REVCOVI- elapegademase-lvlr injection
Revcovi by
Drug Labeling and Warnings
Revcovi by is a Prescription medication manufactured, distributed, or labeled by Leadiant Biosciences, Inc., Exelead, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use REVCOVI® safely and effectively. See full prescribing information for REVCOVI.
REVCOVI (elapegademase-lvlr) injection, for intramuscular use
Initial U.S. Approval: 2018RECENT MAJOR CHANGES
INDICATIONS AND USAGE
REVCOVI is a recombinant adenosine deaminase indicated for the treatment of adenosine deaminase severe combined immune deficiency (ADA-SCID) in pediatric and adult patients. (1)
DOSAGE AND ADMINISTRATION
- Patients transitioning from Adagen to REVCOVI: The starting dose of REVCOVI is 0.2 mg/kg weekly, intramuscularly. See Full Prescribing Information (FPI) for conversion formula from Adagen to REVCOVI. (2.1)
- Adagen-naïve patients: The starting dose of REVCOVI is 0.4 mg/kg weekly based on ideal body weight or actual weight whichever is greater, divided into two doses (0.2 mg/kg twice a week), intramuscularly. (2.1)
- For complete information, maintenance dosing and therapeutic monitoring, see FPI. (2.1, 2.3)
- REVCOVI is for intramuscular injection only. See FPI for administration instructions. (2.2)
DOSAGE FORMS AND STRENGTHS
Injection: 2.4 mg/1.5 mL (1.6 mg/mL) in a single-dose vial. (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Injection Site Bleeding in Patients with Thrombocytopenia: Increased risk of local bleeding in patients with thrombocytopenia; should not be used if thrombocytopenia is severe. (5.1)
- Delay in Improvement of Immune Function: Protect immune deficient patients from infections until improvement in immune function. (5.2)
ADVERSE REACTIONS
The most common adverse reactions reported were cough (50%) and vomiting (33%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Leadiant at toll-free phone 1-888-393-4584 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Administration Instructions
2.3 Therapeutic Monitoring Schedule
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Injection Site Bleeding in Patients with Thrombocytopenia
5.2 Delay in Improvement of Immune Function
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience with ADAGEN
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Study 1
14.2 Study 2
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Patients transitioning from Adagen to REVCOVI
If a patient's weekly Adagen dose is unknown, or a patient's weekly Adagen dose is at or lower than 30 U/kg, the recommended minimum starting dose of REVCOVI is 0.2 mg/kg, intramuscularly, once a week.
If a patient's weekly Adagen dose is above 30 U/kg, an equivalent weekly REVCOVI dose (mg/kg) should be calculated using the following conversion formula:
REVCOVI dose in mg/kg = Adagen dose in U/kg
150Subsequent doses may be increased by increments of 0.033 mg/kg weekly if trough ADA activity is under 30 mmol/hr/L, trough deoxyadenosine nucleotides (dAXP) are above 0.02 mmol/L, and/or the immune reconstitution is inadequate based on the clinical assessment of the patient. The total weekly dose may be divided into multiple intramuscular (IM) administrations during a week.
Adagen-naïve patients
The starting weekly dose of REVCOVI is 0.4 mg/kg based on ideal body weight or actual weight whichever is greater, divided into two doses (0.2 mg/kg twice a week), intramuscularly, for a minimum of 12 to 24 weeks until immune reconstitution is achieved. After that, the dose may be gradually adjusted down to maintain trough ADA activity over 30 mmol/hr/L, trough dAXP level under 0.02 mmol/L, and/or to maintain adequate immune reconstitution based on clinical assessment of the patient.
The optimal long-term dose and schedule of administration should be established by the treating physician for each patient individually and may be adjusted based on the laboratory values for trough ADA activity, trough dAXP level, and/or on the treating physician's medical assessment of the patient's clinical status.
2.2 Administration Instructions
REVCOVI is for IM injection only. Follow sterile IM administration technique guidelines appropriate to the patient's age and anatomy (i.e. choice of needle gauge and length, site of administration). Take precautions not to inject into or near an artery or nerve. Alternate the injection site periodically.
Preparation of Injection and Procedure Instructions
- REVCOVI should not be diluted nor mixed with any other drug prior to administration.
- Visually inspect REVCOVI for particulate matter and discoloration prior to administration. REVCOVI is a clear, colorless solution; discard if solution is discolored, cloudy or contains particulate matter.
