SPORANOX- itraconazole solution
SPORANOX by
Drug Labeling and Warnings
SPORANOX by is a Prescription medication manufactured, distributed, or labeled by Janssen Pharmaceuticals, Inc., Janssen Pharmaceutical Sciences Unlimited Company, Janssen Pharmaceutica, NV. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
BOXED WARNING
(What is this?)
BOXED WARNING
Congestive Heart Failure, Cardiac Effects and Drug Interactions
If signs or symptoms of congestive heart failure occur during administration of SPORANOX® (itraconazole) Oral Solution, continued SPORANOX® use should be reassessed. When itraconazole was administered intravenously to dogs and healthy human volunteers, negative inotropic effects were seen. (See CONTRAINDICATIONS, WARNINGS, PRECAUTIONS).
Drug Interactions, ADVERSE REACTIONS: Post-marketing Experience, and CLINICAL PHARMACOLOGY: Special Populations for more information.)
Drug Interactions
Coadministration of the following drugs are contraindicated with SPORANOX® Oral Solution: methadone, disopyramide, dofetilide, dronedarone, quinidine, isavuconazole, ergot alkaloids (such as dihydroergotamine, ergometrine (ergonovine), ergotamine, methylergometrine (methylergonovine)), irinotecan, lurasidone, oral midazolam, pimozide, triazolam, felodipine, nisoldipine, ivabradine, ranolazine, eplerenone, cisapride, naloxegol, lomitapide, lovastatin, simvastatin, avanafil, ticagrelor. In addition, coadministration with colchicine, fesoterodine and solifenacin is contraindicated in subjects with varying degrees of renal or hepatic impairment, and coadministration with eliglustat is contraindicated in subjects that are poor or intermediate metabolizers of CYP2D6 and in subjects taking strong or moderate CYP2D6 inhibitors. See PRECAUTIONS: Drug Interactions Section for specific examples. Coadministration with itraconazole can cause elevated plasma concentrations of these drugs and may increase or prolong both the pharmacologic effects and/or adverse reactions to these drugs. For example, increased plasma concentrations of some of these drugs can lead to QT prolongation and ventricular tachyarrhythmias including occurrences of torsades de pointes, a potentially fatal arrhythmia. See CONTRAINDICATIONS and WARNINGS Sections, and PRECAUTIONS: Drug Interactions Section for specific examples.
-
DESCRIPTION
SPORANOX® is the brand name for itraconazole, an azole antifungal agent. Itraconazole is a 1:1:1:1 racemic mixture of four diastereomers (two enantiomeric pairs), each possessing three chiral centers. It may be represented by the following structural formula and nomenclature:
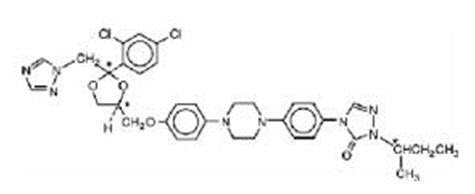
(±)-1-[(R*)-sec-butyl]-4-[p-[4-[p-[[(2R*,4S*)-2-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]-1-piperazinyl]phenyl]-Δ2-1,2,4-triazolin-5-one mixture with (±)-1-[(R*)-sec-butyl]-4-[p-[4-[p-[[(2S*,4R*)-2-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]-1-piperazinyl]phenyl]-Δ2-1,2,4-triazolin-5-one
or
(±)-1-[(RS)-sec-butyl]-4-[p-[4-[p-[[(2R,4S)-2-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]-1-piperazinyl]phenyl]-Δ2-1,2,4-triazolin-5-one.
Itraconazole has a molecular formula of C35H38Cl2N8O4 and a molecular weight of 705.64. It is a white to slightly yellowish powder. It is insoluble in water, very slightly soluble in alcohols, and freely soluble in dichloromethane. It has a pKa of 3.70 (based on extrapolation of values obtained from methanolic solutions) and a log (n-octanol/water) partition coefficient of 5.66 at pH 8.1.
SPORANOX® (itraconazole) Oral Solution contains 10 mg of itraconazole per mL, solubilized by hydroxypropyl-β-cyclodextrin (400 mg/mL) as a molecular inclusion complex. SPORANOX® Oral Solution is clear and yellowish in color with a target pH of 2. Other ingredients are hydrochloric acid, propylene glycol, purified water, sodium hydroxide, sodium saccharin, sorbitol, cherry flavor 1, cherry flavor 2 and caramel flavor.
-
CLINICAL PHARMACOLOGY
Pharmacokinetics and Metabolism
Itraconazole
General Pharmacokinetic Characteristics
Peak plasma concentrations are reached within 2.5 hours following administration of the oral solution. As a consequence of non-linear pharmacokinetics, itraconazole accumulates in plasma during multiple dosing. Steady-state concentrations are generally reached within about 15 days, with Cmax and AUC values 4 to 7-fold higher than those seen after a single dose. Steady-state Cmax values of about 2 µg/mL are reached after oral administration of 200 mg once daily. The terminal half-life of itraconazole generally ranges from 16 to 28 hours after single dose and increases to 34 to 42 hours with repeated dosing. Once treatment is stopped, itraconazole plasma concentrations decrease to an almost undetectable concentration within 7 to 14 days, depending on the dose and duration of treatment. Itraconazole mean total plasma clearance following intravenous administration is 278 mL/min. Itraconazole clearance decreases at higher doses due to saturable hepatic metabolism.
Absorption
Itraconazole is rapidly absorbed after administration of the oral solution. Peak plasma concentrations of itraconazole are reached within 2.5 hours following administration of the oral solution under fasting conditions. The observed absolute bioavailability of itraconazole under fed conditions is about 55% and increases by 30% when the oral solution is taken in fasting conditions. Itraconazole exposure is greater with the oral solution than with the capsule formulation when the same dose of drug is given. (see WARNINGS)
Distribution
Most of the itraconazole in plasma is bound to protein (99.8%), with albumin being the main binding component (99.6% for the hydroxy-metabolite). It has also a marked affinity for lipids. Only 0.2% of the itraconazole in plasma is present as free drug. Itraconazole is distributed in a large apparent volume in the body (>700 L), suggesting extensive distribution into tissues. Concentrations in lung, kidney, liver, bone, stomach, spleen and muscle were found to be two to three times higher than corresponding concentrations in plasma, and the uptake into keratinous tissues, skin in particular, up to four times higher. Concentrations in the cerebrospinal fluid are much lower than in plasma.
Metabolism
Itraconazole is extensively metabolized by the liver into a large number of metabolites. In vitro studies have shown that CYP3A4 is the major enzyme involved in the metabolism of itraconazole. The main metabolite is hydroxy-itraconazole, which has in vitro antifungal activity comparable to itraconazole; trough plasma concentrations of this metabolite are about twice those of itraconazole.
Excretion
Itraconazole is excreted mainly as inactive metabolites in urine (35%) and in feces (54%) within one week of an oral solution dose. Renal excretion of itraconazole and the active metabolite hydroxy-itraconazole account for less than 1% of an intravenous dose. Based on an oral radiolabeled dose, fecal excretion of unchanged drug ranges from 3% to 18% of the dose.
