CALQUENCE- acalabrutinib tablet, film coated
CALQUENCE by
Drug Labeling and Warnings
CALQUENCE by is a Prescription medication manufactured, distributed, or labeled by AstraZeneca Pharmaceuticals LP, AstraZeneca PLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use CALQUENCE safely and effectively. See full prescribing information for CALQUENCE.
CALQUENCE® (acalabrutinib) tablets, for oral use
Initial U.S. Approval: 2017RECENT MAJOR CHANGES
INDICATIONS AND USAGE
CALQUENCE is a kinase inhibitor indicated:
- In combination with bendamustine and rituximab for the treatment of adult patients with previously untreated mantle cell lymphoma (MCL) who are ineligible for autologous hematopoietic stem cell transplantation (HSCT). (1.1)
- For the treatment of adult patients with MCL who have received at least one prior therapy. (1.2)
- For the treatment of adult patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). (1.3)
DOSAGE AND ADMINISTRATION
- Recommended dose is 100 mg orally approximately every 12 hours; swallow whole with water and with or without food. (2.1)
- Advise patients not to chew, crush, dissolve, or cut tablets. (2.1)
- Manage toxicities using treatment interruption, dose reduction, or discontinuation. (2.3)
- Avoid CALQUENCE in patients with severe hepatic impairment. (8.6)
DOSAGE FORMS AND STRENGTHS
Tablets: 100 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Serious and Opportunistic Infections: Monitor for signs and symptoms of infection and treat promptly. (5.1)
- Hemorrhage: Monitor for bleeding and manage appropriately. (5.2)
- Cytopenias: Monitor complete blood counts regularly. (5.3)
- Second Primary Malignancies: Other malignancies have occurred, including skin cancers and other solid tumors. Advise patients to use sun protection. (5.4)
- Cardiac Arrhythmias: Monitor for symptoms of arrhythmias and manage. (5.5)
- Hepatotoxicity, Including Drug Induced Liver Injury: Monitor hepatic function throughout treatment. (5.6)
ADVERSE REACTIONS
The most common adverse reactions (≥ 30%), excluding laboratory abnormalities, are upper respiratory tract infection, diarrhea, headache, and musculoskeletal pain. The most common Grade 3 or 4 laboratory abnormalities (≥ 10%) are absolute neutrophil count decreased, uric acid increased, absolute lymphocyte count decreased, and platelets decreased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Previously Untreated Mantle Cell Lymphoma
1.2 Previously Treated Mantle Cell Lymphoma
1.3 Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Recommended Dosage for Drug Interactions
2.3 Dosage Modifications for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious and Opportunistic Infections
5.2 Hemorrhage
5.3 Cytopenias
5.4 Second Primary Malignancies
5.5 Cardiac Arrhythmias
5.6 Hepatotoxicity, Including Drug-Induced Liver Injury
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on CALQUENCE
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Previously Untreated Mantle Cell Lymphoma DOCVARIABLE vault_nd_4d1e0c80-2ff6-409f-bee7-897264eadbff \* MERGEFORMAT
14.2 Previously Treated Mantle Cell Lymphoma
14.3 Chronic Lymphocytic Leukemia
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Previously Untreated Mantle Cell Lymphoma
CALQUENCE in combination with bendamustine and rituximab is indicated for the treatment of adult patients with previously untreated mantle cell lymphoma (MCL) who are ineligible for autologous hematopoietic stem cell transplantation (HSCT).
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
CALQUENCE Administration Instructions
Advise patients to swallow tablet whole with water. Advise patients not to chew, crush, dissolve, or cut the tablets. CALQUENCE may be taken with or without food. If a dose of CALQUENCE is missed by more than 3 hours, it should be skipped, and the next dose should be taken at its regularly scheduled time. Extra tablets of CALQUENCE should not be taken to make up for a missed dose.
CALQUENCE as Monotherapy
For patients with MCL, CLL or SLL, the recommended dosage of CALQUENCE is 100 mg taken orally approximately every 12 hours until disease progression or unacceptable toxicity.
CALQUENCE in Combination with Bendamustine and Rituximab
For patients with previously untreated MCL, the recommended dosage of CALQUENCE is 100 mg taken orally approximately every 12 hours until disease progression or unacceptable toxicity.
Start CALQUENCE on Day 1 of Cycle 1 (each cycle is 28 days) and administer until disease progression or unacceptable toxicity. Administer bendamustine 90 mg/m2 on Days 1 and 2 and rituximab 375 mg/m2 on Day 1 of Cycle 1 and continue for a total of 6 cycles. Patients achieving a response (PR or CR) after the first 6 cycles may receive maintenance rituximab on Day 1 of every other cycle for a maximum of 12 additional doses, starting on Cycle 8 up to Cycle 30 [see Clinical Studies (14.1)].
CALQUENCE in Combination with Obinutuzumab
For patients with previously untreated CLL or SLL, the recommended dosage of CALQUENCE is 100 mg taken orally approximately every 12 hours until disease progression or unacceptable toxicity. Start CALQUENCE at Cycle 1 (each cycle is 28 days). Start obinutuzumab at Cycle 2 for a total of 6 cycles and refer to the obinutuzumab prescribing information for recommended dosing. Administer CALQUENCE prior to obinutuzumab when given on the same day.
CALQUENCE in Combination with Venetoclax
For patients with previously untreated CLL or SLL, the recommended dosage of CALQUENCE is 100 mg taken orally approximately every 12 hours until disease progression, unacceptable toxicity or completion of 14 cycles of treatment. Start CALQUENCE at Cycle 1 (each cycle is 28 days). Start venetoclax at Cycle 3 for total of 12 cycles. Start venetoclax at 20 mg daily for first week of treatment and increase weekly as per dosing schedule for 5-week ramp up (up to 400 mg daily) as described in the venetoclax USPI. Refer to the venetoclax USPI for additional details.
2.2 Recommended Dosage for Drug Interactions
Dosage Modifications for Use with CYP3A Inhibitors or Inducers
These are described in Table 1 [see Drug Interactions (7)].
Table 1: Recommended Dosage Modifications for Use with CYP3A Inhibitors or Inducers CYP3A
Co-administered Drug
Recommended CALQUENCE use
Inhibition
Strong CYP3A inhibitor
Avoid co-administration.
If these inhibitors will be used short-term (such as anti‑infectives for up to seven days), interrupt CALQUENCE.
After discontinuation of strong CYP3A inhibitor for at least 24 hours, resume previous dosage of CALQUENCE.
Moderate CYP3A inhibitor
Reduce the CALQUENCE 100 mg every 12 hours dosage to 100 mg once daily.
Induction
Strong CYP3A inducer
Avoid co-administration.
If co-administration is unavoidable, increase CALQUENCE dosage to 200 mg approximately every 12 hours.
2.3 Dosage Modifications for Adverse Reactions
Recommended dosage modifications are provided in Table 2, 3 and 4.
Table 2: Recommended Dosage Modifications for Adverse Reactions in Patients Receiving CALQUENCE Monotherapy and CALQUENCE in Combination with Obinutuzumab Event Adverse Reaction Occurrence Dosage Modification
(Starting dose = 100 mg approximately every 12 hours)Grade 3 or greater non-hematologic toxicities,
Grade 3 thrombocytopenia with bleeding,
Grade 4 thrombocytopenia or
Grade 4 neutropenia lasting longer than 7 days
First and Second
Interrupt CALQUENCE.
Once toxicity has resolved to Grade 1 or baseline level, CALQUENCE may be resumed at 100 mg approximately every 12 hours.
Third
Interrupt CALQUENCE.
Once toxicity has resolved to Grade 1 or baseline level, CALQUENCE may be resumed at a reduced frequency of 100 mg once daily.
Fourth
Discontinue CALQUENCE.
Adverse reactions graded by the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE).
Table 3: Recommended Dosage Modifications for Adverse Reactions in Patients Receiving CALQUENCE in Combination with BR Adverse Reaction
Severitya
Dosage Modification
(Starting dosage of CALQUENCE = 100 mg approximately every 12 hours)
Neutropeniab[see Warnings and Precautions (5.3)]
Absolute neutrophil count less than 0.5 x 109/L for greater than 7 days
Interrupt CALQUENCE. Once toxicity has resolved to Grade ≤ 2, resume CALQUENCE at starting dosage.
Upon 2nd or 3rd occurrence, reduce dosage of CALQUENCE to 100 mg once daily.c
Discontinue CALQUENCE at 4th occurrence.
For bendamustineb:
Interrupt bendamustine. Once toxicity has resolved to Grade ≤ 2, resume bendamustine and consider dosage reduction to 70 mg/m2.d,e
Thrombocytopeniaf[see Warnings and Precautions (5.3)]
Platelet count 25 to 50 x 109/L with clinically significant bleeding or platelet count less than 25 x 109/L
Interrupt CALQUENCE. Once toxicity has resolved to Grade ≤ 2 or baseline, resume CALQUENCE at starting dosage.
If recurrence, reduce dosage of CALQUENCE to 100 mg once daily.c
Consider discontinuing CALQUENCE at 3rd occurrence.
For bendamustinef:
Interrupt bendamustine. Once toxicity has resolved to Grade ≤ 2 or baseline, resume bendamustine and consider dose reduction to 70 mg/m2.e
Non-hematologic adverse reactions [see Warnings and Precautions (5)]
Grade 3 or higher
Interrupt CALQUENCE. Once toxicity has resolved to Grade ≤ 2 or baseline, resume CALQUENCE at starting dosage.
If recurrence, reduce dosage of CALQUENCE to 100 mg once daily.c
Discontinue CALQUENCE at 3rd occurrence of Grade 4 toxicity. For Grade 3 toxicity, consider the risks and benefits of continuing CALQUENCE.
For bendamustine:
Interrupt bendamustine. Once toxicity has resolved to Grade ≤ 2 or baseline, resume bendamustine and consider dose reduction to 70 mg/m2.e
a Graded per National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03.
b For neutropenia with ANC less than 1 x 109/L, consideration for bendamustine dose interruption and dosage reduction to 70 mg/m2 may be appropriate in certain circumstances.
c Dose may be re-escalated at the discretion of the physician if patient tolerates a reduced dose for ≥4 weeks.
d Consider use of myeloid growth factors before bendamustine dosage reduction.
e Consider discontinuing bendamustine if additional dosage reduction is required.
f For thrombocytopenia, a platelet count below 50 x 109/L should prompt bendamustine dose interruption even in the absence of clinically significant bleeding.
Table 4: Recommended Dosage Modifications for Adverse Reactions in Patients Receiving CALQUENCE in Combination with Venetoclax Adverse Reactiona
Adverse Reaction Occurrence
Dose Modification
Grade 3 or 4 neutropenia with or without fever and/or infection; Grade 4 neutropenia lasting more than 7 days
First occurrence
Interrupt CALQUENCE and/or venetoclax.b
Once toxicity resolves to Grade ≤ 1 or baseline, restart CALQUENCE and/or venetoclax at same dose.
Second occurrence
Interrupt CALQUENCE and/or venetoclax.b
Once toxicity resolves to Grade ≤ 1 or baseline, restart CALQUENCE at same dose and venetoclax at one lower dose levelc.
Subsequent occurrence
Withhold CALQUENCE and/or venetoclax until toxicity resolves to Grade ≤ 1 or baseline.b,d
Grade 3 or 4 thrombocytopenia and/or bleedingf
First occurrence
Interrupt CALQUENCE and/or venetoclax. When bleeding resolves and thrombocytopenia is Grade ≤ 1 or baseline without transfusion support for 5 consecutive days, restart CALQUENCE and/or venetoclax at same dose.
Second occurrence
Interrupt CALQUENCE and venetoclax until resolution of bleeding and thrombocytopenia resolves to Grade ≤ 1 or baseline.
Restart CALQUENCE at same dose and/or restart venetoclax at one lower dose level.e
Subsequent occurrences of severe thrombocytopenia
Interrupt CALQUENCE and venetoclax until resolution of bleeding and thrombocytopenia resolves to Grade ≤ 1 or baseline.
Restart CALQUENCE at a reduced frequency of 100 mg once daily and/or venetoclax at one lower dose level.c,d,e
Grade 3 or 4 tumour lysis syndrome (TLS)
First and subsequent episodes
If a subject experiences blood chemistry changes suggestive of TLS, the following day’s venetoclax and acalabrutinib dose should be withheld. If resolved within 24–48 hours of last dose, treatment can be resumed at the same dose.
