VENCLEXTA- venetoclax kit VENCLEXTA- venetoclax tablet, film coated
Venclexta by
Drug Labeling and Warnings
Venclexta by is a Prescription medication manufactured, distributed, or labeled by AbbVie Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VENCLEXTA safely and effectively. See full prescribing information for VENCLEXTA.
VENCLEXTA® (venetoclax tablets) for oral use
Initial U.S. Approval: 2016RECENT MAJOR CHANGES
Indications and Usage, CLL (1.1) 05/2019 Indications and Usage, AML (1.2) 11/2018 Dosage and Administration (2.1, 2.2, 2.3, 2.4, 2.5) 07/2019 Warnings and Precautions, Neutropenia (5.2) 11/2018 Warnings and Precautions, Infections (5.3) 05/2019 Warnings and Precautions, Increased Mortality (5.6) 07/2019 INDICATIONS AND USAGE
VENCLEXTA is a BCL-2 inhibitor indicated:
- For the treatment of adult patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). (1.1)
- In combination with azacitidine or decitabine or low-dose cytarabine for the treatment of newly-diagnosed acute myeloid leukemia (AML) in adults who are age 75 years or older, or who have comorbidities that preclude use of intensive induction chemotherapy.
This indication is approved under accelerated approval based on response rates. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. (1.2)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 10 mg, 50 mg, 100 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Tumor Lysis Syndrome (TLS): Anticipate TLS; assess risk in all patients. Premedicate with anti-hyperuricemics and ensure adequate hydration. Employ more intensive measures (intravenous hydration, frequent monitoring, hospitalization) as overall risk increases. (2.2, 5.1)
- Neutropenia: Monitor blood counts and for signs of infection; manage as medically appropriate. (2.3, 5.2)
- Infections: Monitor for signs and symptoms of infection and treat promptly. Withhold treatment for Grade 3 and higher infection until resolution. (5.3)
- Immunization: Do not administer live attenuated vaccines prior to, during, or after VENCLEXTA treatment. (5.4)
- Embryo-Fetal Toxicity: May cause embryo-fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment. (5.5)
- Treatment of patients with multiple myeloma with VENCLEXTA in combination with bortezomib plus dexamethasone is not recommended outside of controlled clinical trials. (5.6)
ADVERSE REACTIONS
In CLL/SLL, the most common adverse reactions (≥20%) for VENCLEXTA when given in combination with obinutuzumab or rituximab or as monotherapy were neutropenia, thrombocytopenia, anemia, diarrhea, nausea, upper respiratory tract infection, cough, musculoskeletal pain, fatigue, and edema. (6.1)
In AML, the most common adverse reactions (≥30%) in combination with azacitidine or decitabine or low-dose cytarabine were nausea, diarrhea, thrombocytopenia, constipation, neutropenia, febrile neutropenia, fatigue, vomiting, peripheral edema, pyrexia, pneumonia, dyspnea, hemorrhage, anemia, rash, abdominal pain, sepsis, back pain, myalgia, dizziness, cough, oropharyngeal pain, and hypotension. (6.2)
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 7/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
1.2 Acute Myeloid Leukemia
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Risk Assessment and Prophylaxis for Tumor Lysis Syndrome
2.3 Dosage Modifications Based on Toxicities
2.4 Dosage Modifications for Concomitant Use with Strong or Moderate CYP3A Inhibitors or P-gp Inhibitors
2.5 Dosage Modifications for Patients with Severe Hepatic Impairment
2.6 Missed Dose
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Tumor Lysis Syndrome
5.2 Neutropenia
5.3 Infections
5.4 Immunization
5.5 Embryo-Fetal Toxicity
5.6 Increased Mortality in Patients with Multiple Myeloma when VENCLEXTA is Added to Bortezomib and Dexamethasone
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience with Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
6.2 Clinical Trial Experience with Acute Myeloid Leukemia
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on VENCLEXTA
7.2 Effect of VENCLEXTA on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
14.2 Acute Myeloid Leukemia
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
VENCLEXTA is indicated for the treatment of adult patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL).
1.2 Acute Myeloid Leukemia
VENCLEXTA is indicated in combination with azacitidine, or decitabine, or low-dose cytarabine for the treatment of newly-diagnosed acute myeloid leukemia (AML) in adults who are age 75 years or older, or who have comorbidities that preclude use of intensive induction chemotherapy.
This indication is approved under accelerated approval based on response rates [see Clinical Studies (14.2)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Assess patient-specific factors for level of risk of tumor lysis syndrome (TLS) and provide prophylactic hydration and anti-hyperuricemics to patients prior to first dose of VENCLEXTA to reduce risk of TLS [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
Instruct patients to take VENCLEXTA tablets with a meal and water at approximately the same time each day. VENCLEXTA tablets should be swallowed whole and not chewed, crushed, or broken prior to swallowing.
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
VENCLEXTA dosing begins with a 5-week ramp-up.
VENCLEXTA 5-week Dose Ramp-Up Schedule
Administer the VENCLEXTA dose according to a weekly ramp-up schedule over 5 weeks to the recommended daily dose of 400 mg as shown in Table 1. The 5-week ramp-up dosing schedule is designed to gradually reduce tumor burden (debulk) and decrease the risk of TLS.
Table 1. Dosing Schedule for Ramp-Up Phase in Patients with CLL/SLL VENCLEXTA
Daily DoseWeek 1 20 mg Week 2 50 mg Week 3 100 mg Week 4 200 mg Week 5 and beyond 400 mg The CLL/SLL Starting Pack provides the first 4 weeks of VENCLEXTA according to the ramp-up schedule. The 400 mg dose is achieved using 100 mg tablets supplied in bottles [see How Supplied/Storage and Handling (16)].
VENCLEXTA in Combination with Obinutuzumab
Start obinutuzumab administration at 100 mg on Cycle 1 Day 1, followed by 900 mg on Cycle 1 Day 2. Administer 1000 mg on Days 8 and 15 of Cycle 1 and on Day 1 of each subsequent 28-day cycle, for a total of 6 cycles. Refer to the obinutuzumab prescribing information for recommended obinutuzumab dosing information.
On Cycle 1 Day 22, start VENCLEXTA according to the 5-week ramp-up schedule (see Table 1). After completing the ramp-up schedule on Cycle 2 Day 28, patients should continue VENCLEXTA 400 mg once daily from Cycle 3 Day 1 until the last day of Cycle 12.
VENCLEXTA in Combination with Rituximab
Start rituximab administration after the patient has completed the 5-week dose ramp-up schedule with VENCLEXTA (see Table 1) and has received the 400 mg dose of VENCLEXTA for 7 days. Administer rituximab on Day 1 of each 28-day cycle for 6 cycles, with rituximab dosed at 375 mg/m2 intravenously for Cycle 1 and 500 mg/m2 intravenously for Cycles 2-6.
Patients should continue VENCLEXTA 400 mg once daily for 24 months from Cycle 1 Day 1 of rituximab.
The recommended dose of VENCLEXTA is 400 mg once daily after the patient has completed the 5-week dose ramp-up schedule. VENCLEXTA should be taken orally once daily until disease progression or unacceptable toxicity is observed.
The dose of VENCLEXTA depends upon the combination agent.
The VENCLEXTA dosing schedule (including ramp-up) is shown in Table 2. Initiate the azacitidine or decitabine or low-dose cytarabine on Day 1.
Table 2. Dosing Schedule for Ramp-up Phase in Patients with AML VENCLEXTA
Daily DoseDay 1 100 mg Day 2 200 mg Day 3 400 mg Days 4 and beyond 400 mg
when dosing in combination with
azacitidine or decitabine600 mg
when dosing in combination with
low-dose cytarabineContinue VENCLEXTA, in combination with azacitidine or decitabine or low-dose cytarabine, until disease progression or unacceptable toxicity is observed.
2.2 Risk Assessment and Prophylaxis for Tumor Lysis Syndrome
Patients treated with VENCLEXTA may develop tumor lysis syndrome. Refer to the appropriate section below for specific details on management. Assess patient-specific factors for level of risk of tumor lysis syndrome (TLS) and provide prophylactic hydration and anti-hyperuricemics to patients prior to first dose of VENCLEXTA to reduce risk of TLS.
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
VENCLEXTA can cause rapid reduction in tumor and thus poses a risk for TLS in the initial 5-week ramp-up phase. Changes in blood chemistries consistent with TLS that require prompt management can occur as early as 6 to 8 hours following the first dose of VENCLEXTA and at each dose increase.
The risk of TLS is a continuum based on multiple factors, including tumor burden and comorbidities. Reduced renal function (creatinine clearance [CLcr] <80 mL/min) further increases the risk. Perform tumor burden assessments, including radiographic evaluation (e.g., CT scan), assess blood chemistry (potassium, uric acid, phosphorus, calcium, and creatinine) in all patients and correct pre-existing abnormalities prior to initiation of treatment with VENCLEXTA. The risk may decrease as tumor burden decreases [see Warnings and Precautions (5.1) and Use in Specific Populations (8.6)].
Table 3 below describes the recommended TLS prophylaxis and monitoring during VENCLEXTA treatment based on tumor burden determination from clinical trial data. Consider all patient comorbidities before final determination of prophylaxis and monitoring schedule.
Table 3. Recommended TLS Prophylaxis Based on Tumor Burden in Patients with CLL/SLL Tumor Burden Prophylaxis Blood Chemistry
Monitoringc,dHydrationa Anti-hyperuricemics Setting and
Frequency of
AssessmentsLow All LN <5 cm AND
ALC <25 x109/LOral
(1.5-2 L)Allopurinolb Outpatient - For first dose of 20 mg and 50 mg: Pre-dose, 6 to 8 hours, 24 hours
- For subsequent ramp-up doses: Pre-dose
Medium Any LN 5 cm to <10 cm
OR
ALC ≥25 x109/LOral
(1.5-2 L)
and consider additional intravenousAllopurinol Outpatient - For first dose of 20 mg and 50 mg: Pre-dose, 6 to 8 hours, 24 hours
- For subsequent ramp-up doses: Pre-dose
- For first dose of 20 mg and 50 mg: Consider hospitalization for patients with CLcr <80ml/min; see below for monitoring in hospital
High Any LN ≥10 cm OR
ALC ≥25 x109/L AND
any LN ≥5 cmOral (1.5-2L)
and intravenous
(150-200 mL/hr
as tolerated)Allopurinol; consider rasburicase if baseline uric acid is elevated In hospital - For first dose of 20 mg and 50 mg: Pre-dose, 4, 8, 12 and 24 hours
- For subsequent ramp-up doses: Pre-dose, 6 to 8 hours, 24 hours
ALC = absolute lymphocyte count; CLcr = creatinine clearance; LN = lymph node.
aAdminister intravenous hydration for any patient who cannot tolerate oral hydration.
bStart allopurinol or xanthine oxidase inhibitor 2 to 3 days prior to initiation of VENCLEXTA.
cEvaluate blood chemistries (potassium, uric acid, phosphorus, calcium, and creatinine); review in real time.
dFor patients at risk of TLS, monitor blood chemistries at 6 to 8 hours and at 24 hours at each subsequent ramp-up dose.- All patients should have white blood cell count less than 25 × 109/L prior to initiation of VENCLEXTA. Cytoreduction prior to treatment may be required.
- Prior to first VENCLEXTA dose, provide all patients with prophylactic measures including adequate hydration and anti-hyperuricemic agents and continue during ramp-up phase.
- Assess blood chemistry (potassium, uric acid, phosphorus, calcium, and creatinine) and correct pre-existing abnormalities prior to initiation of treatment with VENCLEXTA.
- Monitor blood chemistries for TLS at pre-dose, 6 to 8 hours after each new dose during ramp-up and 24 hours after reaching final dose.
- For patients with risk factors for TLS (e.g., circulating blasts, high burden of leukemia involvement in bone marrow, elevated pretreatment lactate dehydrogenase (LDH) levels, or reduced renal function) additional measures should be considered, including increased laboratory monitoring and reducing VENCLEXTA starting dose.
2.3 Dosage Modifications Based on Toxicities
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
Interrupt dosing or reduce dose for toxicities. See Table 4 and Table 5 for recommended dose modifications for toxicities related to VENCLEXTA. For patients who have had a dosing interruption greater than 1 week during the first 5 weeks of ramp-up phase or greater than 2 weeks after completing the ramp-up phase, reassess for risk of TLS to determine if reinitiation with a reduced dose is necessary (e.g., all or some levels of the dose ramp-up schedule) [see Dosage and Administration (2.1, 2.2)].
Table 4. Recommended VENCLEXTA Dose Modifications for Toxicitiesa in CLL/SLL Event Occurrence Action Tumor Lysis Syndrome Blood chemistry changes or symptoms suggestive of TLS Any Withhold the next day’s dose. If resolved within 24 to 48 hours of last dose, resume at the same dose. For any blood chemistry changes requiring more than 48 hours to resolve, resume at a reduced dose (see Table 5) [see Dosage and Administration (2.2)]. For any events of clinical TLS,b resume at a reduced dose following resolution (see Table 5) [see Dosage and Administration (2.2)]. Non-Hematologic Toxicities Grade 3 or 4 non-hematologic toxicities 1st occurrence Interrupt VENCLEXTA.
Once the toxicity has resolved to Grade 1 or baseline level, VENCLEXTA therapy may be resumed at the same dose. No dose modification is required.2nd and subsequent occurrences Interrupt VENCLEXTA.
Follow dose reduction guidelines in Table 5 when resuming treatment with VENCLEXTA after resolution. A larger dose reduction may occur at the discretion of the physician.Hematologic Toxicities Grade 3 neutropenia with infection or fever; or Grade 4 hematologic toxicities (except lymphopenia) [see Warnings and Precautions (5.2)] 1st occurrence Interrupt VENCLEXTA.
To reduce the infection risks associated with neutropenia, granulocyte-colony stimulating factor (G-CSF) may be administered with VENCLEXTA if clinically indicated. Once the toxicity has resolved to Grade 1 or baseline level, VENCLEXTA therapy may be resumed at the same dose.2nd and subsequent occurrences Interrupt VENCLEXTA.
Consider using G-CSF as clinically indicated.