- Do not freeze or shake. REVCOVI should not be used if there are any indications that it may have been frozen. Once removed from refrigeration, allow REVCOVI to equilibrate to room temperature for 30 minutes.
- REVCOVI is to be administered using polypropylene syringes. Draw the solution from the vial with a 25- gauge needle or larger.
- Change the needle to a size and gauge appropriate for the patient's intramuscular administration.
- REVCOVI should be administered immediately after syringe preparation.
- Any remaining medication in the vial must be discarded immediately.
2.3 Therapeutic Monitoring Schedule
The treatment of ADA-SCID with REVCOVI should be monitored by measuring trough plasma ADA activity, trough dAXP levels, and/or total lymphocyte counts. Monitoring should be more frequent if therapy was interrupted or if an enhanced rate of clearance of plasma ADA activity develops.
Collect blood samples for the analysis of trough plasma ADA activity and trough dAXP level prior to the first administration of REVCOVI for the week.
ADA Activity
Once treatment with REVCOVI has been initiated, a target trough plasma ADA activity should be at least 30 mmol/hr/L. In order to determine an effective dose of REVCOVI, trough plasma ADA activity (pre-injection) should be determined every 2 weeks for Adagen-naïve patients and every 4 weeks for patients previously receiving Adagen therapy, during the first 8 - 12 weeks of treatment, and every 3 - 6 months thereafter.
A decrease of ADA activity below this level suggests noncompliance to treatment or a development of antibodies (anti-drug, anti-PEG, and neutralizing antibodies). Antibodies to REVCOVI should be suspected if a persistent fall in pre-injection levels of trough plasma ADA activity below 15 mmol/hr/L occurs. In such patients, testing for antibodies to REVCOVI should be performed.
If a persistent decline in trough plasma ADA activity occurs, immune function and clinical status should be monitored closely and precautions should be taken to minimize the risk of infection. If antibodies to REVCOVI are found to be the cause of a persistent fall in trough plasma ADA activity, then adjustment in the dosage of REVCOVI and other measures may be taken to induce tolerance and restore adequate ADA activity.
Erythrocyte dAXP
Two months after starting REVCOVI treatment, trough erythrocyte dAXP levels should be maintained below 0.02 mmol/L, and monitored at least twice a year.
Immune Function
The degree of immune function may vary from patient to patient. Each patient will require appropriate monitoring consistent with immunologic status. Total and subset lymphocytes should be monitored periodically as follows:
- Adagen-naïve patients: every 4 – 8 weeks for up to 1 year, and every 3 – 6 months thereafter
- Other patients: every 3 - 6 months
Immune function, including the ability to produce antibodies, generally improves after 2 - 6 months of therapy, and matures over a longer period. In general, there is a lag between the correction of the metabolic abnormalities and improved immune function. Improvement in the general clinical status of the patient may be gradual (as evidenced by improvement in various clinical parameters) but should be apparent by the end of the first year of therapy.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
- 5 WARNINGS AND PRECAUTIONS
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
REVCOVI was administered intramuscularly in two prospective, open-label, single-arm, multi‑center studies to evaluate efficacy, safety, tolerability, and pharmacokinetics in patients with ADA-SCID: Study 1 was performed in the US and Study 2 was performed in Japan [see Clinical Studies (14)]. Overall, 10 patients were treated and adverse reactions reported are summarized below.
Study 1
Study 1 is a one-way crossover study, conducted in the US, to evaluate the safety, efficacy, and pharmacokinetics of REVCOVI in patients with ADA‑SCID who were receiving therapy with Adagen. Six patients, 8 to 37 years of age enrolled in the study. Patients' exposure to REVCOVI ranged from 2 weeks to 146 weeks. No deaths were reported and one patient discontinued treatment due to injection site pain associated with an earlier drug product formulation that was consequently modified.
The most common adverse reactions were cough (3/6 patients) and vomiting (2/6 patients). Other adverse reactions that were reported in one patient each were: abdominal pain upper, arthralgia, asthenia, cerumen impaction, conjunctivitis, convulsion, dental caries, diarrhea, ear canal irritation, ear lobe infection, epistaxis, fatigue, fungal skin infection, gait disturbance, gastrointestinal infection, groin abscess, hematochezia, haemophilus infection (pulmonary), hemoptysis, influenza, injection site discomfort, laceration, lymphadenopathy, migraine, nasal edema, nausea, nephrolithiasis, oral candidiasis, oropharyngeal pain, otitis externa, productive cough, rash, stoma site infection, swelling face, tooth abscess, tooth extraction and upper respiratory tract infection, regardless of investigator causality assessment.
