These highlights do not include all the information needed to use TRUSELTIQ safely and effectively. See full prescribing information for TRUSELTIQ. TRUSELTIQ (infigratinib) capsules, for oral use Initial U.S. Approval: 2021
TRUSELTIQ by
Drug Labeling and Warnings
TRUSELTIQ by is a Prescription medication manufactured, distributed, or labeled by QED Therapeutics, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
TRUSELTIQ- infigratinib capsule
QED Therapeutics, Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TRUSELTIQ safely and effectively. See full prescribing information for TRUSELTIQ.
TRUSELTIQ (infigratinib) capsules, for oral use Initial U.S. Approval: 2021 INDICATIONS AND USAGE
TRUSELTIQ is a kinase inhibitor indicated for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement as detected by an FDA-approved test. ( 1, 2.1)
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s). ( 1) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSCapsules: 25 mg and 100 mg. ( 3) CONTRAINDICATIONSNone. ( 4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact QED Therapeutics, Inc. at 1-844-550-BBIO (2246) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for FDA-approved patient labeling. Revised: 5/2021 |
FULL PRESCRIBING INFORMATION
1. INDICATIONS AND USAGE
TRUSELTIQ is indicated for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement as detected by an FDA-approved test [see Dosage and Administration ( 2.1)] .
This indication is approved under accelerated approval based on overall response rate and duration of response [see Clinical Studies ( 14.1)] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
2. DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for the treatment of unresectable locally advanced or metastatic cholangiocarcinoma with TRUSELTIQ based on the presence of an FGFR2 fusion or rearrangement, as detected by an FDA-approved test [ seeClinical Studies ( 14.1) ].
