TAMSULOSIN HYDROCHLORIDE capsule
TAMSULOSIN HYDROCHLORIDE by
Drug Labeling and Warnings
TAMSULOSIN HYDROCHLORIDE by is a Prescription medication manufactured, distributed, or labeled by H.J. Harkins Company, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use tamsulosin hydrochloride safely and effectively. See full prescribing information for tamsulosin hydrochloride.
Tamsulosin Hydrochloride Capsules USP*, 0.4 mg
Initial U.S. Approval: 1997RECENT MAJOR CHANGES
Dosage and Administration (2) 4/2009
Contraindications (4) 12/2009
Warnings and Precautions
Drug Interactions (5.2) 12/2009
Screening for Prostate Cancer (5.4) 12/2009INDICATIONS AND USAGE
- Tamsulosin hydrochloride is an alpha1 adrenoceptor antagonist indicated for treatment of the signs and symptoms of benign prostatic hyperplasia (1)
- Tamsulosin hydrochloride capsules are not indicated for the treatment of hypertension (1)
DOSAGE AND ADMINISTRATION
- 0.4 mg once daily taken approximately one-half hour following the same meal each day (2)
- Can be increased to 0.8 mg once daily for patients who fail to respond to the 0.4 mg dose after 2 to 4 weeks of dosing (2)
- If discontinued or interrupted for several days, therapy should start again with the 0.4 mg once-daily dose (2)
DOSAGE FORMS AND STRENGTHS
- Capsules: 0.4 mg (3)
CONTRAINDICATIONS
- Contraindicated in patients known to be hypersensitive to tamsulosin hydrochloride or any component of tamsulosin hydrochloride capsules (4, 6.2)
WARNINGS AND PRECAUTIONS
- Advise patients about the possibility of symptoms related to postural hypotension and to avoid situations where injury could result should syncope occur (5.1)
- Should not be used in combination with strong inhibitors of CYP3A4 (5.2, 7.1). Use with caution in combination with moderate inhibitors of CYP3A4, with strong or moderate inhibitors of CYP2D6, in patients known to be CYP2D6 poor metabolizers, or in combination with other cytochrome P450 inhibitors. (5.2, 7.1, 12.3)
- Should not be used in combination with other alpha adrenergic blocking agents (5.2, 7.2, 12.3)
- Exercise caution with concomitant administration of warfarin (5.2, 7.4, 12.3)
- Advise patients about the possibility and seriousness of priapism (5.3)
- Intraoperative Floppy Iris Syndrome has been observed during cataract surgery in some patients. Advise patients considering cataract surgery to tell their ophthalmologist that they have taken tamsulosin hydrochloride capsules. (5.5)
- Advise patients to be screened for the presence of prostate cancer prior to treatment and at regular intervals afterwards (5.4)
ADVERSE REACTIONS
- The most common adverse events (≥2% of patients and at a higher incidence than placebo) with the 0.4 mg dose or 0.8 mg dose were headache, dizziness, rhinitis, infection, abnormal ejaculation, asthenia, back pain, diarrhea, pharyngitis, chest pain, cough increased, somnolence, nausea, sinusitis, insomnia, libido decreased, tooth disorder, and blurred vision (6.1)
1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
- Tamsulosin hydrochloride capsules 0.4 mg should not be used with strong inhibitors of CYP3A4 (e.g., ketoconazole). Tamsulosin hydrochloride capsules should be used with caution in combination with moderate inhibitors of CYP3A4 (e.g., erythomycin), in combination with strong (e.g.,paroxetine) or moderate (e.g.,terbinafine) inhibitors of CYP2D6, or in patients known to be CYP2D6 poor metabolizers, particularly at a dose higher than 0.4 mg (e.g., 0.8 mg) (5.2, 7.1, 12.3)
- Concomitant use of PDE5 inhibitors with tamsulosin can potentially cause symptomatic hypotension (5.2, 7.3, 12.3)
USE IN SPECIFIC POPULATIONS
- Pediatric Use: Not indicated for use in pediatric populations (8.4, 12.3)
- Geriatric Use: No overall differences in efficacy or safety vs. younger patients, but greater sensitivity of some older adults cannot be ruled out (8.5, 12.3)
- Renal Impairment: Has not been studied in patients with end stage renal disease (8.6, 12.3)
- Hepatic Impairment: Has not been studied in patients with severe hepatic impairment (8.7, 12.3)
See 17 for PATIENT COUNSELING INFORMATION and PATIENT COUNSELING INFORMATION.
Revised: 9/2011
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Orthostasis
5.2 Drug Interactions
5.3 Priapism
5.4 Screening for Prostate Cancer
5.5 Intraoperative Floppy Iris Syndrome
5.6 Sulfa Allergy
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Cytochrome P450 Inhibition
7.2 Other Alpha Adrenergic Blocking Agents
7.3 PDE5 Inhibitors
7.4 Warfarin
7.5 Nifedipine, Atenolol, Enalapril
7.6 Digoxin and Theophylline
7.7 Furosemide
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Hypotension
17.2 Priapism
17.3 Screening for Prostate Cancer
17.4 Intraoperative Floppy Iris Syndrome
17.5 Administration
17.6 FDA-approved Patient Labeling
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
Tamsulosin hydrochloride capsules 0.4 mg once daily is recommended as the dose for the treatment of the signs and symptoms of BPH. It should be administered approximately one-half hour following the same meal each day.
For those patients who fail to respond to the 0.4 mg dose after 2 to 4 weeks of dosing, the dose of tamsulosin hydrochloride capsules can be increased to 0.8 mg once daily. Tamsulosin hydrochloride capsules 0.4 mg should not be used in combination with strong inhibitors of CYP3A4 (e.g., ketoconazole) [see Warnings and Precautions (5.2)].
If tamsulosin hydrochloride capsules administration is discontinued or interrupted for several days at either the 0.4 mg or 0.8 mg dose, therapy should be started again with the 0.4 mg once-daily dose. - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Orthostasis
The signs and symptoms of orthostasis (postural hypotension, dizziness and vertigo) were detected more frequently in tamsulosin hydrochloride capsules-treated patients than in placebo recipients. As with other alpha adrenergic blocking agents there is a potential risk of syncope [see Adverse Reactions (6.1)]. Patients beginning treatment with tamsulosin hydrochloride capsules should be cautioned to avoid situations where injury could result should syncope occur [see Patient Counseling Information (17.1)].5.2 Drug Interactions
Tamsulosin is extensively metabolized, mainly by CYP3A4 and CYP2D6. Tamsulosin hydrochloride capsules 0.4 mg should not be used in combination with strong inhibitors of CYP3A4 (e.g., ketoconazole) [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
Tamsulosin hydrochloride capsules should be used with caution in combination with moderate inhibitors of CYP3A4 (e.g., erythromycin), in combination with strong (e.g., paroxetine) or moderate (e.g., terbinafine) inhibitors of CYP2D6, in patients known to be CYP2D6 poor metabolizers particularly at a dose higher than 0.4 mg (e.g., 0.8 mg) [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
Tamsulosin hydrochloride capsules should be used with caution in combination with cimetidine, particularly at a dose higher than 0.4 mg (e.g., 0.8 mg) [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)]
Tamsulosin hydrochloride capsules should not be used in combination with other alpha adrenergic blocking agents [see Drug Interactions (7.2) and Clinical Pharmacology (12.3)].
Caution is advised when alpha adrenergic blocking agents including tamsulosin hydrochloride are coadministered with PDE5 inhibitors. Alpha-adrenergic blockers and PDE5 inhibitors are both vasodilators that can lower blood pressure. Concomitant use of these two drug classes can potentially cause symptomatic hypotension [see Drug Interactions (7.3) and Clinical Pharmacology (12.3)].
