REYVOW- lasmiditan tablet
Reyvow by
Drug Labeling and Warnings
Reyvow by is a Prescription medication manufactured, distributed, or labeled by Eli Lilly and Company. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use REYVOW safely and effectively. See full prescribing information for REYVOW.
REYVOW (lasmiditan) tablets, for oral use, CV
Initial U.S. Approval: 2020INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 50 mg, 100 mg (3)
CONTRAINDICATIONS
- None. (4)
WARNINGS AND PRECAUTIONS
- Driving Impairment: Advise patients not to drive or operate machinery until at least 8 hours after taking each dose of REYVOW. Patients who cannot follow this advice should not take REYVOW. Patients may not be able to assess their own driving competence and the degree of impairment caused by REYVOW. (5.1)
- Central Nervous System (CNS) Depression: REYVOW may cause CNS depression and should be used with caution if used in combination with alcohol or other CNS depressants. (5.2, 7.1)
- Serotonin Syndrome: Reactions consistent with serotonin syndrome were reported in patients treated with REYVOW. Discontinue REYVOW if symptoms of serotonin syndrome occur. (5.3)
- Medication Overuse Headache: Detoxification may be necessary. (5.4)
ADVERSE REACTIONS
Most common adverse reactions (≥5% and > placebo) were dizziness, fatigue, paresthesia, and sedation. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eli Lilly and Company at 1-800-LillyRx (1-800-545-5979) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Driving Impairment
5.2 Central Nervous System Depression
5.3 Serotonin Syndrome
5.4 Medication Overuse Headache
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 CNS Depressants
7.2 Serotonergic Drugs
7.3 Heart Rate Lowering Drugs
7.4 P-gp and Breast Cancer Resistant Protein (BCRP)
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
9.2 Abuse
9.3 Dependence
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Migraine
14.2 Effects on Driving
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
The recommended dose of REYVOW is 50 mg, 100 mg, or 200 mg taken orally, as needed. No more than one dose should be taken in 24 hours, and REYVOW should not be taken unless the patient can wait at least 8 hours between dosing and driving or operating machinery [see Warnings and Precautions (5.1)].
A second dose of REYVOW has not been shown to be effective for the same migraine attack.
The safety of treating an average of more than 4 migraine attacks in a 30-day period has not been established.
REYVOW may be taken with or without food.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Driving Impairment
REYVOW may cause significant driving impairment. In a driving study, administration of single 50 mg, 100 mg, or 200 mg doses of REYVOW significantly impaired subjects' ability to drive [see Clinical Studies (14.2)]. Additionally, more sleepiness was reported at 8 hours following a single dose of REYVOW compared to placebo. Advise patients not to engage in potentially hazardous activities requiring complete mental alertness, such as driving a motor vehicle or operating machinery, for at least 8 hours after each dose of REYVOW. Patients who cannot follow this advice should not take REYVOW. Prescribers and patients should be aware that patients may not be able to assess their own driving competence and the degree of impairment caused by REYVOW.
5.2 Central Nervous System Depression
REYVOW may cause central nervous system (CNS) depression, including dizziness and sedation [see Adverse Reactions (6.1)].
Because of the potential for REYVOW to cause sedation, other cognitive and/or neuropsychiatric adverse reactions, and driving impairment, REYVOW should be used with caution if used in combination with alcohol or other CNS depressants [see Drug Interactions (7.1)]. Patients should be warned against driving and other activities requiring complete mental alertness for at least 8 hours after REYVOW is taken [see Warnings and Precautions (5.1)].
5.3 Serotonin Syndrome
In clinical trials, reactions consistent with serotonin syndrome were reported in patients treated with REYVOW who were not taking any other drugs associated with serotonin syndrome. Serotonin syndrome may also occur with REYVOW during coadministration with serotonergic drugs [e.g., selective serotonin reuptake inhibitors (SSRIs), serotonin norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants (TCAs), and monoamine oxidase (MAO) inhibitors]. Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular signs (e.g., hyperreflexia, incoordination), and/or gastrointestinal signs and symptoms (e.g., nausea, vomiting, diarrhea). The onset of symptoms usually occurs within minutes to hours of receiving a new or a greater dose of a serotonergic medication. Discontinue REYVOW if serotonin syndrome is suspected.
5.4 Medication Overuse Headache
Overuse of acute migraine drugs (e.g., ergotamines, triptans, opioids, or a combination of these drugs for 10 or more days per month) may lead to exacerbation of headache (i.e., medication overuse headache). Medication overuse headache may present as migraine-like daily headaches or as a marked increase in frequency of migraine attacks. Detoxification of patients including withdrawal of the overused drugs and treatment of withdrawal symptoms (which often includes a transient worsening of headache) may be necessary.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Driving Impairment [see Warnings and Precautions (5.1)]
- Central Nervous System Depression [see Warnings and Precautions (5.2)]
- Serotonin Syndrome [see Warnings and Precautions (5.3)]
- Medication Overuse Headache [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The safety of REYVOW has been evaluated in 4,878 subjects who received at least one dose of REYVOW. In 2 placebo-controlled, Phase 3 trials in adult patients with migraine (Studies 1 and 2), a total of 3,177 patients received REYVOW 50, 100, or 200 mg [see Clinical Studies (14.1)]. Of the REYVOW-treated patients in these 2 studies, approximately 84% were female, 78% were White, 18% were Black, and 18% were of Hispanic or Latino ethnicity. The mean age at study entry was 42.4 years (range 18 to 81).