For events of clinical TLS or blood chemistry changes requiring more than 48 hours to resolve, venetoclax should be resumed at one lower dose level.c When resuming treatment after interruption due to TLS, monitor for TLS and provide prophylaxis.
Grade 3 other non-hematologic eventsg
First occurrence
Interrupt CALQUENCE and/or venetoclax until toxicity resolves to Grade ≤ 1.
Restart CALQUENCE and/or venetoclax at same dose.
Second occurrence
Interrupt CALQUENCE and/or venetoclax until toxicity resolves to Grade ≤ 1d.
Grade 4 other non-hematologic eventsg
First occurrence
Interrupt CALQUENCE and/or venetoclax until toxicity resolves to Grade ≤ 1. Restart CALQUENCE at a reduced frequency of 100 mg once daily and/or venetoclax at one lower dose level.c, e
Second occurrence
Interrupt CALQUENCE and/or venetoclax until toxicity resolves to Grade ≤ 1d.
a Adverse reactions graded by the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 5.0.
b Growth factor may be used at physician discretion.
c See venetoclax USPI for dose level reductions details.
d Clinical judgment of the treating physician should guide the management plan of each patient based on the individual benefit/risk assessment for treatment with CALQUENCE in combination with venetoclax.
e CALQUENCE dose may be re-escalated at the discretion of the physician if patient tolerates a reduced dose for ≥4 weeks.
f Platelets may be used at physician discretion.
g Certain treatment-emergent non-hematologic AEs (e.g., venous thromboembolic events) may be managed and become clinically stable following medical intervention but may not improve to Grade ≤ 1 according to the NCI CTCAE definitions. In such cases, if a subject is clinically stable, resumption of CALQUENCE may be possible based on clinical judgement of the treating physician.
Refer to the prescribing information of each of the products used in combination with CALQUENCE for additional information for management of toxicities.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious and Opportunistic Infections
Fatal and serious infections, including opportunistic infections, have occurred in patients with hematologic malignancies treated with CALQUENCE.
Serious or Grade 3 or higher infections (bacterial, viral, or fungal) occurred in 29% of 2,055 patients exposed to CALQUENCE in clinical trials, most often due to respiratory tract infections (18% of all patients, including pneumonia in 14%) [see Adverse Reactions (6.1)]. These infections predominantly occurred in the absence of Grade 3 or 4 neutropenia, with neutropenic infection reported in 8% of all patients. Opportunistic infections in recipients of CALQUENCE have included, but are not limited to, hepatitis B virus reactivation, fungal pneumonia, Pneumocystis jirovecii pneumonia, Epstein-Barr virus reactivation, cytomegalovirus, and progressive multifocal leukoencephalopathy (PML). Consider prophylaxis in patients who are at increased risk for opportunistic infections. Monitor patients for signs and symptoms of infection and treat promptly.
In an additional cohort of patients receiving CALQUENCE in combination with venetoclax with obinutuzumab (AVO) (an unapproved regimen for previously untreated CLL/SLL in AMPLIFY), serious or Grade 3 or higher infections occurred in 25% receiving AVO compared to 14% in patients receiving AV. Fatal infections occurred in 6% receiving AVO compared to 3.1% of patients receiving AV, most commonly due to COVID-19. The safety and effectiveness of AVO has not been established in patients with previously untreated CLL/SLL [see Clinical Studies (14.3)].
5.2 Hemorrhage
Fatal and serious hemorrhagic events have occurred in patients treated with CALQUENCE. Major hemorrhage (serious or Grade 3 or higher bleeding or any central nervous system bleeding) occurred in 4.7% of patients, with fatal hemorrhage occurring in 0.1% of 2,055 patients exposed to CALQUENCE in clinical trials. Bleeding events of any grade, excluding bruising and petechiae, occurred in 39% of patients [see Adverse Reactions (6.1)].
Use of antithrombotic agents concomitantly with CALQUENCE may further increase the risk of hemorrhage. In clinical trials, major hemorrhage occurred in 5% of patients taking CALQUENCE without antithrombotic agents and 3.2% of patients taking CALQUENCE with antithrombotic agents. Consider the risks and benefits of antithrombotic agents when co-administered with CALQUENCE. Monitor patients for signs of bleeding.
Consider the benefit-risk of withholding CALQUENCE for 3 to 7 days pre- and post-surgery depending upon the type of surgery and the risk of bleeding.
5.3 Cytopenias
CALQUENCE can cause Grade 3 or 4 cytopenias. Grade 3 or 4 cytopenias included absolute neutrophil count decreased (28%), absolute lymphocyte count decreased (10%), hemoglobin decreased (9%), and platelets decreased (9%) in 1,758 patients treated with CALQUENCE alone and in combination with obinutuzumab or venetoclax; Grade 4 neutropenia developed in 14% [see Adverse Reactions (6.1)].
Monitor complete blood counts regularly during treatment. Interrupt treatment, reduce the dose, or discontinue treatment as warranted [see Dosage and Administration (2.3)].
5.4 Second Primary Malignancies
Second primary malignancies, including skin cancers and other solid tumors, occurred in 16% of 2,055 patients exposed to CALQUENCE in clinical trials [see Adverse Reactions (6.1)]. The most frequent second primary malignancy was non-melanoma skin cancer, reported in 9% of patients, followed by other solid tumors in 8% (including melanoma, lung cancer, gastrointestinal cancers, and genitourinary cancers) and hematologic malignancies (1.1%). Fatal second primary malignancies occurred in 0.8% of patients. Monitor patients for the development of second cancers and advise protection from sun exposure.
5.5 Cardiac Arrhythmias
Fatal and serious cardiac arrhythmias have occurred in patients treated with CALQUENCE. Grade 3 or 4 atrial fibrillation or flutter was reported in 2.2% of 2,055 patients treated with CALQUENCE, with all grades of atrial fibrillation or flutter reported in 7% of all patients [see Adverse Reactions (6.1)]. Grade 3 or higher ventricular arrhythmia events were reported in 0.5% of patients, including fatal cases in 0.3% of all patients. The risk of arrhythmias may be increased in patients with cardiac risk factors, hypertension, previous arrhythmias, and acute infection. Monitor for symptoms of arrhythmia (e.g., palpitations, dizziness, syncope, dyspnea) and manage as appropriate.
5.6 Hepatotoxicity, Including Drug-Induced Liver Injury
Hepatotoxicity, including severe, life-threatening, and potentially fatal cases of drug-induced liver injury (DILI), has occurred in patients treated with Bruton tyrosine kinase inhibitors, including CALQUENCE.
Evaluate bilirubin and transaminases at baseline and throughout treatment with CALQUENCE. For patients who develop abnormal liver tests after CALQUENCE, monitor more frequently for liver test abnormalities and clinical signs and symptoms of hepatic toxicity. If DILI is suspected, withhold CALQUENCE. Upon confirmation of DILI, discontinue CALQUENCE.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Serious and Opportunistic Infections [see Warnings and Precautions (5.1)]
- Hemorrhage [see Warnings and Precautions (5.2)]
- Cytopenias [see Warnings and Precautions (5.3)]
- Second Primary Malignancies [see Warnings and Precautions (5.4)]
- Cardiac Arrhythmias [see Warnings and Precautions (5.5)]
- Hepatotoxicity, including DILI [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the Warnings and Precautions reflect exposure to CALQUENCE 100 mg approximately every 12 hours in 2,055 patients with hematologic malignancies. Treatment includes CALQUENCE monotherapy in 1258 patients in 9 trials, and CALQUENCE combinations in 797 patients in 4 trials. Among these recipients of CALQUENCE, 89% were exposed for at least 6 months and 82% were exposed for at least one year. In this pooled safety population, adverse reactions in ≥ 30% of 2,055 patients, excluding laboratory abnormalities, were upper respiratory tract infection (37%), diarrhea (36%), headache (35%), and musculoskeletal pain (32%). The most common Grade 3 or 4 laboratory abnormalities (≥ 10%) were absolute neutrophil count decreased (32%), uric acid increased (27%), absolute lymphocyte count decreased (21%) and platelets decreased (10%).
Previously Untreated Mantle Cell Lymphoma
The safety data described below reflect exposure to CALQUENCE (100 mg approximately every 12 hours, with or without BR) in patients with MCL [see Clinical Studies (14.1)].
ECHO
The safety of CALQUENCE in combination with bendamustine and rituximab (CALQUENCE plus BR) was evaluated in 297 patients with previously untreated MCL in ECHO [see Clinical Studies (14.1)]. The trial enrolled patients with previously untreated MCL, ≥ 65 years of age with no intention for transplant, total bilirubin ≤ 1.5 × ULN, AST or ALT ≤ 2.5 × ULN, and estimated creatinine clearance of > 50 mL/min. Patients received 6 cycles (as 28-day cycles) of CALQUENCE 100 mg orally twice daily (n = 297) or placebo (n = 297) in combination with bendamustine and rituximab. Patients then received CALQUENCE 100 mg orally twice daily or placebo continuously until progressive disease or unacceptable toxicity, with 12 additional dosages of rituximab every other cycle up to Cycle 30.
The median duration of treatment with CALQUENCE was 28.6 months. A total of 171 (57.6%) patients were treated with CALQUENCE for ˃ 24 months and 122 (41.1%) patients were treated for ˃ 36 months.
Serious adverse reactions occurred in 69% of patients who received CALQUENCE plus BR. Serious adverse reactions reported in ≥ 2% of patients were pneumonia (23%; includes COVID-19 pneumonia), COVID-19 (20%; includes COVID-19 pneumonia), pyrexia (6%), second primary malignancy (7%), rash (3.4%), febrile neutropenia (3.4%), atrial fibrillation (3%), sepsis (2.7%), and anemia (2.4%). Fatal adverse reactions that occurred within 30 days of the last study treatment were reported in 12% who received CALQUENCE plus BR including COVID-19 (6%; includes COVID-19 pneumonia), pneumonia (1%), sepsis (0.3%), second primary malignancy (0.7%), and pneumonitis (0.3%).
Adverse reactions led to permanent discontinuation of CALQUENCE in 43%, dosage interruptions in 74%, and dosage reductions in 10% of patients. Adverse reactions that resulted in dosage modification in > 10% included infections, cytopenias, rashes, and gastrointestinal toxicity. Adverse reactions which resulted in permanent discontinuation of CALQUENCE in ≥ 4% of patients included COVID-19 (includes COVID-19 pneumonia) and neutropenia.
Table 5 and Table 6 summarize select adverse reactions and laboratory abnormalities observed in patients treated in ECHO.
Table 5: Adverse Reactions* (≥ 15%) in Patients with Previously Untreated MCL Who Received CALQUENCE plus BR in ECHO Body System
Adverse Reactions*
CALQUENCE plus BR
N = 297
Placebo plus BR
N = 297
All Grades
(%)
Grade
3 or 4 (%)
All Grades
(%)
Grade
3 or 4 (%)
Skin and subcutaneous tissue disorders
Rasha
47
12
31
3
Infections
COVID-19b
38
13
27
11
Upper respiratory tract infectionc
30
0.7
29
1
Pneumoniadd
31
17
25
14
Gastrointestinal disorders
Diarrhea
37
3
28
2.4
Vomiting
26
0.7
14
1
Constipation
25
1
25
0.3
General disorders
Fatigue
37
3.7
32
4.4
Pyrexia
29
2.4
24
1.3
Edema
20
1.3
19
0
Nervous system disorders
Headache
31
1.7
14
0.7
Dizziness
18
1
17
0.3
Respiratory, thoracic and mediastinal disorders
Cough
27
0
20
0.3
Dyspnea
17
1
11
2.7
Neoplasms
Secondary primary malignancye
19
7
15
7
Musculoskeletal and connective tissue disorders
Arthralgia
18
0.7
16
1
Vascular disorders
Hemorrhagef
20
1.7
11
3
*Excludes laboratory terms.
a Includes rash, dermatitis, and other related terms.
b Includes the following fatal adverse reactions: n=24 for COVID-19.
c Includes upper respiratory tract infection, sinusitis, pharyngitis, and related terms.
d Includes pneumonia, terms containing pneumonia, and related infections. COVID-19 pneumonia is represented under both Pneumonia and COVID-19.
e Includes terms related to malignant neoplasms including cutaneous neoplasms.
f Includes all terms containing hematoma or hemorrhage and related terms indicative of bleeding.