Follow dose reduction guidelines in Table 5 when resuming treatment with VENCLEXTA after resolution. A larger dose reduction may occur at the discretion of the physician.Consider discontinuing VENCLEXTA for patients who require dose reductions to less than 100 mg for more than 2 weeks.
aAdverse reactions were graded using NCI CTCAE version 4.0.
bClinical TLS was defined as laboratory TLS with clinical consequences such as acute renal failure, cardiac arrhythmias, or sudden death and/or seizures [see Adverse Reactions (6.1)].Table 5. Dose Reduction for Toxicity During VENCLEXTA Treatment in CLL/SLL Dose at Interruption, mg Restart Dose, mga 400 300 300 200 200 100 100 50 50 20 20 10 aDuring the ramp-up phase, continue the reduced dose for 1 week before increasing the dose. Monitor blood counts frequently through resolution of cytopenias. Management of some adverse reactions [see Warnings and Precautions (5.2) and Adverse Reactions (6.2)] may require dose interruptions or permanent discontinuation of VENCLEXTA. Table 6 shows the dose modification guidelines for hematologic toxicities.
Table 6. Recommended Dose Modifications for Toxicitiesa in AML Event Occurrence Action Hematologic Toxicities Grade 4 neutropenia with or without fever or infection; or Grade 4 thrombocytopenia [see Warnings and Precautions (5.2)] Occurrence prior to achieving remission Transfuse blood products, administer prophylactic and treatment anti-infectives as clinically indicated.
In most instances, VENCLEXTA and azacitidine, decitabine, or low-dose cytarabine cycles should not be interrupted due to cytopenias prior to achieving remission.First occurrence after achieving remission and lasting at least 7 days Delay subsequent treatment cycle of VENCLEXTA and azacitidine, decitabine, or low-dose cytarabine and monitor blood counts.
Administer granulocyte-colony stimulating factor (G-CSF) if clinically indicated for neutropenia. Once the toxicity has resolved to Grade 1 or 2, resume VENCLEXTA therapy at the same dose in combination with azacitidine or decitabine or low-dose cytarabine.Subsequent occurrences in cycles after achieving remission and lasting 7 days or longer Delay subsequent treatment cycle of VENCLEXTA and azacitidine, or decitabine, or low-dose cytarabine and monitor blood counts.
Administer G-CSF if clinically indicated for neutropenia. Once the toxicity has resolved to Grade 1 or 2, resume VENCLEXTA therapy at the same dose and the duration reduced by 7 days for each subsequent cycle.aAdverse reactions were graded using NCI CTCAE version 4.0. 2.4 Dosage Modifications for Concomitant Use with Strong or Moderate CYP3A Inhibitors or P-gp Inhibitors
Table 7 describes VENCLEXTA contraindication or dosage modification based on concomitant use with a strong or moderate CYP3A inhibitor or P-gp inhibitor [see Drug Interactions (7.1)] at initiation, during, or after the ramp-up phase.
Resume the VENCLEXTA dosage that was used prior to concomitant use of a strong or moderate CYP3A inhibitor or P-gp inhibitor 2 to 3 days after discontinuation of the inhibitor [see Dosage and Administration (2.3) and Drug Interactions (7.1)].
Table 7. Management of Potential VENCLEXTA Interactions with CYP3A and P-gp Inhibitors Coadministered
drugInitiation and
Ramp-Up PhaseSteady Daily Dose
(After Ramp-Up Phase)aPosaconazole CLL/SLL Contraindicated Reduce VENCLEXTA dose to 70 mg. AML Day 1 – 10 mg
Day 2 – 20 mg
Day 3 – 50 mg
Day 4 – 70 mgOther strong CYP3A
inhibitorCLL/SLL Contraindicated Reduce VENCLEXTA dose to 100 mg. AML Day 1 – 10 mg
Day 2 – 20 mg
Day 3 – 50 mg
Day 4 – 100 mgModerate CYP3A
inhibitorReduce the VENCLEXTA dose by at least 50%. P-gp inhibitor aIn patients with CLL/SLL, consider alternative medications or reduce the VENCLEXTA dose as described in Table 7. 2.5 Dosage Modifications for Patients with Severe Hepatic Impairment
Reduce the VENCLEXTA once daily dose by 50% for patients with severe hepatic impairment (Child-Pugh C); monitor these patients more closely for signs of toxicity [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
2.6 Missed Dose
If the patient misses a dose of VENCLEXTA within 8 hours of the time it is usually taken, the patient should take the missed dose as soon as possible and resume the normal daily dosing schedule. If a patient misses a dose by more than 8 hours, the patient should not take the missed dose and should resume the usual dosing schedule the next day.
If the patient vomits following dosing, no additional dose should be taken that day. The next prescribed dose should be taken at the usual time.
-
3 DOSAGE FORMS AND STRENGTHS
Table 8. VENCLEXTA Tablet Strength and Description Tablet Strength Description of Tablet 10 mg Round, biconvex shaped, pale yellow film-coated tablet debossed with “V” on one side and “10” on the other side 50 mg Oblong, biconvex shaped, beige film-coated tablet debossed with “V” on one side and “50” on the other side 100 mg Oblong, biconvex shaped, pale yellow film-coated tablet debossed with “V” on one side and “100” on the other side - 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Tumor Lysis Syndrome
Tumor lysis syndrome (TLS), including fatal events and renal failure requiring dialysis, has occurred in patients with high tumor burden when treated with VENCLEXTA [see Adverse Reactions (6.1, 6.2)].
In patients with CLL who followed the current (5 week) dose ramp-up and the TLS prophylaxis and monitoring measures, the rate of TLS was 2% in the VENCLEXTA CLL monotherapy studies. The rate of TLS remained consistent with VENCLEXTA in combination with obinutuzumab or rituximab. With a 2 to 3 week dose ramp-up and higher starting dose in patients with CLL/SLL, the TLS rate was 13% and included deaths and renal failure [see Adverse Reactions (6.1)].
VENCLEXTA can cause rapid reduction in tumor and thus poses a risk for TLS at initiation and during the ramp-up phase. Changes in blood chemistries consistent with TLS that require prompt management can occur as early as 6 to 8 hours following the first dose of VENCLEXTA and at each dose increase.
The risk of TLS is a continuum based on multiple factors, including tumor burden and comorbidities. Reduced renal function further increases the risk. Patients should be assessed for risk and should receive appropriate prophylaxis for TLS, including hydration and anti-hyperuricemics. Monitor blood chemistries and manage abnormalities promptly. Interrupt dosing if needed. Employ more intensive measures (intravenous hydration, frequent monitoring, hospitalization) as overall risk increases [see Dosage and Administration (2.2, 2.3) and Use in Specific Populations (8.6)].
Concomitant use of VENCLEXTA with P-gp inhibitors or strong or moderate CYP3A inhibitors increases venetoclax exposure, may increase the risk of TLS at initiation and during ramp-up phase and requires VENCLEXTA dose adjustment [see Dosage and Administration (2.4) and Drug Interactions (7.1)].
5.2 Neutropenia
In patients with CLL, Grade 3 or 4 neutropenia developed in 63% to 64% of patients and Grade 4 neutropenia developed in 31% to 33% of patients treated with VENCLEXTA in combination and monotherapy studies (see Tables 10, 12, 14). Febrile neutropenia occurred in 4% to 6% of patients treated with VENCLEXTA in combination and monotherapy studies[see Adverse Reactions (6.1)].
In patients with AML, baseline neutrophil counts worsened in 97% to 100% of patients treated with VENCLEXTA in combination with azacitidine or decitabine or low-dose cytarabine. Neutropenia can recur with subsequent cycles of therapy.
Monitor complete blood counts throughout the treatment period. Interrupt dosing or reduce dose for severe neutropenia. Consider supportive measures including antimicrobials for signs of infection and use of growth factors (e.g., G-CSF) [see Dosage and Administration (2.3)].
5.3 Infections
Fatal and serious infections such as pneumonia and sepsis have occurred in patients treated with VENCLEXTA [see Adverse Reactions (6.1)]. Monitor patients closely for signs and symptoms of infection and treat promptly. Withhold VENCLEXTA for Grade 3 and higher infection [see Dosage and Administration (2.3)].
5.4 Immunization
Do not administer live attenuated vaccines prior to, during, or after treatment with VENCLEXTA until B-cell recovery occurs. The safety and efficacy of immunization with live attenuated vaccines during or following VENCLEXTA therapy have not been studied. Advise patients that vaccinations may be less effective.
5.5 Embryo-Fetal Toxicity
Based on its mechanism of action and findings in animals, VENCLEXTA may cause embryo-fetal harm when administered to a pregnant woman. In an embryo-fetal study conducted in mice, administration of venetoclax to pregnant animals at exposures equivalent to that observed in patients at a dose of 400 mg daily resulted in post-implantation loss and decreased fetal weight. There are no adequate and well-controlled studies in pregnant women using VENCLEXTA. Advise females of reproductive potential to avoid pregnancy during treatment. If VENCLEXTA is used during pregnancy or if the patient becomes pregnant while taking VENCLEXTA, the patient should be apprised of the potential hazard to the fetus [see Use in Specific Populations (8.1)].
5.6 Increased Mortality in Patients with Multiple Myeloma when VENCLEXTA is Added to Bortezomib and Dexamethasone
In a randomized trial (BELLINI; NCT02755597) in patients with relapsed or refractory multiple myeloma, the addition of VENCLEXTA to bortezomib plus dexamethasone, a use for which VENCLEXTA is not indicated, resulted in increased mortality. Treatment of patients with multiple myeloma with VENCLEXTA in combination with bortezomib plus dexamethasone is not recommended outside of controlled clinical trials.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Tumor Lysis Syndrome [see Warnings and Precautions (5.1)]
- Neutropenia [see Warnings and Precautions (5.2)]
- Infections [see Warnings and Precautions (5.3)]
Because clinical trials are conducted under widely variable conditions, adverse event rates observed in clinical trials of a drug cannot be directly compared with rates of clinical trials of another drug and may not reflect the rates observed in practice.
6.1 Clinical Trial Experience with Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
The safety of VENCLEXTA in combination with obinutuzumab (VEN+G) versus obinutuzumab in combination with chlorambucil (GClb) was evaluated in a randomized, open-label, actively controlled trial in patients with previously untreated CLL.
Patients randomized to the VEN+G arm were treated with VENCLEXTA and obinutuzumab in combination for six cycles, then with VENCLEXTA as monotherapy for an additional six cycles. Patients initiated the first dose of the 5 week ramp-up for VENCLEXTA on Day 22 of Cycle 1 and once completed, continued VENCLEXTA 400 mg once daily for a total of 12 cycles. Details of the study treatment are described in Section 14 [see Clinical Studies (14.1)]. The trial required a total Cumulative Illness Rating Scale (CIRS) >6 or CLcr <70 mL/min, hepatic transaminases and total bilirubin ≤2 times upper limit of normal, and excluded patients with any individual organ/system impairment score of 4 by CIRS except eye, ear, nose, and throat organ system.
A total of 426 patients were treated (212 with VEN+G, 214 with GClb). The median duration of exposure to VENCLEXTA was 10.5 months (range: 0 to 13.5 months). The median number of cycles was 6 for obinutuzumab and 12 for chlorambucil.
In the VEN+G arm, fatal adverse reactions that occurred in the absence of disease progression and with onset within 28 days of the last study treatment were reported in 2% (4/212) of patients, most often from infection. Serious adverse reactions were reported in 49% of patients in the VEN+G arm, most often due to febrile neutropenia and pneumonia (5% each).
In the VEN+G arm, adverse reactions led to treatment discontinuation in 16% of patients, dose reduction in 21%, and dose interruption in 74%. In the VEN+G arm, neutropenia led to dose interruption of VENCLEXTA in 41% of patients, reduction in 13%, and discontinuation in 2%.
Table 9 and Table 10 present adverse reactions and laboratory abnormalities identified in the CLL14 trial, respectively. The most common (≥15%) adverse reactions observed with VEN+G were neutropenia, diarrhea, fatigue, nausea, anemia, and upper respiratory tract infection.
Table 9. Common (≥10%) Adverse Reactions in Patients Treated with VEN+G Adverse Reaction by
Body SystemVENCLEXTA + Obinutuzumab
(N = 212)Obinutuzumab + Chlorambucil
(N = 214)All Grades
%Grade ≥3
%All Grades
%Grade ≥3
%Blood & lymphatic system disorders Neutropeniaa 60 56 62 52 Anemiaa 17 8 20 7 Gastrointestinal disorders Diarrhea 28 4 15 1 Nausea 19 0 22 1 Constipation 13 0 9 0 Vomiting 10 1 8 1 General disorders and administration site conditions Fatiguea 21 2 23 1 Infections and Infestations Upper respiratory
tract infectiona17 1 17 1 aIncludes multiple adverse reaction terms. Other clinically important adverse reactions (all Grades) reported in <10% of patients treated with VEN+G are presented below:
Blood & lymphatic system disorders: febrile neutropenia (6%)
Infection and infestations (all include multiple adverse reaction terms): pneumonia (9%), urinary tract infection (6%), sepsis (4%)
Metabolism and nutrition disorder: tumor lysis syndrome (1%)
During treatment with single agent VENCLEXTA after completion of VEN+G combination treatment, the most common all grade adverse reaction (≥10% patients) reported was neutropenia (26%). The most common grade ≥3 adverse reactions (≥2% patients) were neutropenia (23%), and anemia (2%).
Table 10. New or Worsening Clinically Important Laboratory Abnormalities Occurring at ≥10% in Patients Treated with VEN+G VENCLEXTA +
Obinutuzumab
(N = 212)Obinutuzumab +
Chlorambucil
(N = 214)Laboratory
AbnormalityaAll Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)Hematology Leukopenia 90 46 89 41 Lymphopenia 87 57 87 51 Neutropenia 83 63 79 56 Thrombocytopenia 68 28 71 26 Anemia 53 15 46 11 Chemistry Blood creatinine increased 80 6 74 2 Hypocalcemia 67 9 58 4 Hyperkalemia 41 4 35 3 Hyperuricemia 38 38 38 38 aIncludes laboratory abnormalities that were new or worsening, or with worsening from baseline unknown. Grade 4 laboratory abnormalities developing in ≥2% of patients treated with VEN+G include neutropenia (32%), leukopenia and lymphopenia (10%), thrombocytopenia (8%), hypocalcemia (8%), hyperuricemia (7%), blood creatinine increased (3%), hypercalcemia (3%), and hypokalemia (2%).