Information on FDA-approved test(s) for the detection of FGFR2 fusions or rearrangements in cholangiocarcinoma is available at: http://www.fda.gov/CompanionDiagnostics.
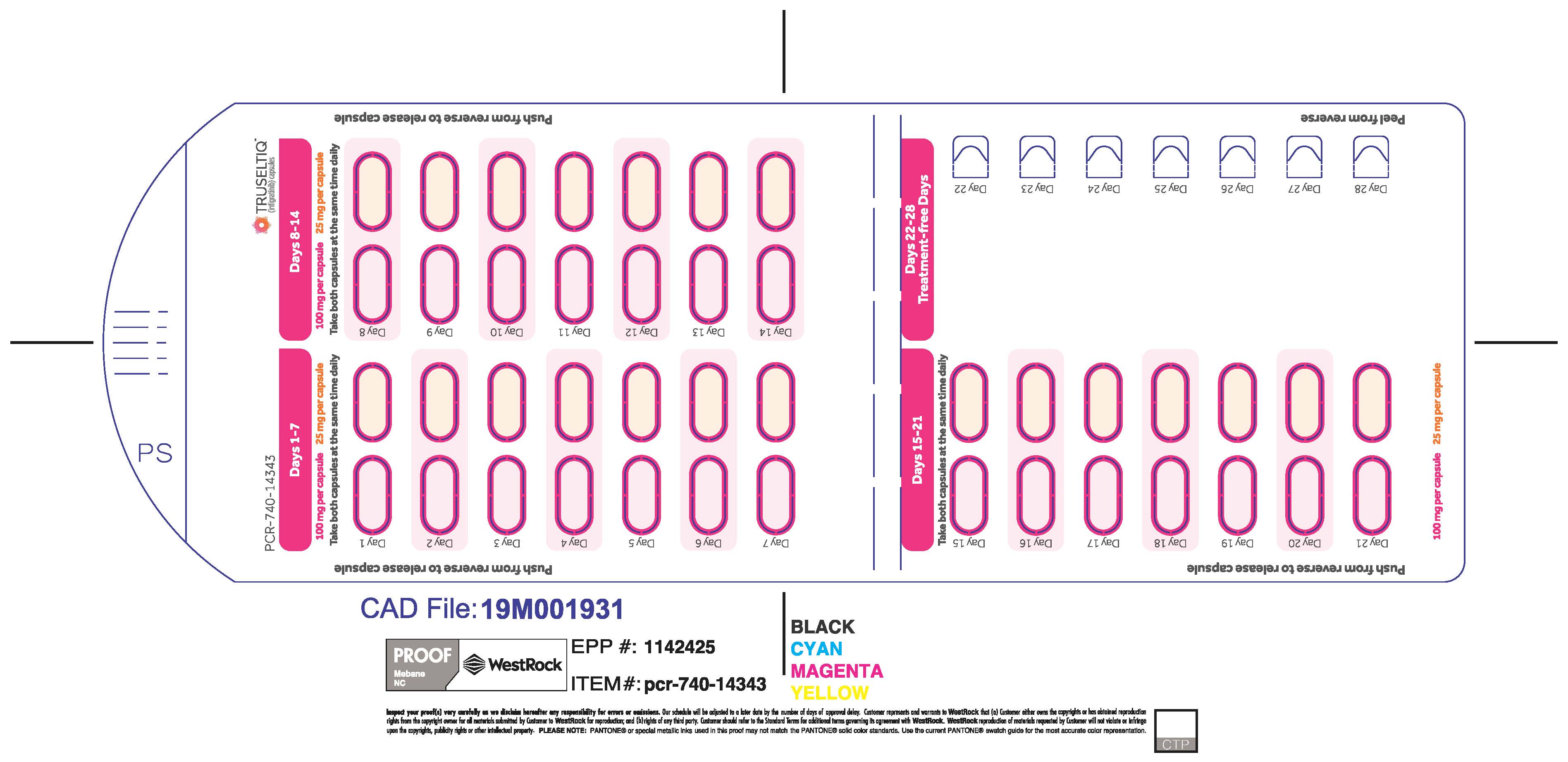
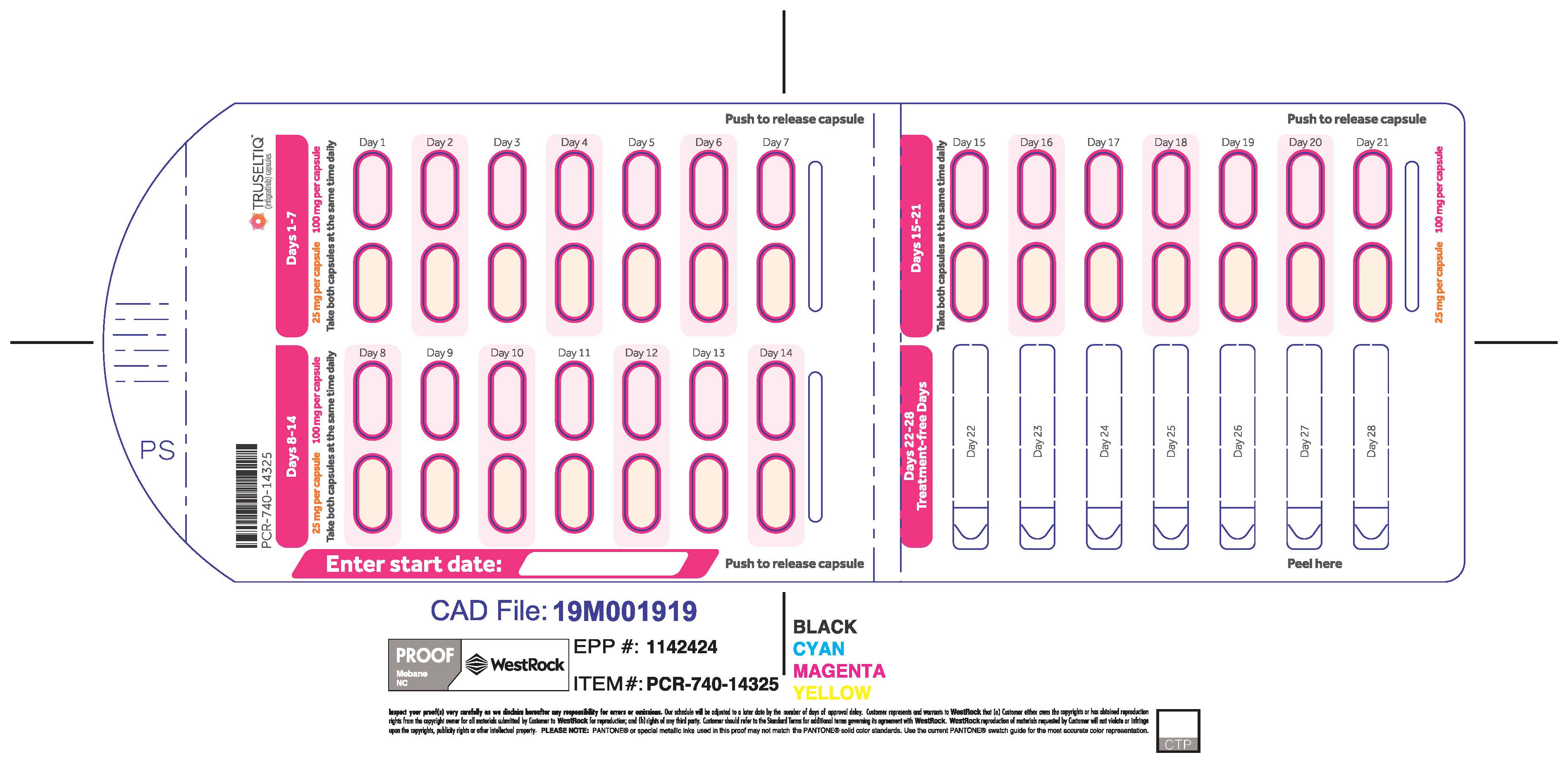
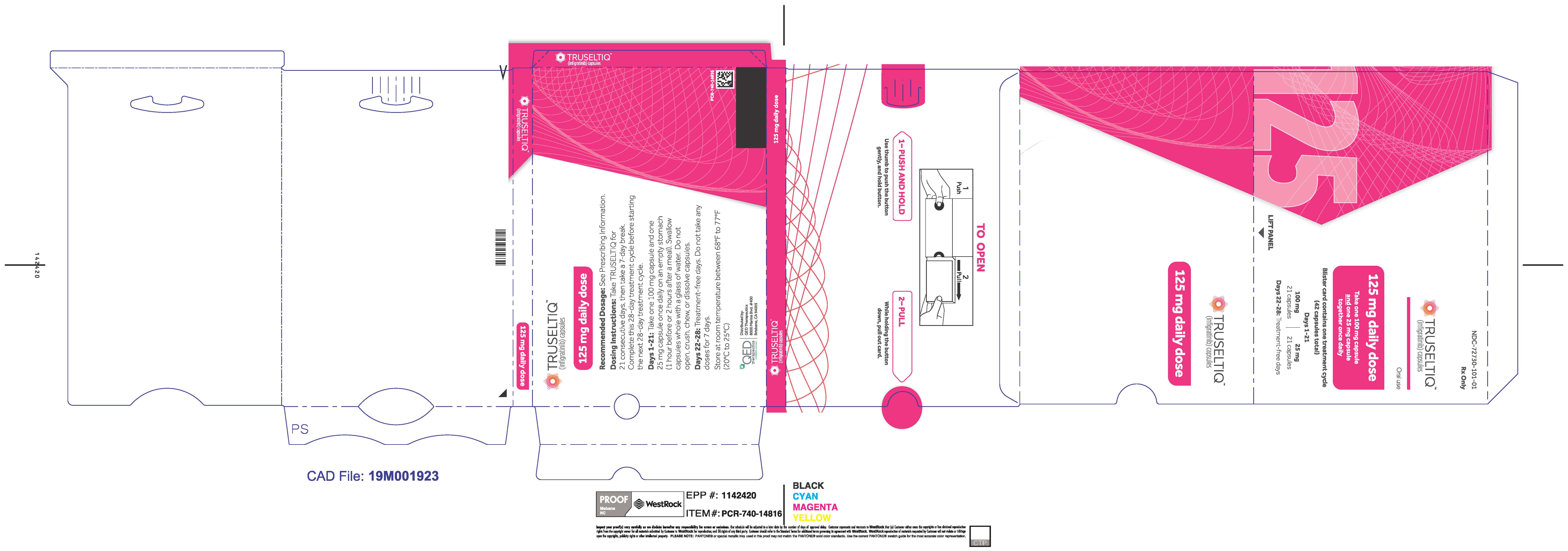
2.2 Recommended Dosage
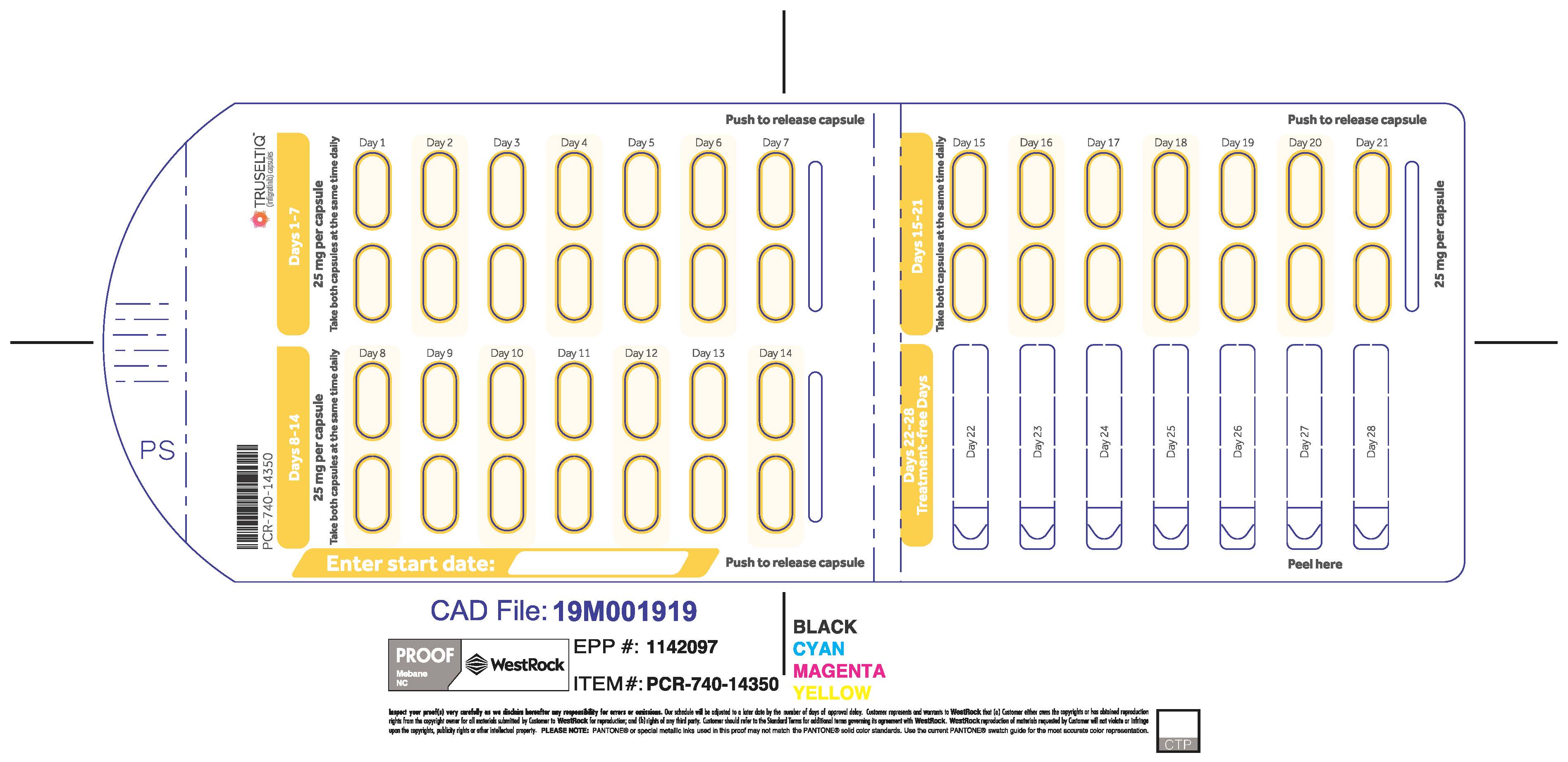
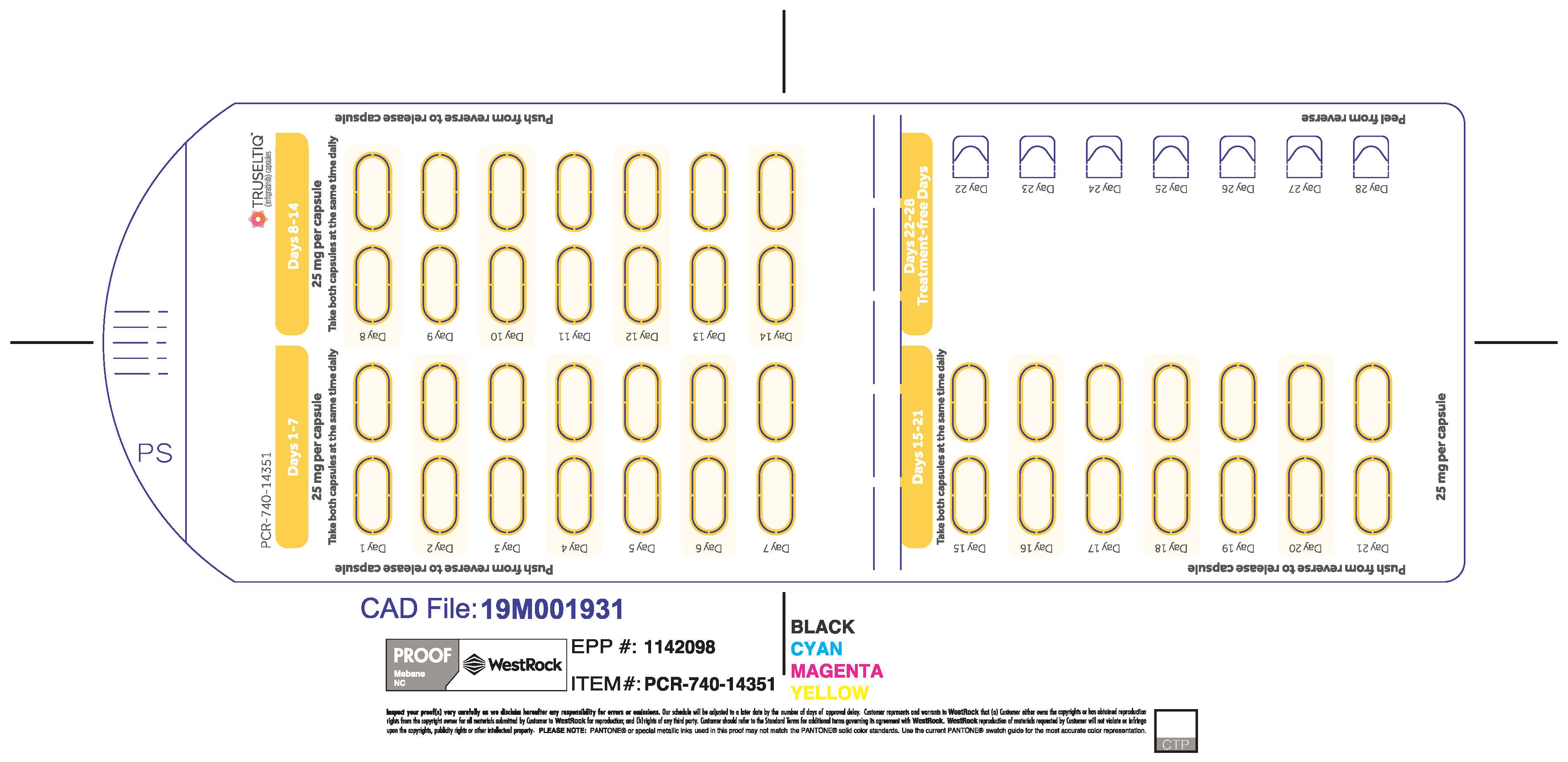
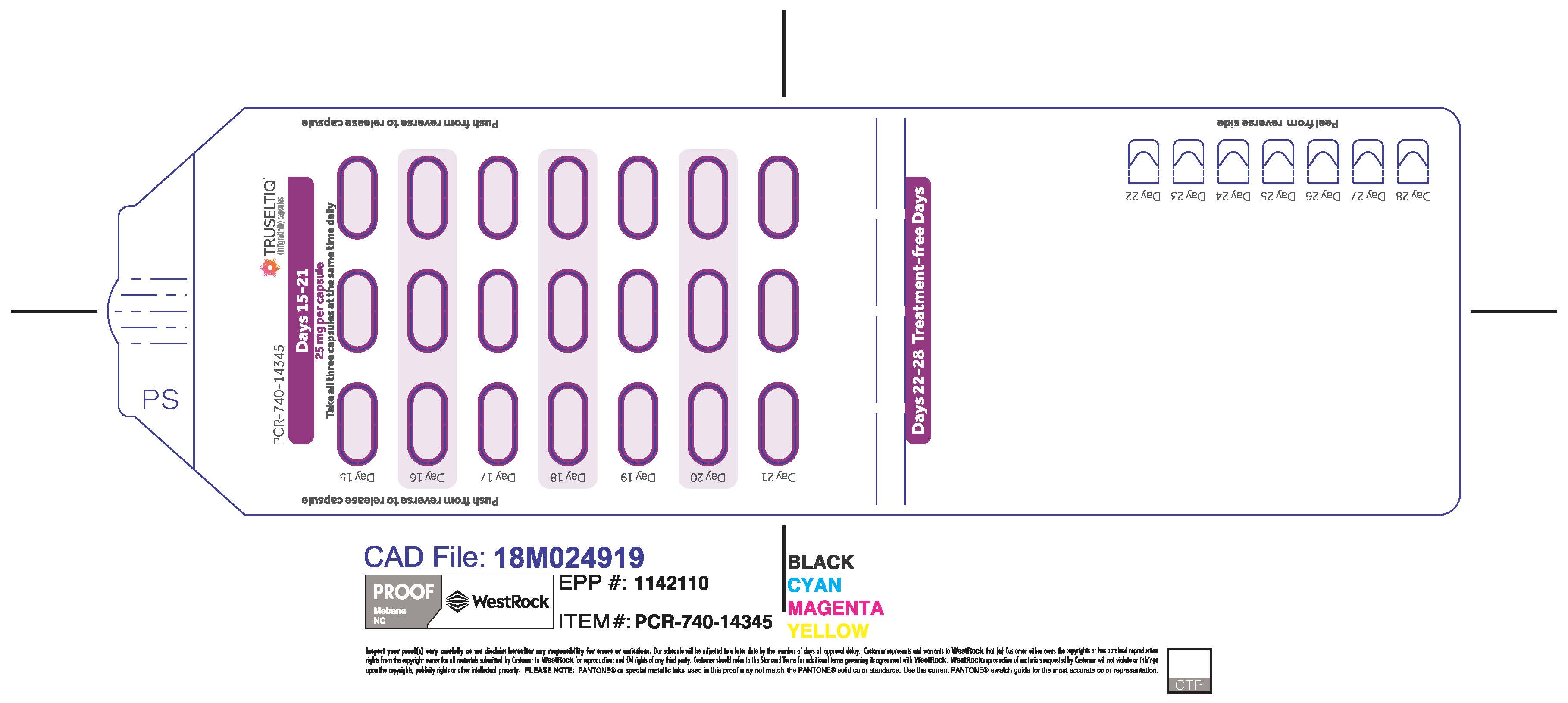
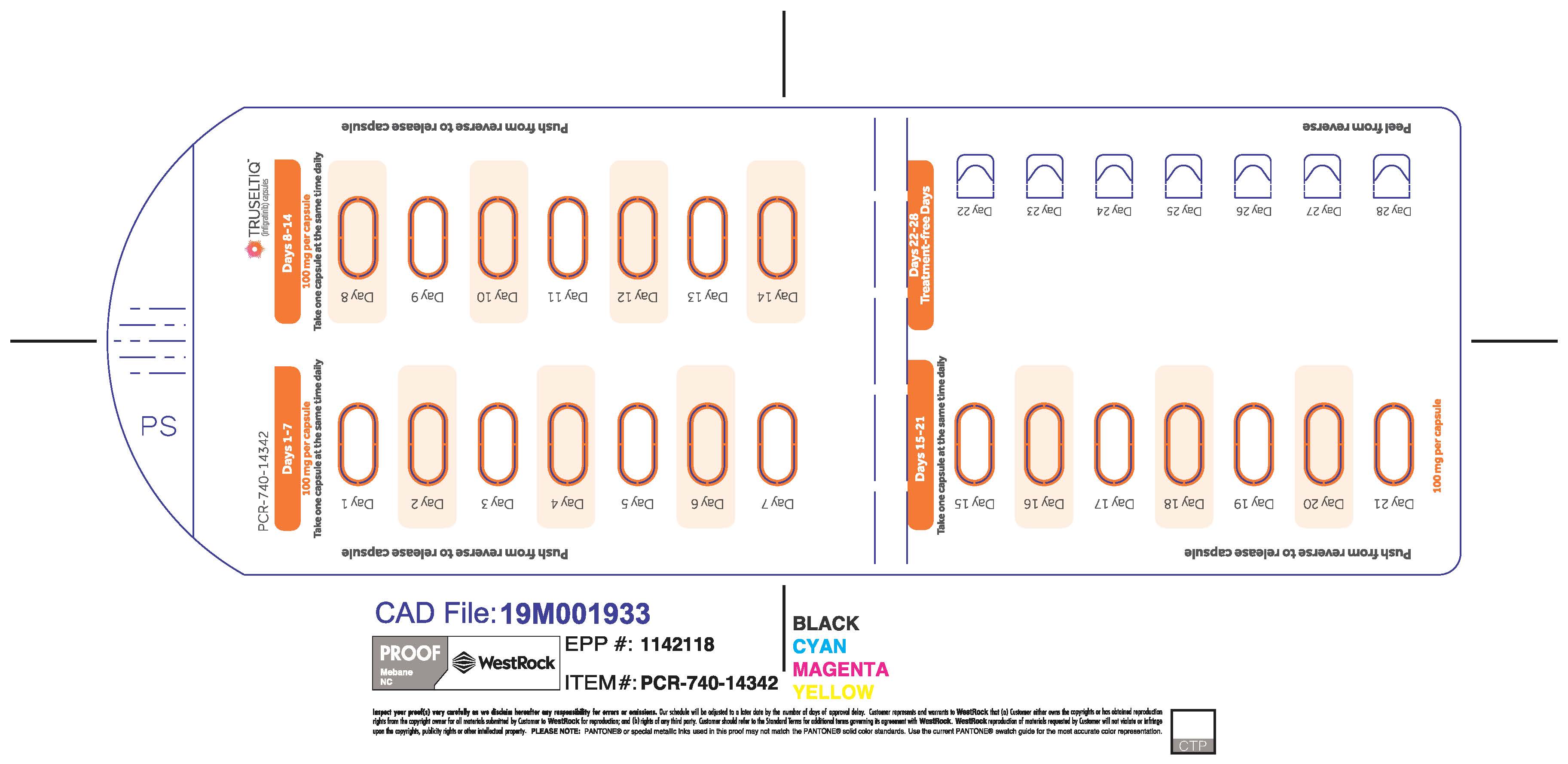
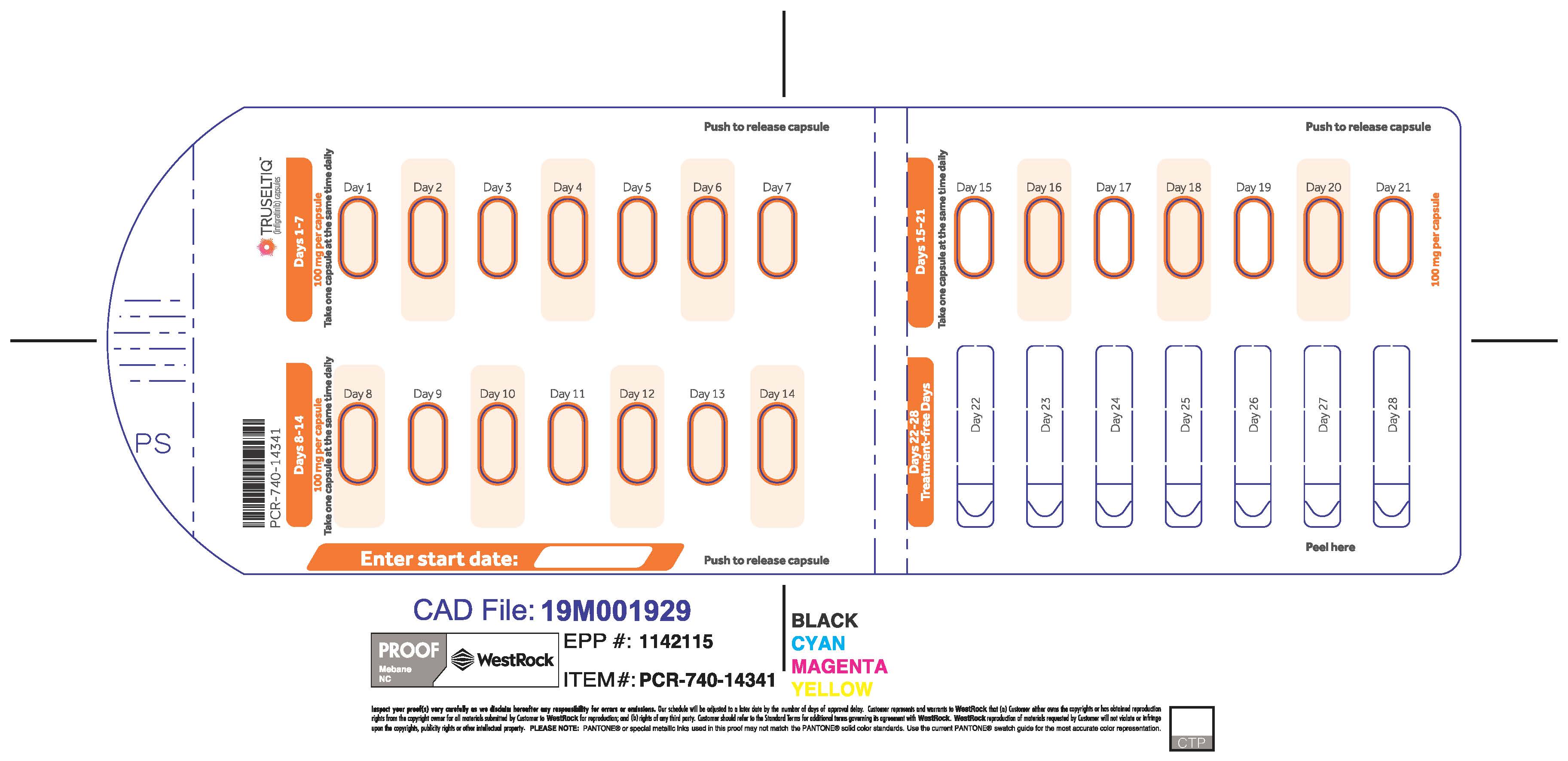
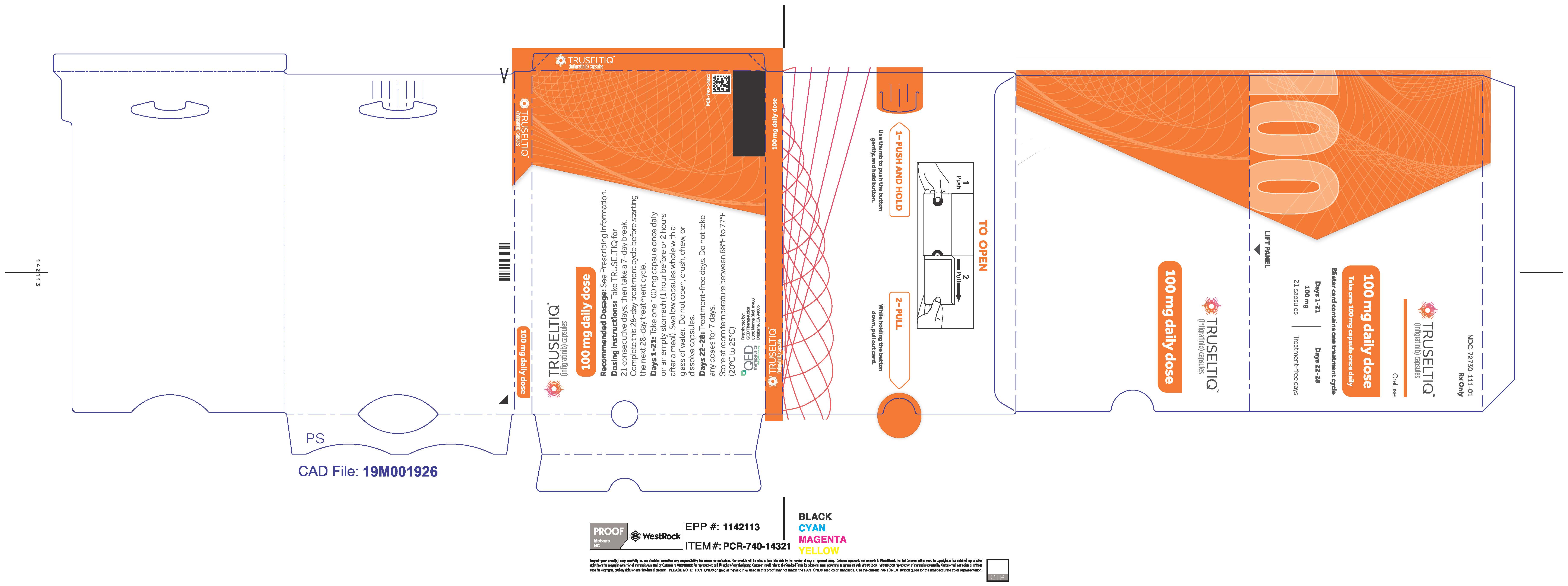
The recommended dosage of TRUSELTIQ is 125 mg (one 100 mg capsule and one 25 mg capsule) orally once daily for 21 consecutive days followed by 7 days off therapy, in 28-day cycles. Continue treatment until disease progression or unacceptable toxicity.