Long-term safety was assessed for 2,030 patients, dosing intermittently for up to 12 months in a long-term safety study. Of these, 728 patients were exposed to 100 mg or 200 mg for at least 3 months, 361 patients were exposed to these doses for at least 6 months, and 180 patients were exposed to these doses for at least 12 months, all of whom treated at least 2 migraine attacks per month on average. In that study, 14% (148 out of 1,039) in the 200 mg dose group, and 11% (112 out of 991) in the 100 mg dose group withdrew from the trial because of an adverse event. The most common adverse event resulting in discontinuation in the long-term safety study (greater than 2%) was dizziness.
Table 1 shows adverse reactions that occurred in at least 2% of patients treated with REYVOW and more frequently than in patients who received placebo in Studies 1 and 2. The most common adverse reactions (at least 5%) were dizziness, fatigue, paresthesia, and sedation.
Table 1: Adverse Reactions Occurring in ≥2% and at a Frequency Greater than Placebo in Studies 1 and 2 a Fatigue includes the adverse reaction related terms asthenia and malaise.
b Paresthesia includes the adverse reaction related terms paresthesia oral, hypoesthesia, and hypoesthesia oral.
c Sedation includes the adverse reaction related term somnolence.
Adverse Reaction REYVOW 50 mg
N=654
%REYVOW 100 mg
N=1265
%REYVOW 200 mg
N=1258
%Placebo
N=1262
%Dizziness 9 15 17 3 Fatiguea 4 5 6 1 Paresthesiab 3 7 9 2 Sedationc 6 6 7 2 Nausea and/or Vomiting 3 4 4 2 Muscle Weakness 1 1 2 0 Less Common Adverse Reactions
The following adverse reactions occurred in less than 2% of REYVOW-treated patients but more frequently than in patients receiving placebo: vertigo, incoordination, lethargy, visual impairment, feeling abnormal, palpitations, anxiety, tremor, restlessness, sleep abnormalities including sleep disturbance and abnormal dreams, muscle spasm, limb discomfort, cognitive changes, confusion, euphoric mood, chest discomfort, speech abnormalities, dyspnea, and hallucinations.
Hypersensitivity
Events of hypersensitivity, including angioedema, rash and photosensitivity reaction, occurred in patients treated with REYVOW. In controlled trials, hypersensitivity was reported in 0.2% of patients treated with REYVOW compared to no patients who received placebo. If a serious or severe hypersensitivity reaction occurs, initiate appropriate therapy and discontinue administration of REYVOW.
Vital Sign Changes
Heart Rate Decrease
REYVOW was associated with mean decreases in heart rate of 5 to 10 beats per minute (bpm) while placebo was associated with mean decreases of 2 to 5 bpm. Consider evaluating heart rate after administration of REYVOW in patients for whom these changes may not be tolerated, including patients taking other medications that lower heart rate [see Drug Interactions (7.3)].
Blood Pressure Increase
REYVOW may increase blood pressure following a single dose. In non-elderly healthy volunteers there was a mean increase from baseline in ambulatory systolic and diastolic blood pressure of approximately 2 to 3 mm Hg one hour after administration of 200 mg REYVOW compared to a mean increase of up to 1 mm Hg for placebo. In healthy volunteers over 65 years of age, there was a mean increase from baseline in ambulatory systolic blood pressure of 7 mm Hg one hour after administration of 200 mg REYVOW compared to a mean increase of 4 mm Hg for placebo. By 2 hours, there were no increases in mean blood pressure with REYVOW compared to placebo. REYVOW has not been well studied in patients with ischemic heart disease. Consider evaluating blood pressure after administration of REYVOW in patients for whom these changes may not be tolerated.
-
7 DRUG INTERACTIONS
7.1 CNS Depressants
Concomitant administration of REYVOW and alcohol or other CNS depressant drugs has not been evaluated in clinical studies. Because of the potential of REYVOW to cause sedation, as well as other cognitive and/or neuropsychiatric adverse reactions, REYVOW should be used with caution if used in combination with alcohol or other CNS depressants [see Warnings and Precautions (5.2)].
7.2 Serotonergic Drugs
Concomitant administration of REYVOW and drugs (e.g., SSRIs, SNRIs, TCAs, MAO inhibitors, trazodone, etc.), over-the counter medications (e.g., dextromethorphan), or herbal supplements (e.g., St. John's Wort) that increase serotonin may increase the risk of serotonin syndrome [see Warnings and Precautions (5.3)]. Use REYVOW with caution in patients taking medications that increase serotonin.
7.3 Heart Rate Lowering Drugs
REYVOW has been associated with a lowering of heart rate [see Adverse Reactions (6.1)]. In a drug interaction study, addition of a single 200 mg dose of REYVOW to propranolol decreased heart rate by an additional 5 beats per minute compared to propranolol alone, for a mean maximum of 19 beats per minute. Use REYVOW with caution in patients taking concomitant medications that lower heart rate if this magnitude of heart rate decrease may pose a concern.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of REYVOW in pregnant women. In animal studies, adverse effects on development (increased incidences of fetal abnormalities, increased embryofetal and offspring mortality, decreased fetal body weight) occurred at maternal exposures less than (rabbit) or greater than (rat) those observed clinically (see Data).
In the U.S. general population, the estimated background risk of major birth defects and of miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The estimated rates of major birth defects (2.2% to 2.9%) and miscarriage (17%) among deliveries to women with migraine are similar to rates reported in women without migraine.
Data
Animal Data
Oral administration of lasmiditan (0, 100, 175, or 250 mg/kg/day) to pregnant rats throughout organogenesis resulted in increases in skeletal variations at the mid and high doses and reduced fetal body weight at the high dose. The high dose was associated with maternal toxicity. At the no-effect dose (100 mg/kg/day) for adverse effects on embryofetal development in rats, plasma exposure (AUC) was approximately 10 times that in humans at the MRHD.