Caution should be exercised with concomitant administration of warfarin and tamsulosin hydrochloride capsules [See Drug Interactions (7.4) and Clinical Pharmacology (12.3)].5.3 Priapism
Rarely (probably less than 1 in 50,000 patients), tamsulosin, like other alpha1 antagonists, has been associated with priapism (persistent painful penile erection unrelated to sexual activity). Because this condition can lead to permanent impotence if not properly treated, patients must be advised about the seriousness of the condition [ see Patient Counseling Information (17.2)].5.4 Screening for Prostate Cancer
Prostate cancer and BPH frequently co-exist; therefore, patients should be screened for the presence of prostate cancer prior to treatment with tamsulosin hydrochloride capsules and at regular intervals afterwards [see Patient Counseling Information (17.3)].
5.5 Intraoperative Floppy Iris Syndrome
Intraoperative Floppy Iris Syndrome (IFIS) has been observed during cataract surgery in some patients treated with alpha1 blockers, including tamsulosin hydrochloride capsules [see Adverse Reactions (6.2)]. Most reports were in patients taking the alpha1 blocker when IFIS occurred, but in some cases, the alpha1 blocker had been stopped prior to surgery. In most of these cases, the alpha1 blocker had been stopped recently prior to surgery (2 to 14 days), but in a few cases, IFIS was reported after the patient had been off the alpha1 blocker for a longer period (5 weeks to 9 months). IFIS is a variant of small pupil syndrome and is characterized by the combination of a flaccid iris that billows in response to intraoperative irrigation currents, progressive intraoperative miosis despite preoperative dilation with standard mydriatic drugs and potential prolapse of the iris toward the phacoemulsification incisions. The patient's ophthalmologist should be prepared for possible modifications to their surgical technique, such as the utilization of iris hooks, iris dilator rings, or viscoelastic substances. The benefit of stopping alpha1 blocker therapy prior to cataract surgery has not been established. -
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reactions rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The incidence of treatment-emergent adverse events has been ascertained from six short-term U.S. and European placebo-controlled clinical trials in which daily doses of 0.1 to 0.8 mg tamsulosin hydrochloride capsules were used. These studies evaluated safety in 1783 patients treated with tamsulosin hydrochloride capsules and 798 patients administered placebo. Table 1 summarizes the treatment-emergent adverse events that occurred in ≥2% of patients receiving either tamsulosin hydrochloride capsules 0.4 mg or 0.8 mg and at an incidence numerically higher than that in the placebo group during two 13-week U.S. trials (US92-03A and US93-01) conducted in 1487 men.
Table 1 Treatment-Emergent1 Adverse Events Occurring in ≥2% of Tamsulosin Hydrochloride Capsules or Placebo Patients in Two U.S. Short-Term Placebo-Controlled Clinical Studies
1A treatment-emergent adverse event was defined as any event satisfying one of the following criteria:BODY SYSTEM/
ADVERSE EVENTTAMSULOSIN HYDROCHLORIDE CAPSULES GROUPS
0.4 mg 0.8 mg
n=502 n=492
PLACEBO
n=493BODY AS WHOLE
Headache
97 (19.3%)
104 (21.1%)
99 (20.1%)
Infection2
45 (9.0%)
53 (10.8%)
37 (7.5%)
Asthenia
39 (7.8%) 42 (8.5%) 27 (5.5)%
Back Pain
35 (7.0%)
41 (8.3%) 27 (5.5%) Chest Pain
20 (4.0%) 20 (4.1%) 18 (3.7%) NERVOUS SYSTEM
Dizziness
75 (14.9%) 84 (17.1%) 50 (10.1%) Somnolence
15 (3.0%) 21 (4.3%) 8 (1.6%) Insomnia
12 (2.4%)
7 (1.4%) 3 (0.6%) Libido Decreased
5 (1.0%)
10 (2.0%)
6 (1.2%) RESPIRATORY SYSTEM
Rhinitis3
66 (13.1%)
88 (17.9%) 41 (8.3%) Pharyngitis
29 (5.8%) 25 (5.1%) 23 (4.7%) Cough Increased
17 (3.4%)
22 (4.5%)
12 (2.4%) Sinusitis
11 (2.2%)
18 (3.7%)
8 (1.6%) DIGESTIVE SYSTEM
Diarrhea
31 (6.2%)
21 (4.3%) 22 (4.5%) Nausea
13 (2.6%)
19 (3.9%) 16 (3.2%)
Tooth Disorder
6 (1.2%)
10 (2.0%) 7 (1.4%) UROGENITAL SYSTEM
Abnormal Ejaculation
42 (8.4%)
89 (18.1%) 1 (0.2%) SPECIAL SENSES
Blurred vision
1 (0.2%)
10 (2.0%) 2 (0.4%)
- The adverse event occurred for the first time after initial dosing with double-blind study medication.
- The adverse event was present prior to or at the time of initial dosing with double-blind study medication and subsequently increased in severity during double-blind treatment; or
- The adverse event was present prior to or at the time of initial dosing with double-blind study medication, disappeared completely, and then reappeared during double-blind treatment.
3Coding preferred terms also include nasal congestion, stuffy nose, runny nose, sinus congestion, and hay fever.
Signs and Symptoms of Orthostasis
In the two U.S. studies, symptomatic postural hypotension was reported by 0.2% of patients (1 of 502) in the 0.4 mg group, 0.4% of patients (2 of 492) in the 0.8 mg group, and by no patients in the placebo group. Syncope was reported by 0.2% of patients (1 of 502) in the 0.4 mg group, 0.4% of patients (2 of 492) in the 0.8 mg group and 0.6% of patients (3 of 493) in the placebo group. Dizziness was reported by 15% of patients (75 of 502) in the 0.4 mg group, 17% of patients (84 of 492) in the 0.8 mg group, and 10% of patients (50 of 493) in the placebo group. Vertigo was reported by 0.6% of patients (3 of 502) in the 0.4 mg group, 1% of patients (5 of 492) in the 0.8 mg group and by 0.6% of patients (3 of 493) in the placebo group.
Multiple testing for orthostatic hypotension was conducted in a number of studies. Such a test was considered positive if it met one or more of the following criteria: (1) a decrease in systolic blood pressure of ≥20 mmHg upon standing from the supine position during the orthostatic tests; (2) a decrease in diastolic blood pressure ≥10 mmHg upon standing, with the standing diastolic blood pressure <65 mmHg during the orthostatic test; (3) an increase in pulse rate of ≥20 bpm upon standing with a standing pulse rate ≥100 bpm during the orthostatic test; and (4) the presence of clinical symptoms (faintness, lightheadedness/lightheaded, dizziness, spinning sensation, vertigo, or postural hypotension) upon standing during the orthostatic test.
Following the first dose of double-blind medication in Study 1, a positive orthostatic test result at 4 hours post-dose was observed in 7% of patients (37 of 498) who received tamsulosin hydrochloride capsules 0.4 mg once daily and in 3% of the patients (8 of 253) who received placebo. At 8 hours post-dose, a positive orthostatic test result was observed for 6% of the patients (31 of 498) who received tamsulosin hydrochloride capsules 0.4 mg once daily and 4% (9 of 250) who received placebo (Note: patients in the 0.8 mg group received 0.4 mg once daily for the first week of Study 1).
In Studies 1 and 2, at least one positive orthostatic test result was observed during the course of these studies for 81 of the 502 patients (16%) in the tamsulosin hydrochloride capsules 0.4 mg once-daily group, 92 of the 491 patients (19%) in the tamsulosin hydrochloride capsules 0.8 mg once-daily group and 54 of the 493 patients (11%) in the placebo group.
Because orthostasis was detected more frequently in tamsulosin hydrochloride capsules-treated patients than in placebo recipients, there is a potential risk of syncope [ see Warnings and Precautions (5.1)].