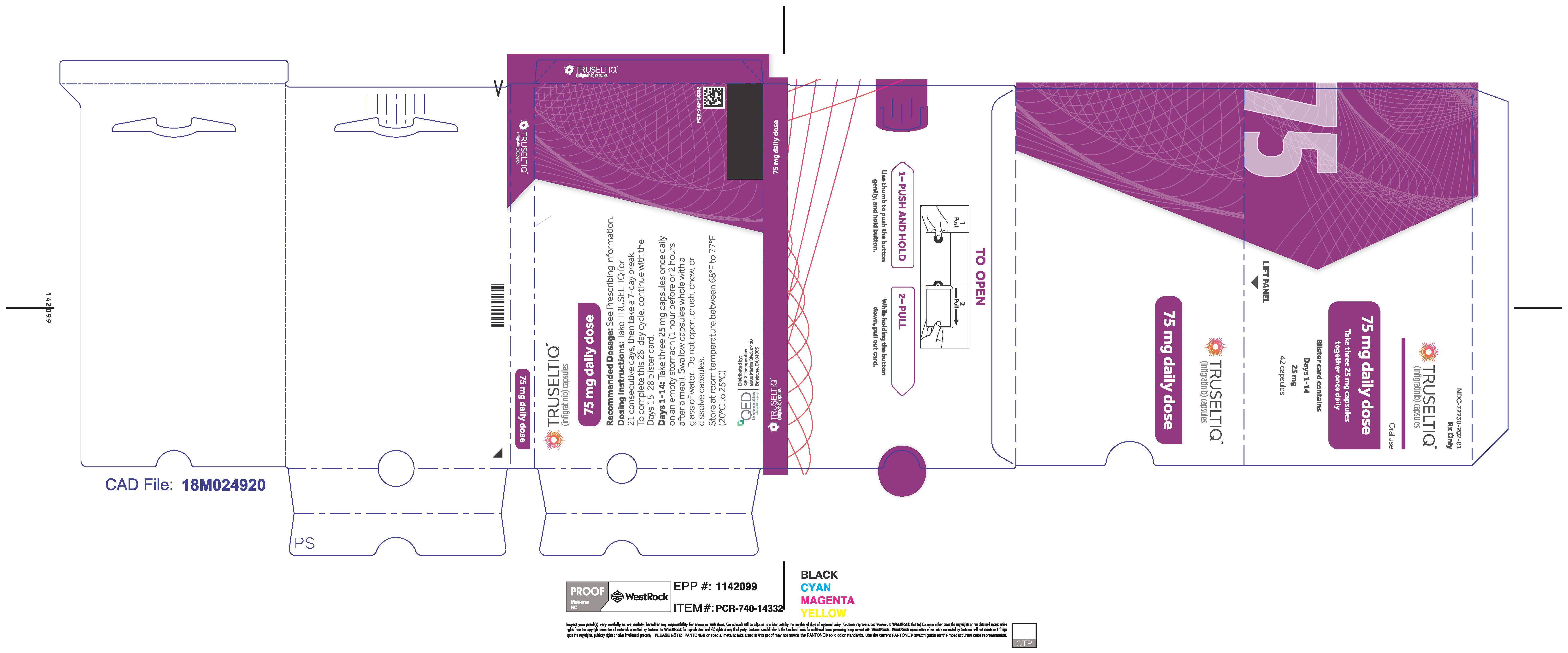
Instruct patients to take TRUSELTIQ on an empty stomach at least 1 hour before or 2 hours after food [ seeClinical Pharmacology( 12.3) ], at approximately the same time each day. Instruct patients to swallow capsules whole with a glass of water. Advise patients to not crush, chew, or dissolve capsules.
If a dose of TRUSELTIQ is missed by ≥4 hours or if vomiting occurs, instruct patients to resume the regular daily dose schedule for TRUSELTIQ the next day.
2.3 Dose Modification for Adverse Reactions
The recommended dose reductions for adverse reactions are listed in Table 1.
| 1 stdose reduction | 2 nddose reduction | 3 rddose reduction |
| 100 mg (one 100 mg capsule) | 75 mg (three 25 mg capsules) | 50 mg (two 25 mg capsules) |
The recommended dosage modifications for adverse reactions are provided in Table 2 .
| Adverse Reaction | Severity a | TRUSELTIQ Dose Modifications |
|
Retinal Pigment Epithelial Detachment (RPED)[ see Warnings and Precautions ( 5.1) ] | Not Applicable |
Continue TRUSELTIQ at the current dose and continue periodic ophthalmic evaluation:
|
| Hyperphosphatemia[ see Warnings and Precautions ( 5.2) ] | Serum phosphate >5.5 – ≤7.5 mg/dL | Continue TRUSELTIQ at the current dose and initiate or dose adjust phosphate binder according to respective label. Monitor serum phosphate weekly. Phosphate binder dosing should be held during the week off TRUSELTIQ therapy each cycle (Days 22-28) and during TRUSELTIQ dose interruptions for non-hyperphosphatemia adverse events. |
|
Serum phosphate >7.5 mg/dL Or Single serum phosphate >9 mg/dL regardless of duration or dose of phosphate lowering therapy. |
Withhold TRUSELTIQ until level returns to serum phosphate ≤5.5 mg/dL. Resume TRUSELTIQ as below, with maximal phosphate binder dosing:
|
|
| Serum phosphate with life-threatening consequences; urgent intervention indicated (e.g., dialysis) | Permanently discontinue TRUSELTIQ. | |
| Other Adverse Reactions | Grade 3 | Withhold dose of TRUSELTIQ until resolved to Grade ≤1, then resume at the next lower dose level of TRUSELTIQ. If not resolved within ≤14 days, permanently discontinue TRUSELTIQ. |
| Grade 4 | Permanently discontinue TRUSELTIQ. |
aSeverity as defined by National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE version 4.03).
2.4 Dose Modification for Moderate Hepatic Impairment
Avoid coadministration of a proton pump inhibitor (PPI), a histamine-2 (H2) receptor antagonist, or a locally-acting antacid with TRUSELTIQ [ see Drug Interactions ( 7.1) ]. If coadministration cannot be avoided:
- H2-antagonist: Separate administration of TRUSELTIQ by 2 hours before or 10 hours after.
- Locally-acting antacid: Separate administration of TRUSELTIQ by 2 hours before or after.
2.5 Recommended Dosage for Mild and Moderate Renal Impairment
The recommended dosage of TRUSELTIQ for patients with mild to moderate renal impairment (creatinine clearance 30 to 89 mL/min, estimated by Cockcroft-Gault) is 100 mg once daily for 21 consecutive days followed by 7 days off therapy, in 28-day cycles [ see Use in Specific Population( 8.6), Clinical Pharmacology ( 12.3) ].
2.6 Recommended Dosage for Mild and Moderate Hepatic Impairment
The recommended dosage of TRUSELTIQ for patients with mild (total bilirubin > upper limit of normal [ULN] to 1.5 × ULN or AST > ULN ) or moderate hepatic impairment (total bilirubin >1.5 to 3 × ULN with any AST) is as follows [ see Use in Specific Population ( 8.7), Clinical Pharmacology ( 12.3) ].
- Mild Hepatic Impairment: 100 mg once daily for 21 consecutive days followed by 7 days off therapy, in 28-day cycles.
- Moderate Hepatic Impairment: 75 mg once daily for 21 consecutive days followed by 7 days off therapy, in 28-day cycles.
3. DOSAGE FORMS AND STRENGTHS
Capsules:
- 25 mg: Hard gelatin capsule with a white opaque body and a gray opaque cap - imprinted with black text on the body – INFI 25mg
- 100 mg: Hard gelatin capsule with a white opaque body and a light orange opaque cap - imprinted with black text on the body – INFI 100mg
5. WARNINGS AND PRECAUTIONS
5.1 Ocular Toxicity
Retinal Pigment Epithelial Detachment (RPED)
TRUSELTIQ can cause RPED, which may cause symptoms such as blurred vision.
Among 351 patients who received TRUSELTIQ across clinical trials [ see Adverse Reactions ( 6.1) ] where ophthalmologic monitoring did not routinely include optical coherence tomography (OCT), RPED occurred in 11% of patients, including patients with asymptomatic RPED. The median time to first onset of RPED was 26 days. RPED led to dose interruption/reduction of TRUSELTIQ in 3.4% of patients, and permanent discontinuation in 0.6% of patients.
Perform a comprehensive ophthalmic examination including OCT prior to initiation of TRUSELTIQ, at 1 month, at 3 months, and then every 3 months thereafter during treatment. Refer patients for ophthalmic evaluation urgently for onset of visual symptoms, and follow-up every 3 weeks until resolution or discontinuation of TRUSELTIQ.
Withhold TRUSELTIQ as recommended [ see Dosage and Administration ( 2.3), Adverse Reactions ( 6.1) ].
Dry Eye
Among 351 patients who received TRUSELTIQ across clinical trials [ see Adverse Reactions ( 6.1) ], dry eye occurred in 29% of patients. Treat patients with ocular demulcents as needed.
5.2 Hyperphosphatemia and Soft Tissue Mineralization
TRUSELTIQ can cause hyperphosphatemia leading to soft tissue mineralization, cutaneous calcinosis, non-uremic calciphylaxis, vascular calcification, and myocardial calcification. Increases in phosphate levels are a pharmacodynamic effect of TRUSELTIQ [ see Clinical Pharmacology ( 12.2) ]. Among 351 patients who received TRUSELTIQ across clinical trials [ see Adverse Reactions ( 6.1) ], hyperphosphatemia was reported in 82% of patients based on laboratory values above the upper limit of normal. The median time to onset of hyperphosphatemia was 8 days (range 1-349). Phosphate binders were received by 83% of patients who received TRUSELTIQ.