The safety of VENCLEXTA in combination with rituximab (VEN+R) versus bendamustine in combination with rituximab (B+R), was evaluated in an open-label randomized study, in patients with CLL who had received at least one prior therapy.
Patients randomized to VEN+R completed the scheduled ramp-up (5 weeks) and received VENCLEXTA 400 mg once daily in combination with rituximab for 6 cycles followed by single agent VENCLEXTA for a total of 24 months after ramp-up. Patients randomized to B+R received 6 cycles (28 days per cycle) for a total of 6 months. Details of the study treatment are described in Section 14 [see Clinical Studies (14.1)].
At the time of analysis, the median duration of exposure was 22 months in the VEN+R arm compared with 6 months in the B+R arm.
In the VEN+R arm, fatal adverse reactions that occurred in the absence of disease progression and within 30 days of the last VENCLEXTA treatment and/or 90 days of last rituximab were reported in 2% (4/194) of patients. Serious adverse reactions were reported in 46% of patients in the VEN+R arm, with most frequent (≥5%) being pneumonia (9%).
In the VEN+R arm, adverse reactions led to treatment discontinuation in 16% of patients, dose reduction in 15%, and dose interruption in 71%. In the B+R arm, adverse reactions led to treatment discontinuation in 10% of patients, dose reduction in 15%, and dose interruption in 40%. In the VEN+R arm, neutropenia led to dose interruption of VENCLEXTA in 46% of patients and discontinuation in 3%, and thrombocytopenia led to discontinuation in 3% of patients.
Table 11 and Table 12 present adverse reactions and laboratory abnormalities, respectively, identified in the MURANO trial. The MURANO trial was not designed to demonstrate a statistically significant difference in adverse reaction rates for VEN+R as compared with B+R, for any specific adverse reaction or laboratory abnormality.
Table 11. Common (≥10%) Adverse Reactions Reported with ≥5% Higher All-Grade or ≥2% Higher Grade ≥3 Incidence in Patients Treated with VEN+R Compared with B+R Adverse Reaction by
Body SystemVENCLEXTA + Rituximab
Followed by Single Agent
VENCLEXTA
(N = 194)Bendamustine + Rituximab
(N = 188)All Grades
(%)Grade ≥3
(%)All Grades
(%)Grade ≥3
(%)Blood & lymphatic system disorders Neutropeniaa 65 62 50 44 Gastrointestinal disorders Diarrhea 40 3 17 1 Infections & infestations Upper respiratory
tract infectiona39 2 23 2 Lower respiratory
tract infectiona18 2 10 2 Musculoskeletal and connective tissue disorders Musculoskeletal paina 19 1 13 0 Metabolism and nutrition disorders Tumor lysis
syndrome3 3 1 1 aIncludes multiple adverse reaction terms. Other adverse reactions (all grades) reported in ≥10% of patients in the VEN+R arm in MURANO, and other important adverse reactions are presented below:
Blood & lymphatic system disorders: anemia (16%), thrombocytopenia (15%), febrile neutropenia (4%)
Gastrointestinal disorders: nausea (21%), constipation (14%), abdominal pain (13%), mucositis (10%), vomiting (8%)
Respiratory disorders: cough (22%)
General disorders and administration site conditions: fatigue (22%), pyrexia (15%)
Nervous system and psychiatric disorders: headache (11%), insomnia (11%)
Infections & infestations: pneumonia (10%), sepsis (1%)
During treatment with single agent VENCLEXTA after completion of VEN+R combination treatment, the most common all grade adverse reactions (≥10% patients) reported were upper respiratory tract infection (21%), diarrhea (19%), neutropenia (16%), and lower respiratory tract infections (11%). The most common Grade 3 or 4 adverse reactions (≥2% patients) were neutropenia (12%) and anemia (3%).
Table 12 describes common treatment-emergent laboratory abnormalities identified in the MURANO trial.
Table 12. Common (≥10%) New or Worsening Laboratory Abnormalities Occurring at ≥5% (Any Grade) or ≥2% (Grade 3 or 4) Higher Incidence with VEN+R Compared with B+R VENCLEXTA + Rituximab
N=194Bendamustine + Rituximab
N=188Laboratory
AbnormalityAll Gradesa
(%)Grade 3 or 4
(%)All Gradesa
(%)Grade 3 or 4
(%)Hematology Leukopenia 89 46 81 35 Lymphopenia 87 56 79 55 Neutropenia 86 64 84 59 Chemistry Hypocalcemia 62 5 51 2 Hypophosphatemia 57 14 35 4 AST/SGOT increased 46 2 31 3 Hyperuricemia 36 36 33 33 Alkaline phosphatase
increased35 1 20 1 Hyperbilirubinemia 33 4 26 3 Hyponatremia 30 6 20 3 Hypokalemia 29 6 18 3 Hyperkalemia 24 3 19 2 Hypernatremia 24 1 13 0 Hypoglycemia 16 2 7 0 aIncludes laboratory abnormalities that were new or worsening, or with worsening from baseline unknown. New Grade 4 laboratory abnormalities reported in ≥2% of patients treated with VEN+R included neutropenia (31%), lymphopenia (16%), leukopenia (6%), thrombocytopenia (6%), hyperuricemia (4%), hypocalcemia (2%), hypoglycemia (2%), and hypermagnesemia (2%).
Monotherapy Studies (M13-982, M14-032, and M12-175)
The safety of single agent VENCLEXTA at the 400 mg recommended daily dose following a dose ramp-up schedule is based on pooled data from three single-arm trials (M13-982, M14-032, and M12-175). In the pooled dataset, consisting of 352 patients with previously treated CLL or SLL, the median age was 66 years (range: 28 to 85 years), 93% were white, and 68% were male. The median number of prior therapies was 3 (range: 0 to 15). The median duration of treatment with VENCLEXTA at the time of data analysis was 14.5 months (range: 0 to 50 months). Fifty-two percent of patients received VENCLEXTA for more than 60 weeks.
Fatal adverse reactions that occurred in the absence of disease progression and within 30 days of venetoclax treatment were reported in 2% of patients in the VENCLEXTA monotherapy studies, most commonly (2 patients) from septic shock. Serious adverse reactions were reported in 52% of patients, with the most frequent (≥5%) being pneumonia (9%), febrile neutropenia (5%), and sepsis (5%).
Adverse reactions led to treatment discontinuation in 9% of patients, dose reduction in 13%, and dose interruption in 36%. The most frequent adverse reactions leading to drug discontinuation were thrombocytopenia and autoimmune hemolytic anemia. The most frequent adverse reaction (≥5%) leading to dose reductions or interruptions was neutropenia (8%).
Adverse reactions identified in these trials of single-agent VENCLEXTA are presented in Table 13.
Table 13. Adverse Reactions Reported in ≥10% (Any Grade) or ≥5% (Grade ≥3) of Patients with Previously Treated CLL/SLL (VENCLEXTA Monotherapy) Body System Adverse Reaction Any Grade
(%)
N=352Grade ≥3
(%)
N=352Blood and lymphatic system
disordersNeutropeniaa 50 45 Anemiaa 33 18 Thrombocytopeniaa 29 20 Lymphopeniaa 11 7 Febrile neutropenia 6 6 Gastrointestinal disorders Diarrhea 43 3 Nausea 42 1 Abdominal paina 18 3 Vomiting 16 1 Constipation 16 <1 Mucositisa 13 <1 General disorders and
administration site conditionsFatiguea 32 4 Edemaa 22 2 Pyrexia 18 <1 Infections and infestations Upper respiratory tract
infectiona36 1 Pneumoniaa 14 8 Lower respiratory tract
infectiona11 2 Musculoskeletal and
connective tissue disordersMusculoskeletal paina 29 2 Arthralgia 12 <1 Nervous system disorders Headache 18 <1 Dizzinessa 14 0 Respiratory, thoracic, and
mediastinal disordersCougha 22 0 Dyspneaa 13 1 Skin and subcutaneous tissue disorders Rasha 18 <1 Adverse Reactions graded using NCI Common Terminology Criteria for Adverse Events version 4.0.
aIncludes multiple adverse reaction terms.Table 14 describes common laboratory abnormalities reported throughout treatment that were new or worsening from baseline. The most common (>5%) Grade 4 laboratory abnormalities observed with VENCLEXTA monotherapy were hematologic laboratory abnormalities, including neutropenia (33%), leukopenia (11%), thrombocytopenia (15%), and lymphopenia (9%).
Table 14. New or Worsening Laboratory Abnormalities with VENCLEXTA Monotherapy (≥40% Any Grade or ≥10% Grade 3 or 4) Laboratory Abnormality All Gradesa
(%)
N=352Grade 3 or 4
(%)
N=352Hematology Leukopenia 89 42 Neutropenia 87 63 Lymphopenia 74 40 Anemia 71 26 Thrombocytopenia 64 31 Chemistry Hypocalcemia 87 12 Hyperglycemia 67 7 Hyperkalemia 59 5 AST increased 53 3 Hypoalbuminemia 49 2 Hypophosphatemia 45 11 Hyponatremia 40 9 aIncludes laboratory abnormalities that were new or worsening, or worsening from baseline unknown. Tumor lysis syndrome is an important identified risk when initiating VENCLEXTA.
The incidence of TLS was 1% (3/212) in patients treated with VEN+G [see Warnings and Precautions (5.1)]. All three events of TLS resolved and did not lead to withdrawal from the study. Obinutuzumab administration was delayed in two cases in response to the TLS events.
In the open-label randomized phase 3 study, the incidence of TLS was 3% (6/194) in patients treated with VEN+R. After 77/389 patients were enrolled in the study, the protocol was amended to incorporate the current TLS prophylaxis and monitoring measures described in sections 2.1 and 2.2 [see Dosage and Administration (2.1, 2.2)]. All events of TLS occurred during the VENCLEXTA ramp-up period and were resolved within two days. All six patients completed the ramp-up and reached the recommended daily dose of 400 mg of VENCLEXTA. No clinical TLS was observed in patients who followed the current 5-week ramp-up schedule and TLS prophylaxis and monitoring measures described in sections 2.1 and 2.2 [see Dosage and Administration (2.1, 2.2)]. Rates of laboratory abnormalities relevant to TLS for patients treated with VEN+R are presented in Table 12.
Monotherapy Studies (M13-982 and M14-032)
In 168 patients with CLL treated according to recommendations described in sections 2.1 and 2.2, the rate of TLS was 2% [see Dosage and Administration (2.1, 2.2)]. All events either met laboratory TLS criteria (laboratory abnormalities that met ≥2 of the following within 24 hours of each other: potassium >6 mmol/L, uric acid >476 µmol/L, calcium <1.75 mmol/L, or phosphorus >1.5 mmol/L); or were reported as TLS events. The events occurred in patients who had a lymph node(s) ≥5 cm and/or ALC ≥25 x 109/L. All events resolved within 5 days. No TLS with clinical consequences such as acute renal failure, cardiac arrhythmias or sudden death and/or seizures was observed in these patients. All patients had CLcr ≥50 mL/min. Laboratory abnormalities relevant to TLS were hyperkalemia (17% all Grades, 1% Grade ≥3), hyperphosphatemia (14% all Grades, 2% Grade ≥3), hypocalcemia (16% all Grades, 2% Grade ≥3), and hyperuricemia (10% all Grades, <1% Grade ≥3).
In the initial Phase 1 dose-finding trials, which had shorter (2-3 week) ramp-up phase and higher starting doses, the incidence of TLS was 13% (10/77; 5 laboratory TLS, 5 clinical TLS), including 2 fatal events and 3 events of acute renal failure, 1 requiring dialysis. After this experience, TLS risk assessment, dosing regimen, TLS prophylaxis and monitoring measures were revised [see Dosage and Administration (2.1, 2.2)].
6.2 Clinical Trial Experience with Acute Myeloid Leukemia
The safety of VENCLEXTA (400 mg daily dose) in combination with azacitidine (n=67) or decitabine (n= 13) and VENCLEXTA (600 mg daily dose) in combination with low-dose cytarabine (n= 61) is based on two non-randomized trials of patients with newly-diagnosed AML [see Clinical Studies (14.2)]. The median duration of exposure for patients taking VENCLEXTA in combination with azacitidine and decitabine was 6.5 months (range: 0.1 to 31.9 months) and 8.4 months (range: 0.5 to 22.3 months), respectively. The median duration of exposure for patients taking VENCLEXTA in combination with low dose cytarabine was 3.9 months (range: 0.2 to 29.2 months).
VENCLEXTA in Combination with Azacitidine or Decitabine
The most common adverse reactions (≥30%) of any grade were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia.
Serious adverse reactions were reported in 75% of patients. The most frequent serious adverse reactions (≥5%) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome.
The incidence of fatal adverse drug reactions was 1.5% within 30 days of starting treatment. No reaction had an incidence of ≥2%.
Discontinuations due to adverse reactions occurred in 21% of patients. The most frequent adverse reactions leading to drug discontinuation (≥2%) were febrile neutropenia and pneumonia (excluding fungal).
Dosage interruptions due to adverse reactions occurred in 61% of patients. The most frequent adverse reactions leading to dose interruption (≥5%) were neutropenia, febrile neutropenia, and pneumonia (excluding fungal).
Dosage reductions due to adverse reactions occurred in 12% of patients. The most frequent adverse reaction leading to dose reduction (≥5%) was neutropenia.
The most common adverse reactions (≥30%) of any grade were febrile neutropenia, constipation, fatigue, thrombocytopenia, abdominal pain, dizziness, hemorrhage, nausea, pneumonia (excluding fungal), sepsis (excluding fungal), cough, diarrhea, neutropenia, back pain, hypotension, myalgia, oropharyngeal pain, peripheral edema, pyrexia, and rash.
Serious adverse reactions were reported in 85% of patients. The most frequent serious adverse reactions (≥5%) were febrile neutropenia, sepsis (excluding fungal), pneumonia (excluding fungal), diarrhea, fatigue, cellulitis, and localized infection.