Clinically relevant adverse reactions in < 15% of patients receiving CALQUENCE plus BR included bruising, abdominal pain, atrial fibrillation or flutter, and tumor lysis syndrome.
Table 6: Select Laboratory Abnormalities (≥ 15%) in Patients with Previously Untreated MCL in ECHO Laboratory Abnormality
CALQUENCE plus BRa
Placebo plus BRa
All grade
(%)
Grade
3 or 4 (%)
All grade
(%)
Grade
3 or 4 (%)
Hematologic Abnormalities
Lymphocytes decreased
98
87
97
89
Hemoglobin decreased
80
11
65
11
Neutrophils decreased
76
56
77
51
Platelets decreased
69
18
60
16
Chemistry Abnormalities
AST increased
53
5
50
3.4
Uric acid increased
45
45
40
40
ALT increased
44
7
41
2.4
Potassium increased
40
2
38
2.7
Creatinine increased
37
3
28
2.4
Phosphate decreased
36
4.4
30
4.7
Potassium decreased
29
7
23
6
Bilirubin increased
19
2
12
2
a The denominator used to calculate the rate varied between 296 and 297 based on the number of patients with a baseline value and at least one post-treatment value.
Grade 4 laboratory abnormalities in > 15% of patients treated with CALQUENCE plus BR include absolute lymphocyte count decreased (26%), absolute neutrophil count decreased (36%), and uric acid increased (17%).
Previously Treated Mantle Cell Lymphoma
ACE-LY-004
The safety data described in this section reflect exposure to CALQUENCE (100 mg approximately every 12 hours) in 124 patients with previously treated MCL in Trial LY-004 [see Clinical Studies (14.2)]. The median duration of treatment with CALQUENCE was 16.6 (range: 0.1 to 26.6) months. A total of 91 (73.4%) patients were treated with CALQUENCE for ≥ 6 months and 74 (59.7%) patients were treated for ≥ 1 year.
The most common adverse reactions (≥ 20%) of any grade were anemia, thrombocytopenia, headache, neutropenia, diarrhea, fatigue, myalgia, and bruising. Grade 1 severity for the non-hematologic, most common events were as follows: headache (25%), diarrhea (16%), fatigue (20%), myalgia (15%), and bruising (19%). The most common Grade ≥ 3 non-hematological adverse reaction (reported in at least 2% of patients) was diarrhea.
Dose reductions and discontinuation due to any adverse reaction were reported in 1.6% and 6.5% of patients, respectively.
Tables 7 and 8 present the frequency category of adverse reactions observed in patients with MCL treated with CALQUENCE.
Table 7: Non-Hematologic Adverse Reactions in ≥ 5% (All Grades) of Patients with MCL in Trial LY-004 Body System
Adverse Reactions*
CALQUENCE Monotherapy
N=124
All Grades (%)
Grade ≥ 3 (%)
Nervous system disorders
Headache
39
1.6
Gastrointestinal disorders
Diarrhea
31
3.2
Nausea
19
0.8
Abdominal pain
15
1.6
Constipation
15
-
Vomiting
13
1.6
General disorders
Fatigue
28
0.8
Musculoskeletal and connective tissue disorders
Myalgia
21
0.8
Skin and subcutaneous tissue disorders
Bruisinga
21
-
Rashb
18
0.8
Vascular disorders
Hemorrhagec
8
0.8
Respiratory, thoracic and mediastinal disorders
Epistaxis
6
-
*Per NCI CTCAE version 4.03.
a Bruising: Includes all terms containing ‘bruise,’ ‘contusion,’ ‘petechiae,’ or ‘ecchymosis’.
b Rash: Includes all terms containing ‘rash’.
c Hemorrhage: Includes all terms containing ‘hemorrhage’ or ‘hematoma’.
Table 8: Hematologic Adverse Reactions Reported in ≥ 20% of Patients with MCL in Trial LY-004 Hematologic
Adverse Reactions*
CALQUENCE Monotherapy
N=124
All Grades (%)
Grade ≥ 3 (%)
Hemoglobin decreased
46
10
Platelets decreased
44
12
Neutrophils decreased
36
15
*Per NCI CTCAE version 4.03; based on laboratory measurements and adverse reactions.
Increases in creatinine to 1.5 to 3 times the upper limit of normal (ULN) occurred in 4.8% of patients.
Chronic Lymphocytic Leukemia
The safety data described below reflect exposure to CALQUENCE (100 mg approximately every 12 hours, with or without obinutuzumab) in 511 patients with CLL from two randomized controlled clinical trials [see Clinical Studies (14.3)].
The most common adverse reactions (≥ 30%) of any grade in patients with CLL were anemia, neutropenia, thrombocytopenia, headache, upper respiratory tract infection, and diarrhea.
ELEVATE-TN
The safety of CALQUENCE plus obinutuzumab (CALQUENCE+G), CALQUENCE monotherapy, and obinutuzumab plus chlorambucil (GClb) was evaluated in a randomized, multicenter, open-label, actively controlled trial in 526 patients with previously untreated CLL [see Clinical Studies (14.3)].
Patients randomized to the CALQUENCE+G arm were treated with CALQUENCE and obinutuzumab in combination for six cycles, then with CALQUENCE as monotherapy until disease progression or unacceptable toxicity. Patients initiated obinutuzumab on Day 1 of Cycle 2, continuing for a total of 6 cycles. Patient randomized to CALQUENCE monotherapy received CALQUENCE approximately every 12 hours until disease progression or unacceptable toxicity. The trial required age ≥ 65 years of age or 18 to < 65 years of age with a total Cumulative Illness Rating Scale (CIRS) > 6 or creatinine clearance of 30 to 69 mL/min, hepatic transaminases ≤ 3 times ULN and total bilirubin ≤ 1.5 times ULN, and allowed patients to receive antithrombotic agents other than warfarin or equivalent vitamin K antagonists.
During randomized treatment, the median duration of exposure to CALQUENCE in the CALQUENCE+G and CALQUENCE monotherapy arms was 27.7 months (range 0.3 to 40 months), with 95% and 92% and 89% and 86% of patients with at least 6 months and 12 months of exposure, respectively. In the obinutuzumab and chlorambucil arm the median number of cycles was 6 with 84% of patients receiving at least 6 cycles of obinutuzumab, 70% of patients received at least 6 cycles of chlorambucil. Eighty-five percent of patients in the CALQUENCE+G arm received at least 6 cycles of obinutuzumab.
In the CALQUENCE+G and CALQUENCE monotherapy arms, fatal adverse reactions that occurred in the absence of disease progression and with onset within 30 days of the last study treatment were reported in 2% for each treatment arm, most often from infection. Serious adverse reactions were reported in 39% of patients in the CALQUENCE+G arm and 32% in the CALQUENCE monotherapy arm, most often due to events of pneumonia (2.8% to 7%).
In the CALQUENCE+G arm, adverse reactions led to treatment discontinuation in 11% of patients and a dose reduction of CALQUENCE in 7% of patients. In the CALQUENCE monotherapy arm, adverse reactions led to discontinuation in 10% and dose reduction in 4% of patients.
Tables 9 and 10 present adverse reactions and laboratory abnormalities identified in the ELEVATE-TN trial.
Table 9: Common Adverse Reactions (≥ 15% Any Grade) with CALQUENCE in Patients with CLL (ELEVATE-TN) Body System
Adverse Reaction*
CALQUENCE plus Obinutuzumab
N=178CALQUENCE Monotherapy
N=179
Obinutuzumab plus Chlorambucil
N=169All Grades (%)
Grade ≥ 3 (%)
All Grades (%)
Grade ≥ 3 (%)
All Grades (%)
Grade ≥ 3 (%)
Infections
Infection†
69
22‡
65
14‡
46
13‡
Upper respiratory tract infection§
39
2.8
35
0
17
1.2
Lower respiratory tract infectiona
24
8
18
4.5
7
1.8
Urinary tract infection
15
1.7
15
2.8
5
0.6
Blood and lymphatic system disordersb
Neutropeniac
53
37
23
13
78
50
Anemiad
52
12
53
10
54
14
Thrombocytopeniae
51
12
32
3.4
61
16
Lymphocytosisf
12
11
16
15
0.6
0.6
Nervous system disorders
Headache
40
1.1
39
1.1
12
0
Dizziness
20
0
12
0
7
0
Gastrointestinal disorders
Diarrhea
39
4.5
35
0.6
21
1.8
Nausea
20
0
22
0
31
0
Musculoskeletal and connective tissue disorders
Musculoskeletal paing
37
2.2
32
1.1
16
2.4
Arthralgia
22
1.1
16
0.6
4.7
1.2
General disorders and administration site conditions
Fatigueh
34
2.2
23
1.1
24
1.2
Skin and subcutaneous tissue disorders
Bruisingi
31
0
21
0
5
0
Rashj
26
2.2
25
0.6
9
0.6
Vascular disorders
Hemorrhagek
20
1.7
20
1.7
6
0
*Per NCI CTCAE version 4.03.
† Includes any adverse reactions involving infection or febrile neutropenia.
‡ Includes 3 fatal cases in the CALQUENCE plus obinutuzumab arm, 3 fatal cases in the CALQUENCE monotherapy arm and 1 fatal case in the obinutuzumab plus chlorambucil arm.
§ Includes upper respiratory tract infection, nasopharyngitis and sinusitis.
a Includes pneumonia, lower respiratory tract infection, bronchitis, bronchiolitis, tracheitis, and lung infection.
b Derived from adverse reaction and laboratory data.
c Includes neutropenia, neutrophil count decreased, and related laboratory data.
d Includes anemia, red blood cell count decreased, and related laboratory data.
e Includes thrombocytopenia, platelet count decreased, and related laboratory data.
f Includes lymphocytosis, lymphocyte count increased, and related laboratory data.
g Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal pain, musculoskeletal discomfort, myalgia, neck pain, pain in extremity and spinal pain.
h Includes asthenia, fatigue, and lethargy.
i Includes bruise, contusion, and ecchymosis.
j Includes rash, dermatitis, and other related terms.
k Includes hemorrhage, hematoma, hemoptysis, hematuria, menorrhagia, hemarthrosis, and epistaxis.
Other clinically relevant adverse reactions (all grades incidence < 15%) in recipients of CALQUENCE (CALQUENCE in combination with obinutuzumab and monotherapy) included:
- Neoplasms: second primary malignancy (10%), non-melanoma skin cancer (5%)
- Cardiac disorders: atrial fibrillation or flutter (3.6%), hypertension (5%)
- Infection: herpesvirus infection (6%)
Table : Select Non-Hematologic Laboratory Abnormalities (≥ 15% Any Grade), New or Worsening from Baseline in Patients Receiving CALQUENCE (ELEVATE-TN) - * Per NCI CTCAE version 4.03
- † Excludes electrolytes
CALQUENCE plus Obinutuzumab
N=178CALQUENCE Monotherapy
N=179
Obinutuzumab plus Chlorambucil
N=169All Grades (%)
Grade ≥ 3 (%)
All Grades (%)
Grade ≥ 3 (%)
All Grades (%)
Grade ≥ 3 (%)
Uric acid increase
29
29
22
22
37
37
ALT increase
30
7
20
1.1
36
6
AST increase
38
5
17
0.6
60
8
Bilirubin increase
13
0.6
15
0.6
11
0.6
Increases in creatinine to 1.5 to 3 times ULN occurred in 3.9% and 2.8% of patients in the CALQUENCE combination arm and monotherapy arm, respectively.
ASCEND
The safety of CALQUENCE in patients with relapsed or refractory CLL was evaluated in a randomized, open-label study (ASCEND) [see Clinical Studies (14.3)]. The trial enrolled patients with relapsed or refractory CLL after at least one prior therapy and required hepatic transaminases ≤ 2 times ULN, total bilirubin ≤ 1.5 times ULN, and an estimated creatinine clearance ≥ 30 mL/min. The trial excluded patients having an absolute neutrophil count < 500/µL, platelet count < 30,000/µL, prothrombin time or activated partial thromboplastin time > 2 times ULN, significant cardiovascular disease, or a requirement for strong CYP3A inhibitors or inducers. Patients were allowed to receive antithrombotic agents other than warfarin or equivalent vitamin K antagonist.
