SACUBITRIL AND VALSARTAN tablet, film coated
Sacubitril and Valsartan by
Drug Labeling and Warnings
Sacubitril and Valsartan by is a Prescription medication manufactured, distributed, or labeled by Laurus Labs Limited, Laurus Labs Limited (VSP2). Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SACUBITRIL AND VALSARTAN TABLETS safely and effectively. See full prescribing information for SACUBITRIL AND VALSARTAN TABLETS.
SACUBITRIL AND VALSARTAN tablets, for oral use
Initial U.S. Approval: 2015
RECENT MAJOR CHANGES
Dosage and Administration.(2.3) 4/2024
INDICATIONS AND USAGE
Sacubitril and valsartan tablets are a combination of sacubitril, a neprilisin inhibitor, and valsartan, an angiotensin II receptor blocker, and is indicated:
- to reduce the risk of cardiovascular death and hospitalization for heart failure in adult patients with chronic heart failure. Benefits are most clearly evident in patients with left ventricular ejection fraction (LVEF) below normal. (1.1)
- for the treatment of symptomatic heart failure with systemic left ventricular systolic dysfunction in pediatric patients aged one year and older. Sacubitril and valsartan tablets reduce NT-proBNP and are expected to improve cardiovascular outcomes. (1.2)
DOSAGE AND ADMINISTRATION
The recommended starting dosage for adults is 49 mg/51 mg orally twice daily. The target maintenance dose is 97 mg/103 mg orally twice daily. (2.2)
Adjust adult doses every 2 to 4 weeks to the target maintenance dose, as tolerated by the patient. (2.2)
For pediatric patients, see the Full Prescribing Information for recommended dosage, titrations, preparation and administration instructions. (2.3, 2.4)
Reduce starting dose to half the usually recommended starting dosage for:
o patients not currently taking an angiotensin-converting enzyme (ACE) inhibitor or angiotensin II receptor blocker (ARB) or previously taking a low dose of these agents. (2.6)
o patients with severe renal impairment. (2.7)
o patients with moderate hepatic impairment. (2.8)DOSAGE FORMS AND STRENGTHS
- Film-coated tablets: 24/26 mg; 49/51 mg; 97/103 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
Adverse reactions occurring greater than or equal to 5% are hypotension, hyperkalemia, cough, dizziness, and renal failure. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Laurus Generics Inc. at 1-833-3-LAURUS (1-833-352-8787) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Avoid concomitant use with aliskiren in patients with estimated glomerular filtration rate (eGFR) less than 60. (7.1)
- Potassium-sparing diuretics: May lead to increased serum potassium. (7.2)
- Nonsteroidal Anti-Inflammatory Drugs (NSAIDs): May lead to increased risk of renal impairment. (7.3)
- Lithium: Increased risk of lithium toxicity. (7.4)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: FETAL TOXICITY
1 INDICATIONS AND USAGE
1.1 Adult Heart Failure
1.2 Pediatric Heart Failure
2 DOSAGE AND ADMINISTRATION
2.1 General Considerations
2.2 Adult Heart Failure
2.3 Pediatric Heart Failure
2.4 Preparation of Oral Suspension Using Tablets
2.6 Dose Adjustment for Patients Not Taking an ACE inhibitor or ARB or Previously Taking Low Doses of These Agents
2.7 Dose Adjustment for Severe Renal Impairment
2.8 Dose Adjustment for Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Fetal Toxicity
5.2 Angioedema
5.3 Hypotension
5.4 Impaired Renal Function
5.5 Hyperkalemia
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Dual Blockade of the Renin-Angiotensin-Aldosterone System
7.2 Potassium-Sparing Diuretics
7.3 Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) Including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
7.4 Lithium
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Adult Heart Failure
14.2 Pediatric Heart Failure
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- BOXED WARNING (What is this?)
-
1 INDICATIONS AND USAGE
1.1 Adult Heart Failure
Sacubitril and valsartan tablets are indicated to reduce the risk of cardiovascular death and hospitalization for heart failure in adult patients with chronic heart failure. Benefits are most clearly evident in patients with left ventricular ejection fraction (LVEF) below normal.