Abnormal Ejaculation
Abnormal ejaculation includes ejaculation failure, ejaculation disorder, retrograde ejaculation and ejaculation decrease. As shown in Table 1, abnormal ejaculation was associated with tamsulosin hydrochloride capsules administration and was dose-related in the U.S. studies. Withdrawal from these clinical studies of tamsulosin hydrochloride capsules because of abnormal ejaculation was also dose-dependent with 8 of 492 patients (1.6%) in the 0.8 mg group, and no patients in the 0.4 mg or placebo groups discontinuing treatment due to abnormal ejaculation.
Laboratory Tests
No laboratory test interactions with tamsulosin hydrochloride capsules are known. Treatment with tamsulosin hydrochloride capsules for up to 12 months had no significant effect on prostate-specific antigen (PSA).6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of tamsulosin hydrochloride capsules. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. Decisions to include these reactions in labeling are typically based on one or more of the following factors:
(1) seriousness of the reaction, (2) frequency of reporting, or (3) strength of causal connection to tamsulosin hydrochloride capsules.
Allergic-type reactions such as skin rash, urticaria, pruritus, angioedema and respiratory symptoms have been reported with positive rechallenge in some cases. Priapism has been reported rarely. Infrequent reports of palpitations, hypotension, skin desquamation, constipation and vomiting have been received during the postmarketing period.
During cataract surgery, a variant of small pupil syndrome known as Intraoperative Floppy Iris Syndrome (IFIS) has been reported in association with alpha1 blocker therapy [ see Warnings and Precautions (5.5)]. -
7 DRUG INTERACTIONS
7.1 Cytochrome P450 Inhibition
Strong and Moderate Inhibitors of CYP3A4 or CYP2D6
Tamsulosin is extensively metabolized, mainly by CYP3A4 and CYP2D6.
Concomitant treatment with ketoconazole (a strong inhibitor of CYP3A4) resulted in an increase in the Cmax and AUC of tamsulosin by a factor of 2.2 and 2.8, respectively [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)]. The effects of concomitant administration of a moderate CYP3A4 inhibitor (e.g., erythromycin) on the pharmacokinetics of tamsulosin hydrochloride have not been evaluated [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].
Concomitant treatment with paroxetine (a strong inhibitor of CYP2D6) resulted in an increase in the Cmax and AUC of tamsulosin by a factor of 1.3 and 1.6, respectively [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)]. A similar increase in exposure is expected in CYP2D6 poor metabolizers (PM) as compared to extensive metabolizers (EM). Since CYP2D6 PMs cannot be readily identified and the potential for significant increase in tamsulosin exposure exists when tamsulosin hydrochloride 0.4 mg is coadministered with strong CYP3A4 inhibitors in CYP2D6 PMs, tamsulosin hydrochloride 0.4 mg capsules should not be used in combination with strong inhibitors of CYP3A4 (e.g., ketoconazole) [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].
The effects of concomitant administration of a moderate CYP2D6 inhibitor (e.g., terbinafine) on the pharmacokinetics of tamsulosin hydrochloride have not been evaluated [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].
The effects of coadministration of both a CYP3A4 and a CYP2D6 inhibitor with tamsulosin hydrochloride capsules have not been evaluated. However, there is a potential for significant increase in tamsulosin exposure when tamsulosin hydrochloride 0.4 mg is coadministered with a combination of both CYP3A4 and CYP2D6 inhibitors [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].
Cimetidine
Treatment with cimetidine resulted in a significant decrease (26%) in the clearance of tamsulosin hydrochloride, which resulted in a moderate increase in tamsulosin hydrochloride AUC (44%) [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].7.2 Other Alpha Adrenergic Blocking Agents
The pharmacokinetic and pharmacodynamic interactions between tamsulosin hydrochloride capsules and other alpha adrenergic blocking agents have not been determined; however, interactions between tamsulosin hydrochloride capsules and other alpha adrenergic blocking agents may be expected [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].
7.3 PDE5 Inhibitors
Caution is advised when alpha adrenergic blocking agents including tamsulosin hydrochloride are coadministered with PDE5 inhibitors Alpha-adrenergic blockers and PDE5 inhibitors are both vasodilators that can lower blood pressure. Concomitant use of these two drug classes can potentially cause symptomatic hypotension [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].7.4 Warfarin
A definitive drug-drug interaction study between tamsulosin hydrochloride and warfarin was not conducted. Results from limited in vitro and in vivo studies are inconclusive. Caution should be exercised with concomitant administration of warfarin and tamsulosin hydrochloride capsules [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].
7.5 Nifedipine, Atenolol, Enalapril
Dosage adjustments are not necessary when tamsulosin hydrochloride capsules are administered concomitantly with nifedipine, atenolol, or enalapril [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].
7.6 Digoxin and Theophylline
Dosage adjustments are not necessary when a tamsulosin hydrochloride capsule is administered concomitantly with digoxin or theophylline [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].7.7 Furosemide
Tamsulosin hydrochloride capsules had no effect on the pharmacodynamics (excretion of electrolytes) of furosemide. While furosemide produced an 11% to 12% reduction in tamsulosin hydrochloride Cmax and AUC, these changes are expected to be clinically insignificant and do not require adjustment of the tamsulosin hydrochloride capsules dosage [ see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)]. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Teratogenic Effects,Pregnancy Category B.
Administration of tamsulosin hydrochloride to pregnant female rats at dose levels up to approximately 50 times the human therapeutic AUC exposure (300 mg/kg/day) revealed no evidence of harm to the fetus. Administration of tamsulosin hydrochloride to pregnant rabbits at dose levels up to 50 mg/kg/day produced no evidence of fetal harm. Tamsulosin hydrochloride capsules are not indicated for use in women.8.4 Pediatric Use
Tamsulosin hydrochloride capsules are not indicated for use in pediatric populations.
A description of the data from pediatric studies of tamsulosin hydrochloride capsules is contained in the approved labeling for Boehringer Ingelheim's Flomax® capsules. However, due to Boehringer Ingelheim's marketing exclusivity rights, a description of these pediatric studies is not contained in the approved labeling for this tamsulosin hydrochloride capsules.
8.5 Geriatric Use
Of the total number of subjects (1783) in clinical studies of tamsulosin, 36% were 65 years of age and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and the other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out [ see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Should overdosage of tamsulosin hydrochloride capsules lead to hypotension [ see Warnings and Precautions (5.1) and Adverse Reactions (6.1)], support of the cardiovascular system is of first importance. Restoration of blood pressure and normalization of heart rate may be accomplished by keeping the patient in the supine position. If this measure is inadequate, then administration of intravenous fluids should be considered. If necessary, vasopressors should then be used and renal function should be monitored and supported as needed. Laboratory data indicate that tamsulosin hydrochloride is 94% to 99% protein bound; therefore, dialysis is unlikely to be of benefit.
-
11 DESCRIPTION
Tamsulosin hydrochloride is an antagonist of alpha1A adrenoceptors in the prostate.
Tamsulosin hydrochloride is (-)-(R)-5-[2-[[2-(o-Ethoxyphenoxy)ethyl]amino]propyl]-2-methoxybenzenesulfonamide, monohydrochloride.
Tamsulosin hydrochloride is a white to almost white powder that melts at approximately 227° to 228°C. It is slightly soluble in water, freely soluble in formic acid.