Study 2
Study 2 is a single-arm clinical study that was conducted to assess the safety, efficacy and pharmacokinetics of REVCOVI in patients with ADA-SCID. Four patients 3.4 months to 25 years of age, all Asian, were enrolled in the study and received REVCOVI. Three patients received REVCOVI for 21 weeks and one patient received REVCOVI for 15 weeks. One death due to CMV pneumonitis and respiratory failure was observed in an infant, who had also experienced pulmonary hemorrhage, respiratory failure and upper respiratory tract infection that represented serious adverse events. Neutropenia was a serious adverse reaction reported by one of the patients. There were 22 reported adverse events for four patients. Most common adverse events were respiratory infections (2/4 patients).
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The immunogenicity results from Study 1 and Study 2 suggest that patients who previously received Adagen may present an immunologic response to REVCOVI. Therefore, monitoring for changes in ADA levels during REVCOVI treatment is recommended. [see Dosage and Administration (2.3)]
The observed incidence of antibodies (including neutralizing antibodies) is dependent on assay sensitivity and specificity, assay methodology, and concomitant medications. Therefore, the comparison of the incidence of antibodies to REVCOVI with the incidence of antibodies to other products may be misleading.
6.3 Postmarketing Experience with ADAGEN
The following postmarketing adverse reactions were voluntarily reported for Adagen, the same class of enzyme replacement therapy used in the treatment of ADA-SCID, and may also be seen with REVCOVI treatment:
- Hematologic: hemolytic anemia, auto-immune hemolytic anemia, thrombocythemia, thrombocytopenia and autoimmune thrombocytopenia
- Dermatological: injection site erythema, urticaria
- Lymphomas
Since these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Adequate and well-controlled studies with REVCOVI have not been conducted in pregnant women to inform a drug-associated risk. Animal reproduction studies have not been conducted with REVCOVI. It is not known whether REVCOVI can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity.
All pregnancies have a risk of birth defect, loss, or other adverse outcomes. In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human
No pregnancy was reported for any patients receiving REVCOVI. There are two reports of confirmed cases of successful pregnancy and delivery in ADA-SCID patients treated with Adagen (the same class of enzyme replacement therapy used in the treatment of ADA-SCID). No teratogenic effects of Adagen were reported.
For patients treated with REVCOVI, more frequent monitoring of the health status for both the mother during pregnancy and the development of the offspring is recommended.
8.2 Lactation
Risk Summary
Human or animal lactation studies have not been conducted to assess the presence of REVCOVI in breast milk, the effects on the breastfed infant, or the effects on milk production for the mother.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for REVCOVI and any potential adverse effects on the breastfed infant from REVCOVI or from the underlying maternal condition.
8.4 Pediatric Use
The safety and efficacy of REVCOVI have been established in pediatric patients [see Clinical Studies (14)].
-
10 OVERDOSAGE
There are no reports of administration of REVCOVI in excess of the prescribed doses. The highest weekly prescribed dose administered in the clinical studies was 0.4 mg/kg. In nonclinical studies, there was no evidence of toxicity related to study drug at doses up to 1.8-fold the clinical dose (based on mean human AUC normalized to the dose of REVCOVI administered per patient), except for a slight increase in activated partial thromboplastin time (APTT).
-
11 DESCRIPTION
Elapegademase-lvlr is a recombinant adenosine deaminase (rADA) based on bovine amino acid sequence, conjugated to monomethoxypolyethylene glycol (mPEG). rADA is manufactured in E. coli and is covalently conjugated to mPEG with a succinimidyl carbamate linker to produce methoxypolyethylene glycol recombinant adenosine deaminase (SC-PEG rADA). The approximate molecular weight of elapegademase-lvlr (SC-PEG rADA) is 113 KDa.