LVEF is a variable measure, so use clinical judgment in deciding whom to treat [see Clinical Studies (14.1)].
1.2 Pediatric Heart Failure
Sacubitril and valsartan tablets are indicated for the treatment of symptomatic heart failure with systemic left ventricular systolic dysfunction in pediatric patients aged one year and older. Sacubitril and valsartan tablets reduce NT-proBNP and are expected to improve cardiovascular outcomes.
-
2 DOSAGE AND ADMINISTRATION
2.1 General Considerations
Sacubitril and valsartan tablets are contraindicated with concomitant use of an angiotensin-converting enzyme (ACE) inhibitor. If switching from an ACE inhibitor to sacubitril and valsartan tablets allow a washout period of 36 hours between administration of the two drugs [see Contraindications (4) andDrug Interactions (7.1)].
2.2 Adult Heart Failure
The recommended starting dose of sacubitril and valsartan tablets is 49/51 mg orally twice-daily.
Double the dose of sacubitril and valsartan tablets after 2 to 4 weeks to the target maintenance dose of 97/103 mg twice daily, as tolerated by the patient.
2.3 Pediatric Heart Failure
For the recommended dosage for pediatric patients aged 1 year and older, refer to Table 1 if using the tablets.
Take the recommended dose orally twice daily. Adjust pediatric patient doses every 2 weeks, as tolerated by the patient.
Table 1: Recommended Dose and Titration for Pediatric Patients Using Tablets
† Use of the oral suspension is recommended in these patients. Recommended mg/kg doses are of the combined amount of both sacubitril and valsartan[see Dosage and Administration (2.4)].
‡ Doses of 72 mg/78 mg can be achieved using three 24 mg/26 mg tablets [see Dosage Forms and Strengths (3)].
Weight (kg)
Titration Step Dose (twice daily)
Starting
Second
Final
Less than 40 kg†
1.6 mg/kg
2.3 mg/kg
3.1 mg/kg
At least 40 kg, less than 50kg
24 mg/26 mg
49 mg/51 mg
72 mg/78 mg‡
At least 50 kg
49 mg/51 mg
72 mg/78 mg‡
97 mg/103 mg
2.4 Preparation of Oral Suspension Using Tablets
Sacubitril and valsartan oral suspension can be substituted at the recommended tablet dosage in patients unable to swallow tablets.
Sacubitril and valsartan 800 mg/200 mL oral suspension can be prepared in a concentration of 4 mg/mL (sacubitril/valsartan 1.96/2.04 mg/mL). Use sacubitril and valsartan 49/51 mg tablets in the preparation of the suspension.
To make an 800 mg/200 mL (4 mg/mL) oral suspension, transfer eight tablets of sacubitril and valsartan 49/51 mg film-coated tablets into a mortar. Crush the tablets into a fine powder using a pestle. Add 60 mL of Ora-Plus® into the mortar and triturate gently with pestle for 10 minutes, to form a uniform suspension. Add 140 mL of Ora-Sweet® SF into mortar and triturate with pestle for another 10 minutes, to form a uniform suspension. Transfer the entire contents from the mortar into a clean 200 mL amber colored PET or glass bottle. Place a press-in bottle adapter and close the bottle with a child resistant cap.
The oral suspension can be stored for up to 15 days. Do not store above 25°C (77°F) and do not refrigerate. Shake before each use.
*Ora-Sweet SF® and Ora-Plus® are registered trademarks of Paddock Laboratories, Inc.2.6 Dose Adjustment for Patients Not Taking an ACE inhibitor or ARB or Previously Taking Low Doses of These Agents
In patients not currently taking an ACE inhibitor or an angiotensin II receptor blocker (ARB) and for patients previously taking low doses of these agents, start sacubitril and valsartan tablets at half the usually recommended starting dose. After initiation, increase the dose every 2 to 4 weeks in adults and every 2 weeks in pediatric patients to follow the recommended dose escalation thereafter [see Dosage and Administration (2.2, 2.3)].
Note: Initiate pediatric patients weighing 40 to 50 kg who meet this criterion at 0.8 mg/kg twice daily using the oral suspension [see Dosage and Administration (2.3, 2.4)].
