-9.0
-10.5
-18.2
Assessment b
(-10.5, -0.8)
(-13.8, -4.2)
(-18.6, -2.5)
(-26.3, -10.0)
Disability
1.64
1.01
1.55
0.96
1.55
1.21
1.67
1.39
1.75
1.34
1.70
1.58
index
-0.18
-0.21
-0.25
-0.34
(HAQ)c
(-0.34, -0.02)
(-0.37, -0.05)
(-0.42, -0.09)
(-0.51, -0.17)
CRP (mg per
2.79
1.17
-1.30
2.61
0.25
-2.1562.36
1.89
3.11
1.77
-1.34
2.80
0.28
-2.52
3.705
3.06
dL)
(-2.0, -0.59)
(-2.86, -1.46)
(-2.5, -0.15)
(-3.72, -1.32)
The percent of ACR 20 responders by visit for Study III is shown in Figure 1. Similar response curves were observed in studies I, II, IV, and V.
Figure 1 Percent of ACR 20 Responders by Visit for Study III (Inadequate Response to MTX)*
*The same patients may not have responded at each timepoint.
Radiographic Response
In Study II, structural joint damage was assessed radiographically and expressed as change in total Sharp-Genant score and its components, the erosion score and joint space narrowing score. Radiographs of hands/wrists and forefeet were obtained at baseline, 24 weeks, 52 weeks, and 104 weeks and scored by readers unaware of treatments group and visit number. The results from baseline to week 52 are shown in Table 7. Tocilizumab 4 mg per kg slowed (less than 75% inhibition compared to the control group) and tocilizumab 8 mg per kg inhibited (at least 75% inhibition compared to the control group) the progression of structural damage compared to placebo plus MTX at week 52.
Table 7 Mean Radiographic Change from Baseline to Week 52 in Study II * Week 52 analysis employs linearly extrapolated data for patients after escape, withdrawal, or loss to follow up.
** Difference between the adjusted means (tocilizumab + MTX – Placebo + MTX) SD = standard deviationPlacebo + MTX
Tocilizumab 4 mg per kg + MTX
Tocilizumab 8 mg per kg + MTX
N=294
N=343
N=353
Week 52*
Total Sharp-Genant Score, Mean (SD)
1.17
(3.14)0.33
(1.30)0.25
(0.98)Adjusted Mean
difference** (95%CI)-0.83
(-1.13, -0.52)-0.90
(-1.20, -0.59)Erosion Score,
Mean (SD)0.76
(2.14)0.20
(0.83)0.15
(0.77)Adjusted Mean
difference** (95%CI)-0.55
(-0.76, -0.34)-0.60
(-0.80, -0.39)Joint Space Narrowing Score, Mean (SD)
0.41
(1.71)0.13
(0.72)0.10
(0.49)Adjusted Mean
difference** (95%CI)-0.28
(-0.44, -0.11)-0.30
(-0.46, -0.14)The mean change from baseline to week 104 in Total Sharp-Genant Score for the tocilizumab 4 mg per kg groups was 0.47 (SD = 1.47) and for the 8 mg per kg groups was 0.34 (SD = 1.24). By the week 104, most patients in the control (placebo + MTX) group had crossed over to active treatment, and results are therefore not included for comparison. Patients in the active groups may have crossed over to the alternate active dose group, and results are reported per original randomized dose group.
In the placebo group, 66% of patients experienced no radiographic progression (Total Sharp-Genant Score change ≤ 0) at week 52 compared to 78% and 83% in the tocilizumab 4 mg per kg and 8 mg per kg, respectively. Following 104 weeks of treatment, 75% and 83% of patients initially randomized to tocilizumab 4 mg per kg and 8 mg per kg, respectively, experienced no progression of structural damage compared to 66% of placebo treated patients.
Health Related Outcomes
In Study II, physical function and disability were assessed using the Health Assessment Questionnaire Disability Index (HAQ-DI). Both dosing groups of tocilizumab demonstrated a greater improvement compared to the placebo group in the AUC of change from baseline in the HAQ-DI through week 52. The mean change from baseline to week 52 in HAQ-DI was 0.6, 0.5, and 0.4 for tocilizumab 8 mg per kg, tocilizumab 4 mg per kg, and placebo treatment groups, respectively. Sixty-three percent (63%) and sixty percent (60%) of patients in the tocilizumab 8 mg per kg and tocilizumab 4 mg per kg treatment groups, respectively, achieved a clinically relevant improvement in HAQ-DI (change from baseline of ≥ 0.3 units) at week 52 compared to 53% in the placebo treatment group.
Other Health-Related Outcomes
General health status was assessed by the Short Form Health Survey (SF-36) in Studies I – V. Patients receiving tocilizumab demonstrated greater improvement from baseline compared to placebo in the Physical Component Summary (PCS), Mental Component Summary (MCS), and in all 8 domains of the SF-36.
Cardiovascular Outcomes
Study WA25204 (NCT01331837) was a randomized, open-label (sponsor-blinded), 2-arm parallel-group, multicenter, non-inferiority, cardiovascular (CV) outcomes trial in patients with a diagnosis of moderate to severe RA. This CV safety study was designed to exclude a moderate increase in CV risk in patients treated with tocilizumab compared with a TNF inhibitor standard of care (etanercept).
The study included 3,080 seropositive RA patients with active disease and an inadequate response to non- biologic disease-modifying anti-rheumatic drugs, who were aged ≥50 years with at least one additional CV risk factor beyond RA. Patients were randomized 1:1 to IV tocilizumab 8 mg/kg Q4W or SC etanercept 50 mg QW and followed for an average of 3.2 years. The primary endpoint was the comparison of the time-to-first occurrence of any component of a composite of major adverse CV events (MACE; non-fatal myocardial infarction, non-fatal stroke, or CV death), with the final intent-to-treat analysis based on a total of 161 confirmed CV events (83/1538 [5.4%] for tocilizumab; 78/1542 [5.1%] for etanercept) reviewed by an independent and blinded adjudication committee.
Non-inferiority of tocilizumab to etanercept for cardiovascular risk was determined by excluding >80% relative increase in the risk of MACE. The estimated hazard ratio (HR) for the risk of MACE comparing tocilizumab to etanercept was 1.05; 95% CI (0.77, 1.43).
14.2 Rheumatoid Arthritis – Subcutaneous Administration
The efficacy and safety of subcutaneously administered tocilizumab was assessed in two double-blind, controlled, multicenter studies in patients with active RA. One study, SC-I (NCT01194414), was a non-inferiority study that compared the efficacy and safety of tocilizumab 162 mg administered every week subcutaneously to 8 mg per kg intravenously every four weeks. The second study, SC-II (NCT01232569), was a placebo-controlled superiority study that evaluated the safety and efficacy of tocilizumab 162 mg administered every other week subcutaneously to placebo. Both SC-I and SC-II required patients to be >18 years of age with moderate to severe active rheumatoid arthritis diagnosed according to ACR criteria who had at least 4 tender and 4 swollen joints at baseline (SC-I) or at least 8 tender and 6 swollen joints at baseline (SC-II), and an inadequate response to their existing DMARD therapy, where approximately 20% also had a history of inadequate response to at least one TNF inhibitor. All patients in both SC studies received background non-biologic DMARD(s).
In SC-I, 1262 patients were randomized 1:1 to receive tocilizumab-SC 162 mg every week or intravenous tocilizumab 8 mg/kg every four weeks in combination with DMARD(s). In SC-II, 656 patients were randomized 2:1 to tocilizumab-SC 162 mg every other week or placebo, in combination with DMARD(s). The primary endpoint in both studies was the proportion of patients who achieved an ACR20 response at Week 24.
The clinical response to 24 weeks of tocilizumab-SC therapy is shown in Table 8. In SC-I, the primary outcome measure was ACR20 at Week 24. The pre-specified non-inferiority margin was a treatment difference of 12%. The study demonstrated non-inferiority of tocilizumab with respect to ACR20 at Week 24; ACR50, ACR70, and DAS28 responses are also shown in Table 8. In SC-II, a greater portion of patients treated with tocilizumab 162 mg subcutaneously every other week achieved ACR20, ACR50, and ACR70 responses compared to placebo-treated patients (Table 8). Further, a greater proportion of patients treated with tocilizumab 162 mg subcutaneously every other week achieved a low level of disease activity as measured by a DAS28-ESR less than 2.6 at Week 24 compared to those treated with placebo (Table 8).
Table 8 Clinical Response at Week 24 in Trials of Subcutaneous Tocilizumab (Percent of Patients) TCZ = tocilizumab
a Per Protocol Population
b Intent To Treat PopulationSC-Ia
SC-IIb
TCZ SC 162 mg every week + DMARD
TCZ IV 8mg/kg + DMARD
TCZ SC 162 mg every other week + DMARD
Placebo + DMARD
N=558
N=537
N=437
N=219
ACR20
Week 24
69%
73.4%
61%
32%
Weighted difference (95% CI)
-4% (-9.2, 1.2)
30% (22.0, 37.0)
ACR50
Week 24
47%
49%
40%
12%
Weighted difference (95% CI)
-2% (-7.5, 4.0)
28% (21.5, 34.4)
ACR70
Week 24
24%
28%
20%
5%
Weighted difference (95% CI)
-4% (-9.0, 1.3)
15% (9.8, 19.9)
Change in DAS28 [Adjusted mean]
Week 24
-3.5
-3.5
-3.1
-1.7
Adjusted mean difference (95% CI)
0 (-0.2, 0.1)
-1.4 (-1.7; -1.1)
DAS28 < 2.6
Week 24
38.4%
36.9%
32.0%
4.0%
Weighted difference (95% CI)
0.9 (-5.0, 6.8)
28.6 (22.0, 35.2)
The results of the components of the ACR response criteria and the percent of ACR20 responders by visit for tocilizumab-SC in Studies SC-I and SC-II were consistent with those observed for tocilizumab-IV.