One (8%) fatal adverse drug reaction of bacteremia occurred within 30 days of starting treatment.
Discontinuations due to adverse reactions occurred in 38% of patients. The most frequent adverse reaction leading to drug discontinuation (≥5%) was pneumonia (excluding fungal).
Dosage interruptions due to adverse reactions occurred in 62% of patients. The most frequent adverse reactions leading to dose interruption (≥5%) were febrile neutropenia, neutropenia, and pneumonia (excluding fungal).
Dosage reductions due to adverse reactions occurred in 15% of patients. The most frequent adverse reaction leading to dose reduction (≥5%) was neutropenia.
Adverse reactions reported in patients with newly-diagnosed AML using VENCLEXTA in combination with azacitidine or decitabine are presented in Table 15.
Table 15. Adverse Reactions Reported in ≥30% (Any Grade) or ≥5% (Grade ≥3) of Patients with AML Treated with VENCLEXTA in Combination with Azacitidine or Decitabine Body System Adverse Reaction VENCLEXTA in
Combination with
AzacitidineVENCLEXTA in
Combination with
DecitabineAny Grade
(%)
N = 67Grade ≥3
(%)
N = 67Any Grade
(%)
N = 13Grade ≥3
(%)
N = 13Blood and
lymphatic system
disordersThrombocytopeniaa 49 45 54 54 Neutropeniaa 49 49 38 38 Febrile neutropenia 36 36 69 69 Anemiaa 30 30 15 15 Gastrointestinal
disordersNausea 58 1 46 0 Diarrhea 54 3 38 8 Constipation 49 3 62 0 Vomitinga 40 0 23 0 Abdominal paina 22 4 46 0 General disorders
and
administration site
conditionsPeripheral edemaa 46 1 31 0 Fatiguea 36 7 62 15 Pyrexia 21 3 31 0 Cachexia 0 0 8 8 Multiple organ
dysfunction
syndrome6 6 0 0 Infections and
infestationsPneumonia
(excluding fungal)a27 25 46 31 Sepsis
(excluding fungal)a13 13 46 46 Urinary tract
infection16 6 23 0 Cellulitis 6 0 15 8 Localized infection 0 0 8 8 Musculoskeletal
and connective
tissue disordersBack pain 15 0 31 0 Myalgiaa 10 0 31 0 Nervous system
disordersDizzinessa 28 1 46 0 Skin and
subcutaneous
tissue disordersRasha 33 1 31 0 Respiratory,
thoracic and
mediastinal
disordersCougha 25 0 38 0 Hypoxia 18 6 15 0 Oropharyngeal
pain9 0 31 0 Vascular
disordersHemorrhagea 46 7 46 0 Hypotensiona 21 6 31 0 Hypertension 12 7 15 8 Adverse Reactions graded using NCI Common Terminology Criteria for Adverse Events version 4.0.
aIncludes multiple adverse reaction terms.Table 16 describes common laboratory abnormalities reported throughout treatment that were new or worsening from baseline.
Table 16. New or Worsening Laboratory Abnormalities with VENCLEXTA Reported in ≥40% (Any Grade) or ≥10% (Grade 3 or 4) of Patients with AML Treated with VENCLEXTA in Combination with Azacitidine or Decitabine Laboratory
AbnormalityVENCLEXTA in
Combination with AzacitidineVENCLEXTA in
Combination with DecitabineAny Gradea
(%)
N = 67Grade 3 or 4a
(%)
N = 67Any Gradea
(%)
N = 13Grade 3 or 4a
(%)
N = 13Hematology Neutropenia 100 100 100 100 Leukopenia 100 98 100 100 Thrombocytopenia 91 78 83 83 Lymphopenia 88 73 100 92 Anemia 57 57 69 69 Chemistry Hyperglycemia 75 12 69 0 Hypocalcemia 58 7 85 0 Hypoalbuminemia 52 4 38 8 Hypokalemia 49 7 46 0 Hyponatremia 49 4 38 0 Hypophosphatemia 46 15 23 8 Hyperbilirubinemia 45 9 46 15 Hypomagnesemia 21 0 54 8 aIncludes laboratory abnormalities that were new or worsening, or worsening from baseline unknown. VENCLEXTA in Combination with Low-Dose Cytarabine
The most common adverse reactions (≥30%) of any grade were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea.
Serious adverse reactions were reported in 95% of patients. The most frequent serious adverse reactions (≥5%) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection.
The incidence of fatal adverse drug reactions was 4.9% within 30 days of starting treatment with no reaction having an incidence of ≥2%.
Discontinuations due to adverse reactions occurred in 33% of patients. The most frequent adverse reactions leading to drug discontinuation (≥2%) were hemorrhage and sepsis (excluding fungal).
Dosage interruptions due to adverse reactions occurred in 52% of patients. The most frequent adverse reactions leading to dose interruption (≥5%) were thrombocytopenia, neutropenia, and febrile neutropenia.
Dosage reductions due to adverse reactions occurred in 8% of patients. The most frequent adverse reaction leading to dose reduction (≥2%) was thrombocytopenia.
Adverse reactions reported in patients with newly-diagnosed AML receiving VENCLEXTA in combination with low-dose cytarabine are presented in Table 17.
Table 17. Adverse Reactions Reported in ≥30% (Any Grade) or ≥5% (Grade ≥3) of Patients with AML Treated with VENCLEXTA in Combination with Low-Dose Cytarabine Body System Adverse Reaction Any Grade
(%)
N = 61Grade ≥3
(%)
N = 61Blood and lymphatic system
disordersThrombocytopeniaa 59 59 Neutropeniaa 46 46 Febrile neutropenia 46 44 Anemiaa 26 26 Gastrointestinal disorders Nausea 64 2 Diarrhea 44 3 Constipation 33 0 General disorders and
administration site conditionsFatiguea 44 10 Infections and infestations Sepsisa 20 18 Pneumoniaa 18 16 Device related infection 13 11 Urinary tract infection 8 7 Metabolic and nutritional
disordersDecreased appetitea 28 7 Respiratory disorders Dyspneaa 31 3 Vascular disorders Hemorrhagea 49 15 Hypotensiona 21 7 Hypertension 15 8 Adverse Reactions graded using NCI Common Terminology Criteria for Adverse Events version 4.0.
aIncludes multiple adverse reaction terms.Table 18 describes common laboratory abnormalities reported throughout treatment that were new or worsening from baseline.
Table 18. New or Worsening Laboratory Abnormalities with VENCLEXTA Reported in ≥40% (Any Grade) or ≥10% (Grade 3 or 4) of Patients with AML Treated with VENCLEXTA in Combination with Low-Dose Cytarabine Laboratory Abnormality All Gradesa
(%)
N = 61Grade 3 or 4a
(%)
N = 61Hematology Thrombocytopenia 100 96 Neutropenia 96 96 Leukopenia 96 96 Lymphopenia 93 66 Anemia 61 59 Chemistry Hyperglycemia 85 8 Hypocalcemia 79 16 Hyponatremia 62 11 Hyperbilirubinemia 57 3 Hypoalbuminemia 59 5 Hypokalemia 56 20 Hypophosphatemia 51 21 Hypomagnesemia 46 0 Blood creatinine increased 46 3 Blood bicarbonate decreased 41 0 aIncludes laboratory abnormalities that were new or worsening, or worsening from baseline unknown. Tumor lysis syndrome is an important risk when initiating treatment in patients with AML. The incidence of TLS was 3% (2/61) with VENCLEXTA in combination with low-dose cytarabine with implementation of dose ramp-up schedule in addition to standard prophylaxis and monitoring measures. All events were laboratory TLS, and all patients were able to reach the target dose.
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on VENCLEXTA
Strong or Moderate CYP3A Inhibitors or P-gp Inhibitors
Concomitant use with a strong or moderate CYP3A inhibitor or a P-gp inhibitor increases venetoclax Cmax and AUCinf[see Clinical Pharmacology (12.3)], which may increase VENCLEXTA toxicities, including the risk of TLS [see Warnings and Precautions (5)].
Concomitant use with a strong CYP3A inhibitor at initiation and during the ramp-up phase in patients with CLL/SLL is contraindicated [see Contraindications (4)].
In patients with CLL/SLL taking a steady daily dosage (after ramp-up phase), consider alternative medications or adjust VENCLEXTA dosage and closely monitor for signs of VENCLEXTA toxicities [see Dosage and Administration (2.3, 2.4)].
In patients with AML, adjust VENCLEXTA dosage and closely monitor for signs of VENCLEXTA toxicities [see Dosage and Administration (2.3, 2.4)].
Resume the VENCLEXTA dosage that was used prior to concomitant use with a strong or moderate CYP3A inhibitor or a P-gp inhibitor 2 to 3 days after discontinuation of the inhibitor [see Dosage and Administration (2.3, 2.4)].
Avoid grapefruit products, Seville oranges, and starfruit during treatment with VENCLEXTA, as they contain inhibitors of CYP3A.
Strong or Moderate CYP3A Inducers
Concomitant use with a strong CYP3A inducer decreases venetoclax Cmax and AUCinf[see Clinical Pharmacology (12.3)], which may decrease VENCLEXTA efficacy. Avoid concomitant use of VENCLEXTA with strong CYP3A inducers or moderate CYP3A inducers.
7.2 Effect of VENCLEXTA on Other Drugs
Concomitant use of VENCLEXTA increases warfarin Cmax and AUCinf[see Clinical Pharmacology (12.3)], which may increase the risk of bleeding. Closely monitor international normalized ratio (INR) in patients using warfarin concomitantly with VENCLEXTA.
Concomitant use of VENCLEXTA increases Cmax and AUCinf of P-gp substrates [see Clinical Pharmacology (12.3)], which may increase toxicities of these substrates. Avoid concomitant use of VENCLEXTA with a P-gp substrate. If a concomitant use is unavoidable, separate dosing of the P-gp substrate at least 6 hours before VENCLEXTA.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
There are no available data on VENCLEXTA use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. Based on toxicity observed in mice, VENCLEXTA may cause fetal harm when administered to pregnant women. In mice, venetoclax was fetotoxic at exposures 1.2 times the human clinical exposure based on AUC at a human dose of 400 mg daily. If VENCLEXTA is used during pregnancy or if the patient becomes pregnant while taking VENCLEXTA, the patient should be apprised of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk in the U.S. general population of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
In embryo-fetal development studies, venetoclax was administered to pregnant mice and rabbits during the period of organogenesis. In mice, venetoclax was associated with increased post-implantation loss and decreased fetal body weight at 150 mg/kg/day (maternal exposures approximately 1.2 times the human AUC exposure at a dose of 400 mg daily). No teratogenicity was observed in either the mouse or the rabbit.
8.2 Lactation
There are no data on the presence of VENCLEXTA in human milk, the effects of VENCLEXTA on the breastfed child, or the effects of VENCLEXTA on milk production. Venetoclax was present in the milk when administered to lactating rats (see Data).
Because many drugs are excreted in human milk and because the potential for serious adverse reactions in a breastfed child from VENCLEXTA is unknown, advise nursing women to discontinue breastfeeding during treatment with VENCLEXTA.
Venetoclax was administered (single dose; 150 mg/kg oral) to lactating rats 8 to 10 days parturition. Venetoclax in milk was 1.6 times lower than in plasma. Parent drug (venetoclax) represented the majority of the total drug-related material in milk, with trace levels of three metabolites.
8.3 Females and Males of Reproductive Potential
VENCLEXTA may cause fetal harm [see Warnings and Precautions (5.5) and Use in Specific Populations (8.1)].
Conduct pregnancy testing in females of reproductive potential before initiation of VENCLEXTA [see Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with VENCLEXTA and for at least 30 days after the last dose [see Use in Specific Populations (8.1)].
Based on findings in animals, male fertility may be compromised by treatment with VENCLEXTA [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness have not been established in pediatric patients.
In a juvenile toxicology study, mice were administered venetoclax at 10, 30, or 100 mg/kg/day by oral gavage from 7 to 60 days of age. Clinical signs of toxicity included decreased activity, dehydration, skin pallor, and hunched posture at ≥30 mg/kg/day. In addition, mortality and body weight effects occurred at 100 mg/kg/day. Other venetoclax-related effects were reversible decreases in lymphocytes at ≥10 mg/kg/day; a dose of 10 mg/kg/day is approximately 0.06 times the clinical dose of 400 mg on a mg/m2 basis for a 20 kg child.
8.5 Geriatric Use
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
Of the 352 patients with previously treated CLL/SLL evaluated for safety from 3 open-label trials of VENCLEXTA monotherapy, 57% (201/352) were ≥65 years of age and 18% (62/352) were ≥75 years of age.
No clinically meaningful differences in safety and effectiveness were observed between older and younger patients in the combination and monotherapy studies.
Of the 67 patients treated with VENCLEXTA in combination with azacitidine in the clinical trial, 96% were ≥65 years of age and 50% were ≥ 75 years of age. Of the 13 patients treated with VENCLEXTA in combination with decitabine in the clinical trial, 100% were ≥65 years of age and 26% were ≥ 75 years of age. Of the 61 patients treated with VENCLEXTA in combination with low-dose cytarabine, 97% were ≥65 years of age and 66% were ≥75 years of age.
The efficacy and safety data presented in the Adverse Reactions and Clinical Studies sections were obtained from these patients [see Adverse Reactions (6.2) and Clinical Studies (14.2)]. There are insufficient patient numbers to show differences in safety and effectiveness between geriatric and younger patients.
8.6 Renal Impairment
Due to the increased risk of TLS, patients with reduced renal function (CLcr <80 mL/min, calculated by Cockcroft-Gault formula) require more intensive prophylaxis and monitoring to reduce the risk of TLS when initiating treatment with VENCLEXTA [see Dosage and Administration (2.2, 2.3) and Warnings and Precautions (5.1)].
No dose adjustment is recommended for patients with mild or moderate renal impairment (CLcr ≥ 30 mL/min [see Clinical Pharmacology (12.3)]. A recommended dose has not been determined for patients with severe renal impairment (CLcr < 30 mL/min) or patients on dialysis.
8.7 Hepatic Impairment
No dose adjustment is recommended for patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment.