2.7 Dose Adjustment for Severe Renal Impairment
In adults and pediatric patients with severe renal impairment estimated glomerular filtration rate (eGFR less than 30 mL/min/1.73 m2), start sacubitril and valsartan tablets at half the usually recommended starting dose. After initiation, increase the dose to follow the recommended dose escalation thereafter [see Dosage and Administration (2.2, 2.3)].
Note: Initiate pediatric patients weighing 40 to 50 kg who meet this criterion at 0.8 mg/kg twice daily using the oral suspension [see Dosage and Administration (2.3, 2.4)].
No starting dose adjustment is needed for mild or moderate renal impairment.2.8 Dose Adjustment for Hepatic Impairment
In adults and pediatric patients with moderate hepatic impairment (Child-Pugh B classification), start sacubitril and valsartan tablets at half the usually recommended starting dose. After initiation, increase the dose to follow the recommended dose escalation thereafter [see Dosage and Administration (2.2, 2.3)].
Note: Initiate pediatric patients weighing 40 to 50 kg who meet this criterion at 0.8 mg/kg twice daily using the oral suspension [see Dosage and Administration (2.3, 2.4)].
No starting dose adjustment is needed for mild hepatic impairment.
Use in patients with severe hepatic impairment is not recommended. -
3 DOSAGE FORMS AND STRENGTHS
Sacubitril and valsartan tablets are supplied in the following strengths:
Sacubitril and valsartan tablets 24/26 mg, (sacubitril 24 mg and valsartan 26 mg) are violet white color, oval shaped, biconvex film-coated tablets debossed with “S5” on one side and plain on the other side.
Sacubitril and valsartan tablets 49/51 mg, (sacubitril 49 mg and valsartan 51 mg) are pale yellow color, oval shaped, biconvex film-coated tablets debossed with “S1” on one side and plain on the other side.
Sacubitril and valsartan tablets 97/103 mg, (sacubitril 97 mg and valsartan 103 mg) are light pink color, oval shaped, biconvex film-coated tablets debossed with “S2” on one side and plain on the other side.
-
4 CONTRAINDICATIONS
Sacubitril and valsartan is contraindicated:
- in patients with hypersensitivity to any component
- in patients with a history of angioedema related to previous ACE inhibitor or ARB therapy [see Warnings and Precautions (5.2)]
- with concomitant use of ACE inhibitors. Do not administer within 36 hours of switching from or to an ACE inhibitor [see Drug Interactions (7.1)]
- with concomitant use of aliskiren in patients with diabetes [see Drug Interactions (7.1)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Fetal Toxicity
Sacubitril and valsartan can cause fetal harm when administered to a pregnant woman. Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. When pregnancy is detected, consider alternative drug treatment and discontinue sacubitril and valsartan. However, if there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system, and if the drug is considered lifesaving for the mother, advise a pregnant woman of the potential risk to the fetus [see Use in Specific Populations (8.1)].
5.2 Angioedema
Sacubitril and valsartan may cause angioedema [see Adverse Reactions (6.1)]. If angioedema occurs, discontinue sacubitril and valsartan immediately, provide appropriate therapy, and monitor for airway compromise. Sacubitril and valsartan must not be re-administered. In cases of confirmed angioedema where swelling has been confined to the face and lips, the condition has generally resolved without treatment, although antihistamines have been useful in relieving symptoms.
Angioedema associated with laryngeal edema may be fatal. Where there is involvement of the tongue, glottis or larynx, likely to cause airway obstruction, administer appropriate therapy, e.g., subcutaneous epinephrine/adrenaline solution 1:1,000 (0.3 mL to 0.5 mL) and take measures necessary to ensure maintenance of a patent airway.
Sacubitril and valsartan has been associated with a higher rate of angioedema in Black than in non-Black patients.