Radiographic Response
In study SC-II, the progression of structural joint damage was assessed radiographically and expressed as a change from baseline in the van der Heijde modified total Sharp score (mTSS). At week 24, significantly less radiographic progression was observed in patients receiving tocilizumab-SC every other week plus DMARD(s) compared to placebo plus DMARD(s); mean change from baseline in mTSS of 0.62 vs. 1.23, respectively, with an adjusted mean difference of -0.60 (-1.1, -0.1). These results are consistent with those observed in patients treated with intravenous tocilizumab.
Health Related Outcomes
In studies SC-I and SC-II, the mean decrease from baseline to week 24 in HAQ-DI was 0.6, 0.6, 0.4 and 0.3, and the proportion of patients who achieved a clinically relevant improvement in HAQ-DI (change from baseline of ≥ 0.3 units) was 65%, 67%, 58% and 47%, for the subcutaneous every week, intravenous 8 mg/kg, subcutaneous every other week, and placebo treatment groups, respectively.
Other Health-Related Outcomes
General health status was assessed by the SF-36 in Studies SC-I and SC-II. In Study SC-II, patients receiving tocilizumab every other week demonstrated greater improvement from baseline compared to placebo in the PCS, MCS, and in all 8 domains of the SF-36. In Study SC-I, improvements in these scores were similar between tocilizumab-SC every week and tocilizumab-IV 8 mg/kg.
14.3 Giant Cell Arteritis – Subcutaneous Administration
The efficacy and safety of subcutaneously administered tocilizumab was assessed in a single, randomized, double-blind, multicenter study in patients with active GCA. In Study WA28119 (NCT01791153), 251 screened patients with new-onset or relapsing GCA were randomized to one of four treatment arms. Two subcutaneous doses of tocilizumab (162 mg every week and 162 mg every other week) were compared to two different placebo control groups (pre-specified prednisone-taper regimen over 26 weeks and 52 weeks) randomized 2:1:1:1. The study consisted of a 52-week blinded period, followed by a 104-week open-label extension.
All patients received background glucocorticoid (prednisone) therapy. Each of the tocilizumab-treated groups and one of the placebo-treated groups followed a pre-specified prednisone-taper regimen with the aim to reach 0 mg by 26 weeks, while the second placebo-treated group followed a pre-specified prednisone-taper regimen with the aim to reach 0 mg by 52 weeks designed to be more in keeping with standard practice.
The primary efficacy endpoint was the proportion of patients achieving sustained remission from Week 12 through Week 52. Sustained remission was defined by a patient attaining a sustained (1) absence of GCA signs and symptoms from Week 12 through Week 52, (2) normalization of erythrocyte sedimentation rate (ESR) (to < 30 mm/hr without an elevation to ≥ 30 mm/hr attributable to GCA) from Week 12 through Week 52, (3) normalization of C-reactive protein (CRP) (to < 1 mg/dL, with an absence of successive elevations to ≥ 1mg/dL) from Week 12 through Week 52, and (4) successful adherence to the prednisone taper defined by not more than 100 mg of excess prednisone from Week 12 through Week 52. Tocilizumab 162 mg weekly and 162 mg every other week + 26 weeks prednisone taper both showed superiority in achieving sustained remission from Week 12 through Week 52 compared with placebo + 26 weeks prednisone taper (Table 9). Both tocilizumab treatment arms also showed superiority compared to the placebo + 52 weeks prednisone taper (Table 9).
Table 9 Efficacy Results from Study WA28119 a Sustained remission was achieved by a patient meeting all of the following components: absence of GCA signs and symptomsb, normalization of ESRc, normalization of CRPd and adherence to the prednisone taper regimene.
b Patients who did not have any signs or symptoms of GCA recorded from Week 12 up to Week 52.
c Patients who did not have an elevated ESR ≥30 mm/hr which was classified as attributed to GCA from Week 12 up to Week 52.
d Patients who did not have two or more consecutive CRP records of ≥ 1mg/dL from Week 12 up to Week 52.
e Patients who did not enter escape therapy and received ≤ 100mg of additional concomitant prednisone from Week 12 up to Week 52.PBO + 26
weeks prednisone taper
N=50PBO + 52
weeks prednisone taper
N=51TCZ 162mg SC QW + 26
weeks prednisone taper
N=100TCZ 162 mg SC Q2W + 26
weeks prednisone taper
N=49Sustained remission a
Responders, n (%)
7 (14.0%)
9 (17.6%)
56 (56.0%)
26 (53.1%)
Unadjusted difference in proportions
vs PBO + 26 weeks taper
(99.5% CI)N/A
N/A
42.0%
(18.0, 66.0)39.1%
(12.5, 65.7)Unadjusted difference in proportions
vs PBO + 52 weeks taper
(99.5% CI)N/A
N/A
38.4%
(14.4, 62.3)35.4%
(8.6, 62.2)Components of Sustained Remission
Sustained absence of GCA signs
and symptomsb, n (%)
Sustained ESR<30 mm/hrc, n (%)
Sustained CRP normalizationd, n (%)
Successful prednisone taperinge, n (%)20 (40.0%)
20 (40.0%)
17 (34.0%)
10 (20.0%)23 (45.1%)
22 (43.1%)
13 (25.5%)
20 (39.2%)69 (69.0%)
83 (83.0%)
72 (72.0%)
60 (60.0%)28 (57.1%)
37 (75.5%)
34 (69.4%)
28 (57.1%)Patients not completing the study to week 52 were classified as non-responders in the primary and key secondary analysis: PBO+26: 6 (12.0%), PBO+52: 5 (9.8%), TCZ QW: 15 (15.0%), TCZ Q2W: 9 (18.4%).
CRP = C-reactive protein
ESR = erythrocyte sedimentation rate
PBO = placebo
Q2W = every other week dose
QW = every week dose
TCZ = tocilizumab
The estimated annual cumulative prednisone dose was lower in the two tocilizumab dose groups (medians of 1887 mg and 2207 mg on tocilizumab QW and Q2W, respectively) relative to the placebo arms (medians of 3804 mg and 3902 mg on placebo + 26 weeks prednisone and placebo + 52 weeks prednisone taper, respectively).
14.4 Giant Cell Arteritis – Intravenous Administration
Intravenously administered tocilizumab in patients with GCA was assessed in WP41152 (NCT03923738), an open-label PK-PD and safety study to determine the appropriate intravenous dose of tocilizumab that achieved comparable PK-PD profiles to the tocilizumab-SC regimen.
At enrollment, all patients (n=24) were in remission on tocilizumab-IV. In Period 1, all patients received open label tocilizumab-IV 7 mg/kg every 4 weeks for 20 weeks. Patients who completed Period 1 and remained in remission (n=22) were eligible to enter Period 2, and received open-label tocilizumab-IV 6 mg/kg every 4 weeks for 20 weeks.
The efficacy of intravenous tocilizumab 6 mg/kg in adult patients with GCA is based on pharmacokinetic exposure and extrapolation to the efficacy established for subcutaneous tocilizumab in patients with GCA [see Clinical Pharmacology (12.3) and Clinical Studies (14.3)].
14.5 Polyarticular Juvenile Idiopathic Arthritis – Intravenous Administration
The efficacy of tocilizumab was assessed in a three-part study, WA19977 (NCT00988221), including an open- label extension in children 2 to 17 years of age with active polyarticular juvenile idiopathic arthritis (PJIA), who had an inadequate response to methotrexate or inability to tolerate methotrexate. Patients had at least 6 months of active disease (mean disease duration of 4.2 ± 3.7 years), with at least five joints with active arthritis (swollen or limitation of movement accompanied by pain and/or tenderness) and/or at least 3 active joints having limitation of motion (mean, 20 ± 14 active joints). The patients treated had subtypes of JIA that at disease onset included Rheumatoid Factor Positive or Negative Polyarticular JIA, or Extended Oligoarticular JIA. Treatment with a stable dose of methotrexate was permitted but was not required during the study. Concurrent use of disease modifying antirheumatic drugs (DMARDs), other than methotrexate, or other biologics (e.g., TNF antagonists or T cell costimulation modulator) were not permitted in the study.
Part I consisted of a 16-week active tocilizumab treatment lead-in period (n=188) followed by Part II, a 24-week randomized double-blind placebo-controlled withdrawal period, followed by Part III, a 64-week open-label period. Eligible patients weighing at or above 30 kg received tocilizumab at 8 mg/kg intravenously once every four weeks. Patients weighing less than 30 kg were randomized 1:1 to receive either tocilizumab 8 mg/kg or 10 mg/kg intravenously every four weeks. At the conclusion of the open-label Part I, 91% of patients taking background MTX in addition to tocilizumab and 83% of patients on tocilizumab monotherapy achieved an ACR 30 response at week 16 compared to baseline and entered the blinded withdrawal period (Part II) of the study.