The molecular formula of tamsulosin hydrochloride is C20H28N2O5SHCl. The molecular weight of tamsulosin hydrochloride is 444.98. Its structural formula is:
Each tamsulosin hydrochloride capsule, USP* for oral administration contains tamsulosin hydrochloride 0.4 mg, and the following inactive ingredients: microcrystalline cellulose, magnesium stearate, methacrylic acid copolymer dispersion, talc, triethyl citrate, FD and C blue # 1, FD and C red # 40, FD and C yellow # 10, titanium dioxide, gelatin, shellac, dehydrated alcohol, isopropyl alcohol, butyl alcohol, propylene glycol, strong ammonia solution, black iron oxide and potassium hydroxide.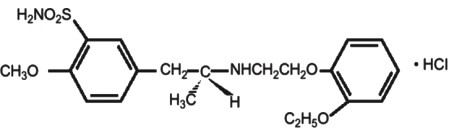
*USP dissolution test is pending. -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The symptoms associated with benign prostatic hyperplasia are related to bladder outlet obstruction, which is comprised of two underlying components: static and dynamic. The static component is related to an increase in prostate size caused, in part, by a proliferation of smooth muscle cells in the prostatic stroma. However, the severity of BPH symptoms and the degree of urethral obstruction do not correlate well with the size of the prostate. The dynamic component is a function of an increase in smooth muscle tone in the prostate and bladder neck leading to constriction of the bladder outlet. Smooth muscle tone is mediated by the sympathetic nervous stimulation of alpha1 adrenoceptors, which are abundant in the prostate, prostatic capsule, prostatic urethra, and bladder neck. Blockade of these adrenoceptors can cause smooth muscles in the bladder neck and prostate to relax, resulting in an improvement in urine flow rate and a reduction in symptoms of BPH.
Tamsulosin, an alpha1 adrenoceptor blocking agent, exhibits selectivity for alpha1 receptors in the human prostate. At least three discrete alpha1 adrenoceptor subtypes have been identified: alpha1A, alpha1B, and alpha1D; their distribution differs between human organs and tissue. Approximately 70% of the alpha1 receptors in human prostate are of the alpha1A subtype.
Tamsulosin hydrochloride capsules are not intended for use as an antihypertensive drug.
12.2 Pharmacodynamics
Urologic pharmacodynamic effects have been evaluated in neurologically impaired pediatric patients and in adults with BPH [see Use in Specific Populations (8.4) and Clinical Studies (14)].12.3 Pharmacokinetics
The pharmacokinetics of tamsulosin hydrochloride have been evaluated in adult healthy volunteers and patients with BPH after single and/or multiple administration with doses ranging from 0.1 mg to 1 mg.
Absorption
Absorption of tamsulosin hydrochloride from tamsulosin hydrochloride capsules 0.4 mg is essentially complete (>90%) following oral administration under fasting conditions. Tamsulosin hydrochloride exhibits linear kinetics following single and multiple dosing, with achievement of steady-state concentrations by the fifth day of once-a-day dosing.
Effect of Food
The time to maximum concentration (Tmax) is reached by four to five hours under fasting conditions and by six to seven hours when tamsulosin hydrochloride capsules are administered with food. Taking tamsulosin hydrochloride under fasted conditions results in a 30% increase in bioavailability (AUC) and 40% to 70% increase in peak concentrations (Cmax) compared to fed conditions (Figure 1).
Figure 1 Mean Plasma Tamsulosin Hydrochloride Concentrations Following Single-Dose Administration of Tamsulosin Hydrochloride Capsules 0.4 mg Under Fasted and Fed Conditions (n=8)
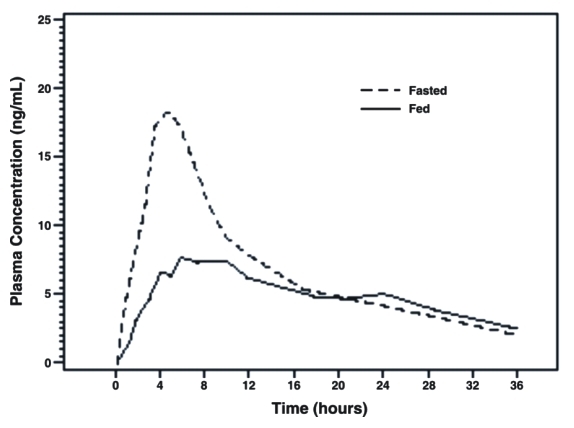
The effects of food on the pharmacokinetics of tamsulosin hydrochloride are consistent regardless of whether a tamsulosin hydrochloride capsule is taken with a light breakfast or a high-fat breakfast (Table 2).
Table 2 Mean (± S.D.) Pharmacokinetic Parameters Following Tamsulosin Hydrochloride Capsules 0.4 mg Once Daily or 0.8 mg Once Daily with a Light Breakfast, High-Fat Breakfast or Fasted
Cmin = observed minimum concentrationPharmacokinetic 0.4 mg QD to healthy volunteers; m=23 0.8 mg QD to healthy volunteers; n=22
Parameter (age range 18 to 32 years) (age range 18 to 32 years)
Light Breakfast Fasted Light Breakfast High-Fast Breakfast Fasted
Cmin (ng/mL)
4.0 ± 2.6
3.8 ± 2.5
12.3 ± 6.7
13.5 ± 7.6
13.3 ± 13.3 Cmax (ng/mL)
10.1 ± 4.8
17.1 ± 17.1
29.8 ± 10.3
29.1 ± 11.0
41.6 ± 15.6 Cmax/Cmin Ratio
3.1 ± 1.0
5.3 ± 2.2
2.7 ± 0.7
2.5 ± 0.8
3.6 ± 1.1 Tmax (hours)
6.0
4.0
7.0
6.6
5.0 T1/2 (hours)
-
-
-
-
14.9 ± 3.9 AUCτ (nghr/mL)
151 ± 81.5
199 ± 94.1
440 ± 195
449 ± 217
557 ± 257
Cmax = observed maximum tamsulosin hydrochloride plasma concentration
Tmax = median time-to-maximum concentration
T1/2 = observed half-life
AUCτ = area under the tamsulosin hydrochloride plasma time curve over the dosing interval
Distribution
The mean steady-state apparent volume of distribution of tamsulosin hydrochloride after intravenous administration to ten healthy male adults was 16 L, which is suggestive of distribution into extracellular fluids in the body.
Tamsulosin hydrochloride is extensively bound to human plasma proteins (94% to 99%), primarily alpha1 acid glycoprotein (AAG), with linear binding over a wide concentration range (20 to 600 ng/mL). The results of two-way in vitro studies indicate that the binding of tamsulosin hydrochloride to human plasma proteins is not affected by amitriptyline, diclofenac, glyburide, simvastatin plus simvastatin-hydroxy acid metabolite, warfarin, diazepam, propranolol, trichlormethiazide, or chlormadinone. Likewise, tamsulosin hydrochloride had no effect on the extent of binding of these drugs.
Metabolism
There is no enantiomeric bioconversion from tamsulosin hydrochloride [R(-) isomer] to the S(+) isomer in humans. Tamsulosin hydrochloride is extensively metabolized by cytochrome P450 enzymes in the liver and less than 10% of the dose is excreted in urine unchanged. However, the pharmacokinetic profile of the metabolites in humans has not been established. Tamsulosin is extensively metabolized, mainly by CYP3A4 and CYP2D6, as well as via some minor participation of other CYP isoenzymes. Inhibition of hepatic drug-metabolizing enzymes may lead to increased exposure to tamsulosin [ see Warnings and Precautions (5.2) and Drug Interactions (7.1)]. The metabolites of tamsulosin hydrochloride undergo extensive conjugation to glucuronide or sulfate prior to renal excretion.
Incubations with human liver microsomes showed no evidence of clinically significant metabolic interactions between tamsulosin hydrochloride and amitriptyline, albuterol (beta agonist), glyburide (glibenclamide) and finasteride (5alpha-reductase inhibitor for treatment of BPH). However, results of the in vitro testing of the tamsulosin hydrochloride interaction with diclofenac and warfarin were equivocal.
Excretion
On administration of the radiolabeled dose of tamsulosin hydrochloride to four healthy volunteers, 97% of the administered radioactivity was recovered, with urine (76%) representing the primary route of excretion compared to feces (21%) over 168 hours.
Following intravenous or oral administration of an immediate-release formulation, the elimination half-life of tamsulosin hydrochloride in plasma ranged from five to seven hours. Because of absorption rate-controlled pharmacokinetics with tamsulosin hydrochloride capsules, the apparent half-life of tamsulosin hydrochloride is approximately 9 to 13 hours in healthy volunteers and 14 to 15 hours in the target population.