In ASCEND, 154 patients received CALQUENCE (100 mg approximately every 12 hours until disease progression or unacceptable toxicity), 118 received idelalisib (150 mg approximately every 12 hours until disease progression or unacceptable toxicity) with up to 8 infusions of a rituximab product, and 35 received up to 6 cycles of bendamustine and a rituximab product. The median age overall was 68 years (range: 32-90); 67% were male; 92% were white; and 88% had an ECOG performance status of 0 or 1.
In the CALQUENCE arm, serious adverse reactions occurred in 29% of patients. Serious adverse reactions in > 5% of patients who received CALQUENCE included lower respiratory tract infection (6%). Fatal adverse reactions within 30 days of the last dose of CALQUENCE occurred in 2.6% of patients, including from second primary malignancies and infection.
In recipients of CALQUENCE, permanent discontinuation due to an adverse reaction occurred in 10% of patients, most frequently due to second primary malignancies followed by infection. Adverse reactions led to dosage interruptions of CALQUENCE in 34% of patients, most often due to respiratory tract infections followed by neutropenia, and dose reduction in 3.9% of patients.
Selected adverse reactions are described in Table 11 and non-hematologic laboratory abnormalities are described in Table 12. These tables reflect exposure to CALQUENCE with median duration of 15.7 months with 94% of patients on treatment for greater than 6 months and 86% of patients on treatment for greater than 12 months. The median duration of exposure to idelalisib was 11.5 months with 72% of patients on treatment for greater than 6 months and 48% of patients on treatment for greater than 12 months. Eighty-three percent of patients completed 6 cycles of bendamustine and rituximab product.
Table 11: Common Adverse Reactions (≥ 15% Any Grade) with CALQUENCE in Patients with CLL (ASCEND) Body System
Adverse Reaction*
CALQUENCE
N=154
Idelalisib plus
Rituximab
Product N=118
Bendamustine plus Rituximab Product
N=35
All Grades (%)
Grade ≥ 3 (%)
All Grades (%)
Grade ≥ 3 (%)
All Grades (%)
Grade ≥ 3 (%)
Infections
Infection†
56
15‡
65
28‡
49
11
Upper respiratory tract infection§
29
1.9
26
3.4
17
2.9
Lower respiratory tract infection a
23
6
26
15
14
6
Blood and lymphatic system disordersb
Neutropeniac
48
23
79
53
80
40
Anemiad
47
15
45
8
57
17
Thrombocytopeniae
33
6
41
13
54
6
Lymphocytosisf
26
19
23
18
2.9
2.9
Nervous system disorders
Headache
22
0.6
6
0
0
0
Gastrointestinal disorders
Diarrheag
18
1.3
49
25
14
0
Vascular disorders
Hemorrhageh
16
1.3
5
1.7
6
2.9
General disorders
Fatiguei
15
1.9
13
0.8
31
6
Musculoskeletal and connective tissue disorders
Musculoskeletal painj
15
1.3
15
1.7
2.9
0
* Per NCI CTCAE version 4.03.
† Includes any adverse reactions involving infection or febrile neutropenia.
‡ Includes 1 fatal case in the CALQUENCE monotherapy arm and 1 fatal case in the Idelalisib plus Rituximab arm.
§ Includes upper respiratory tract infection, rhinitis and nasopharyngitis.
a Includes pneumonia, lower respiratory tract infection, bronchitis, bronchiolitis, tracheitis, and lung infection.
b Derived from adverse reaction and laboratory data.
c Includes neutropenia, neutrophil count decreased, and related laboratory data.
d Includes anemia, red blood cell decreased, and related laboratory data.
e Includes thrombocytopenia, platelet count decreased, and related laboratory data.
f Includes lymphocytosis, lymphocyte count increased and related laboratory data.
g Includes colitis, diarrhea, and enterocolitis.
h Includes hemorrhage, hematoma, hemoptysis, hematuria, menorrhagia, hemarthrosis, and epistaxis.
i Includes asthenia, fatigue, and lethargy.
j Includes back pain, musculoskeletal chest pain, musculoskeletal pain, musculoskeletal discomfort, pain in extremity, myalgia, spinal pain and bone pain.
Other clinically relevant adverse reactions (all grades incidence < 15%) in recipients of CALQUENCE included:
- Skin and subcutaneous disorders: bruising (10%), rash (9%)
- Neoplasms: second primary malignancy (12%), non-melanoma skin cancer (6%)
- Musculoskeletal and connective tissue disorders: arthralgia (8%)
- Cardiac disorders: atrial fibrillation or flutter (5%), hypertension (3.2%)
- Infection: herpesvirus infection (4.5%)
Table 12: Select Non-Hematologic Laboratory Abnormalities (≥ 10% Any Grade), New or Worsening from Baseline in Patients Receiving CALQUENCE (ASCEND) Laboratory Abnormality* CALQUENCE
N=154Idelalisib plus Rituximab Product
N=118Bendamustine plus Rituximab Product
N=35All
Grades
(%)Grade ≥ 3 (%) All
Grades
(%)Grade ≥ 3 (%) All
Grades (%)Grade ≥ 3 (%) - * Excludes electrolytes
Uric acid increase
15
15
11
11
23
23
ALT increase
15
1.9
59
23
26
2.9
AST increase
13
0.6
48
13
31
2.9
Bilirubin increase
13
1.3
16
1.7
26
11
Per NCI CTCAE version 5
Increases in creatinine to 1.5 to 3 times ULN occurred in 1.3% of patients who received CALQUENCE.
AMPLIFY
The safety of CALQUENCE in patients with previously untreated CLL was evaluated in a randomized, multicenter, open-label study (AMPLIFY), in which 291 patients received CALQUENCE plus venetoclax (AV), and 259 patients received Investigator’s choice of FCR/BR (fludarabine plus cyclophosphamide plus rituximab or bendamustine plus rituximab) [see Clinical Studies (14.3)].
Among patients who received AV, 96% were exposed for 6 months or longer and 91% were exposed for greater than one year. The median duration of exposure to CALQUENCE was 12.9 months (range: 1 to 18 months) and to venetoclax was 11.1 months (range: 2 to 14 months).
Serious adverse reactions occurred in 25% of patients receiving AV. The most common serious adverse reactions (≥ 2%) were COVID-19 including COVID-19 pneumonia (9%), second primary malignancies (2.7%), and neutropenia (2.1%). Fatal adverse events occurred in 3.4% of patients. The most common fatal adverse events included COVID-19 and COVID-19 pneumonia.
Treatment discontinuation of CALQUENCE due to adverse reactions occurred in 8% of patients receiving AV. The most common adverse reaction (≥ 2%) leading to treatment discontinuation was COVID-19 pneumonia (2.1%). Dose reduction of CALQUENCE occurred in 6% of patients. Neutropenia was the only adverse reaction leading to dose reduction that occurred in ≥ 1% of patients.
Table 13 and Table 14 summarize select adverse reactions and laboratory abnormalities observed in patients treated in AMPLIFY.
Table 13: Adverse Reactions* (≥ 15% Any Grade) in Patients with Previously Untreated CLL Who Received CALQUENCE plus Venetoclax in AMPLIFY Body System
Adverse Reactions*
CALQUENCE plus Venetoclax
N = 291
Investigator’s choice of FCR or BR
N = 259
All Grades
(%)
Grade
3 or 4 (%)
All Grades
(%)
Grade
3 or 4 (%)
Nervous system disorders
Headache
35
1.4
8
0.4
Gastrointestinal disorders
Diarrhea
33
1.7
11
0.4
Nausea
15
0
36
0
Musculoskeletal and connective tissue disorders
Musculoskeletal pain a
25
0.7
14
0.8
Infections
COVID-19
21
6
3.9
1.5
General disorders
Fatigue b
18
0.3
17
1.5
Skin and subcutaneous tissue disorders
Bruising c
17
0
1.5
0
Rash d
16
1
16
1.5
*Excludes laboratory terms.
a Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal pain, musculoskeletal discomfort, myalgia, neck pain, pain in extremity, spinal pain, non-cardiac chest pain and pain in jaw.
b Includes fatigue and asthenia.
c Includes increased tendency to bruise, contusion, and ecchymosis.
d Includes rash, dermatitis, and other related terms.
Clinically relevant adverse reactions in < 15% of patients receiving CALQUENCE plus Venetoclax included upper respiratory tract infections, lower respiratory tract infection, arthralgia, pneumonia, hemorrhage, dizziness, constipation, vomiting, second primary malignancy and hypertension.
Table 14: Laboratory Abnormalities (≥ 15% Any Grade), New or Worsening from Baseline in in Patients with Previously Untreated CLL Who Received CALQUENCE plus Venetoclax in AMPLIFY Laboratory Abnormality CALQUENCE plus Venetoclax a Investigator’s choice of FCR or BR a All grade
(%)Grade
3 or 4 (%)All grade
(%)Grade
3 or 4 (%)Hematologic Abnormalities
Neutrophils decreased
78
38
80
53
Lymphocytes decreased
56
12
92
73
Platelets decreased
43
5
59
15
Hemoglobin decreased
35
7
56
8
Chemistry Abnormalities
Glucose increased
74
0
84
0
Calcium decreased
30
0.7
25
2.3
ALT increased
26
3.1
28
1.6
Urate increased
25
25
23
23
LDH increased
24
0
40
0
Potassium increased
22
2.4
12
3.1
AST increased
22
1.4
28
1.6
ALP increased
20
0
15
0
Glucose decreased
20
0.3
5
0
Creatinine increased
19
0.3
12
0.8
Sodium increased
15
0.3
9
0.4
a The denominator used to calculate the rate varied between 256 and 290 based on the number of patients with a baseline value and at least one post-treatment value.
Grade 4 laboratory abnormalities in > 15% of patients treated with CALQUENCE plus Venetoclax include absolute neutrophil count decreased (15%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of CALQUENCE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Cardiac disorders: ventricular arrhythmias
- Hepatobiliary disorders: drug induced liver injury
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on CALQUENCE
Strong CYP3A Inhibitors
Clinical Effect
Co-administration of CALQUENCE with a strong CYP3A inhibitor increased acalabrutinib plasma concentrations [see Clinical Pharmacology (12.3)]. Increased acalabrutinib concentrations may result in increased toxicity.
Prevention or Management
Avoid co-administration of CALQUENCE with strong CYP3A inhibitors. Alternatively, if the inhibitor will be used short-term, interrupt CALQUENCE [see Dosage and Administration (2.2)].
Moderate CYP3A Inhibitors
Clinical Effect
Co-administration of CALQUENCE with a moderate CYP3A inhibitor may increase acalabrutinib plasma concentration [see Clinical Pharmacology (12.3)]. Increased acalabrutinib concentrations may result in increased toxicity.
Prevention or Management
Reduce the dosage of CALQUENCE when co-administered with a moderate CYP3A inhibitor [see Dosage and Administration (2.2)].
Strong CYP3A Inducers
Clinical Effect
Co-administration of CALQUENCE with a strong CYP3A inducer decreased acalabrutinib plasma concentration [see Clinical Pharmacology (12.3)]. Decreased acalabrutinib concentrations may reduce CALQUENCE activity.
Prevention or Management
Avoid co-administration of CALQUENCE with strong CYP3A inducers. If co-administration is unavoidable, increase the dosage of CALQUENCE [see Dosage and Administration (2.2)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animals, CALQUENCE may cause fetal harm and dystocia when administered to a pregnant woman. There are no available data in pregnant women to inform the drug-associated risk. In animal reproduction studies, administration of acalabrutinib to animals during organogenesis resulted in dystocia in rats and reduced fetal growth in rabbits at maternal exposures (AUC) 2 times exposures in patients at the recommended dose of 100 mg approximately every 12 hours (see Data). Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In a combined fertility and embryo-fetal development study in female rats, acalabrutinib was administered orally at doses up to 200 mg/kg/day starting 14 days prior to mating through gestational day [GD] 17. No effects on embryo-fetal development and survival were observed. The AUC at 200 mg/kg/day in pregnant rats was approximately 9 times the AUC in patients at the recommended dose of 100 mg approximately every 12 hours. The presence of acalabrutinib and its active metabolite were confirmed in fetal rat plasma.