The proportions of patients with JIA ACR 50/70 responses in Part I were 84.0%, and 64%, respectively for patients taking background MTX in addition to tocilizumab and 80% and 55% respectively for patients on tocilizumab monotherapy.
In Part II, patients (ITT, n=163) were randomized to tocilizumab (same dose received in Part I) or placebo in a 1:1 ratio that was stratified by concurrent methotrexate use and concurrent corticosteroid use. Each patient continued in Part II of the study until Week 40 or until the patient satisfied JIA ACR 30 flare criteria (relative to Week 16) and qualified for escape.
The primary endpoint was the proportion of patients with a JIA ACR 30 flare at week 40 relative to week 16. JIA ACR 30 flare was defined as 3 or more of the 6 core outcome variables worsening by at least 30% with no more than 1 of the remaining variables improving by more than 30% relative to Week 16.
Tocilizumab treated patients experienced significantly fewer disease flares compared to placebo-treated patients (26% [21/82] versus 48% [39/81]; adjusted difference in proportions -21%, 95% CI: -35%, -8%).
During the withdrawal phase (Part II), more patients treated with tocilizumab showed JIA ACR 30/50/70 responses at Week 40 compared to patients withdrawn to placebo.
14.6 Polyarticular Juvenile Idiopathic Arthritis – Subcutaneous Administration
Subcutaneously administered tocilizumab in pediatric patients with polyarticular juvenile idiopathic arthritis (PJIA) was assessed in WA28117 (NCT01904279), a 52-week, open-label, multicenter, PK-PD and safety study to determine the appropriate subcutaneous dose of tocilizumab that achieved comparable PK/PD profiles to the tocilizumab-IV regimen. PJIA patients aged 1 to 17 years with an inadequate response or inability to tolerate MTX, including patients with well-controlled disease on treatment with tocilizumab-IV and tocilizumab-naïve patients with active disease, were treated with subcutaneous tocilizumab based on body weight.
Patients weighing at or above 30 kg (n = 25) were treated with 162 mg of tocilizumab-SC every 2 weeks and patients weighing less than 30 kg (n = 27) received 162 mg of tocilizumab-SC every 3 weeks for 52 weeks. Of these 52 patients, 37 (71%) were naïve to tocilizumab and 15 (29%) had been receiving tocilizumab-IV and switched to tocilizumab-SC at baseline.
The efficacy of subcutaneous tocilizumab in children 2 to 17 years of age is based on pharmacokinetic exposure and extrapolation of the established efficacy of intravenous tocilizumab in polyarticular JIA patients and subcutaneous tocilizumab in patients with RA [see Clinical Pharmacology (12.3) and Clinical Studies (14.2 and 14.6)].
14.7 Systemic Juvenile Idiopathic Arthritis - Intravenous Administration
The efficacy of tocilizumab for the treatment of active SJIA was assessed in WA18221 (NCT00642460), a 12-week randomized, double blind, placebo-controlled, parallel group, 2-arm study. Patients treated with or without MTX, were randomized (tocilizumab:placebo = 2:1) to one of two treatment groups: 75 patients received tocilizumab infusions every two weeks at either 8 mg per kg for patients at or above 30 kg or 12 mg per kg for patients less than 30 kg and 37 were randomized to receive placebo infusions every two weeks. Corticosteroid tapering could occur from week six for patients who achieved a JIA ACR 70 response. After 12 weeks or at the time of escape, due to disease worsening, patients were treated with tocilizumab in the open-label extension phase at weight appropriate dosing.
The primary endpoint was the proportion of patients with at least 30% improvement in JIA ACR core set (JIA ACR 30 response) at Week 12 and absence of fever (no temperature at or above 37.5°C in the preceding 7 days). JIA ACR (American College of Rheumatology) responses are defined as the percentage improvement (e.g., 30%, 50%, 70%) in 3 of any 6 core outcome variables compared to baseline, with worsening in no more than 1 of the remaining variables by 30% or more.
Core outcome variables consist of physician global assessment, parent per patient global assessment, number of joints with active arthritis, number of joints with limitation of movement, erythrocyte sedimentation rate (ESR), and functional ability (childhood health assessment questionnaire-CHAQ).
Primary endpoint result and JIA ACR response rates at Week 12 are shown in Table 10.
Table 10 Efficacy Findings at Week 12 aThe weighted difference is the difference between the tocilizumab and Placebo response rates, adjusted for the stratification factors (weight, disease duration, background oral corticosteroid dose and background methotrexate use).
b CI: confidence interval of the weighted difference.Tocilizumab
N=75Placebo
N=37Primary Endpoint: JIA ACR 30 response + absence of fever
Responders
85%
24%
Weighted difference (95% CI)
62
(45, 78)-
JIA ACR Response Rates at Week 12
JIA ACR 30
Responders Weighted difference a (95% CI) b
91%
67
(51, 83)24%
-JIA ACR 50
Responders Weighted difference a (95% CI) b
85%
74
(58, 90)11%
-JIA ACR 70
Responders Weighted difference a (95% CI) b
71%
63
(46, 80)8%
-The treatment effect of tocilizumab was consistent across all components of the JIA ACR response core variables. JIA ACR scores and absence of fever responses in the open label extension were consistent with the controlled portion of the study (data available through 44 weeks).
Systemic Features
Of patients with fever or rash at baseline, those treated with tocilizumab had fewer systemic features; 35 out of 41 (85%) became fever free (no temperature recording at or above 37.5°C in the preceding 14 days) compared to 5 out of 24 (21%) of placebo-treated patients, and 14 out of 22 (64%) became free of rash compared to 2 out of 18 (11%) of placebo-treated patients. Responses were consistent in the open label extension (data available through 44 weeks).
Corticosteroid Tapering
Of the patients receiving oral corticosteroids at baseline, 8 out of 31 (26%) placebo and 48 out of 70 (69%), tocilizumab patients achieved a JIA ACR 70 response at week 6 or 8 enabling corticosteroid dose reduction. Seventeen (24%) tocilizumab patients versus 1 (3%) placebo patient were able to reduce the dose of corticosteroid by at least 20% without experiencing a subsequent JIA ACR 30 flare or occurrence of systemic symptoms to week 12. In the open label portion of the study, by week 44, there were 44 out of 103 (43%) tocilizumab patients off oral corticosteroids. Of these 44 patients 50% were off corticosteroids 18 weeks or more.
Health Related Outcomes
Physical function and disability were assessed using the Childhood Health Assessment Questionnaire Disability Index (CHAQ-DI). Seventy-seven percent (58 out of 75) of patients in the tocilizumab treatment group achieved a minimal clinically important improvement in CHAQ-DI (change from baseline of ≥ 0.13 units) at week 12 compared to 19% (7 out of 37) in the placebo treatment group.
14.8 Systemic Juvenile Idiopathic Arthritis - Subcutaneous Administration
Subcutaneously administered tocilizumab in pediatric patients with systemic juvenile idiopathic arthritis (SJIA) was assessed in WA28118 (NCT01904292), a 52-week, open-label, multicenter, PK-PD and safety study to determine the appropriate subcutaneous dose of tocilizumab that achieved comparable PK/PD profiles to the tocilizumab-IV regimen.
Eligible patients received tocilizumab subcutaneously dosed according to body weight, with patients weighing at or above 30 kg (n = 26) dosed with 162 mg of tocilizumab every week and patients weighing below 30 kg (n = 25) dosed with 162 mg of tocilizumab every 10 days (n=8) or every 2 weeks (n=17) for 52 weeks. Of these 51 patients, 26 (51%) were naïve to subcutaneous tocilizumab and 25 (49%) had been receiving tocilizumab intravenously and switched to subcutaneous tocilizumab at baseline.
The efficacy of subcutaneous tocilizumab in children 2 to 17 years of age is based on pharmacokinetic exposure and extrapolation of the established efficacy of intravenous tocilizumab in systemic JIA patients [see Clinical Pharmacology (12.3) and Clinical Studies (14.8)].
14.9 Cytokine Release Syndrome – Intravenous Administration
The efficacy of tocilizumab for the treatment of CRS was assessed in a retrospective analysis of pooled outcome data from clinical trials of CAR T-cell therapies for hematological malignancies. Evaluable patients had been treated with tocilizumab 8 mg/kg (12 mg/kg for patients < 30 kg) with or without additional high-dose corticosteroids for severe or life-threatening CRS; only the first episode of CRS was included in the analysis. The study population included 24 males and 21 females (total 45 patients) of median age 12 years (range, 3–23 years); 82% were Caucasian. The median time from start of CRS to first dose of tocilizumab was 4 days (range, 0-18 days). Resolution of CRS was defined as lack of fever and off vasopressors for at least 24 hours. Patients were considered responders if CRS resolved within 14 days of the first dose of tocilizumab, if no more than 2 doses of tocilizumab were needed, and if no drugs other than tocilizumab and corticosteroids were used for treatment. Thirty-one patients (69%; 95% CI: 53%–82%) achieved a response. Achievement of resolution of CRS within 14 days was confirmed in a second study using an independent cohort that included 15 patients (range: 9–75 years old) with CAR T cell-induced CRS.