Tamsulosin hydrochloride undergoes restrictive clearance in humans, with a relatively low systemic clearance (2.88 L/h).
Special Populations
Pediatric Use
Tamsulosin hydrochloride capsules are not indicated for use in pediatric populations [ see Use in Specific Populations (8.4)].
Geriatric (Age) Use
Cross-study comparison of tamsulosin hydrochloride capsules overall exposure (AUC) and half-life indicates that the pharmacokinetic disposition of tamsulosin hydrochloride may be slightly prolonged in geriatric males compared to young, healthy male volunteers. Intrinsic clearance is independent of tamsulosin hydrochloride binding to AAG, but diminishes with age, resulting in a 40% overall higher exposure (AUC) in subjects of age 55 to 75 years compared to subjects of age 20 to 32 years [ see Use in Specific Populations (8.5)].
Renal Impairment
The pharmacokinetics of tamsulosin hydrochloride have been compared in 6 subjects with mild-moderate (30 ≤CLcr <70 mL/min/1.73 m2) or moderate-severe (10 ≤CLcr <30 mL/min/1.73 m2) renal impairment and 6 normal subjects (CLcr >90 mL/min/1.73 m2). While a change in the overall plasma concentration of tamsulosin hydrochloride was observed as the result of altered binding to AAG, the unbound (active) concentration of tamsulosin hydrochloride, as well as the intrinsic clearance, remained relatively constant. Therefore, patients with renal impairment do not require an adjustment in tamsulosin hydrochloride capsules dosing. However, patients with endstage renal disease (CLcr <10 mL/min/1.73 m2) have not been studied [see Use in Specific Populations (8.6)].
Hepatic Impairment
The pharmacokinetics of tamsulosin hydrochloride have been compared in 8 subjects with moderate hepatic impairment (Child-Pugh's classification: Grades A and B) and 8 normal subjects. While a change in the overall plasma concentration of tamsulosin hydrochloride was observed as the result of altered binding to AAG, the unbound (active) concentration of tamsulosin hydrochloride does not change significantly, with only a modest (32%) change in intrinsic clearance of unbound tamsulosin hydrochloride. Therefore, patients with moderate hepatic impairment do not require an adjustment in tamsulosin hydrochloride capsules dosage. Tamsulosin hydrochloride have not been studied in patients with severe hepatic impairment [see Use in Specific Populations (8.7)].
Drug Interactions
Cytochrome P450 Inhibition
Strong and Moderate Inhibitors of CYP3A4 or CYP2D6
The effects of ketoconazole (a strong inhibitor of CYP3A4) at 400 mg once daily for 5 days on the pharmacokinetics of a single tamsulosin hydrochloride capsule 0.4 mg dose was investigated in 24 healthy volunteers (age range 23 to 47 years). Concomitant treatment with ketoconazole resulted in an increase in the Cmax and AUC of tamsulosin by a factor of 2.2 and 2.8, respectively [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)]. The effects of concomitant administration of a moderate CYP3A4 inhibitor (e.g., erythromycin) on the pharmacokinetics of tamsulosin hydrochloride have not been evaluated [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
The effects of paroxetine (a strong inhibitor of CYP2D6) at 20 mg once daily for 9 days on the pharmacokinetics of a single tamsulosin hydrochloride capsule 0.4 mg dose was investigated in 24 healthy volunteers (age range 23 to 47 years). Concomitant treatment with paroxetine resulted in an increase in the Cmax and AUC of tamsulosin by a factor of 1.3 and 1.6, respectively [see Warnings and Precautions (5.2) and Drug Interactions (7.1)]. A similar increase in exposure is expected in CYP2D6 poor metabolizers (PM) as compared to extensive metabolizers (EM). A fraction of the population (about 7% of Caucasians and 2% of African Americans) are CYP2D6 PMs. Since CYP2D6 PMs cannot be readily identified and the potential for significant increase in tamsulosin exposure exists when tamsulosin hydrochloride 0.4 mg is coadministered with strong CYP3A4 inhibitors in CYP2D6 PMs, tamsulosin hydrochloride 0.4 mg capsules should not be used in combination with strong inhibitors of CYP3A4 (e.g., ketoconazole) [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
The effects of concomitant administration of a moderate CYP2D6 inhibitor (e.g., terbinafine) on the pharmacokinetics of tamsulosin hydrochloride have not been evaluated [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
The effects of coadministration of both a CYP3A4 and a CYP2D6 inhibitor with tamsulosin hydrochloride capsules have not been evaluated. However, there is a potential for significant increase in tamsulosin exposure when tamsulosin hydrochloride 0.4 mg is coadministered with a combination of both CYP3A4 and CYP2D6 inhibitors [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
Cimetidine
The effects of cimetidine at the highest recommended dose (400 mg every 6 hours for 6 days) on the pharmacokinetics of a single tamsulosin hydrochloride capsule 0.4 mg dose was investigated in 10 healthy volunteers (age range 21 to 38 years). Treatment with cimetidine resulted in a significant decrease (26%) in the clearance of tamsulosin hydrochloride, which resulted in a moderate increase in tamsulosin hydrochloride AUC (44%) [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
Other Alpha Adrenergic Blocking Agents
The pharmacokinetic and pharmacodynamic interactions between tamsulosin hydrochloride capsules and other alpha adrenergic blocking agents have not been determined; however, interactions between tamsulosin hydrochloride capsules and other alpha adrenergic blocking agents may be expected [see Warnings and Precautions (5.2) and Drug Interactions (7.2)].
PDE5 Inhibitors
Caution is advised when alpha adrenergic blocking agents including tamsulosin hydrochloride are coadministered with PDE5 inhibitors Alpha-adrenergic blockers and PDE5 inhibitors are both vasodilators that can lower blood pressure. Concomitant use of these two drug classes can potentially cause symptomatic hypotension [see Warnings and Precautions (5.2) and Drug Interactions (7.3)]
Warfarin
A definitive drug-drug interaction study between tamsulosin hydrochloride and warfarin was not conducted. Results from limited in vitro and in vivo studies are inconclusive. Therefore, caution should be exercised with concomitant administration of warfarin and tamsulosin hydrochloride capsules [see Warnings and Precautions (5.2) and Drug Interactions (7.4)].
Nifedipine, Atenolol, Enalapril
In three studies in hypertensive subjects (age range 47 to 79 years) whose blood pressure was controlled with stable doses of nifedipine, atenolol, or enalapril for at least 3 months, tamsulosin hydrochloride capsules 0.4 mg for 7 days followed by tamsulosin hydrochloride capsules 0.8 mg for another 7 days (n=8 per study) resulted in no clinically significant effects on blood pressure and pulse rate compared to placebo (n=4 per study). Therefore, dosage adjustments are not necessary when tamsulosin hydrochloride capsules are administered concomitantly with nifedipine, atenolol, or enalapril [see Drug Interactions (7.5)].
Digoxin and Theophylline
In two studies in healthy volunteers (n=10 per study; age range 19 to 39 years) receiving tamsulosin hydrochloride capsules 0.4 mg/day for 2 days, followed by tamsulosin hydrochloride capsules 0.8 mg/day for 5 to 8 days, single intravenous doses of digoxin 0.5 mg or theophylline 5 mg/kg resulted in no change in the pharmacokinetics of digoxin or theophylline. Therefore, dosage adjustments are not necessary when a tamsulosin hydrochloride capsule is administered concomitantly with digoxin or theophylline [see Drug Interactions (7.6)].