Special Populations
Renal Impairment
Limited data are available on the use of oral itraconazole in patients with renal impairment. A pharmacokinetic study using a single 200-mg oral dose of itraconazole was conducted in three groups of patients with renal impairment (uremia: n = 7; hemodialysis: n = 7; and continuous ambulatory peritoneal dialysis: n = 5). In uremic subjects with a mean creatinine clearance of 13 mL/min.×1.73 m2, the exposure, based on AUC, was slightly reduced compared with normal population parameters. This study did not demonstrate any significant effect of hemodialysis or continuous ambulatory peritoneal dialysis on the pharmacokinetics of itraconazole (Tmax, Cmax, and AUC0–8h). Plasma concentration-versus-time profiles showed wide intersubject variation in all three groups.
After a single intravenous dose, the mean terminal half-lives of itraconazole in patients with mild (defined in this study as CrCl 50–79 mL/min), moderate (defined in this study as CrCl 20–49 mL/min), and severe renal impairment (defined in this study as CrCl <20 mL/min) were similar to that in healthy subjects (range of means 42–49 hours vs 48 hours in renally impaired patients and healthy subjects, respectively). Overall exposure to itraconazole, based on AUC, was decreased in patients with moderate and severe renal impairment by approximately 30% and 40%, respectively, as compared with subjects with normal renal function.
Data are not available in renally impaired patients during long-term use of itraconazole. Dialysis has no effect on the half-life or clearance of itraconazole or hydroxy-itraconazole. (See PRECAUTIONS and DOSAGE AND ADMINISTRATION.)
Hepatic Impairment
Itraconazole is predominantly metabolized in the liver. A pharmacokinetic study was conducted in 6 healthy and 12 cirrhotic subjects who were administered a single 100-mg dose of itraconazole as capsule. A statistically significant reduction in mean Cmax (47%) and a twofold increase in the elimination half-life (37 ± 17 hours vs. 16 ± 5 hours) of itraconazole were noted in cirrhotic subjects compared with healthy subjects. However, overall exposure to itraconazole, based on AUC, was similar in cirrhotic patients and in healthy subjects. Data are not available in cirrhotic patients during long-term use of itraconazole. (See CONTRAINDICATIONS, PRECAUTIONS: Drug Interactions and DOSAGE AND ADMINISTRATION.)
Decreased Cardiac Contractility
When itraconazole was administered intravenously to anesthetized dogs, a dose-related negative inotropic effect was documented. In a healthy volunteer study of itraconazole intravenous infusion, transient, asymptomatic decreases in left ventricular ejection fraction were observed using gated SPECT imaging; these resolved before the next infusion, 12 hours later. If signs or symptoms of congestive heart failure appear during administration of SPORANOX® Oral Solution, monitor carefully and consider other treatment alternatives which may include discontinuation of SPORANOX® Oral Solution administration. (See BOXED WARNING, CONTRAINDICATIONS, WARNINGS, PRECAUTIONS: Drug Interactions and ADVERSE REACTIONS: Post-marketing Experience for more information.)
Cystic Fibrosis
Seventeen cystic fibrosis patients, ages 7 to 28 years old, were administered itraconazole oral solution 2.5 mg/kg b.i.d. for 14 days in a pharmacokinetic study. Sixteen patients completed the study. Steady state trough concentrations >250 ng/mL were achieved in 6 out of 11 patients ≥16 years of age but in none of the 5 patients <16 years of age. Large variability was observed in the pharmacokinetic data (%CV for trough concentrations = 98% and 70% for ≥16 and <16 years, respectively; %CV for AUC = 75% and 58% for ≥16 and <16 years, respectively). If a patient with cystic fibrosis does not respond to SPORANOX® Oral Solution, consideration should be given to switching to alternative therapy.
Hydroxypropyl-β-Cyclodextrin
The oral bioavailability of hydroxypropyl-β-cyclodextrin given as a solubilizer of itraconazole in oral solution is on average lower than 0.5% and is similar to that of hydroxypropyl-β-cyclodextrin alone. This low oral bioavailability of hydroxypropyl-β-cyclodextrin is not modified by the presence of food and is similar after single and repeated administrations.
MICROBIOLOGY
Mechanism of Action
In vitro studies have demonstrated that itraconazole inhibits the cytochrome P450-dependent synthesis of ergosterol, which is a vital component of fungal cell membranes.
Antimicrobial Activity
Itraconazole has been shown to be active against most strains of the following microorganism, both in vitro and in clinical infections.
Candida albicans
Susceptibility Testing Methods
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
Drug Resistance
Isolates from several fungal species with decreased susceptibility to itraconazole have been isolated in vitro and from patients receiving prolonged therapy.
Candida krusei, Candida glabrata and Candida tropicalis are generally the least susceptible Candida species, with some isolates showing unequivocal resistance to itraconazole in vitro.
Itraconazole is not active against Zygomycetes (e.g., Rhizopus spp., Rhizomucor spp., Mucor spp. and Absidia spp.), Fusarium spp., Scedosporium spp. and Scopulariopsis spp.
Cross-resistance
In systemic candidosis, if fluconazole-resistant strains of Candida species are suspected, it cannot be assumed that these are sensitive to itraconazole, hence their sensitivity should be tested before the start of itraconazole therapy.
Several in vitro studies have reported that some fungal clinical isolates, including Candida species, with reduced susceptibility to one azole antifungal agent may also be less susceptible to other azole derivatives. The finding of cross-resistance is dependent on a number of factors, including the species evaluated, its clinical history, the particular azole compounds compared, and the type of susceptibility test that is performed.
Studies (both in vitro and in vivo) suggest that the activity of amphotericin B may be suppressed by prior azole antifungal therapy. As with other azoles, itraconazole inhibits the 14C-demethylation step in the synthesis of ergosterol, a cell wall component of fungi. Ergosterol is the active site for amphotericin B. In one study the antifungal activity of amphotericin B against Aspergillus fumigatus infections in mice was inhibited by ketoconazole therapy. The clinical significance of test results obtained in this study is unknown.
-
CLINICAL STUDIES
Oropharyngeal Candidiasis
Two randomized, controlled studies for the treatment of oropharyngeal candidiasis have been conducted (total n = 344). In one trial, clinical response to either 7 or 14 days of itraconazole oral solution, 200 mg/day, was similar to fluconazole tablets and averaged 84% across all arms. Clinical response in this study was defined as cured or improved (only minimal signs and symptoms with no visible lesions). Approximately 5% of subjects were lost to follow-up before any evaluations could be performed. Response to 14 days therapy of itraconazole oral solution was associated with a lower relapse rate than 7 days of itraconazole therapy. In another trial, the clinical response rate (defined as cured or improved) for itraconazole oral solution was similar to clotrimazole troches and averaged approximately 71% across both arms, with approximately 3% of subjects lost to follow-up before any evaluations could be performed. Ninety-two percent of the patients in these studies were HIV seropositive.
In an uncontrolled, open-label study of selected patients clinically unresponsive to fluconazole tablets (n = 74, all patients HIV seropositive), patients were treated with itraconazole oral solution 100 mg b.i.d. (Clinically unresponsive to fluconazole in this study was defined as having received a dose of fluconazole tablets at least 200 mg/day for a minimum of 14 days.) Treatment duration was 14–28 days based on response. Approximately 55% of patients had complete resolution of oral lesions. Of patients who responded and then entered a follow-up phase (n = 22), all relapsed within 1 month (median 14 days) when treatment was discontinued. Although baseline endoscopies had not been performed, several patients in this study developed symptoms of esophageal candidiasis while receiving therapy with itraconazole oral solution. Itraconazole oral solution has not been directly compared to other agents in a controlled trial of similar patients.