Oral administration of lasmiditan (0, 50, 75, or 115 mg/kg/day) to pregnant rabbits throughout organogenesis resulted in malformations (skeletal and visceral), increases in skeletal variations and embryofetal mortality, and decreased fetal body weight at the highest dose tested, which was associated with maternal toxicity. Plasma exposure (AUC) at the no-effect dose (75 mg/kg/day) for adverse effects on embryofetal development in rabbits was less than that in humans at the MRHD.
Oral administration of lasmiditan (0, 100, 150, or 225 mg/kg/day) to rats throughout pregnancy and lactation resulted in increases in stillbirth and neonatal mortality at the highest dose tested, which was associated with maternal toxicity and delayed parturition. Plasma exposure (AUC) at the no-effect dose (150 mg/kg/day) for adverse effects on pre- and postnatal development was approximately 16 times that in humans at the MRHD.
8.2 Lactation
Risk Summary
There are no data on the presence of lasmiditan in human milk, the effects of lasmiditan on the breastfed infant, or the effects of lasmiditan on milk production. Excretion of lasmiditan and/or metabolites into milk, at levels approximately 3 times those in maternal plasma, was observed in lactating rats following oral administration of lasmiditan.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for REYVOW and any potential adverse effects on the breastfed infant from REYVOW or from the underlying maternal condition.
8.5 Geriatric Use
In controlled clinical trials, dizziness occurred more frequently in patients who were at least 65 years of age (19% for REYVOW, 2% for placebo) compared to patients who were less than 65 years of age (14% for REYVOW, 3% for placebo). A larger increase in systolic blood pressure also occurred in patients 65 years of age and older compared to patients who were less than 65 years of age [see Adverse Reactions (6.1)]. Clinical studies of REYVOW did not include sufficient numbers of subjects aged 65 and over to determine whether there is a difference in efficacy in these patients compared to younger subjects. However, in clinical pharmacology studies, no clinically relevant effect on exposure to REYVOW was observed in elderly subjects [see Clinical Pharmacology (12.3)]. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
Abuse is the intentional, non-therapeutic use of a drug, even once, for its desirable psychological or physiological effects. In a human abuse potential (HAP) study in recreational poly-drug users (n=58), single oral therapeutic doses (100 and 200 mg) and a supratherapeutic dose (400 mg) of REYVOW were compared to alprazolam (2 mg) (C-IV) and placebo. With all doses of REYVOW, subjects reported statistically significantly higher “drug liking” scores than placebo, indicating that REYVOW has abuse potential. In comparison to alprazolam, subjects who received REYVOW reported statistically significantly lower “drug liking” scores. In the HAP study, euphoric mood occurred to a similar extent with REYVOW 200 mg, REYVOW 400 mg, and alprazolam 2 mg (43-49%). A feeling of relaxation was noted in more subjects on alprazolam (22.6%) than with any dose of REYVOW (7-11%).
Phase 2 and 3 studies indicate that, at therapeutic doses, REYVOW produced adverse events of euphoria and hallucinations to a greater extent than placebo. However these events occur at a low frequency (about 1% of patients).
Evaluate patients for risk of drug abuse and observe them for signs of lasmiditan misuse or abuse.
-
11 DESCRIPTION
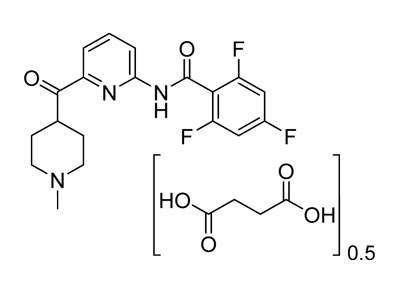
REYVOW (lasmiditan) is a serotonin (5-HT) 1F receptor agonist for oral administration. The chemical name of lasmiditan hemisuccinate is 2,4,6-trifluoro-N-[6-(1-methylpiperidine-4-carbonyl)pyridine-2-yl]benzamide hemisuccinate. It has the empirical formula of C19H18F3N3O20.5[C4H6O4] and a molecular weight of 436.41 (hemisuccinate). Lasmiditan hemisuccinate has the following structural formula:
Lasmiditan hemisuccinate is a white, crystalline powder that is sparingly soluble in water, slightly soluble in ethanol, and soluble in methanol. A 1 mg/mL aqueous solution of lasmiditan hemisuccinate has a pH of 6.8 at ambient conditions.
REYVOW 50 mg tablets contain 50 mg lasmiditan (equivalent to 57.824 mg lasmiditan hemisuccinate) and the inactive ingredients as follows:
Excipients – croscarmellose sodium, magnesium stearate, microcrystalline cellulose, pregelatinized starch, sodium lauryl sulfate.
Color mixture ingredients – black ferric oxide, polyethylene glycol, polyvinyl alcohol, talc, titanium dioxide.
REYVOW 100 mg tablets contain 100 mg lasmiditan (equivalent to 115.65 mg lasmiditan hemisuccinate) and the inactive ingredients as follows:
Excipients – croscarmellose sodium, magnesium stearate, microcrystalline cellulose, pregelatinized starch, sodium lauryl sulfate.
Color mixture ingredients – black ferric oxide, polyethylene glycol, polyvinyl alcohol, red ferric oxide, talc, titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lasmiditan binds with high affinity to the 5-HT1F receptor. Lasmiditan presumably exerts its therapeutic effects in the treatment of migraine through agonist effects at the 5-HT1F receptor; however, the precise mechanism is unknown.
12.3 Pharmacokinetics
Absorption
Following oral administration, lasmiditan is rapidly absorbed with a median tmax of 1.8 hours. In patients with migraine, the absorption or pharmacokinetics of lasmiditan was not different during a migraine attack versus during the interictal period.