In an embryo-fetal development study in rabbits, pregnant animals were administered acalabrutinib orally at doses up to 200 mg/kg/day during the period of organogenesis (from GD 6-18). Administration of acalabrutinib at doses ≥ 100 mg/kg/day produced maternal toxicity and 100 mg/kg/day resulted in decreased fetal body weights and delayed skeletal ossification. The AUC at 100 mg/kg/day in pregnant rabbits was approximately 2 times the AUC in patients at 100 mg approximately every 12 hours.
In a pre- and postnatal development study in rats, acalabrutinib was administered orally to pregnant animals during organogenesis, parturition and lactation, at doses of 50, 100, and 150 mg/kg/day. Dystocia (prolonged or difficult labor) and mortality of offspring were observed at doses ≥ 100 mg/kg/day. The AUC at 100 mg/kg/day in pregnant rats was approximately 2 times the AUC in patients at 100 mg approximately every 12 hours. Underdeveloped renal papilla was also observed in F1 generation offspring at 150 mg/kg/day with an AUC approximately 5 times the AUC in patients at 100 mg approximately every 12 hours.
8.2 Lactation
Risk Summary
No data are available regarding the presence of acalabrutinib or its active metabolite in human milk, its effects on the breastfed child, or on milk production. Acalabrutinib and its active metabolite were present in the milk of lactating rats. Due to the potential for adverse reactions in a breastfed child from CALQUENCE, advise lactating women not to breastfeed while taking CALQUENCE and for 2 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
CALQUENCE may cause embryo-fetal harm and dystocia when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential prior to initiating CALQUENCE therapy.
Contraception
Females
Advise female patients of reproductive potential to use effective contraception during treatment with CALQUENCE and for 1 week following the last dose of CALQUENCE. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be informed of the potential hazard to a fetus.
8.4 Pediatric Use
The safety and efficacy of CALQUENCE in pediatric patients have not been established.
8.5 Geriatric Use
CLL and Previously Treated MCL
Of the 1,758 CALQUENCE-treated patients with B-cell malignancies (excluding previously untreated MCL) in clinical trials, 1,074 (61%) were 65 years of age or older, and 341 (19%) were 75 years of age or older. Among patients 65 years of age or older, 73% had Grade 3 or higher adverse reactions and 55% had serious adverse reactions. Among patients younger than age 65, 58% had Grade 3 or higher adverse reactions and 35% had serious adverse reactions. No clinically relevant differences in efficacy were observed between patients ≥ 65 years and younger.
Of patients that received CALQUENCE in combination with venetoclax in AMPLIFY, 33% (97/291) were ≥ 65 years of age, and 4.5% (13/291) were ≥ 75 years of age. In patients 65 years of age or older and younger than age 65, the fatal adverse reactions were 5% and 2.6% respectively. No clinically relevant differences in efficacy were observed between patients ≥ 65 years of age and younger adults.
Previously Untreated MCL
Of the 297 CALQUENCE-treated patients with previously untreated MCL, 214 (72%) were 65 to 74 years of age and 83 (28%) were 75 years of age and older. No clinically relevant differences in safety or efficacy were observed between patients ages 65 to 74 years and those who were 75 years of age and older.
8.6 Hepatic Impairment
Avoid use of CALQUENCE in patients with severe hepatic impairment (Child-Pugh class C). No dosage adjustment of CALQUENCE is recommended in patients with mild (Child-Pugh class A) or moderate (Child-Pugh class B) hepatic impairment. The safety of CALQUENCE has not been evaluated in patients with moderate or severe hepatic impairment [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
CALQUENCE (acalabrutinib) is a kinase inhibitor. The molecular formula for acalabrutinib maleate is C26H23N7O2C4H4O4.H2O, and the molecular weight is 599.59. The chemical name is 4-{8-Amino-3-[(2S)-1-(but-2-ynoyl) pyrrolidin-2-yl] imidazo[1,5-a]pyrazin-1-yl}-N-(pyridin-2-yl)benzamide (2Z)-2-butenedioic acid hydrate.
The chemical structure of acalabrutinib is shown below:
Acalabrutinib maleate is a white to pale brown powder with pH-dependent solubility. It is freely soluble in water at pH values below 3 and practically insoluble at pH values above 6.
CALQUENCE tablets are for oral administration. Each tablet contains 100 mg of acalabrutinib (equivalent to 129 mg of acalabrutinib maleate). Inactive ingredients in the tablet core are low-substituted hydroxypropyl cellulose, mannitol, microcrystalline cellulose, and sodium stearyl fumarate. The tablet coating consists of copovidone, ferric oxide yellow, ferric oxide red, hypromellose, medium-chain triglycerides, polyethylene glycol 3350, purified water, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Acalabrutinib is a small-molecule inhibitor of Bruton tyrosine kinase (BTK). Acalabrutinib and its active metabolite, ACP-5862, form a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK enzymatic activity. BTK is a signaling molecule of the B cell antigen receptor (BCR) and cytokine receptor pathways. In B cells, BTK signaling results in activation of pathways necessary for B-cell proliferation, trafficking, chemotaxis, and adhesion. In nonclinical studies, acalabrutinib inhibited BTK‑mediated activation of downstream signaling proteins CD86 and CD69 and inhibited malignant B-cell proliferation and tumor growth in mouse xenograft models.
12.2 Pharmacodynamics
In patients with B-cell malignancies dosed with acalabrutinib 100 mg approximately every 12 hours, median steady state BTK occupancy of ≥ 95% in peripheral blood was maintained over 12 hours, resulting in inactivation of BTK throughout the recommended dosing interval.
Cardiac Electrophysiology
At a dose 4 times the approved recommended dosage, CALQUENCE does not prolong the QTc interval to any clinically relevant extent.
12.3 Pharmacokinetics
Acalabrutinib and its active metabolite, ACP-5862, exposures increase proportionally with dose across a dose range of 75 to 250 mg (0.75 to 2.5 times the approved recommended single dosage) in patients with B-cell malignancies. At the recommended dose of 100 mg twice daily, the geometric mean (% coefficient of variation [CV]) daily area under the plasma drug concentration over time curve (AUC24h) and maximum plasma concentration (Cmax) for acalabrutinib were 1843 (38%) ngh/mL and 563 (29%) ng/mL, respectively, and for ACP-5862 were 3947 (43%) ngh/mL and 451 (52%) ng/mL, respectively.
Absorption
The geometric mean absolute bioavailability of acalabrutinib was 25%. Median (min, max) time to peak plasma concentration (Tmax) of acalabrutinib and its active metabolite, ACP-5862 were 0.5 (0.2, 3.0) hours and 0.75 (0.5, 4.0) hours, respectively.
Effect of Food
In healthy subjects, administration of a single 100 mg dose of acalabrutinib with a high-fat, high-calorie meal (approximately 918 calories, 59 grams carbohydrate, 59 grams fat, and 39 grams protein) did not affect the mean AUC as compared to dosing under fasted conditions. Resulting Cmax decreased by 54% and Tmax was delayed 1-2 hours.
Distribution
The geometric mean (% CV) steady-state volume of distribution (Vss) of acalabrutinib and its active metabolite, ACP-5862 was approximately 101 (52%) L and 67 (32%) L, respectively. Reversible binding to human plasma protein of acalabrutinib and its active metabolite, ACP-5862, were 97.5% and 98.6%, respectively. The mean blood-to-plasma ratio of acalabrutinib and its active metabolite, ACP-5862, was 0.8 and 0.7, respectively.
Elimination
The geometric mean (% CV) terminal elimination half-life (t1/2) of acalabrutinib and its active metabolite, ACP-5862, were 1.4 (50%) hours and 6.4 (37%) hours, respectively. The geometric mean (%CV) apparent oral clearance (CL/F) of acalabrutinib and its active metabolite, ACP-5862, were 148 (33%) L/hr and 19 (23%) L/hr, respectively.
Metabolism
Acalabrutinib is predominantly metabolized by CYP3A enzymes, and to a minor extent, by glutathione conjugation and amide hydrolysis, based on in vitro studies. ACP-5862 was identified as the major active metabolite in plasma with a geometric mean exposure (AUC) that was approximately 2- to 3-fold higher than the exposure of acalabrutinib. ACP-5862 is approximately 50% less potent than acalabrutinib with regard to BTK inhibition.
Excretion
Following administration of a single 100 mg radiolabeled acalabrutinib dose in healthy subjects, 84% of the dose was recovered in the feces (< 2% unchanged) and 12% of the dose was recovered in the urine (< 2% unchanged).
Specific Populations
There were no clinically significant differences in the pharmacokinetics of acalabrutinib and its active metabolite, ACP‑5862, based on age (32 to 90 years), sex, race (Caucasian, African American), body weight (40 to 149 kg), or mild to moderate renal impairment (estimated glomerular filtration rate [eGFR] by Modification of Diet in Renal Disease [MDRD] equation: 30 to < 90 mL/min). The effect of severe renal impairment (eGFR < 30 mL/min, MDRD) or renal impairment requiring dialysis on the pharmacokinetics of acalabrutinib is unknown.
Patients with Hepatic Impairment
The AUC of acalabrutinib increased 1.9-fold in subjects with mild hepatic impairment (Child-Pugh class A), 1.5-fold in subjects with moderate hepatic impairment (Child-Pugh class B) and 5.3-fold in subjects with severe hepatic impairment (Child-Pugh class C) compared to subjects with normal liver function. No clinically relevant PK difference in ACP-5862 was observed in subjects with severe hepatic impairment (Child-Pugh Class C) compared to subjects with normal liver function. of acalabrutinib increased 1.9-fold in subjects with mild hepatic impairment (Child-Pugh class A), 1.5-fold in subjects with moderate hepatic impairment (Child-Pugh class B) and 5.3-fold in subjects with severe hepatic impairment (Child-Pugh class C) compared to subjects with normal liver function. No clinically relevant PK difference in ACP-5862 was observed in subjects with severe hepatic impairment (Child-Pugh Class C) compared to subjects with normal liver function. No clinically relevant PK differences in acalabrutinib and ACP-5862 were observed in patients with mild or moderate hepatic impairment (total bilirubin ≤ 3 x ULN and any AST) relative to patients with normal hepatic function (total bilirubin and AST ≤ ULN).
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Strong CYP3A Inhibitors: Co-administration of acalabrutinib with itraconazole (strong CYP3A inhibitor) increase acalabrutinib Cmax by 3.9-fold and AUC by 5.1-fold in healthy subjects.
Moderate CYP3A Inhibitors: Co-administration of acalabrutinib with erythromycin (moderate CYP3A inhibitor), fluconazole (moderate CYP3A inhibitor), diltiazem (moderate CYP3A inhibitor) is predicted to increase acalabrutinib Cmax and AUC by approximately 2- to 3-fold.
Strong CYP3A Inducers: Co-administration of acalabrutinib with rifampin (strong CYP3A inducer) decreased acalabrutinib Cmax by 68% and AUC by 77% in healthy subjects.
Acid-Reducing Agents: No clinically significant differences in the pharmacokinetics of acalabrutinib were observed when co-administered with rabeprazole (proton pump inhibitor).
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Acalabrutinib is an inhibitor of CYP3A4/5, CYP2C8 and CYP2C9, but not CYP1A2, CYP2B6, CYP2C19, or CYP2D6. Acalabrutinib’s active metabolite, ACP-5862, is an inhibitor of CYP2C8, CYP2C9 and CYP2C19, but not CYP1A2, CYP2B6, CYP2D6, or CYP3A4/5. Acalabrutinib is an inducer of CYP1A2, CYP2B6, and CYP3A4. Acalabrutinib’s active metabolite, ACP-5862, is an inducer of CYP3A4.
Uridine diphosphate (UDP)-glucuronosyl transferase (UGT) Enzymes: Acalabrutinib and its active metabolite, ACP‑5862, are not inhibitors of UGT1A1 or UGT2B7.
Transporter System: Acalabrutinib is an inhibitor of breast cancer resistance protein (BCRP), but not multidrug and toxin extrusion protein 1 (MATE1). Acalabrutinib’s active metabolite, ACP-5862, is an inhibitor of MATE1, but not BCRP. Acalabrutinib and its active metabolite, ACP-5862, are not inhibitors of P-glycoprotein (P-gp), organic anion transporter (OAT) 1, OAT3, organic cation transporter 2 (OCT2), organic anion transporting polypeptide (OATP) 1B1, OATP1B3, or MATE2-K.