Reduce the dose of VENCLEXTA for patients with severe hepatic impairment (Child-Pugh C); monitor these patients more closely for signs of toxicity [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There is no specific antidote for VENCLEXTA. For patients who experience overdose, closely monitor and provide appropriate supportive treatment; during ramp-up phase interrupt VENCLEXTA and monitor carefully for signs and symptoms of TLS along with other toxicities [see Dosage and Administration (2.2, 2.3)]. Based on venetoclax large volume of distribution and extensive protein binding, dialysis is unlikely to result in significant removal of venetoclax.
-
11 DESCRIPTION
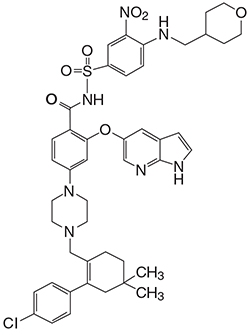
Venetoclax is a selective inhibitor of BCL-2 protein. It is a light yellow to dark yellow solid with the empirical formula C45H50ClN7O7S and a molecular weight of 868.44. Venetoclax has very low aqueous solubility. Venetoclax is described chemically as 4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide) and has the following chemical structure:
VENCLEXTA tablets for oral administration are supplied as pale yellow or beige tablets that contain 10, 50, or 100 mg venetoclax as the active ingredient. Each tablet also contains the following inactive ingredients: copovidone, colloidal silicon dioxide, polysorbate 80, sodium stearyl fumarate, and calcium phosphate dibasic. In addition, the 10 mg and 100 mg coated tablets include the following: iron oxide yellow, polyvinyl alcohol, polyethylene glycol, talc, and titanium dioxide. The 50 mg coated tablets also include the following: iron oxide yellow, iron oxide red, iron oxide black, polyvinyl alcohol, talc, polyethylene glycol and titanium dioxide. Each tablet is debossed with “V” on one side and “10”, “50” or “100” corresponding to the tablet strength on the other side.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Venetoclax is a selective and orally bioavailable small-molecule inhibitor of BCL-2, an anti-apoptotic protein. Overexpression of BCL-2 has been demonstrated in CLL and AML cells where it mediates tumor cell survival and has been associated with resistance to chemotherapeutics. Venetoclax helps restore the process of apoptosis by binding directly to the BCL-2 protein, displacing pro-apoptotic proteins like BIM, triggering mitochondrial outer membrane permeabilization and the activation of caspases. In nonclinical studies, venetoclax has demonstrated cytotoxic activity in tumor cells that overexpress BCL-2.
12.2 Pharmacodynamics
Based on the exposure response analyses for efficacy, a relationship between drug exposure and a greater likelihood of response was observed in clinical studies in patients with CLL/SLL, and in patients with AML. Based on the exposure response analyses for safety, a relationship between drug exposure and a greater likelihood of some safety events was observed in clinical studies in patients with AML. No exposure-safety relationship was observed in patients with CLL/SLL at doses up to 1200 mg given as monotherapy and up to 600 mg given in combination with rituximab.
The effect of multiple doses of VENCLEXTA up to 1200 mg once daily (2 times the maximum approved recommended dosage) on the QTc interval was evaluated in an open-label, single-arm study in 176 patients with previously treated hematologic malignancies. VENCLEXTA had no large effect on QTc interval (i.e., > 20 ms) and there was no relationship between venetoclax exposure and change in QTc interval.
12.3 Pharmacokinetics
Venetoclax mean (± standard deviation) steady state Cmax was 2.1 ± 1.1 mcg/mL and AUC0-24 was32.8 ± 16.9 mcgh/mL following administration of 400 mg once daily with a low-fat meal. Venetoclax steady state AUC increased proportionally over the dose range of 150 to 800 mg (0.25 to 1.33 times the maximum approved recommended dosage). The pharmacokinetics of venetoclax does not change over time.
Maximum plasma concentration of venetoclax was reached 5 to 8 hours following multiple oral administration under fed conditions.
Administration with a low-fat meal (approximately 512 kilocalories, 25% fat calories, 60% carbohydrate calories, and 15% protein calories) increased venetoclax exposure by approximately 3.4-fold and administration with a high-fat meal (approximately 753 kilocalories, 55% fat calories, 28% carbohydrate calories, and 17% protein calories) increased venetoclax exposure by 5.1- to 5.3-fold compared with fasting conditions.
Venetoclax is highly bound to human plasma protein with unbound fraction in plasma <0.01 across a concentration range of 1-30 micromolar (0.87-26 mcg/mL). The mean blood-to-plasma ratio was 0.57. The apparent volume of distribution (Vdss/F) of venetoclax ranged from 256-321 L in patients.
The terminal elimination half-life of venetoclax was approximately 26 hours.
Venetoclax is predominantly metabolized by CYP3A in vitro. The major metabolite identified in plasma, M27, has an inhibitory activity against BCL-2 that is at least 58-fold lower than venetoclax in vitro and its AUC represented 80% of the parent AUC.
After single oral dose of radiolabeled [14C]-venetoclax 200 mg to healthy subjects, > 99.9% of the dose was recovered in feces (20.8% as unchanged) and < 0.1% in urine within 9 days.
No clinically significant differences in the pharmacokinetics of venetoclax were observed based on age (19 to 90 years), sex, race (White, Black, Asians, and Others), weight, mild to moderate renal impairment (CLcr 30 to 89 mL/min, calculated by Cockcroft-Gault), or mild to moderate hepatic impairment (normal total bilirubin and aspartate transaminase (AST) > upper limit of normal (ULN) or total bilirubin 1 to 3 times ULN). The effect of severe renal impairment (CLcr < 30 mL/min) or dialysis on venetoclax pharmacokinetics is unknown.
Patients with Hepatic Impairment
Following a single dose of VENCLEXTA 50 mg, venetoclax systemic exposure (AUCinf) was 2.7-fold higher in subjects with severe hepatic impairment (Child-Pugh C) compared to subjects with normal hepatic function [see Dosage and Administration (2.5) and Use in Specific Populations (8.7)]. No clinically relevant differences in venetoclax systemic exposure were observed between subjects with mild or moderate hepatic impairment and subjects with normal hepatic function.
No clinically significant differences in venetoclax pharmacokinetics were observed when co-administered with azacitidine, azithromycin, cytarabine, decitabine, gastric acid reducing agents, obinutuzumab, or rituximab.
Concomitant use of ketoconazole (a strong CYP3A, P-gp and BCRP inhibitor) 400 mg once daily for 7 days increased venetoclax Cmax by 130% and AUCinf by 540% [see Drug Interactions (7.1)].
Concomitant use of ritonavir (a strong CYP3A, P-gp and OATP1B1/B3 inhibitor) 50 mg once daily for 14 days increased venetoclax Cmax by 140% and AUC by 690% [see Drug Interactions (7.1)].
Concomitant use of posaconazole (a strong CYP3A and P-gp inhibitor) 300 mg with venetoclax 50 mg and 100 mg for 7 days resulted in 61% and 86% higher venetoclax Cmax, respectively, compared with venetoclax 400 mg administered alone. The venetoclax AUC24 was 90% and 144% higher, respectively.
Concomitant use of a single dose of rifampin(an OATP1B1/1B3 and P-gp inhibitor) 600 mg increased venetoclax Cmax by 106% and AUCinf by 78%. Concomitant use of multiple doses of rifampin (as a strong CYP3A inducer) 600 mg once daily for 13 days decreased venetoclax Cmax by 42% and AUCinf by 71% [see Drug Interactions (7.1)].
Concomitant use of a single 400 mg dose of venetoclax with 5 mg warfarin resulted in 18% to 28% increase in Cmax and AUCinf of R-warfarin and S-warfarin [see Drug Interactions (7.2)].
Concomitant use of a single dose of venetoclax 100 mg with digoxin (a P-gp substrate) 0.5 mg increased digoxin Cmax by 35% and AUCinf by 9% [see Drug Interactions (7.2)].
Venetoclax is not an inhibitor or inducer of CYP1A2, CYP2B6, CYP2C19, CYP2D6 or CYP3A4. Venetoclax is a weak inhibitor of CYP2C8, CYP2C9, and UGT1A1.
Venetoclax is not an inhibitor of UGT1A4, UGT1A6, UGT1A9, or UGT2B7.
Venetoclax is an inhibitor and substrate of P-gp and BCRP and weak inhibitor of OATP1B1.
Venetoclax is not an inhibitor of OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1, or MATE2K.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with venetoclax.
Venetoclax was not mutagenic in an in vitro bacterial mutagenicity (Ames) assay, did not induce numerical or structural aberrations in an in vitro chromosome aberration assay using human peripheral blood lymphocytes, and was not clastogenic in an in vivo mouse bone marrow micronucleus assay at doses up to 835 mg/kg. The M27 metabolite was negative for genotoxic activity in in vitro Ames and chromosome aberration assays.
Fertility and early embryonic development studies were conducted in male and female mice. These studies evaluate mating, fertilization, and embryonic development through implantation. There were no effects of venetoclax on estrous cycles, mating, fertility, corpora lutea, uterine implants or live embryos per litter at dosages up to 600 mg/kg/day. However, a risk to human male fertility exists based on testicular toxicity (germ cell loss) observed in dogs at exposures as low as 0.5 times the human AUC exposure at a dose of 400 mg.
13.2 Animal Toxicology and/or Pharmacology
In dogs, venetoclax caused single-cell necrosis in various tissues, including the gallbladder, exocrine pancreas, and stomach with no evidence of disruption of tissue integrity or organ dysfunction; these findings were minimal to mild in magnitude. Following a 4-week dosing period and subsequent 4-week recovery period, minimal single-cell necrosis was still present in some tissues and reversibility has not been assessed following longer periods of dosing or recovery.
In addition, after approximately 3 months of daily dosing in dogs, venetoclax caused progressive white discoloration of the hair coat, due to loss of melanin pigment.
-
14 CLINICAL STUDIES
14.1 Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
CLL14 (BO25323) was a randomized (1:1), multicenter, open label, actively controlled, phase 3 trial (NCT02242942) that evaluated the efficacy and safety of VENCLEXTA in combination with obinutuzumab (VEN+G) versus obinutuzumab in combination with chlorambucil (GClb) for patients with previously untreated CLL with coexisting medical conditions (total Cumulative Illness Rating Scale [CIRS] score > 6 or CLcr < 70 mL/min). The trial required hepatic transaminases and total bilirubin ≤2 times upper limit of normal and excluded patients with Richter’s transformation or any individual organ/system impairment score of 4 by CIRS except eye, ear, nose, and throat organ system.
All patients received obinutuzumab at 1000 mg on Day 1 (the first dose could be split as 100 mg and 900 mg on Days 1 and 2), and on Days 8 and 15 of Cycle 1, and on Day 1 of each subsequent cycle, for a total of 6 cycles. Patients in the VEN+G arm began the 5-week VENCLEXTA ramp-up schedule [see Dosage and Administration (2.1, 2.2)] on Day 22 of Cycle 1, and received VENCLEXTA 400 mg once daily from Cycle 3 Day 1 until the last day of Cycle 12. Patients randomized to the GClb arm received 0.5 mg/kg oral chlorambucil on Day 1 and Day 15 of Cycles 1 to 12. Each cycle was 28 days.
A total of 432 patients were randomized, 216 to each study arm. Baseline demographic and disease characteristics were similar between the study arms. The median age was 72 years (range: 41 to 89 years), 89% were white, 67% were male; 36% and 43% were Binet stage B and C, respectively, and 88% had Eastern Cooperative Oncology Group (ECOG) performance status <2. The median CIRS score was 8.0 (range: 0 to 28) and 58% of patients had CLcr <70 mL/min. A 17p deletion was detected in 8% of patients, TP53 mutations in 7%, 11q deletion in 19%, and unmutated IgVH in 57%.
The major efficacy outcome was progression-free survival (PFS) as assessed by an Independent Review Committee (IRC). The median duration of follow-up for PFS was 28 months (range: 0.1 to 36 months).
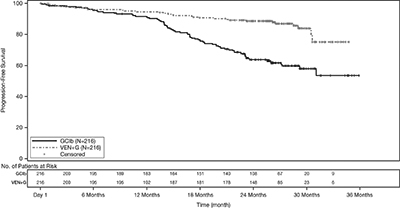
Efficacy results for CLL14 are shown in Table 19. The Kaplan-Meier curve for PFS is shown in Figure 1.
Table 19. Efficacy Results in CLL14 Endpoint VENCLEXTA +
Obinutuzumab
(N = 216)Obinutuzumab +
Chlorambucil
(N = 216)Progression-free survivala Number of events, n (%) 29 (13) 79 (37) Disease progression 14 (6) 71 (33) Death 15 (7) 8 (4) Median, months Not Reached Not Reached HR (95% CI)b 0.33 (0.22, 0.51) p-valueb < 0.0001 Response ratec, n (%) ORRd 183 (85) 154 (71) 95% CI (79, 89) (65, 77) CR 100 (46) 47 (22) CR+CRid 107 (50) 50 (23) PR 76 (35) 104 (48) CI = confidence interval; HR = hazard ratio; CR = complete remission; CRi = complete remission with incomplete marrow recovery; PR = partial remission; ORR = overall response rate (CR + CRi + PR).
aFrom randomization until earliest event of disease progression or death due to any cause. IRC-assessed; Kaplan-Meier estimate.
bHR estimate is based on Cox-proportional hazards model stratified by Binet Stage and geographic region; p-value based on log rank test stratified by the same factors.
cPer 2008 International Workshop for Chronic Lymphocytic Leukemia (IWCLL) guidelines.
dp-values based on Cochran-Mantel-Haenszel test; p=0.0007 for ORR; p <0.0001 for CR+CRi.Figure 1. Kaplan-Meier Curve of IRC-Assessed Progression-free Survival in CLL14
At the time of analysis, median overall survival (OS) had not been reached, with fewer than 10% of patients experiencing an event. The median duration of follow-up for OS was 28 months.
Minimal residual disease (MRD) was evaluated using allele-specific oligonucleotide polymerase chain reaction (ASO-PCR). The definition of negative status was less than one CLL cell per 104 leukocytes. Rates of MRD negativity 3 months after the completion of treatment regardless of response and in patients who achieved CR are shown in Table 20. At this assessment, 134 patients in the VEN+G arm who were MRD negative in peripheral blood had matched bone marrow specimens; of these, 122 patients (91%) were MRD negative in both peripheral blood and bone marrow.