Effect of Food
Coadministration of lasmiditan with a high-fat meal increased the mean lasmiditan Cmax and AUC values by 22% and 19%, respectively, and delayed the median tmax by 1 hour. This difference in exposure is not expected to be clinically significant [see Dosage and Administration (2)]. Lasmiditan was administered without regard to food in clinical efficacy studies.
Distribution
The human plasma protein binding of lasmiditan is approximately 55% to 60% and independent of concentration between 15 and 500 ng/mL.
Elimination
Lasmiditan was eliminated with a geometric mean t½ value of approximately 5.7 hours. No accumulation of lasmiditan was observed with daily dosing. Lasmiditan is primarily eliminated via metabolism, with ketone reduction representing the major pathway. Renal excretion is a minor route of lasmiditan clearance.
Metabolism
Lasmiditan undergoes hepatic and extrahepatic metabolism primarily by non-CYP enzymes. The following enzymes are not involved in metabolism of lasmiditan: MAO-A, MAO-B, flavin monooxygenase 3, CYP450 reductase, xanthine oxidase, alcohol dehydrogenase, aldehyde dehydrogenase, and aldo-keto reductases. Lasmiditan is also metabolized to M7 (oxidation on piperidine ring) and M18 (combination of M7 and M8 pathways). These metabolites are considered pharmacologically inactive.
Specific Populations
Age, Sex, Race/Ethnicity, and Body Weight
Based on a population pharmacokinetic (PK) analysis, age, sex, race/ethnicity, and body weight did not have a significant effect on the PK (Cmax and AUC) of lasmiditan. Therefore, no dose adjustments are warranted based on age, sex, race/ethnicity, or body weight.
Geriatric Use
In a clinical pharmacology study, administration of lasmiditan to subjects 65 years of age or older demonstrated 26% greater exposure in AUC(0-∞) and 21% higher Cmax, compared to subjects 45 years of age or less. This difference in exposure is not expected to be clinically significant [see Use in Specific Populations (8.5)].
Renal Impairment
In a clinical pharmacology study, administration of lasmiditan to subjects with severe renal impairment (eGFR <30 mL/min/1.73 m2) demonstrated 18% greater exposure in AUC(0-∞) and 13% higher Cmax, compared to subjects with normal renal function. No dose adjustment is required based on renal function.
Hepatic Impairment
In a clinical pharmacology study, subjects with mild and moderate hepatic impairment (Child-Pugh Class A and B, respectively) demonstrated 11% and 35%, respectively, greater exposure [AUC(0-∞)] to lasmiditan, compared to subjects with normal hepatic function. The Cmax were higher by 19% and 33%, respectively, for subjects with mild and moderate hepatic impairment. This difference in exposure is not expected to be clinically significant. The use of lasmiditan has not been studied in subjects with severe hepatic impairment [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Potential for Lasmiditan to Affect Other Drugs
Drug Metabolizing Enzymes
Lasmiditan is an in-vitro inhibitor of CYP2D6 but did not significantly inhibit the activity of other CYP450 enzymes. Modeling and simulation of the impact of lasmiditan on the exposure of dextromethorphan, a recognized sensitive CYP2D6 substrate, indicate that lasmiditan is unlikely to exert clinically significant inhibition of CYP2D6. Lasmiditan, M7, S-M8, and [S,R]-M18 are not reversible or time-dependent inhibitors of monoamine oxidase A (MAO-A).
Daily dosing of lasmiditan did not alter the PK of midazolam, caffeine, or tolbutamide, which are substrates of CYP3A, CYP1A2, and CYP2C9, respectively. Coadministration of lasmiditan with sumatriptan, propranolol, or topiramate resulted in no clinically meaningful changes in exposure of these medicinal products.
Drug Transporters
Lasmiditan inhibits P-gp and BCRP in-vitro [see Drug Interactions (7.4)].
Lasmiditan inhibits OCT1 in-vitro. However, in a drug-drug interaction study with lasmiditan and sumatriptan (OCT1 substrate), no change in sumatriptan PK was observed. Lasmiditan inhibits renal efflux transporters, MATE1 and MATE2-K, in-vitro.
Potential for Other Drugs to Affect Lasmiditan
Drug Metabolizing Enzymes
Lasmiditan undergoes hepatic and extrahepatic metabolism primarily by non-CYP enzymes. Therefore, it is unlikely that CYP inhibitors or inducers will affect lasmiditan pharmacokinetics. Clinical studies of lasmiditan with sumatriptan, propranolol, or topiramate did not show any significant drug interaction potential.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No drug-related tumors were observed following oral administration of lasmiditan to TgRasH2 mice at doses of up to 150 (males) or 250 (females) mg/kg/day for 26 weeks or to rats at doses of up to 75 mg/kg/day for 2 years. Plasma exposures (AUC) at the highest dose tested in rats were approximately 15 times that in humans at the maximum recommended human dose (MRHD) of 200 mg/day.
Mutagenesis
Lasmiditan was negative in in vitro (bacterial reverse mutation, chromosomal aberration in mammalian cells) and in vivo (mouse bone marrow micronucleus) assays.
Impairment of Fertility
Oral administration of lasmiditan to male (0, 100, 175, or 200 mg/kg/day) or female (0, 100, 150, or 200 mg/kg/day) rats prior to and during mating and continuing in females to Gestation Day 7 resulted in no adverse effects on fertility or reproductive performance. Plasma exposures (AUC) at the highest dose tested (200 mg/kg/day) were approximately 26 times that in humans at the MRHD.