Acalabrutinib and its active metabolite, ACP-5862, are substrates of P-gp and BCRP. Acalabrutinib is not a substrate of OAT1, OAT3, OCT2, OATP1B1, or OATP1B3. Acalabrutinib’s active metabolite, ACP-5862, is not a substrate of OATP1B1 or OATP1B3.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with acalabrutinib.
Acalabrutinib was not mutagenic in an in vitro bacterial reverse mutation (AMES) assay or clastogenic in an in vitro human lymphocyte chromosomal aberration assay or in an in vivo rat bone marrow micronucleus assay.
In a fertility study in rats, there were no effects of acalabrutinib on fertility in male rats at exposures 11 times, or in female rats at exposures 9 times, the AUC observed in patients at the recommended dose of 100 mg twice daily.
-
14 CLINICAL STUDIES
14.1 Previously Untreated Mantle Cell Lymphoma DOCVARIABLE vault_nd_4d1e0c80-2ff6-409f-bee7-897264eadbff \* MERGEFORMAT
ECHO
The efficacy of CALQUENCE in patients with previously untreated MCL was evaluated in a randomized, double blind, placebo controlled, multicenter study (ECHO; NCT02972840). The study enrolled 598 patients who were ≥ 65 years of age and who had no intention for transplant. The study excluded patients with total bilirubin > 1.5 × upper limit of normal (ULN), AST or ALT > 2.5 × ULN, or estimated creatinine clearance of ≤ 50 mL/min. Patients were randomized in a 1:1 ratio to receive CALQUENCE plus bendamustine and rituximab (CALQUENCE plus BR) or placebo plus BR. Dosing for both arms was administered in 28-day cycles as follows:
- CALQUENCE plus BR was administered for a maximum of 6 treatment cycles. CALQUENCE 100 mg orally was administered twice daily starting on Cycle 1 Day 1. Bendamustine was administered at 90 mg/m2 intravenously over 30 minutes on Days 1 and 2 of each of 6 cycles. Rituximab was administered at 375 mg/m2 intravenously on Day 1 of each cycle for 6 cycles.
- For patients achieving a response (PR or CR), CALQUENCE 100 mg orally twice daily was administered continuously, in combination with rituximab given at 375 mg/m2 on Day 1 every other cycle for a maximum of 12 additional doses up to Cycle 30. After discontinuation of rituximab, patients continued CALQUENCE monotherapy at 100 mg orally twice daily until disease progression or unacceptable toxicity.
Patients on the control arm received the same regimen but placebo in lieu of CALQUENCE. Crossover to CALQUENCE monotherapy was permitted for patients in the placebo plus BR arm at disease progression.
Of all patients randomized, the median age was 71 years (range: 65-86); 71% were male; 78% were White, 16% Asian, 0.5% were Black or African American. In total, 80% had classic histology of MCL, 7.7% had blastoid MCL, and 5.5% had pleomorphic MCL. The simplified MIPI (Mantle Cell Lymphoma International Prognostic Index) score was low in 33%, intermediate in 43%, and high in 24% of patients. A total of 38% of patients had tumor bulk ≥ 5 cm and 86% had Ann Arbor stage IV disease.
The major efficacy outcome was progression-free survival (PFS) as assessed by an Independent Review Committee (IRC) using the Lugano Classification. Efficacy results are presented in Table 15. The Kaplan-Meier curves for PFS are shown in Figure 1. At this prespecified interim analysis, the median follow-up for PFS was 49.8 months in both arms.
Table 15. Efficacy Results in Patients with Previously Untreated MCL in ECHO Outcomes per IRC
CALQUENCE plus BR
N= 299
Placebo plus BR
N= 299
Progression-Free Survivala
- Median (95% CI), months
66.4 (55.1, NE)
49.6 (36.0, 64.1)
- HRb (95% CI)
0.73 (0.57, 0.94)
- P-valuec
0.016
Overall Response Rate (ORR) (CR + PR)
- ORR n (%)
272 (91)
263 (88)
- 95% CI
87, 94
84, 91
- CR n (%)
199 (67)
160 (54)
- PR n (%)
73 (24)
103 (34)
- p-value
0.220
HR = hazard ratio, CR = complete response, PR = partial response, NE – not evaluable
a Stratified by randomization stratification factors: Geographic Regions (North American, Western Europe, Other) and simplified MIPI Score (Low risk [0 to 3], Intermediate risk [4 to 5], High Risk [6 to 11]).
b Estimated based on stratified Cox Proportional Hazards model for hazard ratio (95% CI).
c Estimated based on stratified log-rank test for p-value, with an alpha level of 0.039 derived by the O’Brien-Fleming method.
Figure 1. Kaplan-Meier Curve of IRC-Assessed PFS in Patients with Previously Untreated MCL in ECHO
At the time of the PFS analysis, the median overall survival had not been reached in either arm with a total of 203 deaths: 97 (32%) patients in the CALQUENCE plus BR arm and 106 (35%) patients in the placebo plus BR arm had died.
14.2 Previously Treated Mantle Cell Lymphoma
ACE-LY-004
The efficacy of CALQUENCE was based upon Trial LY-004 titled “An Open-label, Phase 2 Study of ACP-196 in Subjects with Mantle Cell Lymphoma” (NCT02213926). Trial LY-004 enrolled a total of 124 patients with MCL who had received at least one prior therapy.
The median age was 68 (range 42 to 90) years, 80% were male, and 74% were Caucasian. At baseline, 93% of patients had an ECOG performance status of 0 or 1. The median time since diagnosis was 46.3 months and the median number of prior treatments was 2 (range 1 to 5), including 18% with prior stem cell transplant. Patients who received prior treatment with BTK inhibitors were excluded. The most common prior regimens were CHOP-based (52%) and ARA-C (34%). At baseline, 37% of patients had at least one tumor with a longest diameter ≥ 5 cm, 73% had extra nodal involvement including 51% with bone marrow involvement. The simplified Mantle Cell Lymphoma International Prognostic Index (MIPI) score (which includes age, ECOG score, and baseline lactate dehydrogenase and white cell count) was intermediate in 44% and high in 17% of patients.
CALQUENCE was administered orally at 100 mg approximately every 12 hours until disease progression or unacceptable toxicity. The median dose intensity was 98.5%. The major efficacy outcome of Trial LY-004 was overall response rate and the median follow-up was 15.2 months.
Table 16: Efficacy Results in Patients with MCL in Trial LY-004 - * Per 2014 Lugano Classification.
Investigator Assessed
N=124
Independent Review Committee (IRC) Assessed
N=124
Overall Response Rate (ORR)*
ORR (%) [95% CI]
81 [73, 87]
80 [72, 87]
Complete Response (%) [95% CI]
40 [31, 49]
40 [31, 49]
Partial Response (%) [95% CI]
41 [32, 50]
40 [32, 50]
Duration of Response (DoR)
Median DoR in months [range]
NE [1+ to 20+]
NE [0+ to 20+]
CI= Confidence Interval; NE=Not Estimable; + indicates censored observations.
The median time to best response was 1.9 months.
Lymphocytosis
Upon initiation of CALQUENCE, a temporary increase in lymphocyte counts (defined as absolute lymphocyte count increased ≥ 50% from baseline and a post-baseline assessment ≥ 5 x 109/L) in 31.5% of patients in Trial LY-004. The median time to onset of lymphocytosis was 1.1 weeks, and the median duration of lymphocytosis was 6.7 weeks.
14.3 Chronic Lymphocytic Leukemia
The efficacy of CALQUENCE in patients with CLL was demonstrated in two randomized, controlled trials. The indication for CALQUENCE includes patients with SLL because it is the same disease.
ELEVATE-TN
The efficacy of CALQUENCE was evaluated in the ELEVATE-TN trial, a randomized, multicenter, open-label, actively controlled, 3 arm trial of CALQUENCE in combination with obinutuzumab, CALQUENCE monotherapy, and obinutuzumab in combination with chlorambucil in 535 patients with previously untreated chronic lymphocytic leukemia (NCT02475681). Patients 65 years of age or older or between 18 and 65 years of age with a total Cumulative Illness Rating Scale (CIRS) > 6 or creatinine clearance of 30 to 69 mL/min were enrolled. The trial also required hepatic transaminases ≤ 3 times upper limit of normal (ULN) and total bilirubin ≤ 1.5 times ULN, and excluded patients with Richter’s transformation.
Patients were randomized in a 1:1:1 ratio into 3 arms to receive:
- CALQUENCE plus obinutuzumab (CALQUENCE+G): CALQUENCE 100 mg was administered approximately every 12 hours starting on Cycle 1 Day 1 until disease progression or unacceptable toxicity. Obinutuzumab was administered starting on Cycle 2 Day 1 for a maximum of 6 treatment cycles. Obinutuzumab 1,000 mg was administered on Days 1 and 2 (100 mg on Day 1 and 900 mg on Day 2), 8 and 15 of Cycle 2 followed by 1,000 mg on Day 1 of Cycles 3 up to 7. Each cycle was 28 days.
- CALQUENCE monotherapy: CALQUENCE 100 mg was administered approximately every 12 hours until disease progression or unacceptable toxicity.
- Obinutuzumab plus chlorambucil (GClb): Obinutuzumab and chlorambucil were administered for a maximum of 6 treatment cycles. Obinutuzumab 1,000 mg was administered intravenously on Days 1 and 2 (100 mg on Day 1 and 900 mg on Day 2), 8 and 15 of Cycle 1 followed by 1,000 mg on Day 1 of Cycles 2 to 6. Chlorambucil 0.5 mg/kg was administered orally on Days 1 and 15 of Cycles 1 to 6. Each cycle was 28 days.
Randomization was stratified by 17p deletion mutation status, ECOG performance status (0 or 1 versus 2), and geographic region. A total of 535 patients were randomized, 179 to CALQUENCE+G, 179 to CALQUENCE monotherapy, and 177 to GClb. The overall median age was 70 years (range: 41 to 91 years), 47% had Rai stage III or IV disease, 14% had 17p deletion or TP53 mutation, 63% of patients had an unmutated IGVH, and 18% had 11q deletion. Baseline demographic and disease characteristics were similar between treatment arms.
Efficacy was based on progression-free survival (PFS) as assessed by an Independent Review Committee (IRC). The median duration of follow-up was 28.3 months (range: 0.0 to 40.8 months). Efficacy results are presented in Table 17. The Kaplan-Meier curves for PFS are shown in Figure 2.
Table 17. Efficacy Results per IRC in Patients with CLL ‒ ITT population (ELEVATE-TN) - * Per 2008 International Workshop on CLL (IWCLL) criteria.
- † Kaplan-Meier estimate.
- ‡ Based on a stratified Cox-Proportional-Hazards model. Both hazard ratios are compared with the obinutuzumab and chlorambucil arm.
- § Based on a stratified log-rank test, with an alpha level of 0.012 derived from alpha spending function by the O’Brien-Fleming method.
- ¶ Based on a stratified Cochran–Mantel–Haenszel test, for the comparison with the obinutuzumab and chlorambucil arm.
CALQUENCE plus Obinutuzumab
N=179
CALQUENCE Monotherapy
N=179
Obinutuzumab plus Chlorambucil
N=177
Progression-Free Survival*
- Number of events (%)
14 (8)
26 (15)
93 (53)
- PD, n (%)
9 (5)
20 (11)
82 (46)
- Death events, n (%)
5 (3)
6 (3)
11 (6)
- Median (95% CI), months†
NE
NE (34, NE)
22.6 (20, 28)
- HR‡ (95% CI)
0.10 (0.06, 0.17)
0.20 (0.13, 0.30)
-
- p-value§
< 0.0001
< 0.0001
-
Overall Response Rate* (CR + CRi + nPR + PR)
- ORR, n (%)
168 (94)
153 (86)
139 (79)
- (95% CI)
(89, 97)
(80, 90)
(72, 84)
- p-value¶
< 0.0001
0.0763
-
- CR, n (%)
23 (13)
1 (1)
8 (5)
- CRi, n (%)
1 (1)
0
0
- nPR, n (%)
1 (1)
2 (1)
3 (2)
- PR, n (%)
143 (80)
150 (84)
128 (72)
ITT=intent-to-treat; CI=confidence interval; HR=hazard ratio; NE=not estimable; CR=complete response; CRi=complete response with incomplete blood count recovery; nPR=nodular partial response; PR=partial response.