Monitor for hyperphosphatemia throughout treatment. Initiate phosphate lowering therapy when serum phosphate level is >5.5 mg/dL. For serum phosphate level >7.5 mg/dL, withhold TRUSELTIQ and initiate phosphate lowering therapy. Withhold, dose reduce, or permanently discontinue TRUSELTIQ based on duration and severity of hyperphosphatemia [ see Dosage and Administration ( 2.3) ].
5.3 Embryo-Fetal Toxicity
Based on findings in animal studies and its mechanism of action, TRUSELTIQ can cause fetal harm when administered to a pregnant woman. Oral administration of infigratinib to pregnant animals during the period of organogenesis caused malformations, fetal growth retardation, and embryo-fetal death at maternal exposures lower than the human exposure based on area under the curve (AUC) at the clinical dose of 125 mg.
Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with TRUSELTIQ and for 1 month after the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with TRUSELTIQ and for 1 month after the final dose [ see Use in Specific Populations ( 8.1, 8.3) ].
6. ADVERSE REACTIONS
The following adverse reactions are discussed elsewhere in the labeling:
- Ocular Toxicity [ see Warnings and Precautions ( 5.1) ]
- Hyperphosphatemia and Soft Tissue Mineralization [ see Warnings and Precautions ( 5.2) ]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflect exposure to TRUSELTIQ as a single agent at 125 mg orally once daily for 21 consecutive days followed by 7 days off therapy, in 28-day cycles in 351 patients in Study CBGJ398X2204 and in patients with other advanced solid tumors or hematological malignancies. Among 351 patients who received TRUSELTIQ, 27% were exposed for 6 months or longer and 10% were exposed for greater than one year.
Previously Treated, Unresectable Locally Advanced or Metastatic Cholangiocarcinoma
The safety of TRUSELTIQ was evaluated in Study CBGJ398X2204, which included 108 patients with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with an FGFR2 fusion or other rearrangement [ see Clinical Studies ( 14.1) ]. Patients were treated orally with TRUSELTIQ 125 mg once daily for 21 consecutive days followed by 7 days off therapy, in 28-day cycles, until disease progression or unacceptable toxicity. The median duration of treatment was 5.5 months (range: 0.03 to 28.3 months).
The median age of TRUSELTIQ treated patients was 53 years (range 23-81), 62% were females, and 72% were White.
Serious adverse reactions occurred in 32% of patients receiving TRUSELTIQ. Serious adverse reactions in ≥2% of patients who received TRUSELTIQ included infections, anemia, pyrexia, abdominal pain, hypercalcemia, and sepsis. Fatal adverse reactions occurred in 1 (0.9%) patient who received TRUSELTIQ and was due to sepsis.
Permanent discontinuation due to an adverse reaction occurred in 15% of patients who received TRUSELTIQ. Adverse reactions requiring permanent discontinuation in ≥1% of patients were blood creatinine increased, fatigue, subretinal fluid, and calcinosis.
Dosage interruptions due to an adverse reaction occurred in 64% of patients who received TRUSELTIQ. Adverse reactions requiring dosage interruption in ≥5% of patients included hyperphosphatemia, hypercalcemia, palmar-plantar erythrodysesthesia syndrome, stomatitis, diarrhea, and blood creatinine increased.
Dosage reductions due to an adverse reaction occurred in 60% of patients who received TRUSELTIQ. Adverse reactions requiring dosage reductions in ≥2% of patients who received TRUSELTIQ included hyperphosphatemia, stomatitis, palmar-plantar erythrodysesthesia syndrome, increased blood creatinine, increased lipase, hypercalcemia, and onycholysis.
The most common (≥20%) adverse reactions were nail toxicity, stomatitis, dry eye, fatigue, alopecia, palmar-plantar erythrodysesthesia syndrome, arthralgia, dysgeusia, constipation, abdominal pain, dry mouth, eyelash changes, diarrhea, dry skin, decreased appetite, vision blurred and vomiting. The most common laboratory abnormalities (≥20%) were increased creatinine, increased phosphate, decreased phosphate, increased alkaline phosphatase, decreased hemoglobin, increased alanine aminotransferase, increased lipase, increased calcium, decreased lymphocytes, decreased sodium, increased triglycerides, increased aspartate aminotransferase, increased urate, decreased platelets, decreased leukocytes, decreased albumin, increased bilirubin and decreased potassium.
Table 3summarizes the adverse reactions in Study CBGJ398X2204. Table 4summarizes select laboratory abnormalities in Study CBGJ398X2204.
|
TRUSELTIQ N=108 |
||
| Adverse Reaction | All Grades
(%) | Grades 3 or 4
a
(%) |
| Skin and subcutaneous tissue disorders | ||
| Nail toxicity b | 57 | 2* |
| Alopecia | 38 | 0 |
| Palmar-plantar erythrodysesthesia syndrome | 33 | 7* |
|
Dry skin | 23 | 0 |
|
Gastrointestinal disorders |
||
|
Stomatitis c | 56 | 15* |
| Constipation | 30 | 1* |
|
Abdominal pain d | 26 | 5* |
|
Dry mouth | 25 | 0 |
| Diarrhea | 24 | 3* |
| Vomiting | 21 | 1* |
| Nausea | 19 | 1* |
| Dyspepsia | 17 | 0 |
|
Eye disorders e |
||
|
Dry eye f | 44 | 0 |
|
Eyelash changes g | 25 | 0 |
|
Vision blurred | 21 | 0 |
|
General disorders and administrative site conditions |
||
|
Fatigue h | 44 | 4* |
|
Edema i | 17 | 1* |
|
Pyrexia | 15 | 1* |
|
Musculoskeletal and connective tissue disorders |
||
|
Arthralgia | 32 | 0 |
|
Pain in extremity | 17 | 2* |
|
Nervous system disorders |
||
|
Dysgeusia | 32 | 0 |
|
Headache | 17 | 1* |
|
Metabolism and nutrition disorders |
||
|
Decreased appetite | 22 | 1* |
|
Respiratory, thoracic and mediastinal disorders |
||
|
Epistaxis | 18 | 0 |
|
Investigations |
||
|
Weight decreased | 15 | 2* |
Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE 4.03).
aEvents of Grade 3 only (no Grade 4 occurred) are marked with an asterisk.
bIncludes ingrown nail, nail bed bleeding, nail bed disorder, nail bed inflammation, nail bed tenderness, nail discoloration, nail disorder, nail dystrophy, nail hypertrophy, nail infection, nail ridging, onychalgia, onychoclasis, onycholysis, onychomadesis, onychomycosis, and paronychia.
cIncludes mouth ulceration and stomatitis.
dIncludes abdominal pain, abdominal pain upper, abdominal discomfort, and abdominal pain lower.
eSeverity of eye disorders is not represented by CTCAE Grading
fIncludes dry eye, keratitis, lacrimation increased, pinguecula, and punctate keratitis.
gIncludes blepharitis, eyelash changes, eyelash discoloration, growth of eyelashes, trichiasis, and trichomegaly.
hIncludes asthenia and fatigue.
iIncludes edema peripheral and edema.
Clinically relevant adverse reactions occurring in ≤15% of patients included cataracts (12%) and fractures (1%).
|
TRUSELTIQ N=108 |
||
| Laboratory Abnormality |
All Grades (%) |
Grade 3 or 4 (%) |
| Hematology | ||
| Decreased hemoglobin | 53 | 5 |
| Decreased lymphocytes | 43 | 9 |
| Decreased platelets | 37 | 4 |
| Decreased leukocytes | 26 | 3 |
| Decreased neutrophils | 14 | 2 |
| Chemistry | ||
| Increased creatinine | 93 | 7 |
| Increased phosphate a | 90 | 13 |
| Decreased phosphate | 64 | 31 |
| Increased alkaline phosphatase | 54 | 8 |
| Increased alanine aminotransferase | 51 | 6 |
| Increased lipase | 44 | 7 |
| Increased calcium | 43 | 7 |
| Decreased sodium | 41 | 20 |
| Increased triglycerides | 38 | 3 |
| Increased aspartate aminotransferase | 38 | 4 |
| Increased urate | 37 | 37 |
| Decreased albumin | 24 | 1 |
| Increased bilirubin | 24 | 6 |
| Decreased potassium | 21 | 3 |
| Increased cholesterol | 18 | 1 |
| Increased potassium | 17 | 3 |
| Decreased calcium | 10 | 2 |
The denominator used to calculate the rate varied from 104 to 107 based on the number of patients with a baseline value and at least one post-treatment value. These laboratory abnormalities are values that reflect worsening from baseline.
Graded per NCI CTCAE 4.03.
aNCI CTCAE 4.03 does not define grades for increased phosphate. Laboratory value shift table categories were used to assess increased phosphorus levels (Grades ≥3 defined as ≥9mg/dL).
7. DRUG INTERACTIONS
7.1 Effects of Other Drugs on TRUSELTIQ
Strong and Moderate CYP3A Inhibitors
Concomitant use of TRUSELTIQ with a strong or moderate CYP3A inhibitor may increase infigratinib plasma concentrations [ see Clinical Pharmacology ( 12.3) ], which may increase the risk of adverse reactions. Avoid concomitant use of TRUSELTIQ with strong or moderate CYP3A inhibitors.
Strong and Moderate CYP3A Inducers
Concomitant use of TRUSELTIQ with a strong or moderate CYP3A inducer may decrease infigratinib plasma concentrations [ see Clinical Pharmacology ( 12.3) ], which may reduce TRUSELTIQ anti-tumor activity. Avoid concomitant use of TRUSELTIQ with strong or moderate CYP3A inducers.
Gastric Acid Reducing Agents
The coadministration of TRUSELTIQ with a gastric acid reducing agent may decrease infigratinib plasma concentrations [ see Clinical Pharmacology ( 12.3) ], which may reduce TRUSELTIQ anti-tumor activity.
Avoid concomitant use of TRUSELTIQ with proton pump inhibitors (PPIs), H2-antagonists, and locally-acting antacids. If coadministration of H2-antagonists or locally-acting antacids cannot be avoided, stagger administration of TRUSELTIQ [ see Dosage and Administration ( 2.4) ].
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animal studies and its mechanism of action TRUSELTIQ can cause fetal harm or loss of pregnancy when administered to a pregnant woman [ see Clinical Pharmacology ( 13.1) ]. There are no available data on the use of TRUSELTIQ during pregnancy. Oral administration of infigratinib to pregnant animals during the period of organogenesis at maternal exposures below the human exposure at the clinical dose of 125 mg resulted in malformations, fetal growth retardation, and embryo-fetal death (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Once daily oral administration of infigratinib to pregnant rats during organogenesis resulted in an increase in embryo-fetal lethality at 10 mg/kg/day and reductions in fetal body weights at ≥3 mg/kg/day (<0.1 times the human exposure at the clinical dose of 125 mg). Fetal abnormalities (external, soft tissue, and skeletal) were increased at ≥1 mg/kg/day (<0.1 times the human exposure at the clinical dose of 125 mg). In the embryo-fetal portion of a rat fertility study, infigratinib decreased the mean number of embryos and increased nonviable embryos and post-implantation loss in females at 3 mg/kg/day.
Once daily oral administration of infigratinib at ≥0.3 mg/kg/day to pregnant rabbits resulted in maternal toxicity and a corresponding reduction in fetal body weights.