Esophageal Candidiasis
A double-blind randomized study (n = 119, 111 of whom were HIV seropositive) compared itraconazole oral solution (100 mg/day) to fluconazole tablets (100 mg/day). The dose of each was increased to 200 mg/day for patients not responding initially. Treatment continued for 2 weeks following resolution of symptoms, for a total duration of treatment of 3–8 weeks. Clinical response (a global assessment of cured or improved) was not significantly different between the two study arms, and averaged approximately 86% with 8% lost to follow-up. Six of 53 (11%) itraconazole-treated patients and 12/57 (21%) fluconazole-treated patients were escalated to the 200 mg dose in this trial. Of the subgroup of patients who responded and entered a follow-up phase (n = 88), approximately 23% relapsed across both arms within 4 weeks.
-
INDICATIONS AND USAGE
SPORANOX® (itraconazole) Oral Solution is indicated for the treatment of oropharyngeal and esophageal candidiasis.
(See CLINICAL PHARMACOLOGY: Special Populations, WARNINGS, and ADVERSE REACTIONS: Post-marketing Experience for more information.)
-
CONTRAINDICATIONS
Congestive Heart Failure
SPORANOX® (itraconazole) Oral Solution should not be administered to patients with evidence of ventricular dysfunction such as congestive heart failure (CHF) or a history of CHF except for the treatment of life-threatening or other serious infections. (See BOXED WARNING, WARNINGS, PRECAUTIONS: Drug Interactions-Calcium Channel Blockers, ADVERSE REACTIONS: Post-marketing Experience, and CLINICAL PHARMACOLOGY: Special Populations.)
Drug Interactions
Coadministration of a number of CYP3A4 substrates are contraindicated with SPORANOX®. Plasma concentrations increase for the following drugs: levaceytlmethadol (levomethadyl), methadone, disopyramide, dofetilide, dronedarone, quinidine, isavuconazole, ergot alkaloids (such as dihydroergotamine, ergometrine (ergonovine), ergotamine, methylergometrine (methylergonovine)), irinotecan, lurasidone, oral midazolam, pimozide, triazolam, felodipine, nisoldipine, ivabradine, ranolazine, eplerenone, cisapride, naloxegol, lomitapide, lovastatin, simvastatin, avanafil, ticagrelor. In addition, coadministration with colchicine, fesoterodine, and solifenacin is contraindicated in subjects with varying degrees of renal or hepatic impairment, and coadministration with eliglustat is contraindicated in subjects that are poor or intermediate metabolizers of CYP2D6 and in subjects taking strong or moderate CYP2D6 inhibitors. (See PRECAUTIONS: Drug Interactions Section for specific examples.) This increase in drug concentrations caused by coadministration with itraconazole may increase or prolong both the pharmacologic effects and/or adverse reactions to these drugs. For example, increased plasma concentrations of some of these drugs can lead to QT prolongation and ventricular tachyarrhythmias including occurrences of torsade de pointes, a potentially fatal arrhythmia. Specific examples are listed in PRECAUTIONS: Drug Interactions.
SPORANOX® is contraindicated for patients who have shown hypersensitivity to itraconazole. There is limited information regarding cross-hypersensitivity between itraconazole and other azole antifungal agents. Caution should be used when prescribing SPORANOX® to patients with hypersensitivity to other azoles.
-
WARNINGS
Hepatic Effects
SPORANOX® has been associated with rare cases of serious hepatotoxicity, including liver failure and death. Some of these cases had neither pre-existing liver disease nor a serious underlying medical condition, and some of these cases developed within the first week of treatment. If clinical signs or symptoms develop that are consistent with liver disease, treatment should be discontinued and liver function testing performed. Continued SPORANOX® use or reinstitution of treatment with SPORANOX® is strongly discouraged unless there is a serious or life-threatening situation where the expected benefit exceeds the risk. (See PRECAUTIONS: Information for Patients and ADVERSE REACTIONS.)
Cardiac Dysrhythmias
Life-threatening cardiac dysrhythmias and/or sudden death have occurred in patients using drugs such as cisapride, pimozide, methadone, or quinidine concomitantly with SPORANOX® and/or other CYP3A4 inhibitors. Concomitant administration of these drugs with SPORANOX® is contraindicated. (See BOXED WARNING, CONTRAINDICATIONS, and PRECAUTIONS: Drug Interactions.)
Cardiac Disease
SPORANOX® Oral Solution should not be used in patients with evidence of ventricular dysfunction unless the benefit clearly outweighs the risk. For patients with risk factors for congestive heart failure, physicians should carefully review the risks and benefits of SPORANOX® therapy. These risk factors include cardiac disease such as ischemic and valvular disease; significant pulmonary disease such as chronic obstructive pulmonary disease; and renal failure and other edematous disorders. Such patients should be informed of the signs and symptoms of CHF, should be treated with caution, and should be monitored for signs and symptoms of CHF during treatment. If signs or symptoms of CHF appear during administration of SPORANOX® Oral Solution, monitor carefully and consider other treatment alternatives which may include discontinuation of SPORANOX® Oral Solution administration.
Itraconazole has been shown to have a negative inotropic effect. When itraconazole was administered intravenously to anesthetized dogs, a dose-related negative inotropic effect was documented. In a healthy volunteer study of itraconazole intravenous infusion, transient, asymptomatic decreases in left ventricular ejection fraction were observed using gated SPECT imaging; these resolved before the next infusion, 12 hours later.
SPORANOX® has been associated with reports of congestive heart failure. In post-marketing experience, heart failure was more frequently reported in patients receiving a total daily dose of 400 mg although there were also cases reported among those receiving lower total daily doses.
Calcium channel blockers can have negative inotropic effects which may be additive to those of itraconazole. In addition, itraconazole can inhibit the metabolism of calcium channel blockers. Therefore, caution should be used when co-administering itraconazole and calcium channel blockers due to an increased risk of CHF. Concomitant administration of SPORANOX® and felodipine or nisoldipine is contraindicated.
Cases of CHF, peripheral edema, and pulmonary edema have been reported in the post-marketing period among patients being treated for onychomycosis and/or systemic fungal infections. (See CONTRAINDICATIONS, CLINICAL PHARMACOLOGY: Special Populations, PRECAUTIONS: Drug Interactions, and ADVERSE REACTIONS: Post-marketing Experience for more information.)
Interaction potential
SPORANOX® has a potential for clinically important drug interactions. Coadministration of specific drugs with itraconazole may result in changes in efficacy of itraconazole and/or the coadministered drug, life-threatening effects and/or sudden death. Drugs that are contraindicated, not recommended or recommended for use with caution in combination with itraconazole are listed in PRECAUTIONS: Drug Interactions.
Interchangeability
SPORANOX® (itraconazole) Oral Solution and SPORANOX® Capsules should not be used interchangeably. This is because drug exposure is greater with the Oral Solution than with the Capsules when the same dose of drug is given. Only SPORANOX® Oral Solution has been demonstrated effective for oral and/or esophageal candidiasis.
Hydroxypropyl-β-cyclodextrin
SPORANOX® Oral Solution contains the excipient hydroxypropyl-β-cyclodextrin which produced adenocarcinomas in the large intestine and exocrine pancreatic adenocarcinomas in a rat carcinogenicity study. These findings were not observed in a similar mouse carcinogenicity study. The clinical relevance of these adenocarcinomas is unknown. (See PRECAUTIONS: Carcinogenesis, Mutagenesis, and Impairment of Fertility.)