REVCOVI (elapegademase-lvlr) injection is a sterile, preservative free, clear, colorless solution for intramuscular use supplied in single-dose vials. Each vial provides 1.5 mL of solution containing 2.4 mg elapegademase-lvlr (1.6 mg/mL), sodium chloride (12.75 mg), sodium phosphate dibasic heptahydrate (12.7 mg), sodium phosphate monobasic monohydrate (3.81 mg), and Water for Injection, USP. The pH is 6.9.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
SCID associated with a deficiency of ADA enzyme is a rare, inherited, and often fatal disease. ADA enzyme is involved in purine metabolism, catalyzing the irreversible hydrolytic deamination of adenosine or deoxyadenosine to inosine or deoxyinosine, respectively, as well as several naturally occurring methylated adenosine compounds. Maintaining a low level of 2'-deoxyadenosine and adenosine is crucial for proper number and function of immune cells as well as decreasing the frequency of opportunistic infections. Elevated adenosine levels, as occurring in ADA deficiency, contribute to apoptosis and a block in the differentiation of thymocytes, causing severe T‑lymphopenia.
Elapegademase-lvlr provides an exogenous source of ADA enzyme that is associated with a decrease in toxic adenosine and deoxyadenosine nucleotides levels as well as an increase in lymphocyte number [see Clinical Studies (14)].
12.3 Pharmacokinetics
The pharmacokinetics (PK) of REVCOVI were evaluated based on steady state plasma ADA activity in six patients with ADA-SCID (five adults and one pediatric) from two studies (Study 1 and Study 2) who received weekly IM injections at doses ranging from 4.99 to 19.6 mg [see Clinical Studies (14)]. The PK results are summarized in Table 1.
Table 1 Individual Estimates of Steady State Plasma Pharmacokinetic Parameters of REVCOVI Following Weekly IM Administration in ADA-SCID Patients Study Patient's Age (yrs), Sex, Race Weekly Dose
(mg)
[mg/kg]Tmax
(hr)DN AUC0-168hr
(hr*mmol/hr/L)/
(mg/kg)bDN Cmax
(mmol/hr/L)/
(mg/kg)bCtrough
(mmol/hr/L)ca PK data calculated over the dosing interval after weekly IM administration of REVCOVI at a stable REVCOVI dose for at least five consecutive weeks
b Dose-normalized (DN) AUC0-168hr and Cmax estimates based on mg/kg/week dose of REVCOVI
c Non-dose normalized steady state Ctrough ADA activity in plasma at Day 7 prior to administration of next weekly doseStudy 1a
19, Male, Hispanic/Latino
10.0
[0.188]47.7
32710
237
29.0
21, Male, Hispanic/Latino
10.2
[0.224]71.9
31343
219
37.7
37, Male, Black/African American
19.6
[0.2]48.2
42400
292
46.2
30, Female, White/Caucasian
10.0
[0.209]72.0
24564
166
23.5
Study 2a
25, Male, Asian
10.0
[0.167]48.0
37605
251
33.5
16, Female, Asian
4.99
[0.233]27.2
19013
150
20.2
In Study 1, steady state ADA activity levels were reached following seven consecutive once weekly IM doses of REVCOVI. In addition, dAXP activity levels in all patients at the majority of all sampling timepoints in Study 1 were less than 0.02 mmol/L.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Study 1
Study 1, conducted in the US (NCT 01420627), is an ongoing Phase 3, open-label, multicenter, single-arm, one-way crossover study of REVCOVI. The purpose of this clinical study is to evaluate the safety, efficacy, and PK of REVCOVI in 6 patients with ADA-SCID, 4 males and 2 females, who are receiving therapy with Adagen. The study treatment consists of three phases: Adagen Lead-in Phase (minimum of 3 weeks), the REVCOVI Treatment Phase (weeks 1 through 21), and followed by the REVCOVI Maintenance Phase. Six patients treated in the study were 8 to 37 years of age at the start of the study. The starting weekly dose of REVCOVI was calculated based on the last Adagen dose received in the study. Weekly REVCOVI doses ranged from 0.188 mg/kg to 0.292 mg/kg [see Dosage and Administration (2.1)].
The efficacy endpoints assessed were as follows:
- Trough dAXP Level (metabolic detoxification was defined as a trough erythrocyte dAXP concentration equal to or below 0.02 mmol/L)
- Trough plasma ADA activity (adequate trough plasma ADA activity is defined as trough plasma ADA activity equal to or above 15 mmol/hr/L)
- Immune status (lymphocyte and B-, T-, and NK-lymphocyte subset counts as well as quantitative immunoglobulin [Ig] concentration [IgG, IgA, IgM])
A PK assessment was performed during Week 9 of the REVCOVI Treatment phase [see Clinical Pharmacology (12.3)].