14.10 COVID-19 – Intravenous Administration
The efficacy of tocilizumab for the treatment of COVID-19 was based on RECOVERY (NCT04381936), a randomized, controlled, open-label, platform study, and supported by the results from EMPACTA (NCT04372186), a randomized, double-blind, placebo-controlled study. Results of two other randomized, double-blind, placebo-controlled studies, COVACTA (NCT04320615) and REMDACTA (NCT04409262), which evaluated the efficacy of tocilizumab for the treatment of COVID-19 are also summarized.
RECOVERY (Randomised Evaluation of COVID-19 Therapy) Collaborative Group Study in Hospitalized Adults Diagnosed with COVID-19
RECOVERY was a randomized, controlled, open-label, multicenter platform study conducted in the United Kingdom to evaluate the efficacy and safety of potential treatments in hospitalized adult patients with severe COVID-19 pneumonia. Eligible patients for the tocilizumab portion of the study had clinically suspected or laboratory-confirmed SARS-CoV-2 infection and no medical contraindications to any of the treatments and had clinical evidence of progressive COVID-19 (defined as oxygen saturation <92% on room air or receiving oxygen therapy, and CRP ≥75 mg/L). Patients were then randomized to receive either standard of care (SoC) or intravenous tocilizumab at a weight-tiered dosing comparable to the recommended dosage [see Clinical Pharmacology (12.3)] in addition to SoC.
Efficacy analyses were performed in the intent-to-treat (ITT) population comprising 4116 adult patients who were randomized to the tocilizumab + SoC arm (n=2022) or to the SoC arm (n=2094). The mean age of participants was 64 years (range: 20 to 101), and patients were 67% male, 76% White, 11% Asian, 3% Black or African American, and 1% mixed race. At baseline, 0.2% of patients were not on supplemental oxygen, 45% of patients required low flow oxygen, 41% of patients required non-invasive ventilation or high-flow oxygen, and 14% of patients required invasive mechanical ventilation; 82% of patients were reported to be receiving systemic corticosteroids. The primary efficacy endpoint was time to death through Day 28. The results for the overall population and the subgroups of patients who were or were not receiving systemic corticosteroids at time of randomization are summarized in Table 11.
Table 11 Mortality through Day 28 in RECOVERY 1 P-value reflects that the RECOVERY trial primary analysis results were statistically significant at the two-sided significance level of 𝛼 = 0.05. 2 Probabilities of dying by Day 28 were estimated by the Kaplan-Meier method. Tocilizumab +SoC
N=2022
n (%)1SoC
N=2094
n (%)1Hazard Ratio
(95% CI)Risk Difference
(95% CI)Mortality
621 (30.7%)
729 (34.9%)
0.85 (0.76, 0.94)
p= 0.00281-4.1% (-7.0, -1.3)
By baseline receipt of corticosteroid use
Mortality for patients receiving systemic corticosteroids at randomization2
482/1664
(29.0%)600/1721
(34.9%)0.79 (0.70, 0.89)
-5.9% (-9.1, -2.8)
Mortality for patients not receiving systemic corticosteroids at randomization2
139/357
(39.0%)127/367
(34.6%)1.16 (0.91, 1.48)
4.4% (-2.6, 11.5)
EMPACTA
EMPACTA was a randomized, double-blind, placebo-controlled, multicenter study to evaluate intravenous tocilizumab 8 mg/kg in combination with SoC in hospitalized, non-ventilated adult patients with COVID-19 pneumonia. Eligible patients were at least 18 years of age, had confirmed SARS-CoV-2 infection by a positive reverse-transcriptase polymerase chain reaction (RT-PCR) result, had pneumonia confirmed by radiography, and had SpO2 < 94% on ambient air.
Of the 389 patients randomized, efficacy analyses were performed in the modified intent-to-treat (mITT) population comprising 377 patients who were randomized and received study medication (249 in the tocilizumab arm; 128 in the placebo arm). The mean age of participants was 56 years (range: 20 to 95); 59% of patients were male, 56% were of Hispanic or Latino ethnicity, 53% were White, 20% were American Indian/Alaska Native, 15% were Black/African American and 2% were Asian. At baseline, 9% patients were not on supplemental oxygen, 64% patients required low flow oxygen, 27% patients required high-flow oxygen, and 73% were on corticosteroids.
The primary efficacy endpoint evaluated time to progression to mechanical ventilation or death through Day 28. The hazard ratio comparing tocilizumab to placebo was 0.56 (95% CI, 0.33 to 0.97), a statistically significant result (log-rank, p-value = 0.036). The cumulative proportion of patients who required mechanical ventilation or died by Day 28 was 12.0% (95% CI, 8.5% to 16.9%) in the tocilizumab arm and 19.3% (95% CI, 13.3% to 27.4%) in the placebo arm.
Mortality at Day 28 was 10.4% in the tocilizumab arm versus 8.6% in the placebo arm (weighted difference (tocilizumab arm - placebo arm): 2.0% [95% CI, -5.2% to 7.8%]).
COVACTA
COVACTA was a randomized, double-blind, placebo-controlled, multicenter study to evaluate intravenous tocilizumab 8 mg/kg in combination with SoC for the treatment of adult patients hospitalized with severe COVID-19 pneumonia. The study randomized 452 patients who were at least 18 years of age with confirmed SARS-CoV2 infection by a positive RT-PCR result, had pneumonia confirmed by radiography, and had oxygen saturation of 93% or lower on ambient air or a ratio of arterial oxygen partial pressure to fractional inspired oxygen of 300 mmHg or less. At baseline, 3% of patients were not on supplemental oxygen, 28% were on low flow oxygen, 30% were on non-invasive ventilation or high flow oxygen, 38% were on invasive mechanical ventilation, and 22% were on corticosteroids. The primary efficacy endpoint was clinical status on Day 28 assessed on a 7-category ordinal scale that ranged from “discharged” to “death.” There were no statistically significant differences observed in the distributions of clinical status on the 7-category ordinal scale at Day 28 when comparing the tocilizumab arm to the placebo arm.
Mortality at Day 28 was 19.7% in the tocilizumab arm versus 19.4% in the placebo arm (weighted difference (tocilizumab arm - placebo arm): 0.3% [95% CI, -7.6 to 8.2]).
REMDACTA
REMDACTA was a randomized, double-blind, placebo-controlled, multicenter study to evaluate intravenous tocilizumab 8 mg/kg in combination with intravenous remdesivir (RDV) 200 mg on Day 1 followed by 100 mg once daily for a total of 10 days in hospitalized patients with severe COVID-19 pneumonia. The study randomized 649 adult patients with SARS-CoV-2 infection confirmed by a positive polymerase chain reaction (PCR) result, pneumonia confirmed by radiography, and who required supplemental oxygen > 6 L/min to maintain SpO2 >93%. At baseline, 7% of patients were on low flow oxygen, 80% were on non-invasive ventilation or high flow oxygen, 14% were on invasive mechanical ventilation, and 84% were on corticosteroids.
The primary efficacy endpoint was time from randomization to hospital discharge or ‘ready for discharge’ up to Day 28. There was no statistically significant difference between the treatment arms with respect to time to hospital discharge or “ready for discharge” through Day 28.
Mortality at Day 28 was 18.1% in the tocilizumab + RDV arm versus 19.5% in the placebo +RDV arm (weighted difference (tocilizumab arm - placebo arm): -1.3% [95% CI, -7.8% to 5.2%]).
Mortality Across Studies in Patients Receiving Baseline Corticosteroids
A study-level meta-analysis was conducted on EMPACTA, COVACTA, REMDACTA and RECOVERY studies. For each of the four studies, the risk difference through Day 28 was estimated by the Kaplan-Meier method in the subgroup of patients receiving baseline corticosteroids, summarized in Figure 2. Patients from the RECOVERY trial represent 78.8% of the total sample size in this meta-analysis. The combined risk difference showed that tocilizumab treatment (n=2261) resulted in a 4.61% absolute reduction in the risk of death at Day 28 (risk difference=-4.6%; 95% CI: -7.3% to -1.9%) compared to SoC (n=2034).
Figure 2 Risk Differences Through Day 28 for Baseline Corticosteroid Use Subpopulation in RECOVERY, EMPACTA, COVACTA and REMDACTA studies
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied:
TYENNE (tocilizumab-aazg) injection is a preservative-free, sterile clear and colorless to pale yellow solution. The following packaging configurations are available:
For Intravenous Infusion
TYENNE Single-Dose Vial
Each TYENNE carton contains one vial. Each vial is supplied as 80 mg/4 mL (20 mg/mL) (NDC: 65219-590-04), 200 mg/10 mL (20 mg/mL) (NDC: 65219-592-10), and 400 mg/20 mL (20 mg/mL) (NDC: 65219-594-20) TYENNE solution for further dilution prior to intravenous infusion. The vial stopper is not made with natural rubber latex.
For Subcutaneous Injection
TYENNE Prefilled Syringe
Each TYENNE carton contains a single-dose prefilled syringe delivering 162 mg/0.9 mL of TYENNE. The syringe plunger stopper and needle cover are not made with natural rubber latex. The NDC number is 65219-586-04.