Treatment of Severely Neutropenic Patients
SPORANOX® Oral Solution as treatment for oropharyngeal and/or esophageal candidiasis was not investigated in severely neutropenic patients. Due to its pharmacokinetic properties, SPORANOX® Oral Solution is not recommended for initiation of treatment in patients at immediate risk of systemic candidiasis.
-
PRECAUTIONS
Hepatotoxicity
Rare cases of serious hepatotoxicity have been observed with SPORANOX® treatment, including some cases within the first week. It is recommended that liver function monitoring be considered in all patients receiving SPORANOX®. Treatment should be stopped immediately and liver function testing should be conducted in patients who develop signs and symptoms suggestive of liver dysfunction.
Neuropathy
If neuropathy occurs that may be attributable to SPORANOX® Oral Solution, the treatment should be discontinued.
Cystic Fibrosis
If a patient with cystic fibrosis does not respond to SPORANOX® Oral Solution, consideration should be given to switching to alternative therapy (see CLINICAL PHARMACOLOGY: Special Populations).
Hearing Loss
Transient or permanent hearing loss has been reported in patients receiving treatment with itraconazole. Several of these reports included concurrent administration of quinidine which is contraindicated (see BOXED WARNING: Drug Interactions, CONTRAINDICATIONS: Drug Interactions and PRECAUTIONS: Drug Interactions). The hearing loss usually resolves when treatment is stopped, but can persist in some patients.
Information for Patients
- Only SPORANOX® Oral Solution has been demonstrated effective for oral and/or esophageal candidiasis.
- SPORANOX® Oral Solution contains the excipient hydroxypropyl-β-cyclodextrin which produced adenocarcinomas in the large intestine and exocrine pancreatic adenocarcinomas in a rat carcinogenicity study. These findings were not observed in a similar mouse carcinogenicity study. The clinical relevance of these adenocarcinomas is unknown. (See Carcinogenesis, Mutagenesis, and Impairment of Fertility.)
- Taking SPORANOX® Oral Solution under fasted conditions improves the systemic availability of itraconazole. Instruct patients to take SPORANOX® Oral Solution without food, if possible.
- SPORANOX® Oral Solution should not be used interchangeably with SPORANOX® Capsules.
- Instruct patients about the signs and symptoms of congestive heart failure, and if these signs or symptoms occur during SPORANOX® administration, they should discontinue SPORANOX® and contact their healthcare provider immediately.
- Instruct patients to stop SPORANOX® treatment immediately and contact their healthcare provider if any signs and symptoms suggestive of liver dysfunction develop. Such signs and symptoms may include unusual fatigue, anorexia, nausea and/or vomiting, jaundice, dark urine, or pale stools.
- Instruct patients to contact their physician before taking any concomitant medications with itraconazole to ensure there are no potential drug interactions.
- Instruct patients that hearing loss can occur with the use of itraconazole. The hearing loss usually resolves when treatment is stopped, but can persist in some patients. Advise patients to discontinue therapy and inform their physicians if any hearing loss symptoms occur.
- Instruct patients that dizziness or blurred/double vision can sometimes occur with itraconazole. Advise patients that if they experience these events, they should not drive or use machines.
Drug Interactions
Effect of SPORANOX® on Other Drugs
Itraconazole and its major metabolite, hydroxy-itraconazole, are potent CYP3A4 inhibitors. Itraconazole is an inhibitor of the drug transporters P-glycoprotein and breast cancer resistance protein (BCRP). Consequently, SPORANOX® has the potential to interact with many concomitant drugs resulting in either increased or sometimes decreased concentrations of the concomitant drugs. Increased concentrations may increase the risk of adverse reactions associated with the concomitant drug which can be severe or life-threatening in some cases (e.g., QT prolongation, Torsade de Pointes, respiratory depression, hepatic adverse reactions, hypersensitivity reactions, myelosuppression, hypotension, seizures, angioedema, atrial fibrillation, bradycardia, priapism). Reduced concentrations of concomitant drugs may reduce their efficacy. The table below lists examples of drugs that may have their concentrations affected by itraconazole, but is not a comprehensive list. Refer to the approved product labeling to become familiar with the interaction pathways risk potential and specific actions to be taken with regards to each concomitant drug prior to initiating therapy with SPORANOX®.
Although many of the clinical drug interactions in Table 1 below are based on information with a similar azole antifungal, ketoconazole, these interactions are expected to occur with SPORANOX®.
Table 1: Drug Interactions with SPORANOX® that Affect Concomitant Drug Concentrations Concomitant Drug Within Class Prevention or Management - * Based on clinical drug interaction information with itraconazole.
- † Based on 400 mg bedaquiline once daily for 2 weeks.
- ‡ CYP3A4 inhibitors (including itraconazole) may increase systemic contraceptive hormone concentrations.
- § EMs: extensive metabolizers; IMs: intermediate metabolizers, PMs: poor metabolizers
Drug Interactions with SPORANOX® that Increase Concomitant Drug Concentrations and May Increase Risk of Adverse Reactions Associated with the Concomitant Drug Alpha Blockers Alfuzosin
Silodosin
TamsulosinNot recommended during and 2 weeks after SPORANOX® treatment. Analgesics Methadone Contraindicated during and 2 weeks after SPORANOX® treatment. Fentanyl Not recommended during and 2 weeks after SPORANOX® treatment. Alfentanil
Buprenorphine (IV and sublingual)
Oxycodone*
SufentanilMonitor for adverse reactions. Concomitant drug dose reduction may be necessary. Antiarrhythmics Disopyramide
Dofetilide
Dronedarone
Quinidine*Contraindicated during and 2 weeks after SPORANOX® treatment. Digoxin* Monitor for adverse reactions. Concomitant drug dose reduction may be necessary. Antibacterials Bedaquiline† Concomitant SPORANOX® not recommended for more than 2 weeks at any time during bedaquiline treatment. Rifabutin Not recommended 2 weeks before, during, and 2 weeks after SPORANOX® treatment. See also Table 2. Clarithromycin Monitor for adverse reactions. Concomitant drug dose reduction may be necessary. See also Table 2. Trimetrexate Monitor for adverse reactions. Concomitant drug dose reduction may be necessary. Anticoagulants and Antiplatelets Ticagrelor Contraindicated during and 2 weeks after SPORANOX® treatment. Apixaban
Rivaroxaban
VorapaxarNot recommended during and 2 weeks after SPORANOX® treatment. Cilostazol
Dabigatran
WarfarinMonitor for adverse reactions. Concomitant drug dose reduction may be necessary. Anticonvulsants Carbamazepine Not recommended 2 weeks before, during, and 2 weeks after SPORANOX® treatment. See also Table 2. Antidiabetic Drugs Repaglinide*
SaxagliptinMonitor for adverse reactions. Concomitant drug dose reduction may be necessary. Antihelminthics, Antifungals and Antiprotozoals Isavuconazonium Contraindicated during and 2 weeks after SPORANOX® treatment. Praziquantel Monitor for adverse reactions. Concomitant drug dose reduction may be necessary. Artemether-lumefantrine Quinine* Monitor for adverse reactions. Antimigraine Drugs Ergot alkaloids (e.g., dihydroergotamine, ergotamine) Contraindicated during and 2 weeks after SPORANOX® treatment. Eletriptan Monitor for adverse reactions. Concomitant drug dose reduction may be necessary Antineoplastics Irinotecan Contraindicated during and 2 weeks after SPORANOX® treatment. Axitinib
Bosutinib
Cabazitaxel
Cabozantinib
Ceritinib
Cobimetinib*
Crizotinib
Dabrafenib
DasatinibDocetaxel
Ibrutinib
Lapatinib
Nilotinib
Olaparib*
Pazopanib
Sunitinib
Trabectedin
Trastuzumab-emtansine
Vinca alkaloidsNot recommended during and 2 weeks after SPORANOX® treatment. Bortezomib
Brentuximab-vedotin
Busulfan
Erlotinib
Gefitinib*
Idelalisib
Imatinib
IxabepiloneNintedanib
Panobinostat
Ponatinib
Ruxolitinib
Sonidegib
Vandetanib*Monitor for adverse reactions. Concomitant drug dose reduction may be necessary. For idelalisib: see also Table 2. Antipsychotics, Anxiolytics and Hypnotics Alprazolam*
Aripiprazole*
Buspirone*
Cariprazine
Diazepam*
Haloperidol*Midazolam (IV)*
Quetiapine
Ramelteon
Risperidone*
SuvorexantMonitor for adverse reactions. Concomitant drug dose reduction may be necessary. Zopiclone* Monitor for adverse reactions. Concomitant drug dose reduction may be necessary. Lurasidone
Midazolam (oral)*
Pimozide
Triazolam*Contraindicated during and 2 weeks after SPORANOX® treatment. Antivirals Simeprevir Not recommended during and 2 weeks after SPORANOX® treatment. Daclatasvir
Indinavir*
MaravirocMonitor for adverse reactions. Concomitant drug dose reduction may be necessary. For indinavir: see also Table 2. Cobicistat
Elvitegravir (ritonavir-boosted)
Ombitasvir/Paritaprevir/Ritonavir with or without Dasabuvir
Ritonavir
Saquinavir (unboosted)*Monitor for adverse reactions. Elbasvir/grazoprevir Not recommended during and 2 weeks after SPORANOX® treatment. Glecaprevir/pibrentasvir
Tenofovir disoproxil fumarateMonitor for adverse reactions.