Five of six patients reached the 21-week endpoint of the Treatment Phase, and three of six patients received treatment with REVCOVI (elapegademase-lvlr) for over 135 weeks. These patients (except for one value in a patient at Treatment Week 47) had erythrocyte dAXP concentration equal to or below 0.02 mmol/L. These patients had trough plasma ADA activity equal to or above 15 mmol/hr/L at 88/89 timepoints and maintained metabolic detoxification for at least 2 years under REVCOVI treatment. Patients achieved trough plasma ADA activity above 30 mmol/hr/L by week 5, except for one patient who achieved this level at week 1. The mean trough plasma ADA activity for patients receiving REVCOVI at a normalized dose of 0.2 mg/kg/week were 34.3 ± 6.6 mmol/hr/L. The same patients had a mean trough plasma ADA activity of 14.2 ± 5.1 mmol/hr/L when treated with Adagen at a normalized dose of 30 U/kg/week during the Lead-in Phase of the study.
Lymphocyte and subset counts during REVCOVI treatment increased above levels observed during the Adagen Lead-in Phase (i.e., PK day 1 or before the start of REVCOVI treatment): maximum increases of approximately 3-fold at Weeks 60-73 for one patient, maximum increases of approximately 2- to 3-fold at Weeks 73-99 for one patient and approximately 1.5- to 3-fold for the third patient at several timepoints. For these three patients who completed the primary endpoint (21 weeks of treatment) and received REVCOVI for over 135 weeks, a positive trend between high trough plasma ADA activity and increased total lymphocyte counts was observed.
Observations for the other three patients in the study, indicate that these patients also achieved complete detoxification based on trough dAXP level and trough plasma ADA activity, and show stable or slightly increased lymphocyte counts during REVCOVI treatment relative to values recorded during the Adagen Lead-in Phase.
14.2 Study 2
Study 2, conducted in Japan, is a single-arm clinical study that assessed the safety, efficacy and PK of REVCOVI in patients with ADA-SCID. The study includes two phases: 1) Evaluation, consisting of a Dose Adjustment Period (5 weeks) and a Dose Maintenance Period (16 weeks); and 2) Continuous Administration (Extension) Phase, to be continued until the end of the study.
A total of four patients were enrolled in the study: two males (age 25 years and 3.4 months) and two females (age 16 years and 4.3 months). Two patients, who were on Adagen treatment within 4 weeks before entering the study, received a first dose of REVCOVI that was calculated to be equivalent to the last Adagen dose received. One patient, who did not receive Adagen within four weeks prior to entering the study, was given the first dose of REVCOVI at 0.1 mg/kg body weight, followed by second and third doses at 0.133 mg/kg body weight and weekly thereafter. Over the dose adjustment phase of the study, the dose was titrated to meet criteria for dAXP level (equal to or below 0.02 mmol/L) and adequate trough ADA activity (equal to or above 15 mmol/hr/L). These three patients received REVCOVI for at least 21 weeks (having completed the 5-week Dosage Adjustment Period and the 16-week Maintenance Period) before entering the Extension Phase. The fourth patient (newly diagnosed Adagen-naïve patient with CMV pneumonia [see Adverse Reactions (6.1)]) was dosed with REVCOVI at 0.4 mg/kg weekly (divided into two IM administrations) for 16 weeks.
All four of the patients in Study 2 achieved and maintained detoxification (trough dAXP [erythrocyte or blood] ≤0.02 mmol/L) throughout their participation in the Treatment Phase of 21 weeks (Dose Adjustment and Dose Maintenance). Serum ADA activity increased after administering REVCOVI for all four patients, with three patients achieving activity level over 15 mmol/hr/L during the Dose Maintenance Period. Total lymphocyte counts and B-/T-/NK-lymphocyte subset counts for three patients increased from screening to Day 15 during dose adjustment and were stable or increasing during the Maintenance Period.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
REVCOVI (elapegademase-lvlr) injection, 2.4 mg/1.5 mL (1.6 mg/mL), is a sterile, preservative free, clear, colorless solution for intramuscular use available as one single-dose vial per carton (NDC 57665-002-01).
The vial stopper is not made with natural rubber latex.
Single-dose vial; do not re-use the vial. Discard unused portions.