Table 20. Minimal Residual Disease Negativity Rates Three Months After the Completion of Treatment in CLL14 VENCLEXTA +
ObinutuzumabObinutuzumab +
ChlorambucilMRD negativity rate (ITT population) N 216 216 Bone marrow, n (%) 123 (57) 37 (17) 95% CI (50, 64) (12, 23) p-valuea <0.0001 Peripheral blood, n (%) 163 (76) 76 (35) 95% CI (69, 81) (29, 42) p-valuea <0.0001 MRD negativity rate in patients with CR N 100 47 Bone marrow, n (%) 69 (69) 21 (45) 95% CI (59, 78) (30, 60) p-valuea 0.0048 Peripheral blood, n (%) 87 (87) 29 (62) 95% CI (79, 93) (46, 75) p-valuea 0.0005 CI = confidence interval; CR = complete remission.
ap-value based on Chi-square testTwelve months after the completion of treatment, MRD negativity rates in peripheral blood were 58% (126/216) in patients treated with VEN+G and 9% (20/216) in patients treated with GClb.
MURANO was a randomized (1:1), multicenter, open label study (NCT02005471) that evaluated the efficacy and safety of VENCLEXTA in combination with rituximab (VEN+R) versus bendamustine in combination with rituximab (B+R) in patients with CLL who had received at least one line of prior therapy. Patients in the VEN+R arm completed the 5-week ramp-up schedule [see Dosage and Administration (2.1, 2.2)] and received VENCLEXTA 400 mg once daily for 24 months from Cycle 1 Day 1 of rituximab in the absence of disease progression or unacceptable toxicity. Rituximab was initiated intravenously after the 5-week dose ramp-up at 375 mg/m2 on Day 1 of Cycle 1 and 500 mg/m2 on Day 1 of Cycles 2-6. Each cycle was 28 days. Patients randomized to B+R received bendamustine at 70 mg/m2 on Days 1 and 2 for 6 cycles (28-day cycle) and rituximab at the above described dose and schedule.
A total of 389 patients were randomized: 194 to the VEN+R arm and 195 to the B+R arm. Baseline demographic and disease characteristics were similar between the VEN+R and B+R arms. The median age was 65 years (range: 22 to 85 years), 97% were white, 74% were male, and 99% had ECOG performance status <2. Median prior lines of therapy was 1 (range: 1 to 5); 59% had received 1 prior therapy, 26% had received 2 prior therapies, and 16% had received 3 or more prior therapies. Prior therapies included alkylating agents (94%), anti-CD20 antibodies (77%), B-cell receptor pathway inhibitors (2%), and prior purine analogs (81%, including fludarabine/cyclophosphamide/rituximab in 55%). A 17p deletion was detected in 24% of patients, TP53 mutations in 25%, 11q deletion in 32%, and unmutated IgVH in 63%.
Efficacy was based on PFS as assessed by an IRC. The median follow-up for PFS was 23.4 months (range: 0 to 37.4+ months).
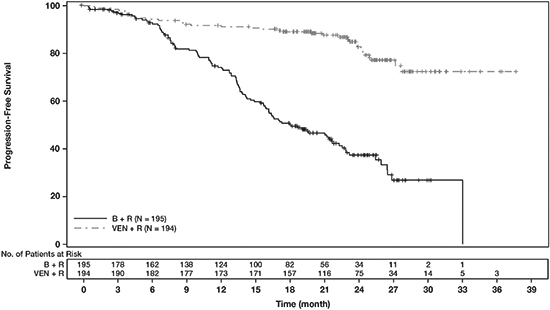
Efficacy results for MURANO are shown in Table 21. The Kaplan-Meier curve for PFS is shown in Figure 2.
Table 21. IRC-Assessed Efficacy Results in MURANO Endpoint VENCLEXTA + Rituximab
(N = 194)Bendamustine + Rituximab
(N = 195)Progression-free survivala Number of events, n (%) 35 (18) 106 (54) Disease progression, n 26 91 Death events, n 9 15 Median, months (95% CI) Not Reached 18.1 (15.8, 22.3) HR (95% CI)b 0.19 (0.13, 0.28) p-valueb <0.0001 Response ratec, n (%) ORR 179 (92) 141 (72) 95% CI (88, 96) (65, 78) CR+CRi 16 (8) 7 (4) nPR 3 (2) 1 (1) PR 160 (82) 133 (68) CI = confidence interval; HR = hazard ratio; CR = complete remission; CRi = complete remission with incomplete marrow recovery; nPR = nodular partial remission; PR = partial remission; ORR = overall response rate (CR + CRi + nPR + PR).
aKaplan-Meier estimate.
bHR estimate is based on Cox-proportional hazards model stratified by 17p deletion, risk status, and geographic region; p-value based on log-rank test stratified by the same factors.
cPer 2008 International Workshop for Chronic Lymphocytic Leukemia (IWCLL) guidelines.
Figure 2. Kaplan-Meier Curve of IRC-Assessed Progression-free Survival in MURANO
At the time of analysis, median overall survival had not been reached in either arm after a median follow-up of 22.9 months.
At 3 months after the last dose of rituximab, the MRD negativity rate in peripheral blood in patients who achieved PR or better was 53% (103/194) in the VEN+R arm and 12% (23/195) in the B+R arm. The MRD-negative CR/CRi rate at this timepoint was 3% (6/194) in the VEN+R arm and 2% (3/195) in the B+R arm.
The efficacy of VENCLEXTA monotherapy in previously-treated CLL or SLL is based on three single-arm studies.
The efficacy of VENCLEXTA was established in study M13-982 (NCT01889186), an open-label, single-arm, multicenter clinical trial of 106 patients with CLL with 17p deletion who had received at least one prior therapy. In the study, 17p deletion was confirmed in peripheral blood specimens from patients using Vysis CLL FISH Probe Kit, which is FDA approved for selection of patients for VENCLEXTA treatment. Patients received VENCLEXTA via a weekly ramp-up schedule starting at 20 mg and ramping to 50 mg, 100 mg, 200 mg and finally 400 mg once daily. Patients continued to receive 400 mg of VENCLEXTA orally once daily until disease progression or unacceptable toxicity.
Efficacy was based on overall response rate (ORR) as assessed by an IRC.
Table 22 summarizes the baseline demographic and disease characteristics of the study population.
Table 22. Baseline Patient Characteristics in Study M13-982 Characteristic N = 106 Age, years; median (range) 67 (37-83) White; % 97 Male; % 65 ECOG performance status; %
0
1
2
40
52
8Tumor burden; %
Absolute lymphocyte count ≥25 x 109/L
One or more nodes ≥5 cm
50
53Number of prior therapies; median (range) 2.5 (1-10) Time since diagnosis, years; median (range)a 6.6 (0.1-32.1) ECOG = Eastern Cooperative Oncology Group.
aN=105.The median time on treatment at the time of evaluation was 12.1 months (range: 0 to 21.5 months). Efficacy results are shown in Table 23.
Table 23. Efficacy Results per IRC for Patients with Previously Treated CLL with 17p Deletion in Study M13-982 Endpoint VENCLEXTA
N=106ORR, n (%)a
(95% CI)85 (80)
(71, 87)CR + CRi, n (%)
CR, n (%)
CRi, n (%)8 (8)
6 (6)
2 (2)nPR, n (%) 3 (3) PR, n (%) 74 (70) CI = confidence interval; CR = complete remission; CRi = complete remission with incomplete marrow recovery; IRC = independent review committee; nPR = nodular partial remission; ORR = overall response rate (CR + CRi + nPR + PR); PR = partial remission.
aPer 2008 IWCLL guidelines.The median time to first response was 0.8 months (range: 0.1 to 8.1 months).
Based on a later data cutoff date and investigator-assessed efficacy, the duration of response (DOR) ranged from 2.9 to 32.8+ months. The median DOR has not been reached with median follow-up of 22 months.
Minimal residual disease was evaluated in peripheral blood and bone marrow for patients who achieved CR or CRi, following treatment with VENCLEXTA. Three percent (3/106) achieved MRD negativity in the peripheral blood and bone marrow (less than one CLL cell per 104 leukocytes).
Study M12-175 (NCT01328626) was a multicenter, open-label trial that enrolled previously treated patients with CLL or SLL, including those with 17p deletion. Efficacy was evaluated in 67 patients (59 with CLL, 8 with SLL) who had received a 400 mg daily dose of VENCLEXTA. Patients continued this dose until disease progression or unacceptable toxicity. The median duration of treatment at the time of evaluation was 22.1 months (range: 0.5 to 71.7 months).
The median age was 65 years (range: 42 to 84 years), 78% were male and 87% were white. The median number of prior treatments was 3 (range: 1 to 11). At baseline, 67% of patients had one or more nodes ≥5 cm, 30% of patients had ALC ≥25 x 109/L, 33% had documented unmutated IgVH, and 21% had documented 17p deletion.
Efficacy was evaluated according to 2008 IWCLL guidelines and assessed by an IRC. The ORR was 76% (95% CI: 64%, 86%), CR + CRi rate was 10%, and PR rate was 66%. The median DOR was 36.2 months (range: 2.4 to 52.4 months).
Study M14-032 (NCT02141282) was an open-label, multicenter, study that evaluated the efficacy of VENCLEXTA in patients with CLL who had been previously treated with and progressed on or after ibrutinib or idelalisib. Patients received a daily dose of 400 mg of VENCLEXTA following the ramp-up schedule. Patients continued to receive VENCLEXTA 400 mg once daily until disease progression or unacceptable toxicity. At the time of analysis, the median duration of treatment was 19.5 months (range: 0.1 to 39.5 months).
Of the 127 patients treated (91 with prior ibrutinib, 36 with prior idelalisib), the median age was 66 years (range: 28 to 85 years), 70% were male and 92% were white. The median number of prior treatments was 4 (range: 1 to 15). At baseline, 41% of patients had one or more nodes ≥5 cm, 31% had an absolute lymphocyte count ≥25 x 109/L, 57% had documented unmutated IgVH, and 39% had documented 17p deletion.
Efficacy was based on 2008 IWCLL guidelines and was assessed by an IRC. The ORR was 70% (95% CI: 61%, 78%), with a CR + CRi rate of 5%, and PR rate of 65%. The median DOR was not reached with a median follow-up time of 19.9 months (range: 2.9 to 36 months).
14.2 Acute Myeloid Leukemia
VENCLEXTA was studied in two open-label non-randomized trials in patients with newly-diagnosed AML who were ≥75 years of age, or had comorbidities that precluded the use of intensive induction chemotherapy based on at least one of the following criteria: baseline ECOG performance status of 2-3, severe cardiac or pulmonary comorbidity, moderate hepatic impairment, CLcr <45 mL/min, or other comorbidity. Efficacy was established based on the rate of complete remission (CR) and the duration of CR.
VENCLEXTA was studied in a non-randomized, open-label clinical trial (NCT02203773) of VENCLEXTA in combination with azacitidine (N=84) or decitabine (N=31) in patients with newly-diagnosed AML. Of those patients, 67 who received azacitidine combination and 13 who received decitabine combination were age 75 years or older, or had comorbidities that precluded the use of intensive induction chemotherapy.
Patients received VENCLEXTA via a daily ramp-up to a final 400 mg once daily dose [see Dosage and Administration (2.1)]. During the ramp-up, patients received TLS prophylaxis and were hospitalized for monitoring. Azacitidine at 75 mg/m2 was administered either intravenously or subcutaneously on Days 1-7 of each 28-day cycle beginning on Cycle 1 Day 1. Decitabine at 20 mg/m2 was administered intravenously on Days 1-5 of each 28-day cycle beginning on Cycle 1 Day 1. Patients continued to receive treatment cycles until disease progression or unacceptable toxicity. Azacitidine dose reduction was implemented in the clinical trial for management of hematologic toxicity, see azacitidine full prescribing information. Dose reductions for decitabine were not implemented in the clinical trial.
Table 24 summarizes the baseline demographic and disease characteristics of the study population.
Table 24. Baseline Patient Characteristics for Patients with AML Treated with VENCLEXTA in Combination with Azacitidine or Decitabine Characteristic VENCLEXTA
in Combination
with Azacitidine
N = 67VENCLEXTA
in Combination
with Decitabine
N = 13Age, years; median (range) 76 (61-90) 75 (68-86) Race White; % 87 77 Black or African American; % 4.5 0 Asian; % 1.5 0 Native Hawaiian or Pacific Islander; % 1.5 15 American Indian/Alaskan Native; % 0 7.7 Unreported/Other; % 6.0 0 Male; % 60 38 ECOG performance status; %
0-1
2
3
64
33
3
92
8
0Disease history; %
De Novo AML
Secondary AML
73
27
85
15Mutation analyses detecteda; % TP53 21 31 IDH1 or IDH2 27 0 FLT-3 16 23 NPM1 19 15 Cytogenetic risk detectedb,c; % Intermediate 64 38 Poor 34 62 Baseline comorbiditiesd, % Severe cardiac disease 4.5 7.7 Severe pulmonary disease 1.5 0 Moderate hepatic impairment 9 0 Creatinine clearance <45 mL/min 13 7.7 ECOG = Eastern Cooperative Oncology Group.
aIncludes 6 patients with insufficient sample for analysis in the azacitidine group and 4 in the decitabine group.
bAs defined by the National Comprehensive Cancer Network (NCCN) risk categorization v2014.
cNo mitosis in 1 patient in azacitidine group (excluded favorable risk by Fluorescence in situ Hybridization [FISH] analysis).
dPatients may have had more than one comorbidity.The efficacy results are shown in Table 25.
Table 25. Efficacy Results for Patients with Newly-Diagnosed AML Treated with VENCLEXTA in Combination with Azacitidine or Decitabine Efficacy Outcomes VENCLEXTA in
Combination with
Azacitidine
N = 67VENCLEXTA in
Combination with
Decitabine
N = 13CR, n (%)
(95% CI)25 (37)
(26, 50)7 (54)
(25, 81)CRh, n (%)
(95% CI)16 (24)
(14, 36)1 (7.7)
(0.2, 36)CI = confidence interval; NR = not reached.