Monitor for adverse reactions.Beta Blockers Nadolol* Monitor for adverse reactions. Concomitant drug dose reduction may be necessary. Calcium Channel Blockers Felodipine*
NisoldipineContraindicated during and 2 weeks after SPORANOX® treatment. Diltiazem
Other dihydropyridines
VerapamilMonitor for adverse reactions. Concomitant drug dose reduction may be necessary. For diltiazem: see also Table 2. Cardiovascular Drugs, Miscellaneous Ivabradine
RanolazineContraindicated during and 2 weeks after SPORANOX® treatment. Aliskiren*
Riociguat
Sildenafil (for pulmonary hypertension)
Tadalafil (for pulmonary hypertension)Not recommended during and 2 weeks after SPORANOX® treatment. For sildenafil and tadalafil, see also Urologic Drugs below. Bosentan
GuanfacineMonitor for adverse reactions. Concomitant drug dose reduction may be necessary. Contraceptives‡ Dienogest
UlipristalMonitor for adverse reactions. Diuretics Eplerenone Contraindicated during and 2 weeks after SPORANOX® treatment. Gastrointestinal Drugs Cisapride
NaloxegolContraindicated during and 2 weeks after SPORANOX® treatment. Aprepitant
Loperamide*Monitor for adverse reactions. Concomitant drug dose reduction may be necessary. Netupitant Monitor for adverse reactions. Immunosuppressants Everolimus
Sirolimus
Temsirolimus (IV)Not recommended during and 2 weeks after SPORANOX® treatment. Budesonide (inhalation)*
Budesonide (non-inhalation)
Ciclesonide (inhalation)
Cyclosporine (IV)*
Cyclosporine (non-IV)
Dexamethasone*Fluticasone (inhalation)*
Fluticasone (nasal)
Methylprednisolone*
Tacrolimus (IV)*
Tacrolimus (oral)Monitor for adverse reactions. Concomitant drug dose reduction may be necessary. Lipid-Lowering Drugs Lomitapide
Lovastatin*
Simvastatin*Contraindicated during and 2 weeks after SPORANOX® treatment. Atorvastatin* Monitor for drug adverse reactions. Concomitant drug dose reduction may be necessary. Respiratory Drugs Salmeterol Not recommended during and 2 weeks after SPORANOX® treatment. SSRIs, Tricyclics and Related Antidepressants Venlafaxine Monitor for adverse reactions. Concomitant drug dose reduction may be necessary. Urologic Drugs Avanafil Contraindicated during and 2 weeks after SPORANOX® treatment. Fesoterodine Patients with moderate to severe renal or hepatic impairment: Contraindicated during and 2 weeks after SPORANOX® treatment.
Other patients: Monitor for adverse reactions. Concomitant drug dose reduction may be necessary.Solifenacin Patients with severe renal or moderate to severe hepatic impairment: Contraindicated during and 2 weeks after SPORANOX® treatment.
Other patients: Monitor for adverse reactions. Concomitant drug dose reduction may be necessary.Darifenacin
VardenafilNot recommended during and 2 weeks after SPORANOX® treatment. Dutasteride
Oxybutynin*
Sildenafil (for erectile dysfunction)
Tadalafil (for erectile dysfunction and benign prostatic hyperplasia)
TolterodineMonitor for adverse reactions. Concomitant drug dose reduction may be necessary. For sildenafil and tadalafil, see also Cardiovascular Drugs above. Miscellaneous Drugs and Other Substances Colchicine Patients with renal or hepatic impairment: Contraindicated during and 2 weeks after SPORANOX® treatment.
Other patients: Not recommended during and 2 weeks after SPORANOX® treatment.Eliglustat CYP2D6 EMs§ taking a strong or moderate CYP2D6 inhibitor, CYP2D6 IMs§, or CYP2D6 PMs§: Contraindicated during and 2 weeks after SPORANOX® treatment.
CYP2D6 EMs§ not taking a strong or moderate CYP2D6 inhibitor: Monitor for adverse reactions. Eliglustat dose reduction may be necessary.Lumacaftor/Ivacaftor Not recommended 2 weeks before, during, and 2 weeks after SPORANOX® treatment. Alitretinoin (oral)
Cabergoline
Cannabinoids
Cinacalcet
Galantamine
IvacaftorMonitor for adverse reactions. Concomitant drug dose reduction may be necessary. Vasopressin Receptor Antagonists Conivaptan
TolvaptanNot recommended during and 2 weeks after SPORANOX® treatment. Drug Interactions with SPORANOX® that Decrease Concomitant Drug Concentrations and May Reduce Efficacy of the Concomitant Drug Antineoplastics Regorafenib Not recommended during and 2 weeks after SPORANOX® treatment. Gastrointestinal Drugs Saccharomyces boulardii Not recommended during and 2 weeks after SPORANOX® treatment. Nonsteroidal Anti-Inflammatory Drugs Meloxicam* Concomitant drug dose increase may be necessary. Effect of Other Drugs on SPORANOX®
Itraconazole is mainly metabolized through CYP3A4. Other substances that either share this metabolic pathway or modify CYP3A4 activity may influence the pharmacokinetics of itraconazole. Some concomitant drugs have the potential to interact with SPORANOX® resulting in either increased or sometimes decreased concentrations of SPORANOX®. Increased concentrations may increase the risk of adverse reactions associated with SPORANOX®. Decreased concentrations may reduce SPORANOX® efficacy.