Store REVCOVI in the refrigerator between 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Do not freeze or shake. REVCOVI should not be used if there are any indications that it may have been frozen.
-
17 PATIENT COUNSELING INFORMATION
Importance of Compliance
Counsel patients and caregivers that continuous therapy and adherence to the recommended drug schedule is important for the success of the treatment.
Manufactured by: Leadiant Biosciences Inc., Gaithersburg, MD 20878, USA, U.S. License No. 2073 at Exelead Inc., 6925 Guion Rd, Indianapolis, IN 46268, USA
Date of Issue: 04/2020
I-004-11-US-C -
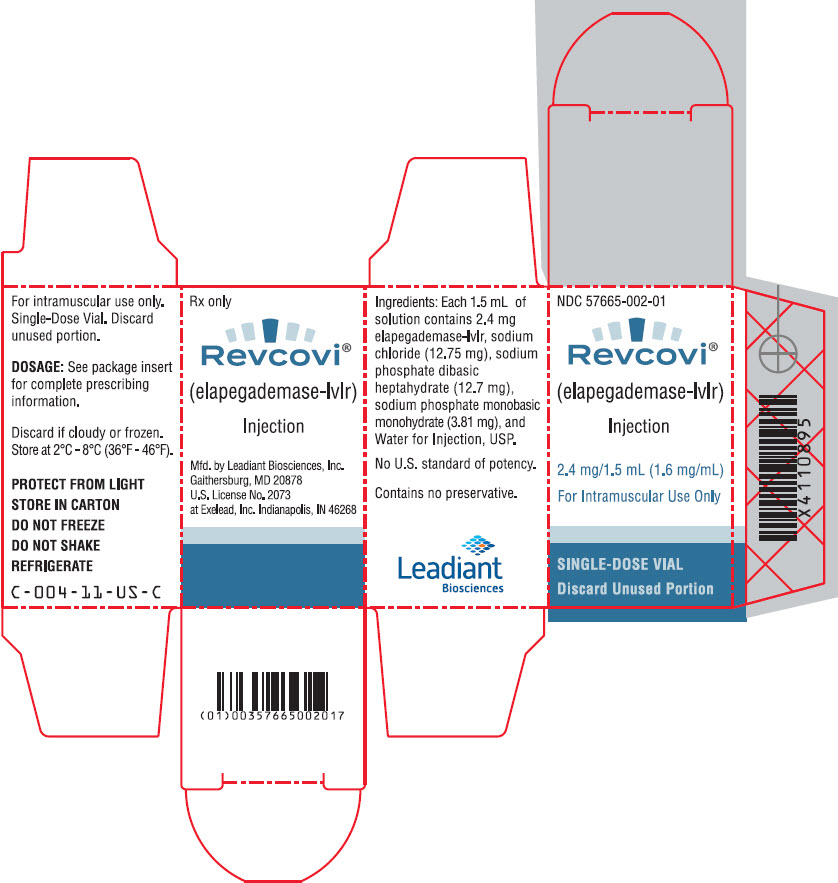
PRINCIPAL DISPLAY PANEL - 2.4 mg/1.5 mL Vial Carton
NDC: 57665-002-01
Revcovi®
(elapegademase-lvlr)
Injection2.4 mg/1.5 mL (1.6 mg/mL)
For Intramuscular Use Only
SINGLE-DOSE VIAL
Discard Unused Portion
-
INGREDIENTS AND APPEARANCE
REVCOVI
elapegademase-lvlr injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 57665-002 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ELAPEGADEMASE (UNII: 9R3D3Y0UHS) (ELAPEGADEMASE - UNII:9R3D3Y0UHS) ELAPEGADEMASE 1.6 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM PHOSPHATE, DIBASIC, HEPTAHYDRATE (UNII: 70WT22SF4B) SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 57665-002-01 1 in 1 CARTON 10/05/2018 1 1.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761092 10/05/2018 Labeler - Leadiant Biosciences, Inc. (068301431) Establishment Name Address ID/FEI Business Operations Exelead, Inc. 961822389 MANUFACTURE(57665-002) , LABEL(57665-002) , PACK(57665-002) , ANALYSIS(57665-002) , RELABEL(57665-002) , REPACK(57665-002)

Trademark Results [Revcovi]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 REVCOVI 87300981 5800819 Live/Registered |
LEADIANT BIOSCIENCES LIMITED 2017-01-13 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.