8.2 Lactation
Risk Summary
There are no data on the presence of infigratinib or its metabolites in human milk, or their effects on either the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed children from TRUSELTIQ, advise women not to breastfeed during treatment and for 1 month after the final dose.
8.3 Females and Males of Reproductive Potential
TRUSELTIQ can cause fetal harm when administered to a pregnant woman [ see Use in Specific Populations ( 8.1) ].
Pregnancy Testing
Verify pregnancy status of females of reproductive potential prior to initiating TRUSELTIQ.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with TRUSELTIQ and for 1 month after the final dose.
Males
Advise males that are partnered with females of reproductive potential to use effective contraception during treatment with TRUSELTIQ and for 1 month after the final dose.
8.4 Pediatric Use
The safety and effectiveness of TRUSELTIQ in pediatric patients have not been established.
Animal Toxicity Data
In rat and dog repeat-dose toxicity studies ≥13 weeks in duration, animals displayed toxicities in bones (rats and dogs) and teeth (rats) at exposures lower than the human exposure at the clinical dose of 125 mg. Once daily oral administration of infigratinib in a 13-week rat study resulted in incisor degeneration (degeneration of enamel and loss of ameloblast layer). Once daily oral administration of infigratinib in a 26-week rat study resulted in bone effects (decreased bone strength consistent with decreases in total bone mineral density in lumbar vertebral bodies). Once daily oral administration of infigratinib in a 39-week dog study resulted in increased growth plate thickness and fractures associated with increased physeal thickness, focal mixed reaction, and bone loss.
8.5 Geriatric Use
Of the 351 patients treated with TRUSELTIQ in clinical studies, 33% were 65 years or older, and 10% were 75 years or older. No overall differences in safety or effectiveness of TRUSELTIQ have been observed between patients 65 years of age and older and younger adult patients.
8.6 Patients with Renal Impairment
Reduce the dosage of TRUSELTIQ for patients with mild or moderate renal impairment (creatinine clearance [CLcr] 30 to 89 mL/min estimated by Cockcroft-Gault). The recommended dosage of TRUSELTIQ has not been established for patients with severe renal impairment (CLcr < 30 mL/min) or for patients with end-stage renal disease receiving intermittent hemodialysis [ see Dosage and Administration ( 2.5) and Clinical Pharmacology ( 12.3) ].
8.7 Patients with Hepatic Impairment
Reduce the dosage of TRUSELTIQ for patients with mild (total bilirubin > upper limit of normal [ULN] to 1.5 × ULN or AST > ULN) or moderate (total bilirubin >1.5 to 3 × ULN with any AST) hepatic impairment. The recommended dosage of TRUSELTIQ has not been established in patients with severe (total bilirubin > 3 × ULN with any AST) hepatic impairment [ see Dosage and Administration ( 2.6), Clinical Pharmacology ( 12.3) ].
11. DESCRIPTION
Infigratinib is a kinase inhibitor. The chemical name is 3-(2,6-dichloro-3,5-dimethoxyphenyl)-1-{6-[4-(4-ethylpiperazin-1-yl)phenylamino]pyrimidin-4-yl}-1-methylurea phosphate (1:1). The molecular formula is C 26H 31Cl 2N 7O 3H 3PO 4and the molecular weight is 560.48 g/mol for the free base, 658.47 g/mol for the phosphate salt.
The chemical structure of infigratinib phosphate is as follows:
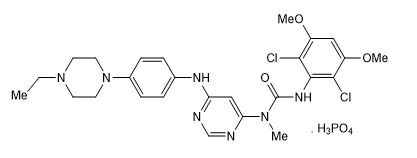
Infigratinib phosphate is a white to off-white powder. It shows adequate solubility in water and 0.1N HCl. It is practically insoluble in pH 6.8 buffer and poorly soluble in common organic solvents. TRUSELTIQ (infigratinib) is supplied as 25 mg and 100 mg hard gelatin capsules for oral administration. Each capsule contains the following inactive ingredients: colloidal silicon dioxide, crospovidone, hypromellose, lactose monohydrate, magnesium stearate (from vegetable source), and microcrystalline cellulose. The capsule shells contain black iron oxide, gelatin, red iron oxide, titanium dioxide, and yellow iron oxide. The printing ink contains black iron oxide, butyl alcohol, dehydrated alcohol, isopropyl alcohol, potassium hydroxide, propylene glycol, shellac, and strong ammonia solution.
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Infigratinib is a small molecule kinase inhibitor of FGFR with IC 50values of 1.1, 1, 2, and 61 nM for FGFR1, FGFR2, FGFR3, and FGFR4, respectively. The major human metabolites of infigratinib, BHS697 and CQM157, have similar in vitro binding affinities for FGFR1, FGFR2, and FGFR3 compared to infigratinib. Infigratinib inhibited FGFR signaling and decreased cell proliferation in cancer cell lines with activating FGFR amplifications, mutations, or fusions. Constitutive FGFR signaling can support the proliferation and survival of malignant cells. Infigratinib had anti-tumor activity in mouse and rat xenograft models of human tumors with activating FGFR2 or FGFR3 alterations, including two patient-derived xenograft models of cholangiocarcinoma that expressed FGFR2-TTC28 or FGFR2-TRA2B fusions. Infigratinib demonstrated brain-to-plasma concentration ratios (based on AUC 0-inf) of 0.682 in rats after a single oral dose.
12.2 Pharmacodynamics
Serum Phosphate
TRUSELTIQ increased serum phosphate levels due to FGFR inhibition. Serum phosphate increased with increasing exposures across the dose range of 20 to 150 mg once daily (0.16 to 1.2 times the approved recommended dosage), with increased risk of hyperphosphatemia with higher exposure to TRUSELTIQ.
Cardiac Electrophysiology
At the recommended dosing regimen, TRUSELTIQ does not result in a large mean increase (i.e., >20 msec) in the QTc interval. The QT effect of infigratinib at higher exposures associated with CYP3A inhibition has not been studied.
12.3 Pharmacokinetics
The infigratinib pharmacokinetic parameters are presented following administration of the approved recommended dosage in cholangiocarcinoma patients, unless otherwise specified.
The mean (coefficient of variation [%CV]) steady-state maximum plasma concentration (C max) and area under the curve over a dosing interval (AUC 0‑24h) of infigratinib and active metabolites, BHS697 and CQM157, are presented in Table 5 .
| Infigratinib | BHS697 | CQM157 | |
| C max | 282.5 ng/mL (54%) | 42.1 ng/mL (65%) | 15.7 ng/mL (92%) |
| AUC 0‑24h | 3780 ngh/mL (59%) | 717 ngh/mL (55%) | 428 ngh/mL (72%) |
Infigratinib C maxand AUC increased more than proportionally across the dose range of 5 to 150 mg (0.04 to 1.2 times the approved recommended dose). Steady state was achieved within 15 days and the mean accumulation ratio was 8- and 5-fold for C maxand AUC, respectively.
Absorption
Median (range) time to achieve peak infigratinib plasma concentration (t max) was 6 hours (2 to 7 hours) at steady state.
Effect of Food
Following administration of TRUSELTIQ with a high-fat and high-calorie meal (800 to 1,000 calories with approximately 50% of total caloric content of the meal from fat) in healthy subjects, the mean AUC infof infigratinib increased by 80%-120% and C maxincreased by 60%-80%, the median T maxshifted from 4 hours to 6 hours. Following administration of TRUSELTIQ with a low-fat low-calorie meal (approximately 330 calories with 20% of total caloric content of the meal from fat), the mean AUC infof infigratinib increased by 70%, C maxincreased by 90%, and the median T maxdid not change.
Distribution
The geometric mean (CV%) apparent volume of distribution of infigratinib was 1600 L (33%) at steady state. The mean infigratinib protein binding was 96.8%, primarily to lipoprotein, and was dependent of drug concentration.
Elimination
The geometric mean (CV%) total apparent clearance (CL/F) of infigratinib was 33.1 L/h (59%) at steady state. The geometric mean (CV%) terminal half-life of infigratinib was 33.5 h (39%) at steady state.
Metabolism
Infigratinib is predominantly metabolized by CYP3A4 (~94%) and to a lesser extent by FMO3 (6%) in vitro. The major drug-related moiety in plasma was unchanged infigratinib (38% of dose) in a human [ 14C] mass balance study, followed by two active metabolites, BHS697 and CQM157 (each at >10% of dose). BHS697 is mainly metabolized by CYP3A4 and CQM157 is metabolized through both Phase I and Phase II biotransformation pathways.
BHS697 and CQM157 contribute about 16% to 33% and 9% to 12% of overall pharmacologic activity, respectively.
Excretion
After a single oral 125 mg dose of radiolabeled infigratinib in healthy subjects, approximately 77% of the dose was recovered in feces (3.4% as unchanged) and 7.2% in urine (1.9% as unchanged).
Specific Populations
No clinically significant differences in the systemic exposure of infigratinib were observed based on age (19‑86 years), sex, race/ethnicity (White 70.7%, Black 15.4%, and Asian 8%), or body weight (36.4-169 kg).
Patients with Renal Impairment
The relative potency adjusted steady state AUC of infigratinib plus its active metabolites (BHS697, CQM157) in plasma increased by 32% and 37% in patients with mild (creatinine clearance [CLcr] 60 to 89 mL/min estimated by Cockcroft-Gault) and moderate renal impairment (CLcr 30 to 59 mL/min), respectively, relative to patients with normal renal function (CLcr ≥ 90 mL/min).
The effect of severe renal impairment (CLcr < 30 mL/min) or renal dialysis in end-stage renal disease on infigratinib exposure is unknown.
Patients with Hepatic Impairment
The relative potency adjusted steady state AUC of infigratinib plus its active metabolites (BHS697, CQM157) in plasma increased by 47%-62% and 99% in patients with mild (total bilirubin > upper limit of normal [ULN] to 1.5 × ULN or AST > ULN) and moderate hepatic impairment (total bilirubin > 1.5 to 3 × ULN with any AST), respectively, relative to patients with normal hepatic function (total bilirubin ≤ ULN and AST ≤ ULN).
The effect of severe hepatic impairment (total bilirubin > 3 × ULN with any AST) on infigratinib exposure is unknown [ see Use in Specific Populations ( 8.7) ].
Drug Interaction Studies
Clinical Studies
Strong CYP3A Inhibitors: Coadministration of multiple doses of itraconazole (strong CYP3A inhibitor) increased infigratinib AUC 0-infby 622% and C maxby 164%, increased BHS697 AUC 0-infby 174%, and decreased CQM157 C maxby 69%, respectively [ see Drug Interactions ( 7.1) ].
Strong CYP3A Inducers: Coadministration of multiple doses of rifampin (strong CYP3A inducer) decreased infigratinib AUC 0-infby 56% and C maxby 44%, decreased BHS697 AUC 0-infby 65% and C maxby 27%, and decreased CQM157 AUC 0-infby 76% and C maxby 50%, respectively [ see Drug Interactions ( 7.1) ].