-
14 CLINICAL STUDIES
14.1 Migraine
The efficacy of REYVOW in the acute treatment of migraine was demonstrated in two randomized, double-blind, placebo-controlled trials [Study 1 (NCT02439320) and Study 2 (NCT02605174)]. These studies enrolled patients with a history of migraine with and without aura according to the International Classification of Headache Disorders (ICHD-II) diagnostic criteria. Patients were predominantly female (84%), and White (78%), with a mean age of 42 years (range 18-81). Twenty-two percent of patients were taking preventive medication for migraine at baseline. Study 1 randomized patients to REYVOW 100 mg (n=744), or 200 mg (n=745) or placebo (n=742) and Study 2 randomized patients to REYVOW 50 mg (n=750), 100 mg (n=754), or 200 mg (n=750) or placebo (n=751). Patients were allowed to take a rescue medication 2 hours after taking study drug; however, opioids, barbiturates, triptans, and ergots were not allowed within 24 hours of study drug administration.
The primary efficacy analyses were conducted in patients that treated a migraine with moderate to severe pain within 4 hours of the onset of the attack. The efficacy of REYVOW was established by an effect on pain freedom at 2 hours and Most Bothersome Symptom (MBS) freedom at 2 hours compared to placebo for Studies 1 and 2. Pain freedom was defined as a reduction of moderate or severe headache pain to no pain, and MBS freedom was defined as the absence of the self-identified MBS (photophobia, phonophobia, or nausea). Among patients who selected an MBS, the most commonly selected MBS was photophobia (54%), followed by nausea (24%), and phonophobia (22%).
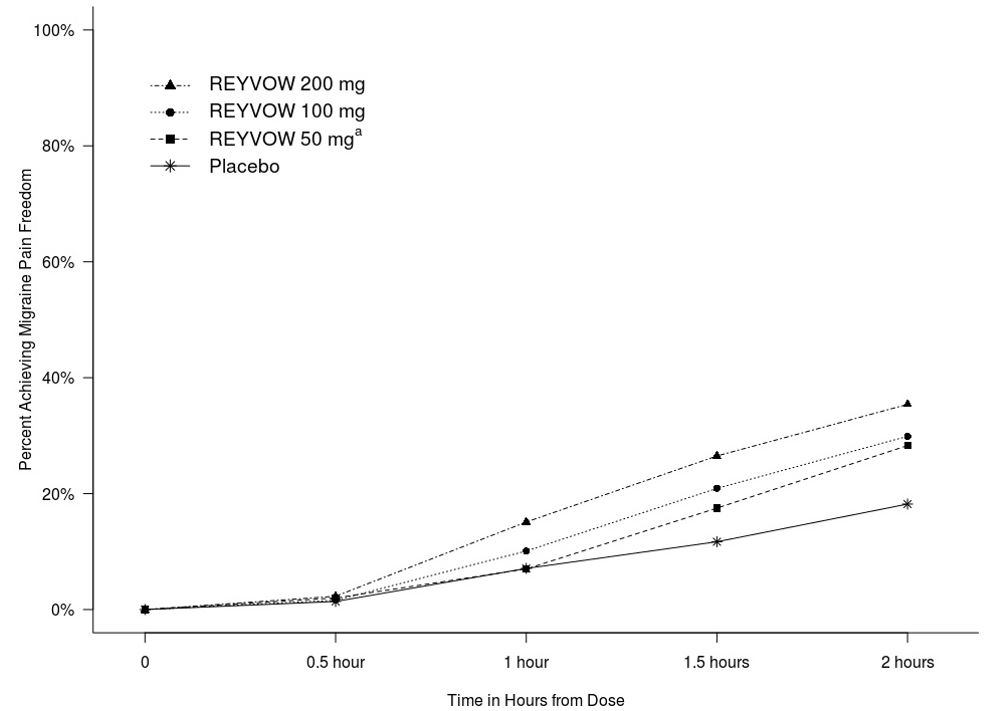
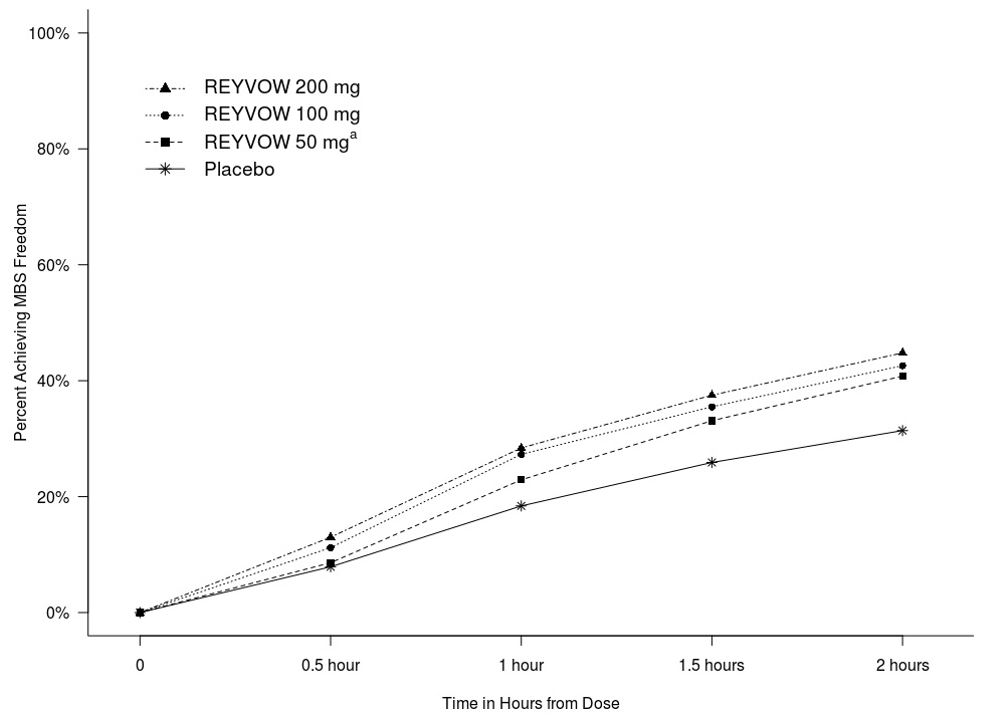
In both studies, the percentage of patients achieving pain freedom and MBS freedom 2 hours after treatment was significantly greater among patients receiving REYVOW at all doses compared to those receiving placebo (see Table 2).