The table below lists examples of drugs that may affect itraconazole concentrations, but is not a comprehensive list. Refer to the approved product labeling to become familiar with the interaction pathways, risk potential and specific actions to be taken with regards to each concomitant drug prior to initiating therapy with SPORANOX®.
Although many of the clinical drug interactions in Table 2 below are based on information with a similar azole antifungal, ketoconazole, these interactions are expected to occur with SPORANOX®.
Table 2. Drug Interactions with Other Drugs that Affect SPORANOX® Concentrations Concomitant Drug Within Class Prevention or Management - * Based on clinical drug interaction information with itraconazole.
Drug Interactions with Other Drugs that Increase SPORANOX® Concentrations and May Increase Risk of Adverse Reactions Associated with SPORANOX® Antibacterials Ciprofloxacin*
Erythromycin*
Clarithromycin*Monitor for adverse reactions. SPORANOX® dose reduction may be necessary. Antineoplastics Idelalisib Monitor for adverse reactions. SPORANOX® dose reduction may be necessary. See also Table 1. Antivirals Cobicistat
Darunavir (ritonavir-boosted)
Elvitegravir (ritonavir-boosted)
Fosamprenavir (ritonavir-boosted)
Indinavir*
Ombitasvir/ Paritaprevir/ Ritonavir with or without Dasabuvir
Ritonavir
SaquinavirMonitor for adverse reactions. SPORANOX® dose reduction may be necessary. For Boceprevir, cobicistat, elvitegravir, indinavir, ombitasvir/ paritaprevir/ ritonavir with or without dasabuvir, ritonavir and saquinavir, see also Table 1. Calcium Channel Blockers Diltiazem Monitor for adverse reactions. SPORANOX® dose reduction may be necessary. See also the table above. Drug Interactions with Other Drugs that Decrease SPORANOX® Concentrations and May Reduce Efficacy of SPORANOX® Antibacterials Isoniazid
Rifampicin*Not recommended 2 weeks before and during SPORANOX® treatment. Rifabutin* Not recommended 2 weeks before, during, and 2 weeks after SPORANOX® treatment. See also Table 1. Anticonvulsants Phenobarbital
Phenytoin*Not recommended 2 weeks before and during SPORANOX® treatment. Carbamazepine Not recommended 2 weeks before, during, and 2 weeks after SPORANOX® treatment. See also Table 1. Antivirals Efavirenz*
Nevirapine*Not recommended 2 weeks before and during SPORANOX® treatment. Miscellaneous Drugs and Other Substances Lumacaftor/Ivacaftor Not recommended 2 weeks before, during, and 2 weeks after SPORANOX® treatment. Carcinogenesis, Mutagenesis, and Impairment of Fertility
Itraconazole
Itraconazole showed no evidence of carcinogenicity potential in mice treated orally for 23 months at dosage levels up to 80 mg/kg/day (approximately 10 times the maximum recommended human dose [MRHD]). Male rats treated with 25 mg/kg/day (3.1 times the MRHD) had a slightly increased incidence of soft tissue sarcoma. These sarcomas may have been a consequence of hypercholesterolemia, which is a response of rats, but not dogs or humans, to chronic itraconazole administration. Female rats treated with 50 mg/kg/day (6.25 times the MRHD) had an increased incidence of squamous cell carcinoma of the lung (2/50) as compared to the untreated group. Although the occurrence of squamous cell carcinoma in the lung is extremely uncommon in untreated rats, the increase in this study was not statistically significant.
Itraconazole produced no mutagenic effects when assayed in DNA repair test (unscheduled DNA synthesis) in primary rat hepatocytes, in Ames tests with Salmonella typhimurium (6 strains) and Escherichia coli, in the mouse lymphoma gene mutation tests, in a sex-linked recessive lethal mutation (Drosophila melanogaster) test, in chromosome aberration tests in human lymphocytes, in a cell transformation test with C3H/10T½ C18 mouse embryo fibroblasts cells, in a dominant lethal mutation test in male and female mice, and in micronucleus tests in mice and rats.
Itraconazole did not affect the fertility of male or female rats treated orally with dosage levels of up to 40 mg/kg/day (5 times the MRHD), even though parental toxicity was present at this dosage level. More severe signs of parental toxicity, including death, were present in the next higher dosage level, 160 mg/kg/day (20 times the MRHD).
Hydroxypropyl-β-cyclodextrin (HP-β-CD)
Hydroxypropyl-β-cyclodextrin (HP-β-CD) is the solubilizing excipient used in SPORANOX® Oral Solution.
Hydroxypropyl-β-cyclodextrin (HP-β-CD) was found to produce neoplasms in the large intestine at 5000 mg/kg/day in rat carcinogenicity study. This dose was about 6 times amount contained in the recommended clinical dose of SPORANOX® Oral Solution based on body surface area comparisons. The clinical relevance of this finding is unknown. The slightly higher incidence of adenocarcinomas in the large intestines was linked to the hypertrophic/hyperplastic and inflammatory changes in the colonic mucosa brought about by HP-β-CD-induced increased osmotic forces.
In addition, HP-β-CD was found to produce pancreatic exocrine hyperplasia and neoplasia when administered orally to rats at doses of 500, 2000 or 5000 mg/kg/day for 25 months. Adenocarcinomas of the exocrine pancreas produced in the treated animals were not seen in the untreated group and are not reported in the historical controls. The recommended clinical dose of SPORANOX® Oral Solution contains approximately 1.7 times the amount of HP-β-CD as was in the 500mg/kg/day dose, based on body surface area comparisons. This finding was not observed in the mouse carcinogenicity study at doses of 500, 2000 or 5000 mg/kg/day for 22–23 months. This finding was also not observed in a 12-month toxicity study in dogs or in a 2-year toxicity study in female cynomolgus monkeys.
Since the development of the pancreatic tumors may be related to a mitogenic action of cholecystokinin and since there is no evidence that cholecystokinin has a mitogenic action in man, the clinical relevance of these findings is unknown.
HP-β-CD has no antifertile effect, and is not mutagenic.
Pregnancy
Teratogenic effects
Itraconazole was found to cause a dose-related increase in maternal toxicity, embryotoxicity, and teratogenicity in rats at dosage levels of approximately 40–160 mg/kg/day (5–20 times the MRHD), and in mice at dosage levels of approximately 80 mg/kg/day (10 times the MRHD). Itraconazole has been shown to cross the placenta in a rat model. In rats, the teratogenicity consisted of major skeletal defects; in mice, it consisted of encephaloceles and/or macroglossia.
SPORANOX® Oral Solution contains the excipient hydroxypropyl-β-cyclodextrin (HP-β-CD). HP-β-CD has no direct embryotoxic and no teratogenic effect.
There are no studies in pregnant women. SPORANOX® should be used in pregnancy only if the benefit outweighs the potential risk. Highly effective contraception should be continued throughout SPORANOX® therapy and for 2 months following the end of treatment.
During post-marketing experience, cases of congenital abnormalities have been reported. (See ADVERSE REACTIONS: Post-marketing Experience.)
Nursing Mothers
Itraconazole is excreted in human milk; therefore, the expected benefits of SPORANOX® therapy for the mother should be weighed against the potential risk from exposure of itraconazole to the infant. The U.S. Public Health Service Centers for Disease Control and Prevention advises HIV-infected women not to breast-feed to avoid potential transmission of HIV to uninfected infants.
Pediatric Use
The efficacy and safety of SPORANOX® have not been established in pediatric patients.