Furosemide
The pharmacokinetic and pharmacodynamic interaction between tamsulosin hydrochloride capsules 0.8 mg/day (steady-state) and furosemide 20 mg intravenously (single dose) was evaluated in ten healthy volunteers (age range 21 to 40 years). Tamsulosin hydrochloride capsules had no effect on the pharmacodynamics (excretion of electrolytes) of furosemide. While furosemide produced an 11% to 12% reduction in tamsulosin hydrochloride Cmax and AUC, these changes are expected to be clinically insignificant and do not require adjustment of the tamsulosin hydrochloride capsules dosage [see Drug Interactions (7.7)]. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Rats administered doses up to 43 mg/kg/day in males and 52 mg/kg/day in females had no increases in tumor incidence, with the exception of a modest increase in the frequency of mammary gland fibroadenomas in female rats receiving doses ≥5.4 mg/kg (P<0.015). The highest doses of tamsulosin hydrochloride evaluated in the rat carcinogenicity study produced systemic exposures (AUC) in rats 3 times the exposures in men receiving the maximum therapeutic dose of 0.8 mg/day.
Mice were administered doses up to 127 mg/kg/day in males and 158 mg/kg/day in females. There were no significant tumor findings in male mice. Female mice treated for 2 years with the two highest doses of 45 and 158 mg/kg/day had statistically significant increases in the incidence of mammary gland fibroadenomas (P<0.0001) and adenocarcinomas (P<0.0075). The highest dose levels of tamsulosin hydrochloride evaluated in the mice carcinogenicity study produced systemic exposures (AUC) in mice 8 times the exposures in men receiving the maximum therapeutic dose of 0.8 mg/day.
The increased incidences of mammary gland neoplasms in female rats and mice were considered secondary to tamsulosin hydrochloride-induced hyperprolactinemia. It is not known if tamsulosin hydrochloride capsules elevate prolactin in humans. The relevance for human risk of the findings of prolactin-mediated endocrine tumors in rodents is not known.
Tamsulosin hydrochloride produced no evidence of mutagenic potential in vitro in the Ames reverse mutation test, mouse lymphoma thymidine kinase assay, unscheduled DNA repair synthesis assay, and chromosomal aberration assays in Chinese hamster ovary cells or human lymphocytes. There were no mutagenic effects in the in vivo sister chromatid exchange and mouse micronucleus assay.
Studies in rats revealed significantly reduced fertility in males dosed with single or multiple daily doses of 300 mg/kg/day of tamsulosin hydrochloride (AUC exposure in rats about 50 times the human exposure with the maximum therapeutic dose). The mechanism of decreased fertility in male rats is considered to be an effect of the compound on the vaginal plug formation possibly due to changes of semen content or impairment of ejaculation. The effects on fertility were reversible showing improvement by 3 days after a single dose and 4 weeks after multiple dosing. Effects on fertility in males were completely reversed within nine weeks of discontinuation of multiple dosing. Multiple doses of 10 and 100 mg/kg/day tamsulosin hydrochloride (1/5 and 16 times the anticipated human AUC exposure) did not significantly alter fertility in male rats. Effects of tamsulosin hydrochloride on sperm counts or sperm function have not been evaluated.
Studies in female rats revealed significant reductions in fertility after single or multiple dosing with 300 mg/kg/day of the R-isomer or racemic mixture of tamsulosin hydrochloride, respectively. In female rats, the reductions in fertility after single doses were considered to be associated with impairments in fertilization. Multiple dosing with 10 or 100 mg/kg/day of the racemic mixture did not significantly alter fertility in female rats. -
14 CLINICAL STUDIES
Four placebo-controlled clinical studies and one active-controlled clinical study enrolled a total of 2296 patients (1003 received tamsulosin hydrochloride capsules 0.4 mg once daily, 491 received tamsulosin hydrochloride capsules 0.8 mg once daily, and 802 were control patients) in the U.S. and Europe.
In the two U.S. placebo-controlled, double-blind, 13-week, multicenter studies [Study 1 (US92-03A) and Study 2 (US93-01)], 1486 men with the signs and symptoms of BPH were enrolled. In both studies, patients were randomized to either placebo, tamsulosin hydrochloride capsules 0.4 mg once daily, or tamsulosin hydrochloride capsules 0.8 mg once daily. Patients in tamsulosin hydrochloride capsules 0.8 mg once-daily treatment groups received a dose of 0.4 mg once daily for one week before increasing to the 0.8 mg once-daily dose. The primary efficacy assessments included: 1) total American Urological Association (AUA) Symptom Score questionnaire, which evaluated irritative (frequency, urgency, and nocturia), and obstructive (hesitancy, incomplete emptying, intermittency, and weak stream) symptoms, where a decrease in score is consistent with improvement in symptoms; and 2) peak urine flow rate, where an increased peak urine flow rate value over baseline is consistent with decreased urinary obstruction.
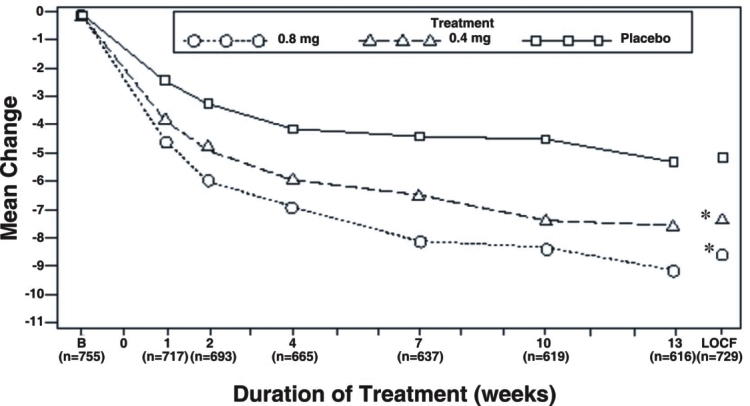
Mean changes from baseline to Week 13 in total AUA Symptom Score were significantly greater for groups treated with tamsulosin hydrochloride capsules 0.4 mg and 0.8 mg once daily compared to placebo in both U.S. studies (Table 3, Figures 2A and 2B). The changes from baseline to Week 13 in peak urine flow rate were also significantly greater for the tamsulosin hydrochloride capsules 0.4 mg and 0.8 mg once-daily groups compared to placebo in Study 1, and for the tamsulosin hydrochloride capsules 0.8 mg once-daily group in Study 2 (Table 3, Figures 3A and 3B). Overall there were no significant differences in improvement observed in total AUA Symptom Scores or peak urine flow rates between the 0.4 mg and the 0.8 mg dose groups with the exception that the 0.8 mg dose in Study 1 had a significantly greater improvement in total AUA Symptom Score compared to the 0.4 mg dose.
Table 3 Mean (±S.D.) Changes from Baseline to Week 13 in Total AUA Symptom Score** and Peak Urine Flow Rate (mL/sec)
* Statistically significant difference from placebo (p-value ≤0.050; Bonferroni-Holm multiple test procedure).
Total AUA Symptom Score
Mean Baseline Value
Mean ChangePeak Urine Flow Rate
Mean Baseline Value
Mean ChangeStudy 1 †
Tamsulosin hydrochloride capsules
0.8 mg once daily19.9 ± 4.9
n=247-9.6* ± 6.7
n=2379.57 ± 2.51
n=2471.78* ± 3.35
n=247Tamsulosin hydrochloride capsules
0.4 mg once daily19.8 ± 5.0
n=254-8.3* ± 6.5
n=2469.46 ± 2.49
n=2541.75* ± 3.57
n=254Placebo 19.6 ± 4.9
n=254-5.5 ± 6.6
n=2469.75 ± 2.54
n=2540.52 ± 3.39
n=253Study 2 ‡
Tamsulosin hydrochloride capsules
0.8 mg once daily18.2 ± 5.6
n=244-5.8* ± 6.4
n=2389.96 ± 3.16
n=2441.79* ± 3.36
n=237Tamsulosin hydrochloride capsules
0.4 mg once daily17.9 ± 5.8
n=248-5.1* ± 6.4
n=2449.94 ± 3.14
n=2481.52 ± 3.64
n=244Placebo 19.2 ± 6.0
n=239-3.6 ± 5.7
n=2359.95 ± 3.12
n=2390.93 ± 3.28
n=235
** Total AUA Symptom Scores ranged from 0 to 35.
† Peak urine flow rate measured 4 to 8 hours post dose at Week 13.