Table 2: Migraine Efficacy Endpoints after Treatment for Studies 1 and 2 Study 1 Study 2 REYVOW
100 mgREYVOW
200 mgPlacebo REYVOW
50 mgREYVOW
100 mgREYVOW
200 mgPlacebo Pain Free at 2 hours N 498 503 515 544 523 521 534 % Responders 28.3 31.8 15.3 28.3 31.4 38.8 21.0 Difference from placebo (%) 13 16.5 7.3 10.4 17.8 p-value <0.001 <0.001 0.006 <0.001 <0.001 MBS Free at 2 hours N 464 467 480 502 491 478 509 % Responders 41.2 40.7 29.6 40.8 44.0 48.7 33.2 Difference from placebo (%) 11.6 11.1 7.6 10.8 15.5 p-value <0.001 <0.001 0.014 <0.001 <0.001 Pain relief at 2 hours, defined as a reduction in migraine pain from moderate or severe to mild or none, was also evaluated (see Table 3).
Table 3: Additional Migraine Efficacy Endpoint after Treatment for Studies 1 and 2 a The analysis of pain relief was descriptive and was not controlled for Type I error.
Study 1 Study 2 REYVOW
100 mgREYVOW
200 mgPlacebo REYVOW
50 mgREYVOW
100 mgREYVOW
200 mgPlacebo Pain Relief at 2 hoursa N 498 503 515 544 523 521 534 % Responders 54.0 55.3 40.0 55.9 61.4 61.0 45.1 Difference from placebo (%) 14.0 15.3 10.8 16.3 15.9 Figure 1 presents the percentage of patients achieving migraine pain freedom within 2 hours following treatment in Studies 1 and 2.
Figure 1: Percentage of Patients Achieving Migraine Pain Freedom within 2 Hours in Pooled Studies 1 and 2
a The 50 mg arm was only included in Study 2.
Figure 2 presents the percentage of patients achieving MBS freedom within 2 hours in Studies 1 and 2.
Figure 2: Percentage of Patients Achieving MBS Freedom within 2 Hours in Pooled Studies 1 and 2
a The 50 mg arm was only included in Study 2.
14.2 Effects on Driving
Driving performance was assessed at 90 minutes after administration of REYVOW 50 mg, 100 mg, 200 mg, alprazolam 1 mg, and placebo in a randomized, double-blind, placebo- and active-controlled, five-period crossover study in 90 healthy volunteers (mean age 34.9 years) using a computer-based driving simulation. Driving performance was evaluated using a validated threshold established in a population with blood alcohol concentration of 0.05%. The primary outcome measure was the difference from placebo in the Standard Deviation of Lateral Position (SDLP), a measure of driving performance. A dose-dependent impairment of computer-based simulated driving performance was seen with all doses of REYVOW at 90 minutes after administration.
Driving performance was also assessed at 8, 12, and 24 hours after administration of REYVOW 100 mg or 200 mg, in a separate randomized, double-blind, placebo- and active-controlled, four-period crossover study in 67 healthy volunteers (mean age 32.8 years) evaluating computer-based simulated driving performance using SDLP as the primary endpoint. Diphenhydramine 50 mg was used as a positive control. The mean SDLP did not reach the threshold for driving impairment at 8 hours or later after administration of REYVOW 100 or 200 mg.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
REYVOW (lasmiditan) 50 mg tablets are light gray, oval, film coated, tablets with “L-50” debossed on one side and “4312” on the other.
REYVOW (lasmiditan) 100 mg tablets are light purple, oval, film coated, tablets with “L-100” debossed on one side and “4491” on the other.
Strengths 50 mg 100 mg Tablet color Light gray Light purple Imprint (debossed) L-50
4312L-100
4491Carton of 8 NDC: 0002-4312-08 NDC: 0002-4491-08 -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Driving Impairment – Advise patients not to engage in potentially hazardous activities requiring complete mental alertness, such as driving a motor vehicle or operating machinery for at least 8 hours after taking each dose of REYVOW. Patients who cannot follow this advice should not take REYVOW. Patients may not be able to assess their own driving competence and the degree of impairment caused by REYVOW [see Warnings and Precautions (5.1)].
CNS Depression – Inform patients that REYVOW may cause dizziness and sedation. Advise patients to use caution if taking REYVOW in combination with alcohol or other CNS depressants [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
Serotonin Syndrome – Caution patients about the risk of serotonin syndrome with the use of REYVOW, particularly during combined use with serotonergic medications such as SSRIs, SNRIs, TCAs, or MAO inhibitors [see Warnings and Precautions (5.3) and Drug Interactions (7.2)].
Medication Overuse Headache – Inform patients that use of drugs to treat migraine attacks for 10 or more days per month may lead to an exacerbation of headache, and encourage patients to record headache frequency and drug use (e.g., by keeping a headache diary) [see Warnings and Precautions (5.4)].
Hypersensitivity – Advise patients to seek immediate medical attention if they experience any symptoms of serious or severe hypersensitivity reaction [see Adverse Reactions (6.1)].
Abuse and Dependence – Advise patients that REYVOW is a federally controlled substance because it has the potential to be abused [see Drug Abuse and Dependence (9)]. Advise patients to keep their medication secure.
Pregnancy – Advise patients to notify their healthcare provider if they become pregnant during treatment or plan to become pregnant [see Use in Specific Populations (8.1)].