CR (complete remission) was defined as absolute neutrophil count >1,000/microliter, platelets >100,000/microliter, red blood cell transfusion independence, and bone marrow with <5% blasts. Absence of circulating blasts and blasts with Auer rods; absence of extramedullary disease.
CRh (complete remission with partial hematological recovery) was defined as <5% of blasts in the bone marrow, no evidence of disease, and partial recovery of peripheral blood counts (platelets >50,000/microliter and ANC >500/microliter).The median follow-up was 7.9 months (range: 0.4 to 36 months) for VENCLEXTA in combination with azacitidine. At the time of analysis, for patients who achieved a CR, the median observed time in remission was 5.5 months (range: 0.4 to 30 months). The observed time in remission is the time from the start of CR to the time of data cut-off date or relapse from CR.
The median follow-up was 11 months (range: 0.7 to 21 months) for VENCLEXTA in combination with decitabine. At the time of analysis, for patients who achieved a CR, the median observed time in remission was 4.7 months (range: 1.0 to 18 months). The observed time in remission is the time from the start of CR to the time of data cut-off date or relapse from CR.
Median time to first CR or CRh for patients treated with VENCLEXTA in combination with azacitidine was 1.0 month (range: 0.7 to 8.9 months).
Median time to first CR or CRh for patients treated with VENCLEXTA in combination with decitabine was 1.9 months (range: 0.8 to 4.2 months).
Of patients treated with VENCLEXTA in combination with azacitidine, 7.5% (5/67) subsequently received stem cell transplant.
The study enrolled 35 additional patients (age range: 65 to 74 years) who did not have known comorbidities that preclude the use of intensive induction chemotherapy and were treated with VENCLEXTA in combination with azacitidine (N=17) or decitabine (N=18).
For the 17 patients treated with VENCLEXTA in combination with azacitidine, the CR rate was 35% (95% CI: 14%, 62%). The CRh rate was 41% (95% CI: 18%, 67%). Seven (41%) patients subsequently received stem cell transplant.
For the 18 patients treated with VENCLEXTA in combination with decitabine, the CR rate was 56% (95% CI: 31%, 79%). The CRh rate was 22% (95% CI: 6.4%, 48%). Three (17%) patients subsequently received stem cell transplant.
VENCLEXTA was studied in a non-randomized, open-label clinical trial (NCT02287233) of VENCLEXTA in combination with low dose cytarabine (N=82) in patients with newly-diagnosed AML, including patients with previous exposure to a hypomethylating agent for an antecedent hematologic disorder. Of those patients, 61 were age 75 years or older, or had comorbidities that precluded the use of intensive induction chemotherapy based on at least one of the following criteria: baseline ECOG performance status of 2-3, severe cardiac or pulmonary comorbidity, moderate hepatic impairment, CLcr ≥30 to <45 mL/min, or other comorbidity.
Patients initiated VENCLEXTA via daily ramp-up to a final 600 mg once daily dose [see Dosage and Administration (2.1)]. During the ramp-up, patients received TLS prophylaxis and were hospitalized for monitoring. Cytarabine at a dose of 20 mg/m2 was administered subcutaneously once daily on Days 1-10 of each 28-day cycle beginning on Cycle 1 Day 1. Patients continued to receive treatment cycles until disease progression or unacceptable toxicity. Dose reduction for low-dose cytarabine was not implemented in the clinical trial.
Table 26 summarizes the baseline demographic and disease characteristics of the study population.
Table 26. Baseline Patient Characteristics for Patients with AML Treated with VENCLEXTA in Combination with Low-Dose Cytarabine Characteristic VENCLEXTA in Combination
with Low-Dose Cytarabine
N = 61Age, years; median (range) 76 (63-90) Race White; % 92 Black or African American; % 1.6 Asian; % 1.6 Unreported; % 4.9 Male; % 74 ECOG performance status; %
0-1
2
3
66
33
1.6Disease history, %
De novo AML
Secondary AML
54
46Mutation analyses detecteda; % TP53 8 IDH1 or IDH2 23 FLT-3 21 NPM1 9.8 Cytogenetic risk detectedb; % Intermediate 59 Poor 34 No mitoses 6.6 Baseline comorbiditiesc, % Severe cardiac disease 9.8 Moderate hepatic impairment 4.9 Creatinine clearance ≥30 or <45 mL/min 3.3 ECOG = Eastern Cooperative Oncology Group.
aIncludes 7 patients with insufficient sample for analysis.
bAs defined by the National Comprehensive Cancer Network (NCCN) risk categorization v2014
cPatients may have had more than one comorbidity.Efficacy results are shown in Table 27.
Table 27. Efficacy Results for Patients with Newly-Diagnosed AML Treated with VENCLEXTA in Combination with Low-Dose Cytarabine Efficacy Outcomes VENCLEXTA in Combination
with Low-Dose
Cytarabine
N = 61CR, n (%)
(95% CI)13 (21)
(12, 34)CRh, n (%)
(95% CI)13 (21)
(12, 34)CI = confidence interval; NR = not reached.
CR (complete remission) was defined as absolute neutrophil count >1,000/microliter, platelets >100,000/microliter, red blood cell transfusion independence, and bone marrow with <5% blasts. Absence of circulating blasts and blasts with Auer rods; absence of extramedullary disease.
CRh (complete remission with partial hematological recovery) was defined as <5% of blasts in the bone marrow, no evidence of disease, and partial recovery of peripheral blood counts (platelets >50,000/microliter and ANC >500/microliter).The median follow-up was 6.5 months (range: 0.3 to 34 months). At the time of analysis, for patients who achieved a CR, the median observed time in remission was 6.0 months (range: 0.03 to 25 months). The observed time in remission is the time from the start of CR to the time of data cut-off date or relapse from CR.
Median time to first CR or CRh for patients treated with VENCLEXTA in combination with low-dose cytarabine was 1.0 month (range: 0.8 to 9.4 months).
The study enrolled 21 additional patients (age range: 67 to 74 years) who did not have known comorbidities that preclude the use of intensive induction chemotherapy and were treated with VENCLEXTA in combination with low-dose cytarabine. The CR rate was 33% (95% CI: 15%, 57%). The CRh rate was 24% (95% CI: 8.2%, 47%). One patient (4.8%) subsequently received stem cell transplant.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
VENCLEXTA is dispensed as follows:
Packaging Presentation Number of Tablets National Drug Code (NDC) CLL/SLL Starting Pack Each pack contains four weekly wallet blister packs: - Week 1 (14 x 10 mg tablets)
- Week 2 (7 x 50 mg tablets)
- Week 3 (7 x 100 mg tablets)
- Week 4 (14 x 100 mg tablets)
0074-0579-28 Wallet containing 10 mg tablets 14 x 10 mg tablets 0074-0561-14 Wallet containing 50 mg tablets 7 x 50 mg tablets 0074-0566-07 Unit dose blister containing 10 mg tablets 2 x 10 mg tablets 0074-0561-11 Unit dose blister containing 50 mg tablet 1 x 50 mg tablet 0074-0566-11 Unit dose blister containing 100 mg tablet 1 x 100 mg tablet 0074-0576-11 Bottle containing 100 mg tablets 120 x 100 mg tablets 0074-0576-22 Bottle containing 100 mg tablets 180 x 100 mg tablets 0074-0576-34 VENCLEXTA 10 mg film-coated tablets are round, biconvex shaped, pale yellow debossed with “V” on one side and “10” on the other side.
VENCLEXTA 50 mg film-coated tablets are oblong, biconvex shaped, beige debossed with “V” on one side and “50” on the other side.
VENCLEXTA 100 mg film-coated tablets are oblong, biconvex shaped, pale yellow debossed with “V” on one side and “100” on the other side.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
-
Tumor Lysis Syndrome
Advise patients of the potential risk of TLS, particularly at treatment initiation and during ramp-up phase, and to immediately report any signs and symptoms associated with this event (fever, chills, nausea, vomiting, confusion, shortness of breath, seizure, irregular heartbeat, dark or cloudy urine, unusual tiredness, muscle pain, and/or joint discomfort) to their health care provider (HCP) for evaluation [see Warnings and Precautions (5.1)].
Advise patients to be adequately hydrated every day when taking VENCLEXTA to reduce the risk of TLS. The recommended volume is 6 to 8 glasses (approximately 56 ounces total) of water each day. Patients should drink water starting 2 days before and on the day of the first dose, and every time the dose is increased [see Dosage and Administration (2.2)].
Advise patients of the importance of keeping scheduled appointments for blood work or other laboratory tests [see Dosage and Administration (2.2)].
Advise patients that it may be necessary to take VENCLEXTA in the hospital or medical office setting to allow monitoring for TLS.
-
Neutropenia
Advise patients to contact their HCP immediately if they develop a fever or any signs of infection. Advise patients of the need for periodic monitoring of blood counts [see Warnings and Precautions (5.2)].
-
Infections
Advise patients to contact their HCP immediately if they develop a fever or any signs of infection [see Warnings and Precautions (5.3)].
-
Drug Interactions
Advise patients to avoid consuming grapefruit products, Seville oranges, or starfruit during treatment with VENCLEXTA. Advise patients that VENCLEXTA may interact with some drugs; therefore, advise patients to inform their health care provider of the use of any prescription medication, over-the-counter drugs, vitamins and herbal products [see Contraindications (4) and Drug Interactions (7.1)].
-
Immunizations
Advise patients to avoid vaccination with live vaccines because they may not be safe or effective during treatment with VENCLEXTA [see Warnings and Precautions (5.4)].
-
Pregnancy and Lactation
Advise women of the potential risk to the fetus and to avoid pregnancy during treatment with VENCLEXTA. Advise female patients of reproductive potential to use effective contraception during therapy and for at least 30 days after completing of therapy. Advise females to contact their HCP if they become pregnant, or if pregnancy is suspected, during treatment with VENCLEXTA. Also advise patients not to breastfeed while taking VENCLEXTA [see Warnings and Precautions (5.5), and Use in Specific Populations (8.1, 8.2, and 8.3)].
-
Male Infertility
Advise patients of the possibility of infertility and possible use of sperm banking for males of reproductive potential [see Use in Specific Populations (8.3)].
Instructions for Taking VENCLEXTA
Advise patients to take VENCLEXTA exactly as prescribed and not to change their dose or to stop taking VENCLEXTA unless they are told to do so by their HCP. Advise patients to take VENCLEXTA orally once daily, at approximately the same time each day, according to their HCP's instructions and that the tablets should be swallowed whole with a meal and water without being chewed, crushed, or broken [see Dosage and Administration (2.1)].
Advise patients with CLL/SLL to keep VENCLEXTA in the original packaging during the first 4 weeks of treatment, and not to transfer the tablets to a different container.
Advise patients that if a dose of VENCLEXTA is missed by less than 8 hours, to take the missed dose right away and take the next dose as usual. If a dose of VENCLEXTA is missed by more than 8 hours, advise patients to wait and take the next dose at the usual time [see Dosage and Administration (2.6)].
Advise patients not to take any additional dose that day if they vomit after taking VENCLEXTA, and to take the next dose at the usual time the following day.
Manufactured and Marketed by:
AbbVie Inc.
North Chicago, IL 60064Marketed by:
Genentech USA, Inc.
A Member of the Roche Group
South San Francisco, CA 94080-4990
-
Tumor Lysis Syndrome
-
MEDICATION GUIDE
MEDICATION GUIDE
VENCLEXTA® (ven-KLEKS-tuh)
(venetoclax tablets)
What is the most important information I should know about VENCLEXTA?
VENCLEXTA can cause serious side effects, including:
Tumor lysis syndrome (TLS). TLS is caused by the fast breakdown of cancer cells. TLS can cause kidney failure, the need for dialysis treatment, and may lead to death. Your healthcare provider will do tests to check your risk of getting TLS before you start taking VENCLEXTA. You will receive other medicines before starting and during treatment with VENCLEXTA to help reduce your risk of TLS. You may also need to receive intravenous (IV) fluids into your vein. Your healthcare provider will do blood tests to check for TLS when you first start treatment and during treatment with VENCLEXTA. It is important to keep your appointments for blood tests. Tell your healthcare provider right away if you have any symptoms of TLS during treatment with VENCLEXTA, including:
- fever
- chills
- nausea
- vomiting
- confusion
- shortness of breath
- seizures
- irregular heartbeat
- dark or cloudy urine
- unusual tiredness
- muscle or joint pain
Drink plenty of water during treatment with VENCLEXTA to help reduce your risk of getting TLS.
Drink 6 to 8 glasses (about 56 ounces total) of water each day, starting 2 days before your first dose, on the day of your first dose of VENCLEXTA, and each time your dose is increased.Your healthcare provider may delay, decrease your dose, or stop treatment with VENCLEXTA if you have side effects.
See "What are the possible side effects of VENCLEXTA?" for more information about side effects.
VENCLEXTA is a prescription medicine used:
- to treat adults with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL).
- in combination with azacitidine, or decitabine, or low-dose cytarabine to treat adults with newly-diagnosed acute myeloid leukemia (AML) who:
- are 75 years of age or older, or
- have other medical conditions that prevent the use of standard chemotherapy.
It is not known if VENCLEXTA is safe and effective in children.
Who should not take VENCLEXTA?
Certain medicines must not be taken when you first start taking VENCLEXTA and while your dose is being slowly increased because of the risk of increased tumor lysis syndrome (TLS).
- Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. VENCLEXTA and other medicines may affect each other causing serious side effects.
- Do not start new medicines during treatment with VENCLEXTA without first talking with your healthcare provider.
Before taking VENCLEXTA, tell your healthcare provider about all of your medical conditions, including if you:
- have kidney problems
- have liver problems
- have problems with your body salts or electrolytes, such as potassium, phosphorus, or calcium
- have a history of high uric acid levels in your blood or gout
- are scheduled to receive a vaccine. You should not receive a “live vaccine” before, during, or after treatment with VENCLEXTA, until your healthcare provider tells you it is okay. If you are not sure about the type of immunization or vaccine, ask your healthcare provider. These vaccines may not be safe or may not work as well during treatment with VENCLEXTA.
- are pregnant or plan to become pregnant. VENCLEXTA may harm your unborn baby.