TYENNE Autoinjector
Each TYENNE carton contains a single-dose autoinjector delivering 162 mg/0.9 mL of TYENNE. The syringe plunger stopper and needle cover are not made with natural rubber latex. The NDC number is 65219-584-01.
Storage and Handling: Do not use beyond expiration date on the container, package, prefilled syringe, or autoinjector. TYENNE must be refrigerated at 36°F to 46°F (2ºC to 8ºC). Do not freeze. A single prefilled syringe (or autoinjector) may be stored at room temperature at or below 77°F (25°C) for a single period of up to 14 days. Protect the vials, syringes and autoinjectors from light by storage in the original package until time of use, and keep syringes and autoinjectors dry. Do not shake.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
- Serious Infections
Inform patients that TYENNE may lower their resistance to infections [see Warnings and Precautions (5.1)]. Instruct the patient of the importance of contacting their doctor immediately when symptoms suggesting infection appear in order to assure rapid evaluation and appropriate treatment.
- Gastrointestinal Perforation
Inform patients that some patients who have been treated with TYENNE have had serious side effects in the stomach and intestines [see Warnings and Precautions (5.2)]. Instruct the patient of the importance of contacting their doctor immediately when symptoms of fever, severe, persistent abdominal pain, and change in bowel habits appear to assure rapid evaluation and appropriate treatment.
- Hypersensitivity and Serious Allergic Reactions
Inform patients that some patients who have been treated with TYENNE have developed serious allergic reactions, including anaphylaxis, as well as serious skin reactions [see Warnings and Precautions (5.6)]. Advise patients to stop taking TYENNE and seek immediate medical attention if they experience any symptom of serious allergic reactions (including rash, hives, and swelling of the face, lips, tongue, and throat that may cause difficulty in breathing or swallowing).
Instruction on Injection Technique
Assess patient suitability for home use for subcutaneous injection. Perform the first injection under the supervision of a qualified healthcare professional. If a patient or caregiver is to administer subcutaneous TYENNE, instruct him/her in injection techniques and assess his/her ability to inject subcutaneously to ensure proper administration of subcutaneous TYENNE and the suitability for home use [See Instructions for Use].
Prior to use, remove the prefilled syringe (PFS) or autoinjector from the refrigerator and allow to sit at room temperature outside of the carton for 30 minutes (PFS) or 45 minutes (autoinjector), out of the reach of children.
Do not warm TYENNE in any other way.
Advise patients to consult their healthcare provider if the full dose is not received.
A puncture-resistant container for disposal of needles, syringes and autoinjectors should be used and should be kept out of the reach of children. Instruct patients or caregivers in the technique as well as proper needle, syringe and autoinjector disposal, and caution against reuse of these items.
Pregnancy
Inform female patients of reproductive potential that TYENNE may cause fetal harm and to inform their prescriber of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
TYENNE (tocilizumab-aazg)
Manufactured by:
Fresenius Kabi USA, LLC
Lake Zurich, IL 60047, U.S.A.U.S. License Number: 2146
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: 02/2025 Medication Guide
TYENNE® (tye en')
(tocilizumab-aazg)
injection for intravenous use
TYENNE® (tye en')
(tocilizumab-aazg)
injection for subcutaneous use
What is the most important information I should know about TYENNE?
TYENNE can cause serious side effects including:
- 1. Serious Infections. TYENNE is a medicine that affects your immune system. TYENNE can lower the ability of your immune system to fight infections. Some people have serious infections while taking TYENNE, including tuberculosis (TB), and infections caused by bacteria, fungi, or viruses that can spread throughout the body. Some people have died from these infections. Your healthcare provider should assess you for TB before starting TYENNE (except if you have COVID-19).
If you have COVID-19, your healthcare provider should monitor you for signs and symptoms of new infections during and after treatment with TYENNE.
Your healthcare provider should monitor you closely for signs and symptoms of TB during and after treatment with
TYENNE- You should not start taking TYENNE if you have any kind of infection unless your healthcare provider says it is okay.
Before starting TYENNE, tell your healthcare provider if you:
- think you have an infection or have symptoms of an infection, with or without a fever, such as:
-
- o sweating or chills
- o shortness of breath
- o warm, red, or painful skin or sores on your body
-
- o feel very tired
- o muscle aches
- o blood in phlegm
- o diarrhea or stomach pain
-
- o cough
- o weight loss
- o burning when you urinate more often than normal
- are being treated for an infection.
- get a lot of infections or have infections that keep coming back.
- have diabetes, HIV, or a weak immune system. People with these conditions have a higher chance for infections.
- have TB or have been in close contact with someone with TB.
- live or have lived or have traveled to certain parts of the country (such as the Ohio and Mississippi River valleys and the Southwest) where there is an increased chance for getting certain kinds of fungal infections (histoplasmosis, coccidiomycosis, or blastomycosis). These infections may happen or become more severe if you use TYENNE. Ask your healthcare provider if you do not know if you have lived in an area where these infections are common.
- have or have had hepatitis B.
After starting TYENNE, call your healthcare provider right away if you have any symptoms of an infection. TYENNE can make you more likely to get infections or make worse any infection that you have.
- 2.
Tears (perforation) of the stomach or intestines.
- Tell your healthcare provider if you have had diverticulitis (inflammation in parts of the large intestine) or ulcers in your stomach or intestines. Some people taking TYENNE get tears in their stomach or intestine. This happens most often in people who also take nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, or methotrexate.
- Tell your healthcare provider right away if you have fever and stomach-area pain that does not go away, and a change in your bowel habits.
- 3. Liver problems (Hepatotoxicity): Some people have experienced serious life-threatening liver problems, which required a liver transplant or led to death. Your healthcare provider may tell you to stop taking TYENNE if you develop new or worse liver problems during treatment with TYENNE. Tell your healthcare provider right away if you have any of the following symptoms:
-
- o feeling tired (fatigue)
- o lack of appetite for several days or longer (anorexia)
- o yellowing of your skin or the whites of your eyes (jaundice)
- o abdominal swelling and pain on the right side of your stomach-area
- o light colored stools
-
- o Weakness
- o nausea and vomiting
- o confusion
- o dark “tea-colored” urine
- 4.
Changes in certain laboratory test results. Your healthcare provider should do blood tests before you start receiving TYENNE. If you have rheumatoid arthritis (RA) or giant cell arteritis (GCA) your healthcare provider should do blood tests every 4 to 8 weeks after you start receiving TYENNE for the first 6 months and then every 3 months after that. If you have polyarticular juvenile idiopathic arthritis (PJIA) you will have blood tests done every 4 to 8 weeks during treatment. If you have systemic juvenile idiopathic arthritis (SJIA) you will have blood tests done every 2 to 4 weeks during treatment. These blood tests are to check for the following side effects of TYENNE:
- low neutrophil count. Neutrophils are white blood cells that help the body fight off bacterial infections.
- low platelet count. Platelets are blood cells that help with blood clotting and stop bleeding.
- increase in certain liver function tests.
- increase in blood cholesterol levels. You may also have changes in other laboratory tests, such as your blood cholesterol levels. Your healthcare provider should do blood tests to check your cholesterol levels 4 to 8 weeks after you start receiving TYENNE.
Your healthcare provider will determine how often you will have follow-up blood tests. Make sure you get all your follow-up blood tests done as ordered by your healthcare provider.
You should not receive TYENNE if your neutrophil or platelet counts are too low, or your liver function tests are too high.
Your healthcare provider may stop your TYENNE treatment for a period of time or change your dose of medicine if needed because of changes in these blood test results.
- 5.
Cancer. TYENNE may increase your risk of certain cancers by changing the way your immune system works. Tell your healthcare provider if you have ever had any type of cancer.
See “What are the possible side effects with TYENNE?” for more information about side effects.
What is TYENNE?
TYENNE is a prescription medicine called an Interleukin-6 (IL-6) receptor antagonist. TYENNE is used:
- To treat adults with moderately to severely active rheumatoid arthritis (RA), after at least one other medicine called a Disease-Modifying Anti-Rheumatic Drug (DMARD) has been used and did not work well.
- To treat adults with giant cell arteritis (GCA).
- To treat people with active PJIA ages 2 and above.
- To treat people with active SJIA ages 2 and above.
- To treat people age 2 years and above who experience severe or life-threatening Cytokine Release Syndrome (CRS) following chimeric antigen receptor (CAR) T cell treatment.
- To treat hospitalized adults with coronavirus disease 2019 (COVID-19) receiving systemic corticosteroids and requiring supplemental oxygen or mechanical ventilation.
- TYENNE is not approved for subcutaneous use in people with CRS or COVID-19.
It is not known if TYENNE is safe and effective in children with PJIA or SJIA or CRS under 2 years of age or in children with conditions other than PJIA or SJIA or CRS.
Do not take TYENNE: if you are allergic to tocilizumab products, or any of the ingredients in TYENNE. See the end of this Medication Guide for a complete list of ingredients in TYENNE.
Before you receive TYENNE, tell your healthcare provider about all of your medical conditions, including if you:
- have an infection. See “What is the most important information I should know about TYENNE?”
- have liver problems.