Nursing Mothers – Inform patients to notify their healthcare provider if they are breastfeeding or plan to breastfeed [see Use in Specific Populations (8.2)].
Literature revised January 2020
Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA
Copyright © 2019, 2020, Eli Lilly and Company. All rights reserved.REY-0001-USPI-202001
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration
Revised: 01/2020
Medication Guide
REYVOW™ (RAY-vow)
(lasmiditan)
tablets, for oral use, CVWhat is the most important information I should know about REYVOW?
- Do not drive or operate machinery for at least 8 hours after you take REYVOW, even if you feel well enough.
- You should not take REYVOW if you cannot wait at least 8 hours between taking REYVOW and driving or operating machinery.
What is REYVOW?
REYVOW is a prescription medicine used for the acute treatment of migraine attacks with or without aura in adults.
- REYVOW is not used as a preventive treatment of migraine.
- It is not known if REYVOW is safe and effective in children.
- REYVOW is a federally controlled substance (CV) because it contains lasmiditan that can be a target for people who abuse prescription medicines or street drugs. Keep REYVOW in a safe place to protect it from theft. Never give your REYVOW to anyone else, because it may harm them. Selling or giving away REYVOW is against the law. Tell your healthcare provider if you have ever abused or been dependent on alcohol, prescription medicines or street drugs.
Before you take REYVOW, tell your healthcare provider about all of your medical conditions, including if you:
- have liver problems
- have high blood pressure
- have a low heart rate
- are allergic to lasmiditan
- are pregnant or plan to become pregnant. It is not known if REYVOW will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if REYVOW passes into your breastmilk.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Your healthcare provider will decide if you can take REYVOW with your other medicines.
Especially, tell your healthcare provider if you take:
- propranolol or other medicines that can lower your heart rate
- any medicines that can increase your blood pressure
- any medicines that make you sleepy
- anti-depressant medicines called:
- selective serotonin reuptake inhibitors (SSRIs)
- serotonin norepinephrine reuptake inhibitors (SNRIs)
- tricyclic anti-depressants (TCAs)
- monoamine oxidase inhibitors (MAOIs)
Ask your healthcare provider or pharmacist for a list of these medicines if you are not sure.
Know the medicines you take. Keep a list of them and show it to your healthcare provider or pharmacist when you get a new medicine.How should I take REYVOW?
- Take REYVOW exactly as your healthcare provider tells you to take it.
- Your healthcare provider may change your dose. Do not change your dose without first talking to your healthcare provider.
- Take REYVOW tablets by mouth with or without food.
- Do not take more than one dose in a 24-hour period.
- If you take REYVOW 50 mg, 100 mg, or 200 mg, and your headache goes away but comes back, you should not take a second dose within 24 hours.
- Some people who take too many REYVOW tablets may have worse headaches (medication overuse headache). If your headaches get worse, your healthcare provider may decide to stop your treatment with REYVOW.
- You should write down when you have headaches and when you take REYVOW so you can talk to your healthcare provider about how REYVOW is working for you.
What should I avoid while taking REYVOW?
- Do not drive or operate machinery for at least 8 hours after taking REYVOW.
- You should not drink alcohol or take other medicines that make you drowsy while taking REYVOW.
What are the possible side effects of REYVOW?
REYVOW can cause serious side effects including:
-
See “What is the most important information I should know about REYVOW?”
-
serotonin syndrome. Serotonin syndrome is a rare but serious problem that can happen in people using REYVOW, especially if REYVOW is used with anti-depressant medicines called SSRIs or SNRIs. Call your healthcare provider right away if you have any of the following symptoms of serotonin syndrome:
- mental changes such as seeing things that are not there (hallucinations), agitation, or coma
- fast heartbeat
- changes in blood pressure
- high body temperature
- tight muscles
- trouble walking
- nausea, vomiting, or diarrhea
- medication overuse headache. Some people who take medicines like REYVOW for the acute treatment of migraine attacks for 10 or more days each month may have worse headaches (medication overuse headache). If your headaches get worse, your healthcare provider may decide to stop your treatment with REYVOW.
-
serotonin syndrome. Serotonin syndrome is a rare but serious problem that can happen in people using REYVOW, especially if REYVOW is used with anti-depressant medicines called SSRIs or SNRIs. Call your healthcare provider right away if you have any of the following symptoms of serotonin syndrome:
The most common side effects of REYVOW include: - dizziness
- sleepiness
- numbness
- feeling tired
- tingling
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all of the possible side effects of REYVOW. For more information ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Lilly at 1-800-LillyRx (1-800-545-5979).How should I store REYVOW?
- Store REYVOW at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep REYVOW and all medicines out of the reach of children.
General information about the safe and effective use of REYVOW.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use REYVOW for a condition for which it was not prescribed. Do not give REYVOW to other people, even if they have the same symptoms you have. It may harm them.
You can ask your pharmacist or healthcare provider for information about REYVOW that is written for health professionals.What are the ingredients in REYVOW?
Active ingredient: lasmiditan hemisuccinate
Inactive ingredients: croscarmellose sodium, magnesium stearate, microcrystalline cellulose, pregelatinized starch, sodium lauryl sulfate.
Color mixture ingredients: black ferric oxide, polyethylene glycol, polyvinyl alcohol, talc, titanium dioxide. 100 mg tablets also contain red ferric oxide.
REYVOW is a trademark of Eli Lilly and Company.
Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA
www.REYVOW.com
Copyright © 2019, 2020, Eli Lilly and Company. All rights reserved.