Figure 2: Kaplan-Meier Curve of IRC-Assessed PFS in Patients with CLL in ELEVATE-TN
With a median follow-up of 28.3 months, median overall survival was not reached in any arm, with fewer than 10% of patients experiencing an event.
ASCEND
The efficacy of CALQUENCE in patients with relapsed or refractory CLL was based upon a multicenter, randomized, open-label trial (ASCEND; NCT02970318). The trial enrolled 310 patients with relapsed or refractory CLL after at least 1 prior systemic therapy. The trial excluded patients with transformed disease, prolymphocytic leukemia, or previous treatment with venetoclax, a Bruton tyrosine kinase inhibitor, or a phosphoinositide-3 kinase inhibitor.
Patients were randomized in a 1:1 ratio to receive either:
- CALQUENCE 100 mg approximately every 12 hours until disease progression or unacceptable toxicity, or
-
Investigator’s choice:
- ∘ Idelalisib plus a rituximab product (IR): Idelalisib 150 mg orally approximately every 12 hours until disease progression or unacceptable toxicity, in combination with 8 infusions of a rituximab product (375 mg/m2 intravenously on Day 1 of Cycle 1, followed by 500 mg/m2 every 2 weeks for 4 doses and then every 4 weeks for 3 doses), with a 28-day cycle length.
- ∘ Bendamustine plus a rituximab product (BR): Bendamustine 70 mg/m2 intravenously (Day 1 and 2 of each 28-day cycle), in combination with a rituximab product (375 mg/m2 intravenously on Day 1 of Cycle 1, then 500 mg/m2 on Day 1 of subsequent cycles), for up to 6 cycles.
Randomization was stratified by 17p deletion mutation status, ECOG performance status (0 or 1 versus 2), and number of prior therapies (1 to 3 versus ≥ 4). Of 310 patients total, 155 were assigned to CALQUENCE monotherapy, 119 to IR, and 36 to BR. The median age overall was 67 years (range: 32 to 90 years), 42% had Rai stage III or IV disease, 28% had 17p deletion or TP53 mutation, 78% of patients had an unmutated IGVH, and 27% had a 11q deletion. The CALQUENCE arm had a median of 1 prior therapy (range: 1 to 8), with 47% having at least 2 prior therapies. The investigator’s choice arm had a median of 2 prior therapies (range: 1 to 10), with 57% having at least 2 prior therapies.
In the CALQUENCE arm, the median treatment duration was 15.7 months, with 94% of patients treated for at least 6 months and 86% of patients treated for at least 1 year. In the investigator’s choice arm, the median treatment duration was 8.4 months, with 59% of patients treated for at least 6 months and 37% treated for at least 1 year.
Efficacy was based on PFS as assessed by an IRC, with a median follow-up of 16.1 months (range 0.03 to 22.4 months). Efficacy results are presented in Table 18. The Kaplan-Meier curve for PFS is shown in Figure 3. There was no statistically significant difference in overall response rates between the two treatment arms.
Table 18: Efficacy Results per IRC in Patients with Relapsed or Refractory CLL – ITT Population (ASCEND) - * Per 2008 IWCLL criteria.
- † Kaplan-Meier estimate.
- ‡ Based on a stratified Cox-Proportional-Hazards model.
- § Based on a stratified Log-rank test. The pre-specified type I error rate (α) for this interim analysis is 0.012 derived from a Lan-DeMets alpha spending function with O’Brien-Fleming boundary.
- ¶ Through a hierarchical testing procedure, the difference in ORR was not statistically significant, based on a Cochran-Mantel-Haenszel test with adjustment for randomization stratification factors.
CALQUENCE Monotherapy
N=155
Investigator’s Choice of Idelalisib + Rituximab Product or Bendamustine + Rituximab Product
N=155
Progression-Free Survival*
- Number of events, n (%)
27 (17)
68 (44)
- Disease progression, n
19
59
- Death, n
8
9
- Median (95% CI), months†
NE (NE, NE)
16.5 (14.0, 17.1)
- HR (95% CI)‡
0.31 (0.20, 0.49)
- P-value§
< 0.0001
- ORR, n (%)¶
126 (81)
117 (75)
- (95% CI)
(74, 87)
(68, 82)
- CR, n (%)
0
2 (1)
- CRi, n (%)
0
0
- nPR, n (%)
0
0
- PR, n (%)
126 (81)
115 (74)
- ITT=intent-to-treat; CI=confidence interval; HR=hazard ratio; NE=not estimable; CR=complete response; CRi=complete response with incomplete blood count recovery; nPR=nodular partial response; PR=partial response
Figure 3: Kaplan-Meier Curve of IRC-Assessed PFS in Patients with CLL in ASCEND
With a median follow-up of 16.1 months, median overall survival was not reached in either arm, with fewer than 11% of patients experiencing an event.
AMPLIFY
The efficacy of CALQUENCE in combination with venetoclax in previously untreated CLL patients was evaluated in AMPLIFY, a randomized, multi-center, open-label study (NCT03836261). The study included patients previously untreated for CLL without del(17p) or TP53 mutation that were 18 years of age and older. Patients were randomized to receive:
- CALQUENCE plus venetoclax (AV): CALQUENCE 100 mg was administered twice daily starting on Cycle 1 Day 1 for a total of 14 cycles or until disease progression or unacceptable toxicity. On Cycle 3 Day 1 patients started the venetoclax 5-week dose-titration schedule, starting at 20 mg and increasing weekly to 50 mg, 100 mg, 200 mg and finally 400 mg once daily. Venetoclax was administered for a total of 12 cycles. Each cycle was 28 days.
-
Investigator’s choice of chemoimmunotherapy (FCR/BR):
- o Fludarabine plus cyclophosphamide plus rituximab (FCR): Fludarabine (25 mg/m2 ) and cyclophosphamide (250 mg/m2) were administered on Days 1–3 up to a maximum of 6 cycles. Rituximab was administered at a dose of 375 mg/m2 on Day 1 Cycle 1 and 500 mg/m2 on Day 1 of Cycles 2 up to 6. Each cycle was 28 days.
- o Bendamustine plus rituximab (BR): Bendamustine 90 mg/m2 was administered on Days 1 and 2 up to maximum of 6 cycles. Rituximab was administered at a dose of 375 mg/m2 on Day 1 Cycle 1 and 500 mg/m2 on Day 1 of Cycles 2 up to 6. Each cycle was 28 days.
- An additional investigational combination regimen [see Warnings and Precautions (5.1)].
Patients were stratified by age (> 65 years or ≤ 65), IGHV mutational status (mutated versus unmutated), Rai stage (high risk [≥ 3] versus non-high risk) and geographic region (North America and Europe versus other). In the efficacy population described in Table 19, overall median age was 61 years (range: 26 to 86 years) and 62% were males; 89% were White, 3.8% Asian, 1.7% were Black or African American, 0.3% American Indian or Alaska Native, 0.3% Native Hawaiian or Other Pacific Islander, and 4.8% not reported; 86% were not Hispanic or Latino, 7% Hispanic or Latino, and 7% not reported. The ECOG performance was 0-1 in 90%, bulky diseases with nodes ≥ 5 cm was seen in 41%, 45% had Rai stage III or IV disease, 17% had 11q deletion and 58% had unmutated IGHV.
The major efficacy outcome was IRC-assessed PFS of AV arm versus Investigator’s choice arm (FCR/BR). The median duration of PFS follow-up was 42.6 months. Efficacy results are presented in Table 19. The Kaplan-Meier curve for IRC-assessed PFS is shown in Figure 4.
Table 19. Efficacy Results per IRC in Patients with previously untreated CLL ‒ ITT population (AMPLIFY) CALQUENCE plus venetoclax
N=291
FCR/BRb
N=290
Progression-free survivala
- Number of events (%)
89 (31)
95 (33)
- PD, n (%)
77 (26)
66 (23)
- Death events (%)
12 (4)
29 (10)
- Median (95% CI), months
NE (51.1, NE)
47.6 (43.3, NE)
- HRc (95% CI)
0.65 (0.49, 0.87)
- P-valued
0.0038
Overall Response Rate (CR, CRi, nPR, PR)e
- ORR n (%)
270 (93)
218 (75)
- (95% CI)
(89, 95)
(70, 80)
- CR
26 (9)
15 (5)
- CRi, n (%)
0
1 (0.3)
- nPR, n (%)
1 (0.3)
1 (0.3)
- PR n (%)
243 (84)
201 (69)
NE= Not estimable; CR = complete response; CRi=complete response with incomplete blood count recovery; nPR=nodular partial response; PR=partial response
a Per IRC assessment.
b Approximately 50% of patients were treated with FCR and 50% were treated with BR per investigator's choice.
c Based on stratified Cox-Proportional-Hazards model.
d Based on a stratified Log-rank test. The pre-specified type I error rate (α) for this interim analysis is 0.0469 derived from a Lan-DeMets alpha spending function with O’Brien-Fleming boundary.
e Per iwCLL 2018 criteria.
Figure 4: Kaplan-Meier Curve of IRC-Assessed PFS in Patients with previously untreated CLL in AMPLIFY
With a median follow-up of 41.0 months, a total of 60 death events were reported; 18 (6%) in the AV arm and 42 (14%) in the FCR/BR arm.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Pack Size
Contents
NDC Number
60-count bottle
Bottle containing 60 tablets with a child-resistant closure
100 mg, orange, oval, biconvex tablet, with debossment ‘ACA100’ on one side and plain on the reverse
0310-3512-60
Storage
Store at 20°C-25°C (68°F-77°F); excursions permitted to 15°C-30°C (59°F-86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Serious and Opportunistic Infections
Inform patients of the possibility of serious infection and to immediately report signs or symptoms suggestive of infection [see Warnings and Precautions (5.1)].
Hemorrhage
Inform patients to immediately report signs or symptoms of bleeding. Inform patients that CALQUENCE may need to be interrupted for major surgeries [see Warnings and Precautions (5.2)].
Cytopenias
Inform patients that they will need periodic blood tests to check blood counts during treatment with CALQUENCE [see Warnings and Precautions (5.3)].
Second Primary Malignancies
Inform patients that other malignancies have been reported in patients who have been treated with CALQUENCE, including skin cancer and other solid tumors. Advise patients to use sun protection [see Warnings and Precautions (5.4)].
Cardiac Arrhythmias
Counsel patients to immediately report any signs of palpitations, dizziness, fainting, chest discomfort, and shortness of breath [see Warnings and Precautions (5.5)].
Hepatotoxicity, Including Drug-Induced Liver Injury:
Inform patients that liver problems, including drug-induced liver injury and abnormalities in liver tests, may develop during CALQUENCE treatment. Advise patients to contact their healthcare provider immediately if they experience abdominal discomfort, dark urine, or jaundice [see Warnings and Precautions (5.6)].
Pregnancy Complication
CALQUENCE may cause fetal harm and dystocia. Advise women to use effective contraception during treatment and for 1 week after the last dose of CALQUENCE [see Use in Specific Populations (8.3)].
Lactation
Advise females not to breastfeed during treatment with CALQUENCE and for 2 weeks after the last dose [see Use in Specific Populations (8.2)].
Dosing Instructions
Instruct patients to take CALQUENCE orally twice daily, about 12 hours apart. CALQUENCE may be taken with or without food. Advise patients that CALQUENCE tablets should be swallowed whole with a glass of water, without chewing, crushing, dissolving, or cutting [see Dosage and Administration (2.1)].
Missed Dose
Advise patients that if they miss a dose of CALQUENCE, they may still take it up to 3 hours after the time they would normally take it. If more than 3 hours have elapsed, they should be instructed to skip that dose and take their next dose of CALQUENCE at the usual time. Warn patients they should not take extra tablets to make up for the dose that they missed [see Dosage and Administration (2.1)].
Drug Interactions
Advise patients to inform their healthcare providers of all concomitant medications, including over‑the‑counter medications, vitamins and herbal products [see Drug Interactions (7)].
Distributed by:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
CALQUENCE is a registered trademark of the AstraZeneca group of companies. ©AstraZeneca 2026
-
PATIENT INFORMATION
- PATIENT INFORMATION
- CALQUENCE® (KAL-kwens)
- (acalabrutinib)
- tablets
What is CALQUENCE?
CALQUENCE is a prescription medicine used to treat adults with:
- Mantle cell lymphoma (MCL) in combination with bendamustine and rituximab in people who have not had prior treatment for MCL and who cannot receive a type of stem cell transplant that uses their own blood stem cells (autologous hematopoietic stem cell transplantation).