The long-term effects of itraconazole on bone growth in children are unknown. In three toxicology studies using rats, itraconazole induced bone defects at dosage levels as low as 20 mg/kg/day (2.5 times the MRHD). The induced defects included reduced bone plate activity, thinning of the zona compacta of the large bones, and increased bone fragility. At a dosage level of 80 mg/kg/day (10 times the MRHD) over 1 year or 160 mg/kg/day (20 times the MRHD) for 6 months, itraconazole induced small tooth pulp with hypocellular appearance in some rats.
Geriatric Use
Clinical studies of SPORANOX® Oral Solution did not include sufficient numbers of subjects aged 65 years and over to determine whether they respond differently from younger subjects. It is advised to use SPORANOX® Oral Solution in these patients only if it is determined that the potential benefit outweighs the potential risks. In general, it is recommended that the dose selection for an elderly patient should be taken into consideration, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Transient or permanent hearing loss has been reported in elderly patients receiving treatment with itraconazole. Several of these reports included concurrent administration of quinidine which is contraindicated (see BOXED WARNING: Drug Interactions, CONTRAINDICATIONS: Drug Interactions and PRECAUTIONS: Drug Interactions).
Renal Impairment
Limited data are available on the use of oral itraconazole in patients with renal impairment. The exposure of itraconazole may be lower in some patients with renal impairment. Caution should be exercised when itraconazole is administered in this patient population and dose adjustment may be needed. (See CLINICAL PHARMACOLOGY: Special Populations and DOSAGE AND ADMINISTRATION.)
Hepatic Impairment
Limited data are available on the use of oral itraconazole in patients with hepatic impairment. Caution should be exercised when this drug is administered in this patient population. It is recommended that patients with impaired hepatic function be carefully monitored when taking SPORANOX®. It is recommended that the prolonged elimination half-life of itraconazole observed in the single oral dose clinical trial with itraconazole capsules in cirrhotic patients be considered when deciding to initiate therapy with other medications metabolized by CYP3A4.
In patients with elevated or abnormal liver enzymes or active liver disease, or who have experienced liver toxicity with other drugs, treatment with SPORANOX® is strongly discouraged unless there is a serious or life-threatening situation where the expected benefit exceeds the risk. It is recommended that liver function monitoring be done in patients with pre-existing hepatic function abnormalities or those who have experienced liver toxicity with other medications. (See CLINICAL PHARMACOLOGY: Special Populations and DOSAGE AND ADMINISTRATION.)
-
ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
SPORANOX® has been associated with rare cases of serious hepatotoxicity, including liver failure and death. Some of these cases had neither pre-existing liver disease nor a serious underlying medical condition. If clinical signs or symptoms develop that are consistent with liver disease, treatment should be discontinued and liver function testing performed. The risks and benefits of SPORANOX® use should be reassessed. (See WARNINGS: Hepatic Effects and PRECAUTIONS: Hepatotoxicity and Information for Patients.)
Adverse Events Reported in Oropharyngeal or Esophageal Candidiasis Trials
U.S. adverse experience data are derived from 350 immunocompromised patients (332 HIV seropositive/AIDS) treated for oropharyngeal or esophageal candidiasis. Table 3 below lists adverse events reported by at least 2% of patients treated with SPORANOX® Oral Solution in U.S. clinical trials. Data on patients receiving comparator agents in these trials are included for comparison.
Table 3: Summary of Adverse Events Reported by ≥2% of SPORANOX® Treated Patients in U.S. Clinical Trials (Total) Itraconazole Body System/ Adverse Event Total
(n = 350*) %All controlled studies
(n = 272) %Fluconazole
(n = 125†) %Clotrimazole (n = 81‡) % - * Of the 350 patients, 209 were treated for oropharyngeal candidiasis in controlled studies, 63 were treated for esophageal candidiasis in controlled studies and 78 were treated for oropharyngeal candidiasis in an open study.
- † Of the 125 patients, 62 were treated for oropharyngeal candidiasis and 63 were treated for esophageal candidiasis.
- ‡ All 81 patients were treated for oropharyngeal candidiasis.
Gastrointestinal disorders Nausea 11 10 11 5 Diarrhea 11 10 10 4 Vomiting 7 6 8 1 Abdominal pain 6 4 7 7 Constipation 2 2 1 0 Body as a whole Fever 7 6 8 5 Chest pain 3 3 2 0 Pain 2 2 4 0 Fatigue 2 1 2 0 Respiratory disorders Coughing 4 4 10 0 Dyspnea 2 3 5 1 Pneumonia 2 2 0 0 Sinusitis 2 2 4 0 Sputum increased 2 3 3 1 Skin and appendages disorders Rash 4 5 4 6 Increased sweating 3 4 6 1 Skin disorder unspecified 2 2 2 1 Central/peripheral nervous system Headache 4 4 6 6 Dizziness 2 2 4 1 Resistance mechanism disorders Pneumocystis carinii infection 2 2 2 0 Psychiatric disorders Depression 2 1 0 1 Adverse events reported by less than 2% of patients in U.S. clinical trials with SPORANOX® included: adrenal insufficiency, asthenia, back pain, dehydration, dyspepsia, dysphagia, flatulence, gynecomastia, hematuria, hemorrhoids, hot flushes, implantation complication, infection unspecified, injury, insomnia, male breast pain, myalgia, pharyngitis, pruritus, rhinitis, rigors, stomatitis ulcerative, taste perversion, tinnitus, upper respiratory tract infection, vision abnormal, and weight decrease. Edema, hypokalemia and menstrual disorders have been reported in clinical trials with itraconazole capsules.
Adverse Events Reported from Other Clinical Trials
A comparative clinical trial in patients who received intravenous itraconazole followed by SPORANOX® Oral Solution or received Amphotericin B reported the following adverse events in the itraconazole intravenous/SPORANOX® Oral Solution treatment arm which are not listed above in the subsection "Adverse Events Reported in Oropharnyngeal or Esophageal Candidiasis Trials" or listed below as postmarketing reports of adverse drug reactions: serum creatinine increased, blood urea nitrogen increased, renal function abnormal, hypocalcemia, hypomagnesemia, hypophosphatemia, hypotension, tachycardia and pulmonary infiltration.
In addition, the following adverse drug reactions were reported in patients who participated in SPORANOX® Oral Solution clinical trials:
Cardiac Disorders: cardiac failure;
General Disorders and Administration Site Conditions: edema;
Hepatobiliary Disorders: hepatic failure, hyperbilirubinemia;
Metabolism and Nutrition Disorders: hypokalemia;
Reproductive System and Breast Disorders: menstrual disorder
The following is a list of additional adverse drug reactions associated with itraconazole that have been reported in clinical trials of SPORANOX® Capsules and itraconazole IV excluding the adverse reaction term "Injection site inflammation" which is specific to the injection route of administration:
Cardiac Disorders: left ventricular failure;
Gastrointestinal Disorders: gastrointestinal disorder;
General Disorders and Administration Site Conditions: face edema;
Hepatobiliary Disorders: jaundice, hepatic function abnormal;
Investigations: alanine aminotransferase increased, aspartate aminotransferase increased, blood alkaline phosphatase increased, blood lactate dehydrogenase increased, gamma-glutamyltransferase increased, urine analysis abnormal;
Metabolism and Nutrition Disorders: hyperglycemia, hyperkalemia;
Nervous System Disorders: somnolence;
Psychiatric Disorders: confusional state;
Renal and Urinary Disorders: renal impairment;
Respiratory, Thoracic and Mediastinal Disorders: dysphonia;
Skin and Subcutaneous Tissue Disorders: rash erythematous;
Vascular Disorders: hypertension
In addition, the following adverse drug reaction was reported in children only who participated in SPORANOX® Oral Solution clinical trials: mucosal inflammation.