Gastric Acid-Lowering Agents: Coadministration of multiple doses of lansoprazole (proton pump inhibitor) decreased infigratinib AUC 0-infby 45% and C maxby 49%, decreased BHS697 AUC 0-infby 32% and C maxby 44%, and decreased CQM157 AUC 0-infby 72% and C maxby 55%, respectively [ see Drug Interactions ( 7.1) ].
CYP3A4 Substrates: No clinically significant differences in pharmacokinetics of midazolam (sensitive CYP3A4 substrate) were observed when co-administered with TRUSELTIQ [ see Drug Interactions ( 7.1) ].
In Vitro Studies
Cytochrome P450 Enzymes: Infigratinib does not induce CYP1A2, CYP2B6, CYP2C9, or CYP3A4. Infigratinib, BHS697, and CQM157 do not inhibit major CYP450 isozymes at clinically relevant concentrations.
Transporter Systems: Infigratinib inhibits MATE1 and BCRP. Infigratinib has a low potential to inhibit P-gp, BSEP, OCT1, OCT2 and MATE-2K at clinically relevant concentrations. Infigratinib is a substrate for P-gp and BCRP. The metabolites BHS697 and CQM157 have a low potential to inhibit OATP1B1, OATP1B3, P-gp, or BCRP at clinically relevant concentrations. The effect of these metabolites to inhibit MATE or OCT at clinically relevant concentrations is unknown.
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with infigratinib.
Infigratinib was not mutagenic in a bacterial reverse mutation (Ames) assay and was not clastogenic in an in vitro human peripheral blood lymphocyte chromosome aberration assay. Infigratinib did not induce micronuclei in an in vivo rat bone marrow micronucleus assay.
In a rat fertility study, there were no effects on mating or fertility, reproductive organ weights, or sperm motility, density, or morphology in males, and no effect on estrous cycling, mating, or fertility in females administered ≤3 mg/kg/day infigratinib.
14. CLINICAL STUDIES
14.1 Cholangiocarcinoma
Study CBGJ398X2204 (NCT02150967), a multicenter open-label single-arm trial, evaluated the efficacy of TRUSELTIQ in 108 patients with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with an FGFR2 fusion or rearrangement as determined for enrollment by local (89%) or central testing (11%). Qualifying in-frame fusions and other rearrangements were predicted to have a breakpoint within intron 17/exon 18 of the FGFR2 gene that leaves the FGFR2 kinase domain intact.
Patients received TRUSELTIQ at a dosage of 125 mg orally once daily for 21 consecutive days followed by 7 days off therapy, in 28-day cycles until disease progression or unacceptable toxicity. The major efficacy outcome measures were overall response rate (ORR) and duration of response (DoR), as determined by blinded independent central review (BICR) according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1.
The median age was 53 years (range: 23 to 81 years), 62% were female, 72% were White, 3.7% were Black or African American, 10% were Asian, and 99% had a baseline Eastern Cooperative Oncology Group (ECOG) performance status of 0 (42%) or 1 (57%). The presence of FGFR2 fusions or other rearrangements was determined in 104 enrolled patients (96%) with Next Generation Sequencing (NGS) testing. Eighty-eight (81%) patients had in-frame FGFR2 fusions, and BICC1 the most commonly reported fusion partner (n=27, 25%). Twenty (19%) patients had other FGFR2 rearrangements that may not be in-frame with the partner gene or the partner gene was not identifiable.
Ninety-nine percent of patients had metastatic (Stage IV) disease at the time of study entry. All patients had received at least 1 prior line of systemic therapy, 32% had 2 prior lines of therapy, and 29% had 3 or more prior lines of therapy. Ninety-nine percent of patients received prior gemcitabine-based therapy and most (88%) had progressed on their prior gemcitabine-based therapy.
Efficacy results are summarized in Table 6 . The median time to response was 3.6 months (range 1.4 – 7.4 months).
|
TRUSELTIQ N=108 |
|
| Efficacy Parameter | BICR Assessment |
| ORR(95% CI) | 23% (16, 32) |
| Complete Response, n (%) | 1 (1%) |
| Partial Response, n (%) | 24 (22%) |
| Median DoR(months) (95% CI) | 5.0 (3.7, 9.3) |
| Patients with DoR ≥6 months, n (%) | 8 (32%) |
| Patients with DoR ≥12 months, n (%) | 1 (4%) |
Abbreviations: BICR= blinded independent central review; CI=confidence interval; DoR=duration of response; ORR=overall response rate.
Note: Data are according to RECIST v1.1.
16. HOW SUPPLIED/STORAGE AND HANDLING
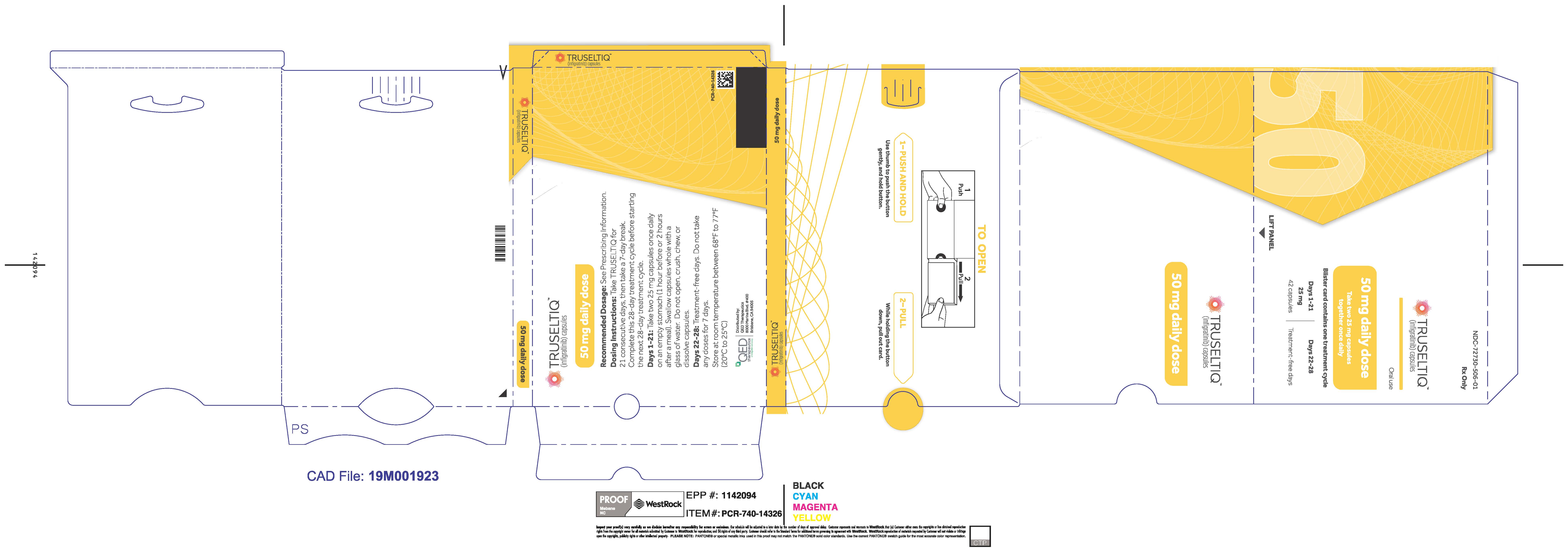
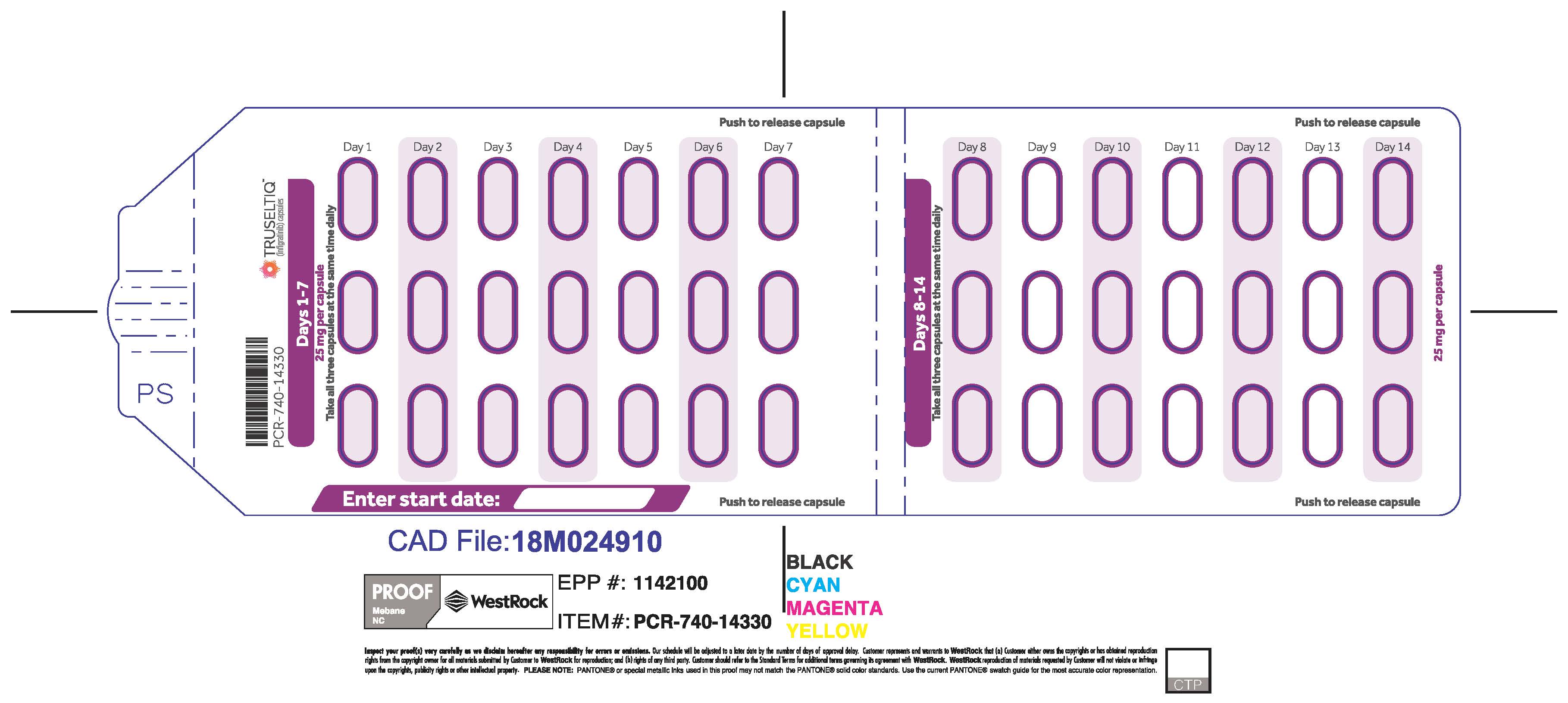
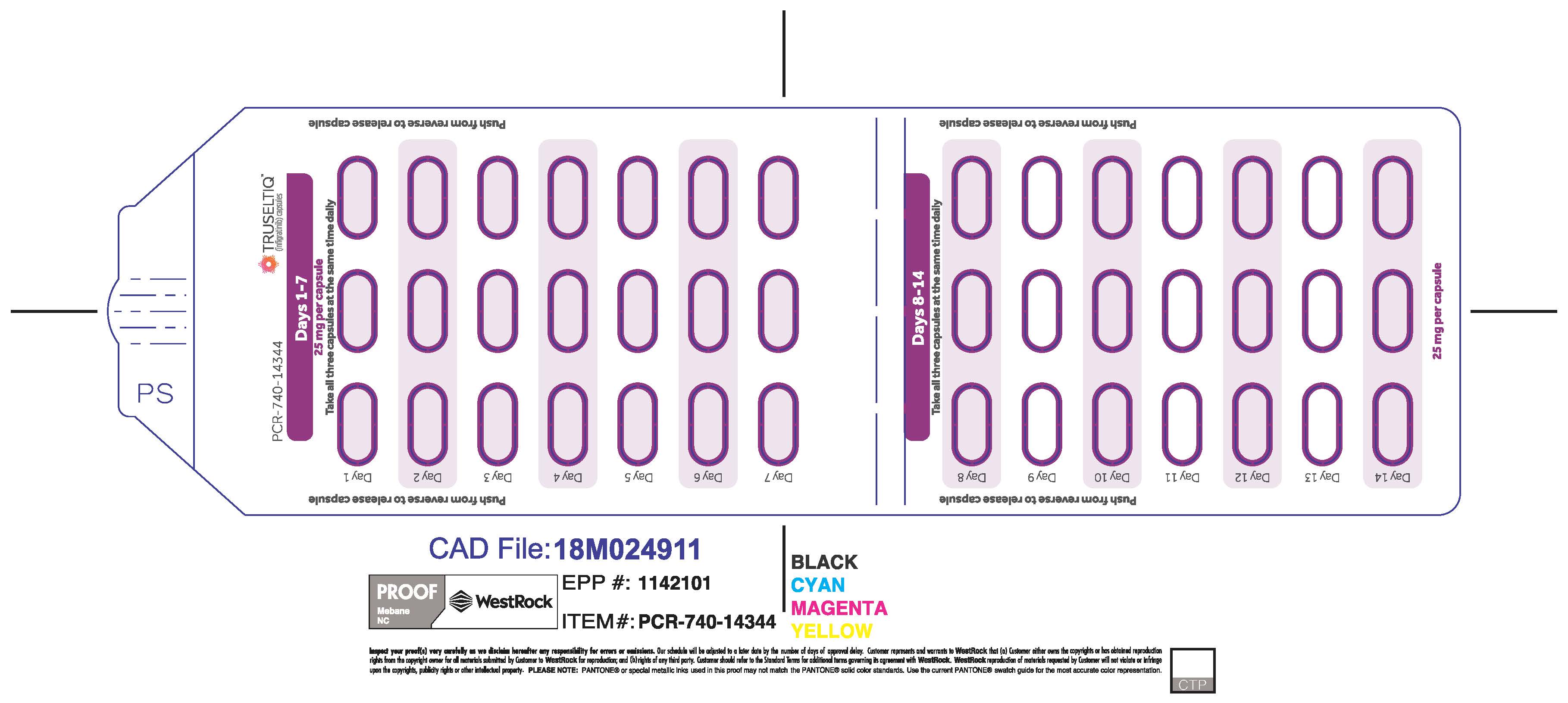
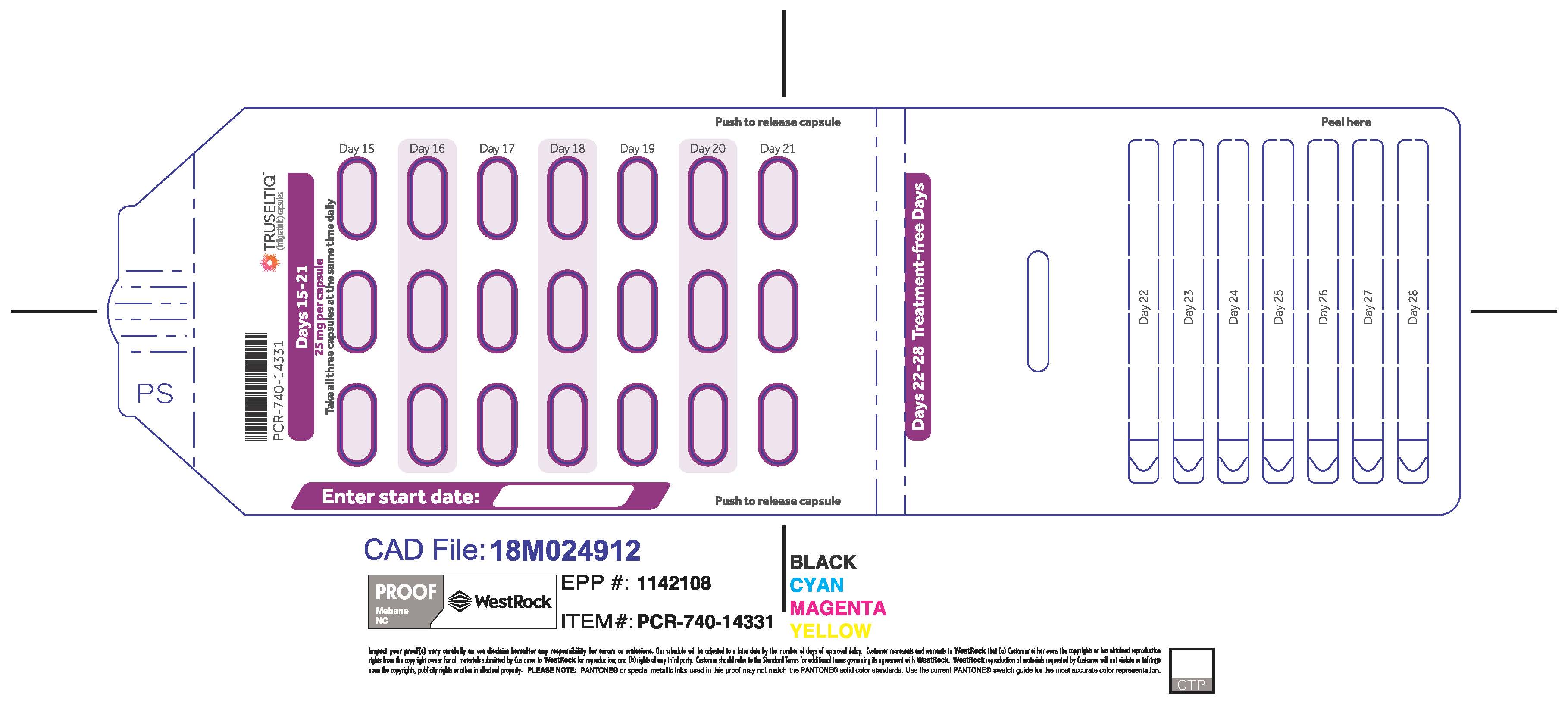