- have any stomach-area (abdominal) pain or been diagnosed with diverticulitis or ulcers in your stomach or intestines.
- have had a reaction to tocilizumab or any of the ingredients in TYENNE before.
- have or had a condition that affects your nervous system, such as multiple sclerosis.
-
have recently received or are scheduled to receive a vaccine:
- o All vaccines should be brought up-to-date before starting TYENNE, unless urgent treatment initiation is required.
- o People who take TYENNE should not receive live vaccines.
- o People taking TYENNE can receive non-live vaccines.
- plan to have surgery or a medical procedure.
- are pregnant or plan to become pregnant or are pregnant. TYENNE may harm your unborn baby. Tell your healthcare provider if you become pregnant or think you may be pregnant during treatment with TYENNE.
-
are breastfeeding or plan to breastfeed. It is not known if TYENNE passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby if you take TYENNE.
Tell your healthcare provider about all of the medicines you take, including prescription, over-the-counter medicines, vitamins, and herbal supplements. TYENNE and other medicines may affect each other causing side effects.
Especially tell your healthcare provider if you take:
- any other medicines to treat your RA. You should not take etanercept (Enbrel), adalimumab (Humira), infliximab (Remicade), rituximab (Rituxan), abatacept (Orencia), anakinra (Kineret), certolizumab (Cimzia), or golimumab (Simponi) while you are taking TYENNE. Taking TYENNE with these medicines may increase your risk of infection.
- medicines that affect the way certain liver enzymes work. Ask your healthcare provider if you are not sure if your medicine is one of these.
Know the medicines you take. Keep a list of them to show to your healthcare provider and pharmacist when you get a new medicine.
How will I receive TYENNE?
Into a vein (IV or intravenous infusion) for Rheumatoid Arthritis, Giant Cell Arteritis, PJIA, SJIA, CRS, or COVID-19:
- If your healthcare provider prescribes TYENNE as an IV infusion, you will receive TYENNE from a healthcare provider through a needle placed in a vein in your arm. The infusion will take about 1 hour to give you the full dose of medicine.
- For rheumatoid arthritis, giant cell arteritis or PJIA you will receive a dose of TYENNE about every 4 weeks.
- For SJIA you will receive a dose of TYENNE about every 2 weeks.
- For CRS you will receive a single dose of TYENNE, and if needed, additional doses.
- For COVID-19, you will receive a single dose of TYENNE, and if needed one additional dose.
- While taking TYENNE, you may continue to use other medicines that help treat your rheumatoid arthritis, PJIA, SJIA or COVID-19 such as methotrexate, non-steroidal anti-inflammatory drugs (NSAIDs) and prescription steroids, as instructed by your healthcare provider.
- Keep all of your follow-up appointments and get your blood tests as ordered by your healthcare provider.
Under the skin (SC or subcutaneous injection) for Rheumatoid Arthritis, Giant Cell Arteritis, PJIA or SJIA:
- See the Instructions for Use at the end of this Medication Guide for instructions about the right way to prepare and give your TYENNE injections at home.
- TYENNE is available as a single-dose prefilled syringe or single-dose prefilled autoinjector.
- You may also receive TYENNE as an injection under your skin (subcutaneous). If your healthcare provider decides that you or a caregiver can give your injections of TYENNE at home, you or your caregiver should receive training on the right way to prepare and inject TYENNE. Do not try to inject TYENNE until you have been shown the right way to give the injections by your healthcare provider.
- For PJIA or SJIA, you may self-inject with the prefilled syringe or prefilled autoinjector, or your caregiver can give you TYENNE, if both your healthcare provider and parent/legal guardian find it appropriate.
- Your healthcare provider will tell you how much TYENNE to use and when to use it.
What are the possible side effects with TYENNE?
TYENNE can cause serious side effects, including:- See “What is the most important information I should know about TYENNE?”
- Hepatitis B infection in people who carry the virus in their blood. If you are a carrier of the hepatitis B virus (a virus that affects the liver), the virus may become active while you use TYENNE. Your healthcare provider may do blood tests before you start treatment with TYENNE and while you are using TYENNE. Tell your healthcare provider if you have any of the following symptoms of a possible hepatitis B infection:
-
- o feel very tired
- o vomiting
- o chills
- o dark urine
-
- o skin or eyes look yellow
- o clay-colored bowel movements
- o stomach discomfort
- o skin rash
-
- o little or no appetite
- o fevers
- o muscle aches
-
Serious Allergic Reactions: Serious: Allergic Reactions. Serious allergic reactions, including death, can happen with TYENNE. These reactions can happen with any infusion or injection of TYENNE, even if they did not occur with an earlier infusion or injection. Stop taking TYENNE, contact your healthcare provider, and get emergency help right away if you have any of the following signs of a serious allergic reaction:
- o swelling of your face, lips, mouth, or tongue
- o trouble breathing
- o wheezing
- o severe itching
- o skin rash, hives, redness, or swelling outside of the injection site are
- o dizziness or fainting
- o fast heartbeat or pounding in your chest (tachycardia)
- o sweating
-
Nervous system problems. While rare, Multiple Sclerosis has been diagnosed in people who take TYENNE. It is not known what effect TYENNE may have on some nervous system disorders.
The most common side effects of TYENNE include:
- upper respiratory tract infections (common cold, sinus infections)
- headache
- increased blood pressure (hypertension)
- injection site reactions
Tell your healthcare provider about any side effect that bothers you or does not go away. These are not all the possible side effects of TYENNE. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to Fresenius Kabi USA, LLC at 1-800-551-7176.
General information about the safe and effective use of TYENNE.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not give TYENNE to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TYENNE that is written for health professionals.
For more information go to www.TYENNE.com or you can enroll in a patient support program by calling 1-833-522-4227 or visiting the patient support program website: www.kabicare.com.
What are the ingredients in TYENNE?
Active ingredient: tocilizumab-aazg.
Inactive ingredients of Intravenous and Subcutaneous TYENNE: arginine, histidine, hydrochloric acid, lactic acid, polysorbate 80, sodium chloride, sodium hydroxide and Water for Injection.
TYENNE is a registered trademark of Fresenius Kabi
Manufactured by: Fresenius Kabi USA LLC, Lake Zurich, IL 60047, U.S.A
U.S License Number 2146
-
INSTRUCTIONS FOR USE
Read and follow the Instructions for Use that come with your TYENNE autoinjector before you start using it and each time you get a refill. There may be new information. This information does not replace talking to your healthcare provider about your medical condition or treatment.
If you have any questions about using your TYENNE autoinjector, please call your healthcare provider or Company at 1-833-522-4227.
Important information
- Read the Medication Guide that comes with your autoinjector for important information you need to know before using it.
- Before you use the autoinjector for the first time, make sure your healthcare provider shows you the right way to use it.
- Do not try to take apart the TYENNE autoinjector at any time.
- Always inject TYENNE the way your healthcare provider taught you to.
Using the TYENNE autoinjector
- The autoinjector is for self-injection or administration with the help of a caregiver.
- The autoinjector is for use at home.
- When injecting TYENNE, children may self-inject if both the healthcare provider and caregiver find it appropriate.
- Do not reuse the autoinjector. The autoinjector is for single-dose (1-time) use only.
- Do not share your autoinjector with another person. You may give another person an infection or get an infection from them.
- Do not remove the autoinjector clear cap until you are ready to inject.
- Do not use the autoinjector if it shows any signs of damage or if it has been dropped.
Storing TYENNE autoinjectors
- Store TYENNE in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Store unused autoinjectors in the original carton to protect from light.
- Do not freeze TYENNE. Do not use TYENNE if frozen. Throw away (dispose of) in a sharps disposal container.
- Do not expose TYENNE to heat or direct sunlight.
- Keep the autoinjector out of the reach of children.
-
TYENNE may be stored at room temperature at or below 77°F (25°C) in the original carton for up to 14 days. Throw away (dispose of) TYENNE in a sharp disposal container if it has been out of the refrigerator more than 14 days. After being stored at room temperature, do not place it back in the refrigerator.
See the Medication Guide for more information.
Traveling with TYENNE autoinjector
- When travelling on an airplane, always check with your airline and your healthcare provider about bringing injectable medicine with you. Always carry TYENNE in your carry-on luggage because the airplane luggage area can be very cold and TYENNE could freeze.
Your TYENNE autoinjector
Before Use
After Use
1.1. Prepare a clean, flat surface, such as a table or countertop, in a well-lit area.
1.2. Gather the following supplies (not included) (see Figure A):
- A sterile cotton ball or gauze
- An alcohol pad
- An FDA-cleared sharps disposal container (see Step 8, “Throw away your autoinjector”).
1.3. Remove the carton containing the autoinjector from the refrigerator.
1.6. Remove the tray from the carton.
2.1 Check the autoinjector to make sure it is not cracked or damaged (see Figure E).
Do not use if the autoinjector shows signs of damage or if it has been dropped.
4.1 If you are giving yourself the injection, you can use:
If a caregiver is giving the injection, they can use the outer area of the upper arm (see Figure I).
Note: Choose a different site for each injection to reduce redness, irritation, or other skin problems.
Do not inject into skin that is sore (tender), bruised, red, hard, scaly, or has lesions, moles, scars, or stretch marks or tattoos.