Post-marketing Experience
Adverse drug reactions that have been first identified during post-marketing experience with SPORANOX® (all formulations) are listed in Table 4 below. Because these reactions are reported voluntarily from a population of uncertain size, reliably estimating their frequency or establishing a causal relationship to drug exposure is not always possible.
Table 4: Postmarketing Reports of Adverse Drug Reactions Blood and Lymphatic System Disorders: Leukopenia, neutropenia, thrombocytopenia Immune System Disorders: Anaphylaxis; anaphylactic, anaphylactoid and allergic reactions; serum sickness; angioneurotic edema Metabolism and Nutrition Disorders: Hypertriglyceridemia Nervous System Disorders: Peripheral neuropathy, paresthesia, hypoesthesia, tremor Eye Disorders: Visual disturbances, including vision blurred and diplopia Ear and Labyrinth Disorders: Transient or permanent hearing loss Cardiac Disorders: Congestive heart failure Respiratory, Thoracic and Mediastinal Disorders: Pulmonary edema Gastrointestinal Disorders: Pancreatitis Hepatobiliary Disorders: Serious hepatotoxicity (including some cases of fatal acute liver failure), hepatitis, reversible increases in hepatic enzymes Skin and Subcutaneous Tissue Disorders: Toxic epidermal necrolysis, Stevens-Johnson syndrome, acute generalized exanthematous pustulosis, erythema multiforme, exfoliative dermatitis, leukocytoclastic vasculitis, alopecia, photosensitivity, urticaria Musculoskeletal and Connective Tissue Disorders: Arthralgia Renal and Urinary Disorders: Urinary incontinence, pollakiuria Reproductive System and Breast Disorders: Erectile dysfunction General Disorders and Administration Site Conditions: Peripheral edema Investigations: Blood creatine phosphokinase increased There is limited information on the use of SPORANOX® during pregnancy. Cases of congenital abnormalities including skeletal, genitourinary tract, cardiovascular and ophthalmic malformations as well as chromosomal and multiple malformations have been reported during post-marketing experience. A causal relationship with SPORANOX® has not been established. (See CLINICAL PHARMACOLOGY: Special Populations, CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS: Drug Interactions for more information.)
-
OVERDOSAGE
Itraconazole is not removed by dialysis. In the event of accidental overdosage, supportive measures should be employed. Contact a certified poison control center for the most up to date information on the management of SPORANOX® (itraconazole) Oral Solution overdosage (1-800-222-1222 or www.poison.org). In general, adverse events reported with overdose have been consistent with adverse drug reactions already listed in this package insert for itraconazole. (See ADVERSE REACTIONS.)
-
DOSAGE AND ADMINISTRATION
Treatment of Oropharyngeal and Esophageal Candidiasis
The solution should be vigorously swished in the mouth (10 mL at a time) for several seconds and swallowed.
The recommended dosage of SPORANOX® (itraconazole) Oral Solution for oropharyngeal candidiasis is 200 mg (20 mL) daily for 1 to 2 weeks. Clinical signs and symptoms of oropharyngeal candidiasis generally resolve within several days.
For patients with oropharyngeal candidiasis unresponsive/refractory to treatment with fluconazole tablets, the recommended dose is 100 mg (10 mL) b.i.d. For patients responding to therapy, clinical response will be seen in 2 to 4 weeks. Patients may be expected to relapse shortly after discontinuing therapy. Limited data on the safety of long-term use (>6 months) of SPORANOX® Oral Solution are available at this time.
The recommended dosage of SPORANOX® Oral Solution for esophageal candidiasis is 100 mg (10 mL) daily for a minimum treatment of three weeks. Treatment should continue for 2 weeks following resolution of symptoms. Doses up to 200 mg (20 mL) per day may be used based on medical judgment of the patient's response to therapy.
SPORANOX® Oral Solution and SPORANOX® Capsules should not be used interchangeably. Patients should be instructed to take SPORANOX® Oral Solution without food, if possible. Only SPORANOX® Oral Solution has been demonstrated effective for oral and/or esophageal candidiasis.
Use in Patients with Renal Impairment
Limited data are available on the use of oral itraconazole in patients with renal impairment. Caution should be exercised when this drug is administered in this patient population. (See CLINICAL PHARMACOLOGY: Special Populations and PRECAUTIONS.)
Use in Patients with Hepatic Impairment
Limited data are available on the use of oral itraconazole in patients with hepatic impairment. Caution should be exercised when this drug is administered in this patient population. (See CLINICAL PHARMACOLOGY: Special Populations, WARNINGS, and PRECAUTIONS.)
-
HOW SUPPLIED
SPORANOX® (itraconazole) Oral Solution is available in 150 mL amber glass bottles (NDC: 50458-295-15) containing 10 mg of itraconazole per mL.
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 150 mL Bottle Label
NDC 50458-295-15
150 mLSPORANOX®
(ITRACONAZOLE)
ORAL SOLUTION10 mg/mL
Each 1 mL contains:
10 mg of itraconazole in an aqueous solution.Dosage: For information concerning dosage and
administration of SPORANOX® (itraconazole) oral
solution, please read accompanying Package Insert.Keep out of reach of children.
Store bottle at or below
25°C (77°F). Do not freeze.672269
Product of Belgium
Manufactured by:
Janssen Pharmaceutica NV
Beerse, BelgiumManufactured for:
Janssen Pharmaceuticals, Inc.
Titusville, NJ 08560janssen

-
INGREDIENTS AND APPEARANCE
SPORANOX
itraconazole solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50458-295 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength itraconazole (UNII: 304NUG5GF4) (itraconazole - UNII:304NUG5GF4) itraconazole 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength HYDROXYPROPYL BETADEX (UNII: 1I96OHX6EK) 400 mg in 1 mL HYDROCHLORIC ACID (UNII: QTT17582CB) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) SACCHARIN SODIUM (UNII: SB8ZUX40TY) SORBITOL (UNII: 506T60A25R) CHERRY (UNII: BUC5I9595W) CARAMEL (UNII: T9D99G2B1R) Product Characteristics Color YELLOW (clear and yellowish) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50458-295-15 150 mL in 1 BOTTLE; Type 1: Convenience Kit of Co-Package 02/21/1997 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020657 02/21/1997 Labeler - Janssen Pharmaceuticals, Inc. (063137772) Establishment Name Address ID/FEI Business Operations Janssen Pharmaceutical Sciences Unlimited Company 985639841 API MANUFACTURE(50458-295) Establishment Name Address ID/FEI Business Operations Janssen Pharmaceutica, NV 374747970 API MANUFACTURE(50458-295) Establishment Name Address ID/FEI Business Operations Janssen Pharmaceutica, NV 370005019 MANUFACTURE(50458-295) , ANALYSIS(50458-295)
Trademark Results [SPORANOX]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 SPORANOX 76036408 2882031 Live/Registered |
Johnson & Johnson 2000-04-28 |
 SPORANOX 76036406 2882030 Dead/Cancelled |
Johnson & Johnson 2000-04-28 |
 SPORANOX 74049328 1635945 Live/Registered |
Johnson & Johnson 1990-04-16 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.