TRUSELTIQ (infigratinib) capsules are available in the strengths and packages listed below:
- 25 mg: Hard gelatin capsule with a white opaque body and a gray opaque cap - imprinted with black text on the body – INFI 25mg
- 100 mg: Hard gelatin capsule with a white opaque body and a light orange opaque cap - imprinted with black text on the body – INFI 100mg
TRUSELTIQ capsules are supplied in 21-day blister pack dose presentations as follows:
- 50 mg daily dose: Each carton contains 1 blister card containing a 21-day supply (42 capsules; 25 mg infigratinib per capsule). [NDC-72730-506-01].
- 75 mg daily dose: Each carton contains 2 blister cards containing a 21-day supply (63 capsules; 25 mg infigratinib per capsule). [NDC-72730-202-01].
- 100 mg daily dose: Each carton contains 1 blister card containing a 21-day supply (21 capsules; 100 mg infigratinib per capsule). [NDC-72730-111-01].
- 125 mg daily dose: Each carton contains 1 blister card containing a 21-day supply (21 capsules, 100 mg infigratinib per capsule and 21 capsules; 25 mg infigratinib per capsule). [NDC-72730-101-01].
Store TRUSELTIQ at 20°C to 25°C (68°F to 77°F), with excursions permitted between 15°C and 30°C (59°F and 86°F).
17. PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Ocular Toxicity
Advise patients that TRUSELTIQ may cause ocular toxicity including RPED and to immediately inform their healthcare provider if they experience any visual changes. Advise patients to use artificial tear substitutes or hydrating or lubricating eye gels to prevent or treat dry eyes. Inform patients that their healthcare provider will closely monitor for eye disorders, with ophthalmic examinations performed by an ophthalmologist, and will manage them as clinically indicated [ see Warnings and Precautions ( 5.1) ].
Hyperphosphatemia and Soft Tissue Mineralization
Inform patients that TRUSELTIQ may cause hyperphosphatemia and soft tissue mineralization and to immediately inform their healthcare provider of any symptoms related to acute changes in phosphate levels such as muscle cramps, numbness, or tingling around the mouth [ see Warnings and Precautions ( 5.2) ].
Nail Disorders
Advise patients that TRUSELTIQ may cause nail disorders [ see Adverse Reactions ( 6.1) ].
Embryo-Fetal Toxicity
- Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform females of the risk to a fetus and potential loss of pregnancy [ see Warnings and Precautions ( 5.3) and Use in Specific Populations ( 8.1) ].
- Advise females of reproductive potential to use effective contraception while on TRUSELTIQ and for 1 month after the final dose [ see Use in Specific Populations ( 8.1) and ( 8.3) ].
- Advise males with female partners of reproductive potential or who are pregnant to use effective contraception during treatment and for 1 month after the final dose of TRUSELTIQ [ see Use in Specific Populations ( 8.3) ].
Lactation
Advise patients not to breastfeed during treatment with TRUSELTIQ and for 1 month after the final dose [ see Use in Specific Populations ( 8.2) ].
Administration
- Instruct patients to take TRUSELTIQ on an empty stomach at least 1 hour before, or 2 hours after food, at approximately the same time each day.
- Instruct patients to swallow the capsules whole with a glass of water. Advise patients to not crush, chew, or dissolve capsules.
- Instruct patients that if a dose cannot be administered within 4 hours of the regularly scheduled time or if vomiting occurs, the dose should not be made up later that day. Dosing should resume at the usual time on the next day [ see Dose and Administration ( 2.2) ].
Drug Interactions
Advise patients to inform their healthcare providers of all concomitant medications, including prescription medicines, over-the-counter drugs, and herbal products. Advise patients to avoid grapefruit products during treatment with TRUSELTIQ [ see Drug Interactions ( 7.1) ].




| TRUSELTIQ
infigratinib capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| TRUSELTIQ
infigratinib capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| TRUSELTIQ
infigratinib capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| TRUSELTIQ
infigratinib capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - QED Therapeutics, Inc. (081223065) |
| Registrant - Helsinn Healthcare SA (481466936) |
Trademark Results [TRUSELTIQ]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TRUSELTIQ 90975538 not registered Live/Pending |
QED Therapeutics, Inc. 2020-10-19 |
 TRUSELTIQ 90262906 not registered Live/Pending |
QED Therapeutics, Inc. 2020-10-19 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.