- Mantle cell lymphoma (MCL) who have received at least one prior therapy.
- Chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL).
It is not known if CALQUENCE is safe and effective in children.
Before taking CALQUENCE, tell your healthcare provider about all of your medical conditions, including if you:
- have had recent surgery or plan to have surgery. Your healthcare provider may stop CALQUENCE for any planned medical, surgical, or dental procedure.
- have bleeding problems.
- have or had heart rhythm problems.
- have an infection.
- have or had liver problems, including hepatitis B virus (HBV) infection.
- are pregnant or plan to become pregnant. CALQUENCE may harm your unborn baby and cause problems during childbirth (dystocia).
Females who are able to become pregnant:
-
- ∘ Your healthcare provider may do a pregnancy test before you start treatment with CALQUENCE.
- ∘ Use effective birth control (contraception) during treatment with CALQUENCE and for 1 week after the last dose of CALQUENCE.
- are breastfeeding or plan to breastfeed. It is not known if CALQUENCE passes into your breast milk. Do not breastfeed during treatment with CALQUENCE and for 2 weeks after your last dose of CALQUENCE.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Taking CALQUENCE with certain other medications may affect how CALQUENCE works and can cause side effects. Especially tell your healthcare provider if you take a blood thinner medicine.
How should I take CALQUENCE?
- Take CALQUENCE exactly as your healthcare provider tells you to take it.
- Do not change your dose or stop taking CALQUENCE unless your healthcare provider tells you to.
- Your healthcare provider may tell you to decrease your dose, temporarily stop, or completely stop taking CALQUENCE if you develop certain side effects.
- Do not switch (interchange) your CALQUENCE tablets with CALQUENCE capsules.
- Take CALQUENCE 2 times a day (about 12 hours apart).
- Take CALQUENCE with or without food.
- Swallow CALQUENCE tablets whole with a glass of water. Do not chew, crush, dissolve, or cut tablets.
- If you miss a dose of CALQUENCE, take it as soon as you remember. If it is more than 3 hours past your usual dosing time, skip the missed dose and take your next dose of CALQUENCE at your regularly scheduled time. Do not take an extra dose to make up for a missed dose.
What are the possible side effects of CALQUENCE?
CALQUENCE can cause serious side effects, including:
- Serious infections have happened in people treated with CALQUENCE and may lead to death. Your healthcare provider may prescribe certain medicines if you have an increased risk of getting infections. Tell your healthcare provider right away if you have any signs or symptoms of an infection, including fever, chills, or flu-like symptoms.
- Bleeding problems (hemorrhage) have happened in people treated with CALQUENCE and can be serious and may lead to death. Your risk of bleeding may increase if you are also taking a blood thinner medicine. Tell your healthcare provider right away if you have any signs or symptoms of bleeding, including:
-
- ∘ blood in your stools or black stools (looks like tar)
- ∘ pink or brown urine
- ∘ unexpected bleeding, or bleeding that is severe or you cannot control
- ∘ vomit blood or vomit that looks like coffee grounds
- ∘ cough up blood or blood clots
-
- ∘ dizziness
- ∘ weakness
- ∘ confusion
- ∘ changes in your speech
- ∘ headache that lasts a long time
- ∘ bruising or red or purple skin marks
- Decrease in blood cell counts. Decreased white blood cell counts, red blood cell counts, and platelet counts can be severe during treatment with CALQUENCE. Your healthcare provider should do blood tests to check your blood counts regularly during treatment with CALQUENCE.
- Second primary cancers. New cancers have happened in people during treatment with CALQUENCE, including cancers of the skin or other organs. Your healthcare provider will check you for skin cancers during treatment with CALQUENCE. Use sun protection when you are outside in sunlight.
- Heart rhythm problems (cardiac arrhythmias) have happened in people treated with CALQUENCE, and can be serious or lead to death. Tell your healthcare provider right away if you have any of the following signs or symptoms:
-
- ∘ fast or irregular heartbeat
- ∘ dizziness
- ∘ feeling faint
-
- ∘ chest discomfort
- ∘ shortness of breath
- Liver problems. Liver problems have happened in people treated with CALQUENCE, and can be severe or life-threatening, or lead to death. Tell your healthcare provider right away if you experience stomach pain or discomfort, urine of dark color or yellowing of your skin. Your healthcare provider will request tests to monitor your liver function during treatment with CALQUENCE.
The most common side effects of CALQUENCE include:
- upper respiratory infection
- diarrhea
- headache
- muscle and joint pain
- decreased white blood cell counts
- decreased platelet counts
These are not all of the possible side effects of CALQUENCE.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store CALQUENCE?
- Store CALQUENCE at room temperature between 68°F to 77°F (20°C to 25°C).
Keep CALQUENCE and all medicines out of the reach of children.
General information about the safe and effective use of CALQUENCE.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use CALQUENCE for a condition for which it was not prescribed. Do not give CALQUENCE to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for more information about CALQUENCE that is written for health professionals.
What are the ingredients in CALQUENCE?
Active ingredient: acalabrutinib
Inactive ingredients:
Tablet core: low-substituted hydroxypropyl cellulose, mannitol, microcrystalline cellulose, and sodium stearyl fumarate.
Tablet coating: copovidone, ferric oxide yellow, ferric oxide red, hypromellose, medium-chain triglycerides, polyethylene glycol 3350, purified water, and titanium dioxide.
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
CALQUENCE is a registered trademark of the AstraZeneca group of companies. ©AstraZeneca 2026
For more information, go to www.CALQUENCE.com or call 1-800-236-9933.
This Patient Information has been approved by the U.S. Food and Drug Administration Revised: 2/2026
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 0310-3512-60
CALQUENCE®
(acalabrutinib) tablets
100 mg
Rx only
Do not chew, crush, dissolve, or cut tablets
Manufactured for:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
By: AstraZeneca AB
SE-151 85 Södertälje Sweden
Product of Switzerland
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 0310-4513-60
CALQUENCE®
(acalabrutinib) tablets
100 mg
Rx only
Do not chew, crush, dissolve, or cut
tablets
Manufactured for:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
AstraZeneca
60 tablets
-
INGREDIENTS AND APPEARANCE
CALQUENCE
acalabrutinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-3512 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ACALABRUTINIB (UNII: I42748ELQW) (ACALABRUTINIB - UNII:I42748ELQW) ACALABRUTINIB 100 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) MANNITOL (UNII: 3OWL53L36A) MEDIUM-CHAIN TRIGLYCERIDES (UNII: C9H2L21V7U) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) WATER (UNII: 059QF0KO0R) Product Characteristics Color ORANGE Score no score Shape OVAL (biconvex) Size 13mm Flavor Imprint Code ACA100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-3512-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 08/04/2022 2 NDC: 0310-3512-95 60 in 1 BLISTER PACK; Type 0: Not a Combination Product 08/04/2022 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216387 08/04/2022 CALQUENCE
acalabrutinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-4513 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ACALABRUTINIB (UNII: I42748ELQW) (ACALABRUTINIB - UNII:I42748ELQW) ACALABRUTINIB 100 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) MANNITOL (UNII: 3OWL53L36A) MEDIUM-CHAIN TRIGLYCERIDES (UNII: C9H2L21V7U) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) WATER (UNII: 059QF0KO0R) Product Characteristics Color ORANGE Score no score Shape OVAL (biconvex) Size 13mm Flavor Imprint Code ACA100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-4513-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 02/19/2025 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216387 08/04/2022 Labeler - AstraZeneca Pharmaceuticals LP (054743190)
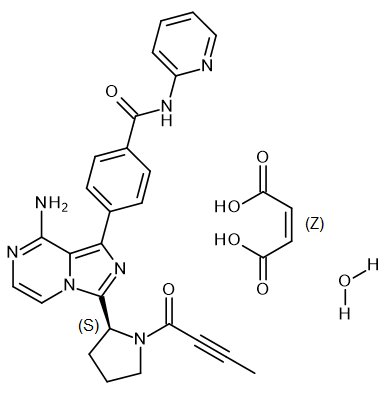
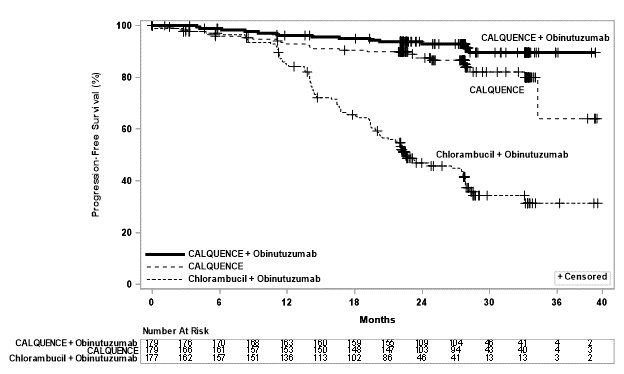
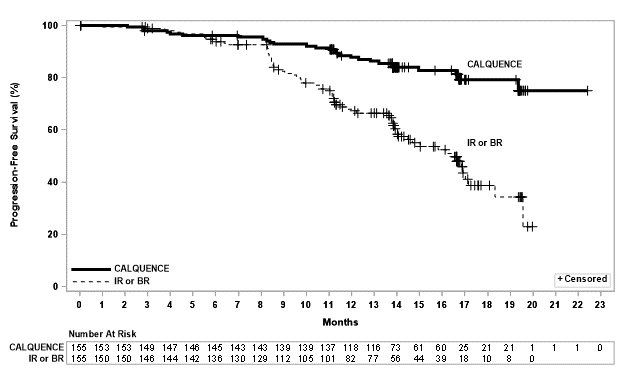
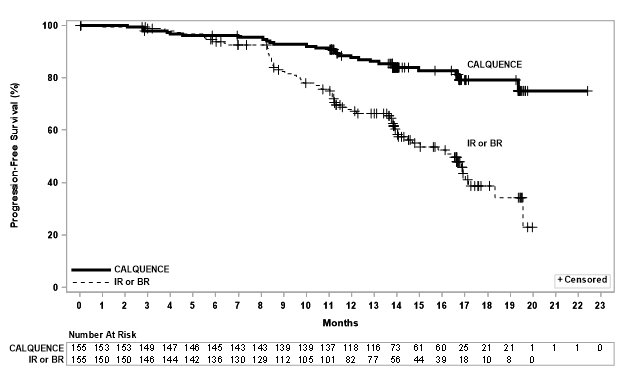
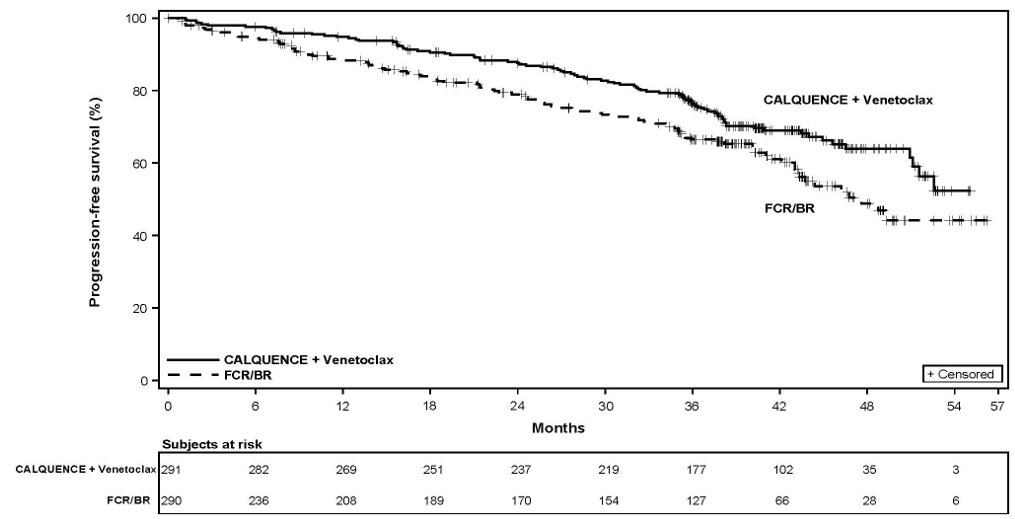
Trademark Results [CALQUENCE]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 CALQUENCE 87072359 5221345 Live/Registered |
AstraZeneca AB 2016-06-15 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.