- If you are able to become pregnant, your healthcare provider should do a pregnancy test before you start treatment with VENCLEXTA.
- Females who are able to become pregnant should use effective birth control during treatment and for at least 30 days after the last dose of VENCLEXTA.
- If you become pregnant or think you are pregnant, tell your healthcare provider right away.
- are breastfeeding or plan to breastfeed. It is not known if VENCLEXTA passes into your breast milk. Do not breastfeed during treatment with VENCLEXTA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. VENCLEXTA and other medicines may affect each other causing serious side effects. See “Who should not take VENCLEXTA?”
- Take VENCLEXTA exactly as your healthcare provider tells you to take it. Do not change your dose of VENCLEXTA or stop taking VENCLEXTA unless your healthcare provider tells you to.
- When you first take VENCLEXTA:
- You may need to take VENCLEXTA at a hospital or clinic to be monitored for TLS.
- If you are taking VENCLEXTA for CLL or SLL, your healthcare provider will start VENCLEXTA at a low-dose. Your dose will be slowly increased weekly over 5 weeks up to the full dose. Read the Quick Start Guide that comes with VENCLEXTA before your first dose.
- If you are taking VENCLEXTA for AML, your healthcare provider will start VENCLEXTA at a low-dose. Your dose will be slowly increased daily up to the full dose. Follow your healthcare provider’s instructions carefully while increasing to the full dose.
- Follow the instructions about drinking water described in the section of this Medication Guide about TLS called “What is the most important information I should know about VENCLEXTA?” and also in the Quick Start Guide.
- Take VENCLEXTA 1 time a day with a meal and water at about the same time each day.
- Swallow VENCLEXTA tablets whole. Do not chew, crush, or break the tablets.
- If you miss a dose of VENCLEXTA and it has been less than 8 hours, take your dose as soon as possible. If you miss a dose of VENCLEXTA and it has been more than 8 hours, skip the missed dose and take the next dose at your usual time.
- If you vomit after taking VENCLEXTA, do not take an extra dose. Take the next dose at your usual time the next day.
What should I avoid while taking VENCLEXTA?
You should not drink grapefruit juice, eat grapefruit, Seville oranges (often used in marmalades), or starfruit while you are taking VENCLEXTA. These products may increase the amount of VENCLEXTA in your blood.
What are the possible side effects of VENCLEXTA?
VENCLEXTA can cause serious side effects, including:
- See "What is the most important information I should know about VENCLEXTA?"
- Low white blood cell count (neutropenia). Low white blood cell counts are common with VENCLEXTA but can also be severe. Your healthcare provider will do blood tests to check your blood counts during treatment with VENCLEXTA.
- Infections. Death and serious infections such as pneumonia and blood infection (sepsis) have happened during treatment with VENCLEXTA. Your healthcare provider will closely monitor and treat you right away if you have fever or any signs of infection during treatment with VENCLEXTA.
Tell your healthcare provider right away if you have a fever or any signs of an infection during treatment with VENCLEXTA.
The most common side effects of VENCLEXTA when used in combination with obinutuzumab or rituximab or alone in people with CLL or SLL include:
- low platelet counts
- low red blood cell counts
- diarrhea
- nausea
- upper respiratory tract infection
- cough
- muscle and joint pain
- tiredness
- swelling of your arms, legs, hands, and feet
The most common side effects of VENCLEXTA in combination with azacitidine or decitabine or low-dose cytarabine in people with AML include:- nausea
- diarrhea
- low platelet counts
- constipation
- fever with low white blood cell counts
- low red blood cell counts
- infection in blood
- rash
- dizziness
- low blood pressure
- fever
- swelling of your arms, legs, hands, and feet
- vomiting
- tiredness
- shortness of breath
- bleeding
- infection in lung
- stomach (abdominal) pain
- pain in muscles or back
- cough
- sore throat
VENCLEXTA may cause fertility problems in males. This may affect your ability to father a child. Talk to your healthcare provider if you have concerns about fertility.
These are not all the possible side effects of VENCLEXTA. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
- Store VENCLEXTA at or below 86°F (30°C).
- For people with CLL/SLL, keep VENCLEXTA tablets in the original package during the first 4 weeks of treatment. Do not transfer the tablets to a different container.
General information about the safe and effective use of VENCLEXTA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use VENCLEXTA for a condition for which it was not prescribed. Do not give VENCLEXTA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about VENCLEXTA that is written for health professionals.
What are the ingredients in VENCLEXTA?
Inactive ingredients: copovidone, colloidal silicon dioxide, polysorbate 80, sodium stearyl fumarate, and calcium phosphate dibasic.
The 10 mg and 100 mg coated tablets also include: iron oxide yellow, polyvinyl alcohol, polyethylene glycol, talc, and titanium dioxide. The 50 mg coated tablets also include: iron oxide yellow, iron oxide red, iron oxide black, polyvinyl alcohol, talc, polyethylene glycol, and titanium dioxide.
Manufactured and Marketed by:
AbbVie Inc.
North Chicago, IL 60064
© 2019 AbbVie Inc.
03-B947Marketed by:
Genentech USA, Inc.
A Member of the Roche Group
South San Francisco, CA 94080-4990
© 2019 Genentech, Inc.For more information go to www.venclexta.com or call 1-800-633-9110 This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 07/2019 -
PRINCIPAL DISPLAY PANEL
NDC: 0074-0579-28
Contact your doctor when you receive this medication.
It may be necessary to take your first dose in the presence of your doctor to prevent a potential serious side effect.
DISPENSER: Each time VENCLEXTA is dispensed give the patient the enclosed Medication Guide.
Dispense the accompanying Medication Guide to each patient.

NDC: 0074-0566-11
Dispense the accompanying Medication Guide to each patient.
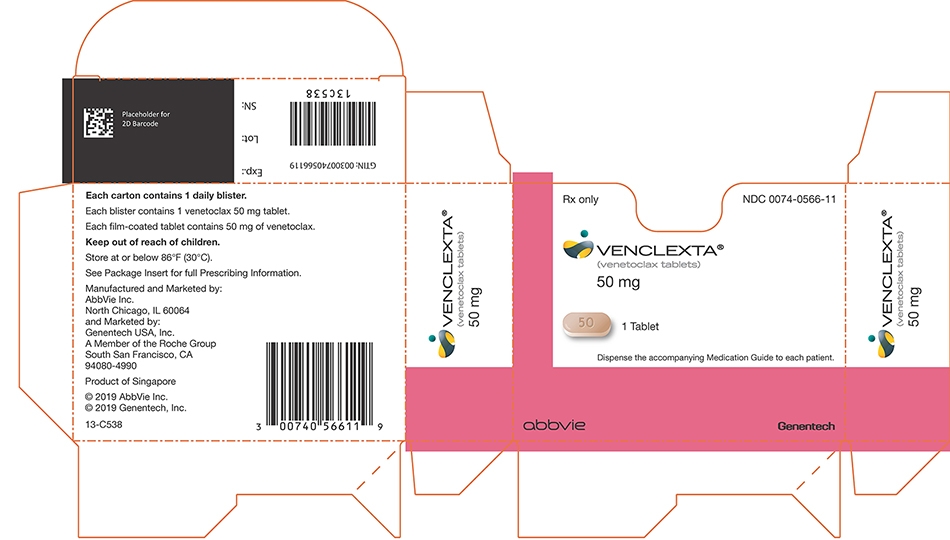
NDC: 0074-0576-22
Do not accept if seal over bottle opening is broken or missing.
Each film-coated tablet contains 100 mg of venetoclax.
Keep out of reach of children.
Store at or below 86°F (30°C).

NDC: 0074-0576-22
Dispense the accompanying Medication Guide to each patient.

Dispense the accompanying Medication Guide to each patient.
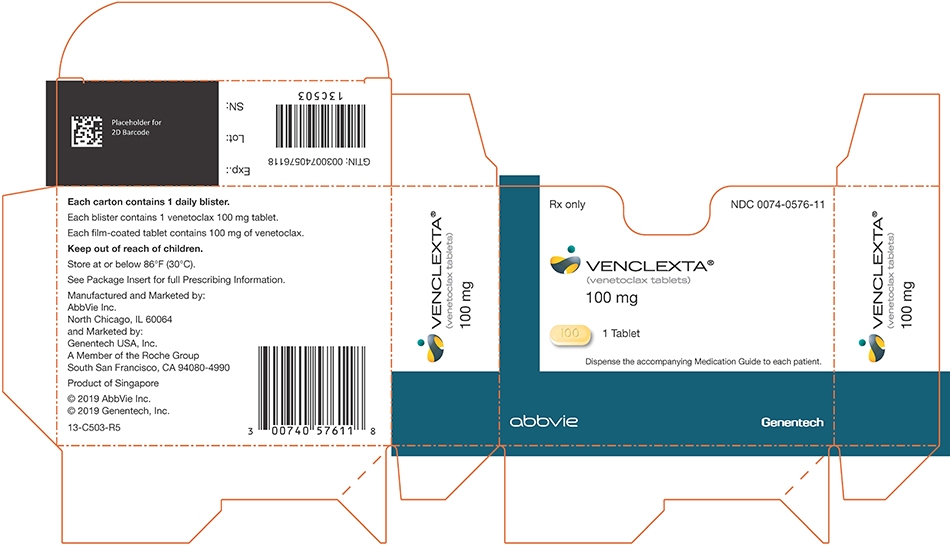
-
INGREDIENTS AND APPEARANCE
VENCLEXTA
venetoclax kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0074-0579 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0074-0579-28 1 in 1 CARTON 04/11/2016 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BLISTER PACK 14 Part 2 1 BLISTER PACK 7 Part 3 1 BLISTER PACK 7 Part 4 1 BLISTER PACK 14 Part 1 of 4 VENCLEXTA
venetoclax tablet, film coatedProduct Information Item Code (Source) NDC: 0074-0561 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Venetoclax (UNII: N54AIC43PW) (Venetoclax - UNII:N54AIC43PW) Venetoclax 10 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) ANHYDROUS DIBASIC CALCIUM PHOSPHATE (UNII: L11K75P92J) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Talc (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) Product Characteristics Color YELLOW (pale yellow) Score no score Shape ROUND (round, biconvex) Size 6mm Flavor Imprint Code V;10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 14 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208573 Part 2 of 4 VENCLEXTA
venetoclax tablet, film coatedProduct Information Item Code (Source) NDC: 0074-0566 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Venetoclax (UNII: N54AIC43PW) (Venetoclax - UNII:N54AIC43PW) Venetoclax 50 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) ANHYDROUS DIBASIC CALCIUM PHOSPHATE (UNII: L11K75P92J) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Talc (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) Product Characteristics Color BROWN (beige) Score no score Shape OVAL (oblong, biconvex) Size 14mm Flavor Imprint Code V;50 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208573 Part 3 of 4 VENCLEXTA
venetoclax tablet, film coatedProduct Information Item Code (Source) NDC: 0074-0576 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Venetoclax (UNII: N54AIC43PW) (Venetoclax - UNII:N54AIC43PW) Venetoclax 100 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) ANHYDROUS DIBASIC CALCIUM PHOSPHATE (UNII: L11K75P92J) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Talc (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) Product Characteristics Color YELLOW (pale yellow) Score no score Shape OVAL (oblong, biconvex) Size 17mm Flavor Imprint Code V;100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208573 Part 4 of 4 VENCLEXTA
venetoclax tablet, film coatedProduct Information Item Code (Source) NDC: 0074-0576 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Venetoclax (UNII: N54AIC43PW) (Venetoclax - UNII:N54AIC43PW) Venetoclax 100 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) ANHYDROUS DIBASIC CALCIUM PHOSPHATE (UNII: L11K75P92J) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Talc (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) Product Characteristics Color YELLOW (pale yellow) Score no score Shape OVAL (oblong, biconvex) Size 17mm Flavor Imprint Code V;100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 14 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208573 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208573 04/11/2016 VENCLEXTA
venetoclax tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0074-0576 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Venetoclax (UNII: N54AIC43PW) (Venetoclax - UNII:N54AIC43PW) Venetoclax 100 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) ANHYDROUS DIBASIC CALCIUM PHOSPHATE (UNII: L11K75P92J) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Talc (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) Product Characteristics Color YELLOW (pale yellow) Score no score Shape OVAL (oblong, biconvex) Size 17mm Flavor Imprint Code V;100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0074-0576-22 120 in 1 BOTTLE; Type 0: Not a Combination Product 04/11/2016 2 NDC: 0074-0576-11 1 in 1 CARTON 04/11/2016 2 1 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 0074-0576-34 1 in 1 CARTON 11/21/2018 3 180 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208573 04/11/2016 VENCLEXTA
venetoclax tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0074-0561 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Venetoclax (UNII: N54AIC43PW) (Venetoclax - UNII:N54AIC43PW) Venetoclax 10 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) ANHYDROUS DIBASIC CALCIUM PHOSPHATE (UNII: L11K75P92J) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Talc (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) Product Characteristics Color YELLOW (pale yellow) Score no score Shape ROUND (round, biconvex) Size 6mm Flavor Imprint Code V;10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0074-0561-14 1 in 1 CARTON 04/11/2016 1 14 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 0074-0561-11 1 in 1 CARTON 04/11/2016 2 2 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208573 04/11/2016 VENCLEXTA
venetoclax tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0074-0566 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Venetoclax (UNII: N54AIC43PW) (Venetoclax - UNII:N54AIC43PW) Venetoclax 50 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) ANHYDROUS DIBASIC CALCIUM PHOSPHATE (UNII: L11K75P92J) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Talc (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) Product Characteristics Color BROWN (beige) Score no score Shape OVAL (oblong, biconvex) Size 14mm Flavor Imprint Code V;50 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0074-0566-07 1 in 1 CARTON 04/11/2016 1 7 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 0074-0566-11 1 in 1 CARTON 04/11/2016 2 1 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208573 04/11/2016 Labeler - AbbVie Inc. (078458370)
Trademark Results [Venclexta]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VENCLEXTA 86855625 5079634 Live/Registered |
AbbVie Inc. 2015-12-21 |
 VENCLEXTA 86560752 5078740 Live/Registered |
AbbVie Inc. 2015-03-11 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.