‡ Peak urine flow rate measured 24 to 27 hours post dose at Week 13.
Week 13: For patients not completing the 13-week study, the last observation was carried forward.
Mean total AUA Symptom Scores for both tamsulosin hydrochloride capsules 0.4 mg and 0.8 mg once-daily groups showed a rapid decrease starting at one week after dosing and remained decreased through 13 weeks in both studies (Figures 2A and 2B).
In Study 1, 400 patients (53% of the originally randomized group) elected to continue in their originally assigned treatment groups in a double-blind, placebo-controlled, 40-week extension trial (138 patients on 0.4 mg, 135 patients on 0.8 mg and 127 patients on placebo). Three hundred twenty-three patients (43% of the originally randomized group) completed one year. Of these, 81% (97 patients) on 0.4 mg, 74% (75 patients) on 0.8 mg and 56% (57 patients) on placebo had a response ≥25% above baseline in total AUA Symptom Score at one year.
Figure 2A Mean Change from Baseline in Total AUA Symptom Score (0-35) Study 1
* indicates significant difference from placebo (p-value ≤0.050).
B = Baseline determined approximately one week prior to the initial dose of double-blind medication at Week 0.
Subsequent values are observed cases.
LOCF = Last observation carried forward for patients not completing the 13-week study.
Note: Patients in the 0.8 mg treatment group received 0.4 mg for the first week.
Note: Total AUA Symptom Scores range from 0 to 35.
Figure 2B Mean Change from Baseline in Total AUA Symptom Score (0-35) Study 2
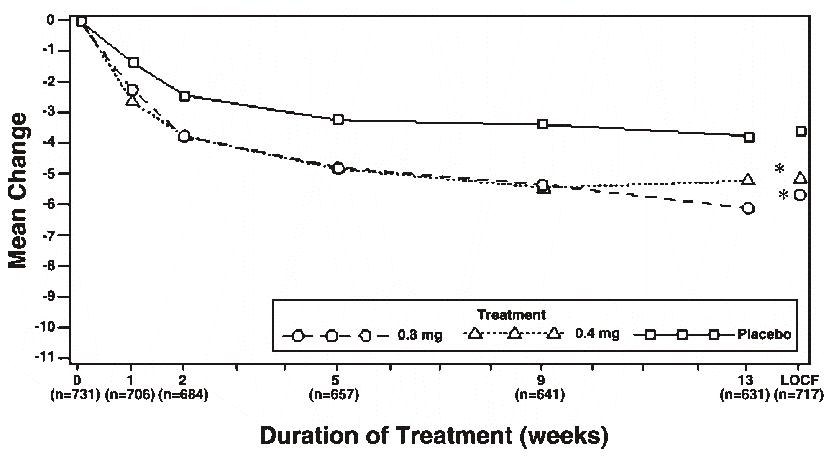
* indicates significant difference from placebo (p-value ≤0.050).
Baseline measurement was taken Week 0. Subsequent values are observed cases.
LOCF = Last observation carried forward for patients not completing the 13-week study.
Note: Patients in the 0.8 mg treatment group received 0.4 mg for the first week.
Note: Total AUA Symptom Scores range from 0 to 35.
Figure 3A Mean Increase in Peak Urine Flow Rate (mL/sec) Study 1
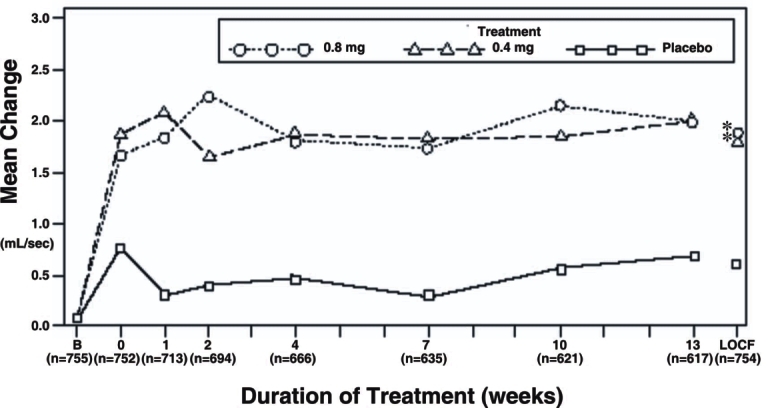
* indicates significant difference from placebo (p-value ≤0.050).
B = Baseline determined approximately one week prior to the initial dose of double-blind medication at Week 0.
Subsequent values are observed cases.
LOCF = Last observation carried forward for patients not completing the 13-week study.
Note: The uroflowmetry assessments at Week 0 were recorded 4-8 hours after patients received the first dose of double-blind medication.
Measurements at each visit were scheduled 4-8 hours after dosing (approximate peak plasma tamsulosin concentration).
Note: Patients in the 0.8 mg treatment groups received 0.4 for the first week.
Figure 3B Mean Increase in Peak Urine Flow Rate (mL/sec) Study 2
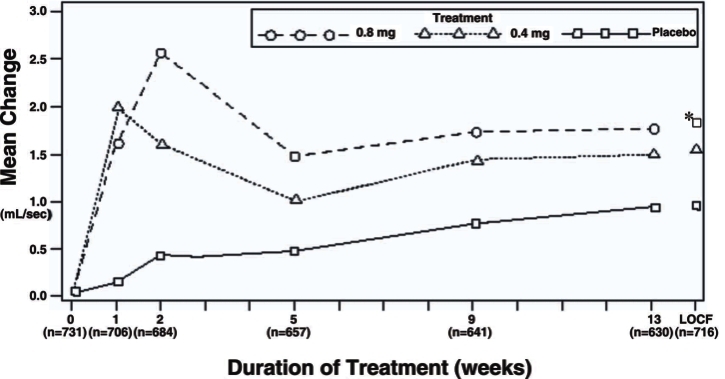
* indicates significant difference from placebo (p-value ≤0.050).
Baseline measurement was taken Week 0. Subsequent values are observed cases.
LOCF = Last observation carried forward for patients not completing the 13-week study.
Note: Patients in the 0.8 mg treatment group received 0.4 mg for the first week.
Note: Week 1 and Week 2 measurements were scheduled 4-8 hours after dosing (approximate peak plasma tamsulosin concentration).
All other visits were scheduled 24-27 hours after dosing (approximate trough tamsulosin concentration). -
16 HOW SUPPLIED/STORAGE AND HANDLING
Tamsulosin hydrochloride capsules USP, 0.4 mg are supplied in high density polyethylene bottles containing 30 or 500 size '2', hard gelatin capsules with olive green opaque cap and orange opaque body, filled with white to off-white pellets. The capsules are imprinted with “W” on the cap with black ink. 516
Tamsulosin hydrochloride capsules USP, 0.4 mg, 30 capsules (NDC: 64679-516-01)
Tamsulosin hydrochloride capsules USP, 0.4 mg, 100 capsules (NDC: 64679-516-02)
Tamsulosin hydrochloride capsules USP, 0.4 mg, 500 capsules (NDC: 64679-516-03)
Tamsulosin hydrochloride capsules USP, 0.4 mg, 16 (2 x 8) unit dose blister carton (NDC: 64679-516-04)
Tamsulosin hydrochloride capsules USP, 0.4 mg, 30 (1 x 30) unit dose blister carton (NDC: 64679-516-05)
Store at 20° to 25°C (68°C to 77°F) [See USP Controlled Room Temperature].
Keep tamsulosin hydrochloride capsules, USP and all medicines out of reach of children. -
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling (17.6).