For more information go to www.REYVOW.com or call 1-800-LillyRx (1-800-545-5979).REY-0001-MG-202001
-
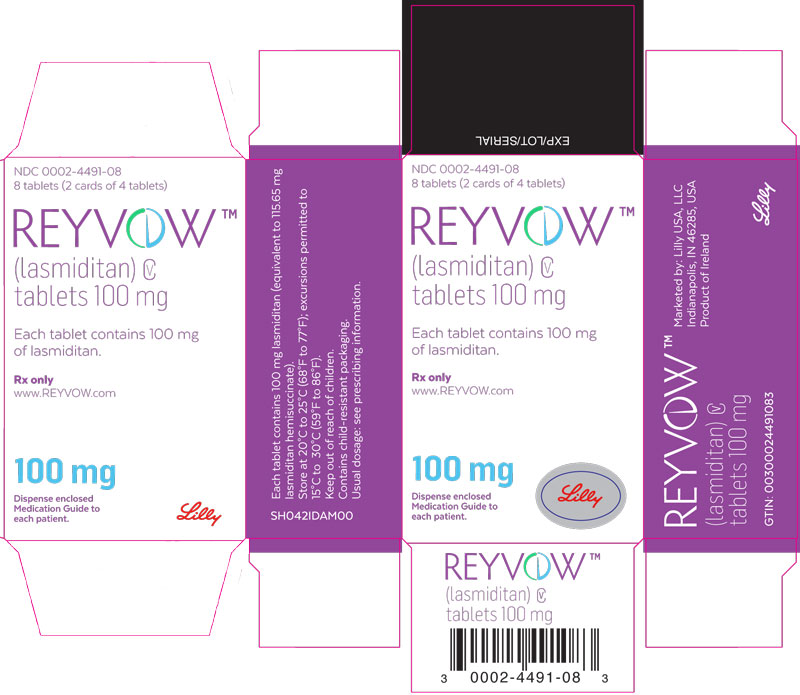
PRINCIPAL DISPLAY PANEL
PACKAGE LABEL - REYVOW 50 mg 8ct carton
NDC: 0002-4312-08
8 tablets (2 cards of 4 tablets)
REYVOW™
(lasmiditan) CV
tablets 50 mg
Each tablet contains 50 mg of lasmiditan.
Rx only
www.REYVOW.com
50 mg
Dispense enclosed Medication Guide to each patient.
Lilly
-
PRINCIPAL DISPLAY PANEL
PACKAGE LABEL - REYVOW 100 mg 8ct carton
NDC: 0002-4491-08
8 tablets (2 cards of 4 tablets)
REYVOW™
(lasmiditan) CV
tablets 100 mg
Each tablet contains 100 mg of lasmiditan.
Rx only
www.REYVOW.com
100 mg
Dispense enclosed Medication Guide to each patient.
Lilly
-
INGREDIENTS AND APPEARANCE
REYVOW
lasmiditan tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0002-4312 Route of Administration ORAL DEA Schedule CV Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength lasmiditan (UNII: 760I9WM792) (lasmiditan - UNII:760I9WM792) lasmiditan 50 mg Inactive Ingredients Ingredient Name Strength Microcrystalline cellulose (UNII: OP1R32D61U) Starch, corn (UNII: O8232NY3SJ) Croscarmellose Sodium (UNII: M28OL1HH48) Sodium Lauryl Sulfate (UNII: 368GB5141J) Magnesium Stearate (UNII: 70097M6I30) Polyvinyl Alcohol, Unspecified (UNII: 532B59J990) Titanium dioxide (UNII: 15FIX9V2JP) Polyethylene Glycol 3350 (UNII: G2M7P15E5P) Talc (UNII: 7SEV7J4R1U) Ferrosoferric Oxide (UNII: XM0M87F357) Product Characteristics Color gray (light gray) Score no score Shape OVAL (oval) Size 9mm Flavor Imprint Code L50;4312 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0002-4312-08 2 in 1 CARTON 01/31/2020 1 4 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211280 10/11/2019 REYVOW
lasmiditan tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0002-4491 Route of Administration ORAL DEA Schedule CV Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength lasmiditan (UNII: 760I9WM792) (lasmiditan - UNII:760I9WM792) lasmiditan 100 mg Inactive Ingredients Ingredient Name Strength Microcrystalline cellulose (UNII: OP1R32D61U) Starch, corn (UNII: O8232NY3SJ) Croscarmellose Sodium (UNII: M28OL1HH48) Sodium Lauryl Sulfate (UNII: 368GB5141J) Magnesium Stearate (UNII: 70097M6I30) Polyvinyl Alcohol, Unspecified (UNII: 532B59J990) Titanium dioxide (UNII: 15FIX9V2JP) Polyethylene Glycol 3350 (UNII: G2M7P15E5P) Talc (UNII: 7SEV7J4R1U) Ferrosoferric Oxide (UNII: XM0M87F357) Ferric Oxide Red (UNII: 1K09F3G675) Product Characteristics Color purple (light purple) Score no score Shape OVAL Size 12mm Flavor Imprint Code L100;4491 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0002-4491-08 2 in 1 CARTON 01/31/2020 1 4 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 0002-4491-61 1 in 1 CARTON 01/31/2020 2 4 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211280 10/11/2019 Labeler - Eli Lilly and Company (006421325)
Trademark Results [Reyvow]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 REYVOW 88035791 not registered Live/Pending |
Eli Lilly and Company 2018-07-12 |
 REYVOW 87446502 not registered Live/Pending |
Eli Lilly and Company 2017-05-11 |
 REYVOW 86926214 not registered Dead/Abandoned |
ELANCO US INC. 2016-03-02 |
 REYVOW 85685418 not registered Dead/Abandoned |
Eli Lilly and Company 2012-07-24 |
 REYVOW 85042374 not registered Dead/Abandoned |
Eli Lilly and Company 2010-05-19 |
 REYVOW 77141452 not registered Dead/Abandoned |
Eli Lilly and Company 2007-03-27 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.