Patients with a prior history of angioedema may be at increased risk of angioedema with sacubitril and valsartan [see Adverse Reactions (6.1)]. Sacubitril and valsartan must not be used in patients with a known history of angioedema related to previous ACE inhibitor or ARB therapy [see Contraindications (4)]. Sacubitril and valsartan should not be used in patients with hereditary angioedema.5.3 Hypotension
Sacubitril and valsartan lowers blood pressure and may cause symptomatic hypotension [see Adverse Reactions (6.1)]. Patients with an activated renin-angiotensin system, such as volume- and/or salt-depleted patients (e.g., those being treated with high doses of diuretics), are at greater risk. Correct volume or salt depletion prior to administration of sacubitril and valsartan or start at a lower dose. If hypotension occurs, consider dose adjustment of diuretics, concomitant antihypertensive drugs, and treatment of other causes of hypotension (e.g., hypovolemia). If hypotension persists despite such measures, reduce the dosage or temporarily discontinue sacubitril and valsartan. Permanent discontinuation of therapy is usually not required.
5.4 Impaired Renal Function
As a consequence of inhibiting the renin-angiotensin-aldosterone system (RAAS), decreases in renal function may be anticipated in susceptible individuals treated with sacubitril and valsartan [see Adverse Reactions (6.1)]. In patients whose renal function depends upon the activity of the renin-angiotensin-aldosterone system (e.g., patients with severe congestive heart failure), treatment with ACE inhibitors and angiotensin receptor antagonists has been associated with oliguria, progressive azotemia and, rarely, acute renal failure and death. Closely monitor serum creatinine, and down-titrate or interrupt sacubitril and valsartan in patients who develop a clinically significant decrease in renal function [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
As with all drugs that affect the RAAS, sacubitril and valsartan may increase blood urea and serum creatinine levels in patients with bilateral or unilateral renal artery stenosis. In patients with renal artery stenosis, monitor renal function.
5.5 Hyperkalemia
Through its actions on the RAAS, hyperkalemia may occur with sacubitril and valsartan [see Adverse Reactions (6.1)]. Monitor serum potassium periodically and treat appropriately, especially in patients with risk factors for hyperkalemia such as severe renal impairment, diabetes, hypoaldosteronism, or a high potassium diet. Dosage reduction or interruption of sacubitril and valsartan may be required [see Dosage and Administration (2.7)].
-
6 ADVERSE REACTIONS
Clinically significant adverse reactions that appear in other sections of the labeling include:
- Angioedema [see Warnings and Precautions (5.2)]
- Hypotension [see Warnings and Precautions (5.3)]
- Impaired Renal Function [see Warnings and Precautions (5.4)]
- Hyperkalemia [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 6,622 heart failure patients were treated with sacubitril and valsartan in the PARADIGM-HF (vs. enalapril) and PARAGON-HF (vs. valsartan) clinical trials. Of these, 5,085 were exposed for at least 1 year.
Adult Heart Failure
In PARADIGM-HF, patients were required to complete sequential enalapril and sacubitril and valsartan run-in periods of (median) 15 and 29 days, respectively, prior to entering the randomized double-blind period comparing sacubitril and valsartan and enalapril. During the enalapril run-in period, 1,102 patients (10.5%) were permanently discontinued from the study, 5.6% because of an adverse event, most commonly renal dysfunction (1.7%), hyperkalemia (1.7%) and hypotension (1.4%). During the sacubitril and valsartan run-in period, an additional 10.4% of patients permanently discontinued treatment, 5.9% because of an adverse event, most commonly renal dysfunction (1.8%), hypotension (1.7%) and hyperkalemia (1.3%). Because of this run-in design, the adverse reaction rates described below are lower than expected in practice.
In the double-blind period, safety was evaluated in 4,203 patients treated with sacubitril and valsartan and 4,229 treated with enalapril. In PARADIGM-HF, patients randomized to sacubitril and valsartan received treatment for up to 4.3 years, with a median duration of exposure of 24 months; 3,271 patients were treated for more than one year. Discontinuation of therapy because of an adverse event during the double-blind period occurred in 450 (10.7%) of sacubitril and valsartan-treated patients and 516 (12.2%) of patients receiving enalapril.
Adverse reactions occurring at an incidence of greater than or equal to 5% in patients who were treated with sacubitril and valsartan in the double-blind period of PARADIGM-HF are shown in Table 3.