17.1 Hypotension
Patients should be told about the possible occurrence of symptoms related to postural hypotension, such as dizziness, when taking tamsulosin hydrochloride capsules, USP and they should be cautioned about driving, operating machinery or performing hazardous tasks [ see Warnings and Precautions (5.1)].17.2 Priapism
Patients should be advised about the possibility of priapism as a result of treatment with tamsulosin hydrochloride capsules, USP and other similar medications. Patients should be informed that this reaction is extremely rare, but if not brought to immediate medical attention, can lead to permanent erectile dysfunction (impotence) [ see Warnings and Precautions (5.3)].17.3 Screening for Prostate Cancer
Prostate cancer and BPH frequently co-exist; therefore, patients should be screened for the presence of prostate cancer prior to treatment with tamsulosin hydrochloride capsules, USP and at regular intervals afterwards [ see Warnings and Precautions (5.4)].17.4 Intraoperative Floppy Iris Syndrome
Patients considering cataract surgery should be advised to tell their ophthalmologist that they have taken tamsulosin hydrochloride capsules, USP [ see Warnings and Precautions (5.5)].
17.5 Administration
Patients should be advised not to crush or chew the tamsulosin hydrochloride capsules, USP.17.6 FDA-approved Patient Labeling
Patient labeling is provided as a stand alone leaflet and is also provided at the end of this prescribing information.
Procardia XL®, is a registered trademark of Pfizer Inc. for Nifedipine Extended-Release Tablets.
Manufactured by:
Wockhardt Limited,
Mumbai, India
Distributed by:
Wockhardt USA LLC.
20 Waterview Blvd.
Parsippany, NJ 07054
USA.
Iss.080310
-
INFORMATION FOR PATIENTS
Patient Information
Tamsulosin Hydrochloride Capsules USP*, 0.4 mg
Read the Patient Information that comes with tamsulosin hydrochloride capsules before you start taking it and each time you refill your prescription. The information may have changed. This leaflet does not take the place of discussions with your doctor about your medical condition or your treatment.
What is tamsulosin hydrochloride?
Tamsulosin hydrochloride is a prescription alpha blocker medicine used to treat the signs and symptoms of benign prostatic hyperplasia (BPH), a condition your doctor may refer to as an enlarged prostate.
- Tamsulosin hydrochloride capsules are not for women.
- Tamsulosin hydrochloride capsules are not for children.
Do not take tamsulosin hydrochloride capsules if you are allergic to any of its ingredients. See the end of this leaflet for a complete list of ingredients in tamsulosin hydrochloride capsules.
What should I tell my doctor before using tamsulosin hydrochloride capsules, USP?
Before taking tamsulosin hydrochloride capsules, USP tell your doctor about all your medical conditions including:
- any kidney or liver problems.
- any history of low blood pressure.
- any allergies to sulfa or any other medicines.
- if you are planning to have cataract surgery.
- any prescription medicines, including blood pressure medicines.
- any non-prescription medicines, including vitamins and herbal supplements.
Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist when you get a new medicine.
How should I take tamsulosin hydrochloride capsules, USP?
- Take tamsulosin hydrochloride capsules exactly as prescribed by your doctor.
- Do not crush, chew, or open tamsulosin hydrochloride capsules.
- Take tamsulosin hydrochloride capsules one time each day, about 30 minutes after the same meal each day. For example, you may take tamsulosin hydrochloride capsules 30 minutes after dinner each day.
- If you miss a dose of tamsulosin hydrochloride capsules, take it as soon as you remember. If you miss your dose for the whole day, continue with your next dose on your regular schedule. Do not take two doses at the same time.
- If you stop or forget to take tamsulosin hydrochloride capsules for several days, talk with your doctor before starting again.
- If you take more tamsulosin hydrochloride capsules than prescribed, call your doctor right away.
Possible side effects of tamsulosin hydrochloride capsules may include:
- Decreased blood pressure when changing positions. Tamsulosin hydrochloride capsules may cause a sudden drop in blood pressure upon standing, especially after the first dose or when changing doses.
- fainting
- dizziness
- lightheadedness
- Allergic reactions. Make your doctor aware of any allergic reactions you may experience while taking tamsulosin hydrochloride.
- rash
- itching
- hives
- swelling of face, tongue, or throat
- difficulty breathing
- A painful erection that will not go away.Tamsulosin hydrochloride capsules can cause a painful erection (priapism), which cannot be relieved by having sex. If this happens, get medical help right away. If priapism is not treated, you may not be able to get an erection in the future.
- Eye problems during cataract surgery. During cataract surgery, a condition called intraoperative floppy iris syndrome (IFIS) can happen if you take or have taken tamsulosin hydrochloride capsules. If you need to have cataract surgery, be sure to tell your surgeon if you take or have taken tamsulosin hydrochloride capsules.
- runny nose
- dizziness
- decreased semen
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088, or by visiting www.fda.gov/medwatch.
What should I avoid while taking tamsulosin hydrochloride capsules, USP?
Avoid driving, operating machinery, or other dangerous activities, until you know how tamsulosin hydrochloride affects you. Tamsulosin hydrochloride capsules may cause a sudden drop in blood pressure upon standing, especially after the first dose or when changing doses. See "What are the possible side effects of tamsulosin hydrochloride capsules, USP?"
How do I store tamsulosin hydrochloride capsules, USP?
Store tamsulosin hydrochloride capsules, USP at 20° to 25°C (68°C to 77°F) [See USP Controlled Room Temperature]. Short-term exposure to higher or lower temperatures [from 59°F (15°C) to 86°F (30°C)] is acceptable. Ask your doctor or pharmacist if you have any questions about storing your capsules.
Keep tamsulosin hydrochloride capsules, USP and all medicines out of the reach of children.
General information
This medicine was prescribed for you by your doctor for your condition. Do not use it for another condition. Do not give tamsulosin hydrochloride capsules to other people, even if they have the same symptoms that you have. It may harm them.
While taking tamsulosin hydrochloride capsules, you must have regular checkups. Follow your doctor's advice about when to have these checkups.
BPH can occur with other more serious conditions, including prostate cancer. Therefore, ask your doctor about screening for prostate cancer prior to treatment with tamsulosin hydrochloride capsules and at regular intervals afterwards.
This patient information leaflet summarizes the most important information about tamsulosin hydrochloride capsules. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about tamsulosin hydrochloride capsules that is written for health professionals. For more information call Wockhardt USA LLC. at 1-800-346-6854.
What are the ingredients in tamsulosin hydrochloride capsules, USP?
- Active Ingredient: tamsulosin hydrochloride
- Inactive Ingredients: microcrystalline cellulose, magnesium stearate, methacrylic acid copolymer dispersion, talc, triethyl citrate, FD and C blue # 1, FD and C red # 40, FD and C yellow # 10, titanium dioxide, gelatin, shellac, dehydrated alcohol, isopropyl alcohol, butyl alcohol, propylene glycol, strong ammonia solution, black iron oxide and potassium hydroxide
*USP dissolution test is pending.
Wockhardt Limited,
Mumbai, India
Distributed by:
Wockhardt USA LLC.
20 Waterview Blvd.
Parsippany, NJ 07054
USA.
Repacked by:
H.J. Harkins Company Inc.
513 Sandydale Drive
Nipomo, CA 93444
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
TAMSULOSIN HYDROCHLORIDE
tamsulosin hydrochloride capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 52959-034(NDC:64679-516) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TAMSULOSIN HYDROCHLORIDE (UNII: 11SV1951MR) (TAMSULOSIN - UNII:G3P28OML5I) TAMSULOSIN HYDROCHLORIDE 0.4 mg Product Characteristics Color orange (orange opaque body ) , green (olive green opaque cap) Score no score Shape CAPSULE (Capsule) Size 2mm Flavor Imprint Code W;516 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 52959-034-30 30 in 1 BOTTLE 2 NDC: 52959-034-60 60 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078938 04/27/2010 Labeler - H.J. Harkins Company, Inc. (147681894) Registrant - H.J. Harkins Company, Inc. (147681894) Establishment Name Address ID/FEI Business Operations H.J. Harkins Company, Inc. 147681894 repack, relabel
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.