Do not use the autoinjector through clothing.5.1 Wipe the skin where you want to inject with an alcohol pad to clean it (see Figure J). Let the skin dry.
Do not blow on or touch the site after cleaning.
6.2 Throw away the clear cap in a sharps disposal container.
6.3 Rotate the autoinjector so that the orange needle cover points downwards.
6.4 Position your hand on the autoinjector so that you can see the window.
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at http://www.fda.gov/safesharpsdisposal
Do not throw away (dispose of) your used sharps disposal container in your household trash unless your community guidelines permit this.
Do not recycle your used sharps disposal container.
Always keep the sharps disposal container out of reach of children.
Manufactured by:
Fresenius Kabi USA, LLC
Lake Zurich, Illinois 60047
U.S. License No. 2146This Instructions for Use has been approved by the U.S. Food and Drug Administration Revised: 02/2025
Read and follow the Instructions for Use that come with your TYENNE prefilled syringe before you start using it and each time you get a refill. There may be new information. This does not replace talking to your healthcare provider about your medical condition or treatment.
If you have any questions about using your TYENNE prefilled syringe, please call your healthcare provider or Company at 1-833-522-4227.
Important Information
- Read the Medication Guide that comes with your TYENNE prefilled syringe for important information you need to know before using it.
- Before you use the prefilled syringe for the first time, make sure your healthcare provider shows you the right way to use it.
- Do not remove the needle cap until you are ready to inject TYENNE.
- Do not try to take apart the prefilled syringe at any time.
- Always inject TYENNE the way your healthcare provider taught you.
Using TYENNE prefilled syringe
- The prefilled syringe is for self-injection or administration with the help of a caregiver.
- Do not use the prefilled syringe if the carton is open or damaged.
-
Do not use the prefilled syringe if it has been dropped on a hard surface.
The prefilled syringe may be broken even if you cannot see the break.
Storing TYENNE prefilled syringes
- Store TYENNE in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Store unused prefilled syringes in the original carton to protect from light.
- Keep the prefilled syringes out of the reach of children.
- Do not freeze TYENNE. Do not use TYENNE if frozen. Throw away (dispose of) in a sharps disposal container.
- Do not expose TYENNE to heat or direct sunlight.
- If needed, for example when traveling, TYENNE may be stored at room temperature at or below 77°F (25°C) in the original carton for up to 14 days. Throw away (dispose of) TYENNE in a sharps disposal container if it has been out of the refrigerator for more than 14 days. After being stored at room temperature, do not place it back in the refrigerator.
See the Medication Guide for more information.
Your TYENNE prefilled syringe (see Figure A)
Do not try to activate the clear needle guard before injecting.
Do not insert your fingers into the opening of the clear needle guard because the needle could injure you.
1.1 Find a comfortable space with a clean, flat working surface.
1.2 Supplies needed for your TYENNE prefilled syringe injection (see Figure B):
- TYENNE prefilled syringe
- An alcohol pad
- A sterile cotton ball or gauze
- Puncture-resistant container or sharps container for safe disposal of used syringe (see Step 7 “Throw away used prefilled syringe”)
1.3 Take the box containing the syringe out of the refrigerator and open the box (see Figure C).
1.4 Remove the plastic tray out of the box and let it warm up for 30 minutes to reach room temperature (see Figure D). If the syringe does not reach room temperature, this could cause your injection to feel uncomfortable and make it difficult to push the plunger in.
- Do not speed up the warming process in any way, such as in a microwave or placing the syringe in hot water.
- Prepare and check your records of previous injection sites. This will help you choose the appropriate injection site for this injection (see Step 8 “Record your injection”).
Remove TYENNE prefilled syringe from the plastic tray
- Place two fingers on either side, in the middle of the clear needle guard.
- Pull the prefilled syringe straight up and out of the tray (see Figure E).
- Do not pick up the prefilled syringe by the plunger or the needle cap. Doing so could damage the prefilled syringe or activate the clear needle guard.
2.1 Check the expiration date on the TYENNE prefilled syringe (see Figure F).
Do not use it if the expiration date has passed because it may not be safe to use. If the expiration date has passed, safely dispose of the syringe in a sharps container and get a new one.
2.2 Check the name on the prefilled syringe says TYENNE (see Figure F).
2.3 Check the liquid in the TYENNE prefilled syringe. It should be clear and colorless to pale yellow (see Figure G).
Do not inject TYENNE if the liquid is cloudy, discolored or has lumps or particles in it because it may not be safe to use. Safely dispose of the syringe in a sharps container and get a new one.
2.4 Check the prefilled syringe to make sure that:
- The prefilled syringe, the clear needle guard, and the needle cap are not cracked or damaged (see Figure H).
- The needle cap is securely attached (see Figure I).
- The needle guard spring is not extended (see Figure J).
- Do not use the syringe if it shows any sign of damage. If damaged, call your healthcare provider or pharmacist right away and throw away the syringe in your sharps disposal container (see Step 7 “Throw away used prefilled syringe”).
Wash your hands well with soap and water (see Figure K).
4.1 Choose an injection site
- The front of the thighs and your belly (abdomen) except for 2-inch area around your navel are the recommended injection sites (see Figure L).
- The outer area of the upper arms may also be used only if the injection is being given by a caregiver. Do not attempt to use the upper arm area by yourself (see Figure M).
4.2 Rotate injection site
- Choose a different injection site for each new injection.
- Do not inject into moles, scars, bruises, or areas where the skin is tender, red, hard or not intact.
5.1 Clean the injection site
- Wipe the injection site with an alcohol pad in a circular motion and let it air dry to reduce the chance of getting an infection. Do not touch the injection site again before giving the injection.
- Do not fan or blow on the clean area.
6.1 Remove the needle cap.
-
Hold the TYENNE prefilled syringe by the clear needle guard with 1 hand and pull the needle cap straight off with your other hand (see Figure N).
- Do not hold the plunger while you remove the needle cap. If you cannot remove the needle cap, you should ask a caregiver for help or contact your healthcare provider.
- Throw away the needle cap in your sharps container.
- There may be a small air bubble in the TYENNE prefilled syringe. You do not need to remove it.
- You may see drops of liquid at the end of the needle. This is normal and will not affect your dose.
- Do not touch the needle or let it touch any surfaces.
- Do not use the prefilled syringe if it is dropped.
- If it is not used within 5 minutes of needle cap removal, the syringe should be disposed of in the puncture resistant container or sharps container and a new syringe should be used.
- Never reattach the needle cap after removal.
6.2 Pinch the skin
- Hold the TYENNE prefilled syringe in 1 hand between the thumb and index finger (see Figure O).
- Do not pull back on the plunger of the syringe.
- Use your other hand and gently pinch the area of skin firmly (see Figure P). Pinching the skin is important to make sure that you inject under the skin (into fatty tissue) but not any deeper (into muscle). Injection into muscle could cause the injection to feel uncomfortable.
6.3 Insert the needle
- Use a quick, dart-like motion to insert the needle all the way into the pinched skin at an angle between 45° to 90° (see Figure Q). It is important to use the correct angle to make sure the medicine is delivered under the skin (into fatty tissue), or the injection could be painful, and the medicine may not work.
- Keep the syringe in position.
- Do not hold or push on the plunger while inserting the needle into the skin.
6.4 Inject
- Slowly inject all of the medicine by gently pushing the plunger all the way down (see Figure R).
- You must press the plunger all the way down to get the full dose of medicine until you cannot press any more (see Figure S).
- Do not pull the needle out of the skin when the plunger is pushed all the way down.
6.5 Finish Injection
- After the plunger is pushed all the way down, hold the syringe firmly without moving it, at the same angle as inserted.
- Slowly release your thumb off the plunger. The plunger will move up. The safety system will remove the needle from the skin and cover the needle (see Figure T).
- Release the pinched skin
Important: Call your healthcare provider right away if you did not inject the full dose. Injecting an incorrect amount of medicine could affect your treatment.
Do not reuse a syringe even if all of the medicine was not injected.
Do not try to recap the needle as it could lead to needle stick injury.
6.6 After the injection
- There may be a little bleeding at the injection site. You can press a cotton ball or gauze on the injection site.
- Do not rub the injection site.
- If needed, you may cover the injection site with a small bandage.
- The TYENNE prefilled syringe should not be reused.
- Do not put the needle cap back on the needle
- If your injection is given by another person, this person must also be careful when removing the syringe and disposing of the syringe to prevent accidental needle stick injury and passing injection.
How do I throw away used syringes?
Put your used syringe in an FDA-cleared sharps disposal container right away after use (see Figure U). Do not throw away (dispose of) loose needles and syringes in your household trash.
-
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant and
-
properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal
- Do not throw away (dispose of) your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Keep TYENNE prefilled syringes and disposal container out of the reach of children.
Write the date, time, and specific part of your body where you injected yourself (see Figure V).
It may also be helpful to write any questions or concerns about the injection, so you can ask your healthcare provider.
Manufactured by:
Fresenius Kabi USA, LLC
Lake Zurich, Illinois 60047U.S. License No. 2146
This Instructions for Use has been approved by the U.S. Food and Drug Administration Revised: 02/2025
-
Principal Display Panel – 0.9 mL Prefilled syringe – Primary Label
NDC: 65219-586-04 Rx only
Tyenne®
(tocilizumab-aazg)
Injection162 mg / 0.9 mL
For Subcutaneous
Use Only
Fresenius Kabi USA, LLC -
Principal Display Panel – 0.9 mL Prefilled syringe - Carton
NDC: 65219-586-04 Rx only
Tyenne®
tocilizumab-aazgInjection
162 mg / 0.9 mL
For Subcutaneous Use Only
1 single-dose prefilled syringe 0.9 mL
Carton contains:
1 single-dose prefilled syringe - Discard Unused Portion
1 Prescribing Information
1 Medication Guide
1 Instruction for UseDispense the enclosed
Medication Guide to each patient
Sterile Solution - No PreservativeRefrigerate Immediately
-
Principal Display Panel – 0.9 mL Autoinjector – Primary Label
NDC: 65219-584-01 Rx only
Tyenne®
(tocilizumab-aazg)
Injection162 mg / 0.9 mL
For Subcutaneous Use Only
Fresenius Kabi USA, LLC
-
Principal Display Panel – 0.9 mL Autoinjector - Carton
NDC: 65219-584-01 Rx only
Tyenne®
tocilizumab-aazgInjection
162 mg / 0.9 mL
For Subcutaneous Use Only
1 single-dose autoinjector 0.9 mL
Carton contains:
1 single-dose autoinjector - Discard Unused Portion
1 Prescribing Information
1 Medication Guide
1 Instruction for UseDispense the enclosed Medication Guide to each patient
Refrigerate Immediately
Sterile Solution - No Preservative -
INGREDIENTS AND APPEARANCE
TYENNE
tocilizumab-aazg injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65219-586 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TOCILIZUMAB (UNII: I031V2H011) (TOCILIZUMAB - UNII:I031V2H011) TOCILIZUMAB 162 mg in 0.9 mL Inactive Ingredients Ingredient Name Strength Arginine (UNII: 94ZLA3W45F) Histidine (UNII: 4QD397987E) Lactic acid, L- (UNII: F9S9FFU82N) Polysorbate 80 (UNII: 6OZP39ZG8H) Sodium chloride (UNII: 451W47IQ8X) Water (UNII: 059QF0KO0R) Hydrochloric acid (UNII: QTT17582CB) Sodium hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65219-586-04 1 in 1 CARTON 07/01/2024 12/31/2027 1 0.9 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761275 07/01/2024 TYENNE
tocilizumab-aazg injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65219-584 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TOCILIZUMAB (UNII: I031V2H011) (TOCILIZUMAB - UNII:I031V2H011) TOCILIZUMAB 162 mg in 0.9 mL Inactive Ingredients Ingredient Name Strength Arginine (UNII: 94ZLA3W45F) Histidine (UNII: 4QD397987E) Lactic acid, L- (UNII: F9S9FFU82N) Polysorbate 80 (UNII: 6OZP39ZG8H) Sodium chloride (UNII: 451W47IQ8X) Water (UNII: 059QF0KO0R) Hydrochloric acid (UNII: QTT17582CB) Sodium hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65219-584-01 1 in 1 CARTON 07/01/2024 01/31/2028 1 0.9 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761275 07/01/2024 Labeler - Fresenius Kabi USA, LLC (013547657) Establishment Name Address ID/FEI Business Operations Fresenius Kabi Austria GmbH 300206604 MANUFACTURE(65219-586, 65219-584) Establishment Name Address ID/FEI Business Operations MERCK SERONO S.p.A 437803088 MANUFACTURE(65219-586, 65219-584)
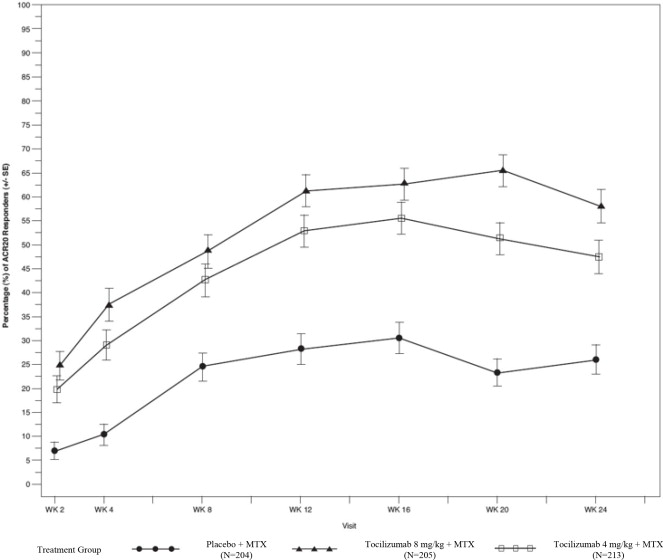
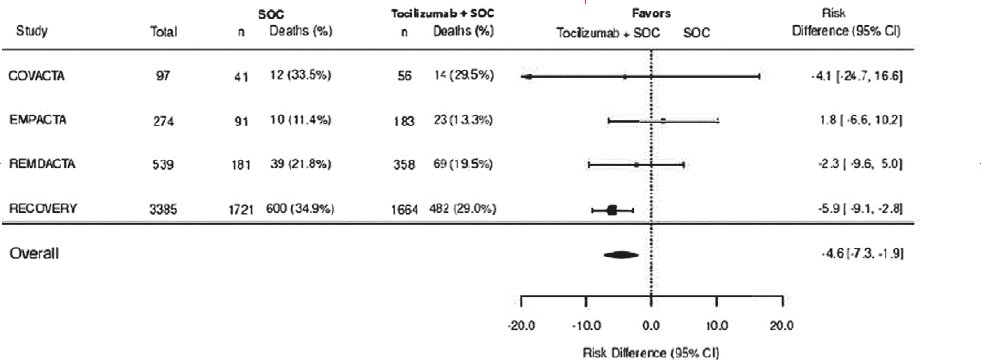

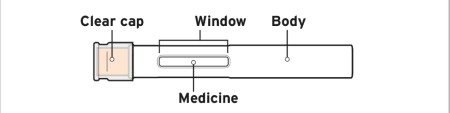
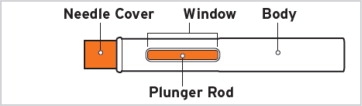


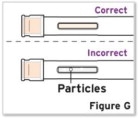

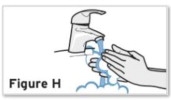

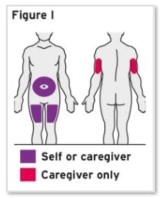


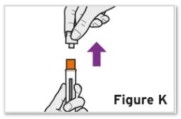
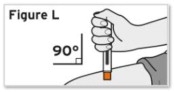

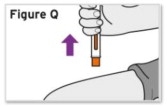
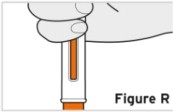
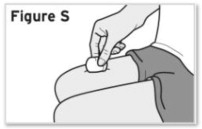

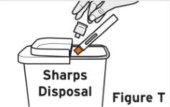



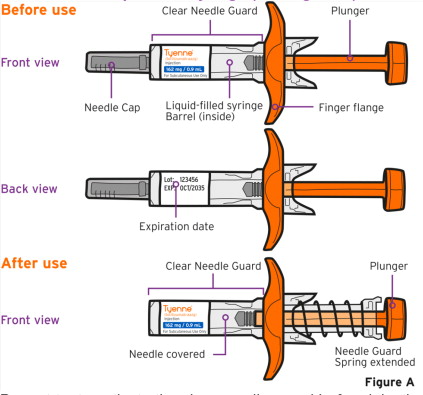






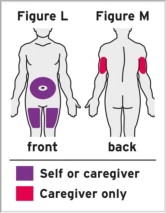


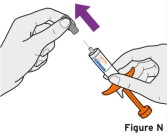
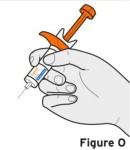
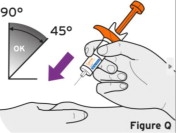
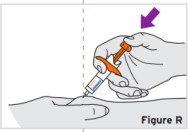
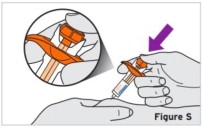
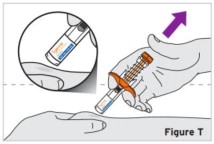

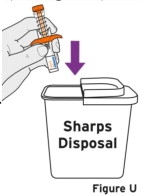




Trademark Results [TYENNE]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TYENNE 97186903 not registered Live/Pending |
Fresenius Kabi Deutschland GmbH 2021-12-23 |
 TYENNE 85912824 not registered Dead/Abandoned |
Ares Trading S.A. 2013-04-23 |
 TYENNE 78856760 3232929 Dead/Cancelled |
Ares Trading S.A. 2006-04-07 |
 TYENNE 77617491 not registered Dead/Abandoned |
Ares Trading S.A. 2008-11-19 |
 TYENNE 77615844 not registered Dead/Abandoned |
Ares Trading S.A. 2008-11-17 |
 TYENNE 74680373 not registered Dead/Abandoned |
ARES TRADING S.A. 1995-05-26 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.