In PARADIGM-HF, the incidence of angioedema was 0.1% in both the enalapril and sacubitril and valsartan run-in periods. In the double-blind period, the incidence of angioedema was higher in patients treated with sacubitril and valsartan than enalapril (0.5% and 0.2%, respectively). The incidence of angioedema in Black patients was 2.4% with sacubitril and valsartan and 0.5% with enalapril [see Warnings and Precautions (5.2)].
Orthostasis was reported in 2.1% of patients treated with sacubitril and valsartan compared to 1.1% of patients treated with enalapril during the double-blind period of PARADIGM-HF. Falls were reported in 1.9% of patients treated with sacubitril and valsartan compared to 1.3% of patients treated with enalapril.
Table 3: Adverse Reactions Reported in greater than or equal to 5% of Patients Treated with Sacubitril and Valsartan in the Double-Blind Period of PARADIGM-HF Sacubitril and Valsartan
(n = 4,203)
%
Enalapril
(n = 4,229)
%
Hypotension
18
12
Hyperkalemia
12
14
Cough
9
13
Dizziness
6
5
Renal failure/acute renal failure
5
5
In PARAGON-HF, no new adverse reactions were identified.
Pediatric Heart Failure
The adverse reactions observed in pediatric patients 1 year to less than 18 years old who received treatment with sacubitril and valsartan were consistent with those observed in adult patients.
Laboratory Abnormalities
Hemoglobin and Hematocrit
Decreases in hemoglobin/hematocrit of greater than 20% were observed in approximately 5% of both sacubitril and valsartan- and enalapril-treated patients in the double-blind period in PARADIGM-HF. Decreases in hemoglobin/hematocrit of greater than 20% were observed in approximately 7% of sacubitril and valsartan-treated patients and 9% of valsartan-treated patients in the double-blind period in PARAGON-HF.
Serum Creatinine
During the double-blind period in PARADIGM-HF, approximately 16% of both sacubitril and valsartan- and enalapril-treated patients had increases in serum creatinine of greater than 50%. During the double-blind period in PARAGON-HF, approximately 17% of sacubitril and valsartan-treated patients and 21% of valsartan-treated patients had increases in serum creatinine of greater than 50%.
Serum Potassium
During the double-blind period of PARADIGM-HF, approximately 16% of both sacubitril and valsartan- and enalapril-treated patients had potassium concentrations greater than 5.5 mEq/L. During the double-blind period of PARAGON-HF, approximately 18% of sacubitril and valsartan-treated patients and 20% of valsartan-treated patients had potassium concentrations greater than 5.5 mEq/L.
6.2 Postmarketing Experience
The following additional adverse reactions have been reported in postmarketing experience. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hypersensitivity including rash, pruritus, and anaphylactic reaction
-
7 DRUG INTERACTIONS
7.1 Dual Blockade of the Renin-Angiotensin-Aldosterone System
Concomitant use of sacubitril and valsartan with an ACE inhibitor is contraindicated because of the increased risk of angioedema [see Contraindications (4)].
Avoid use of sacubitril and valsartan with an ARB, because sacubitril and valsartan contains the angiotensin II receptor blocker valsartan.
The concomitant use of sacubitril and valsartan with aliskiren is contraindicated in patients with diabetes [see Contraindications (4)]. Avoid use with aliskiren in patients with renal impairment (eGFR less than 60 mL/min/1.73 m²).
7.2 Potassium-Sparing Diuretics
As with other drugs that block angiotensin II or its effects, concomitant use of potassium-sparing diuretics (e.g., spironolactone, triamterene, amiloride), potassium supplements, or salt substitutes containing potassium may lead to increases in serum potassium [see Warnings and Precautions (5.5)].
7.3 Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) Including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, concomitant use of NSAIDs, including COX-2 inhibitors, with sacubitril and valsartan may result in worsening of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Sacubitril and valsartan can cause fetal harm when administered to a pregnant woman. Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death (see Clinical Considerations). Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin-angiotensin system from other antihypertensive agents. In animal reproduction studies, sacubitril and valsartan treatment during organogenesis resulted in increased embryo-fetal lethality in rats and rabbits and teratogenicity in rabbits (see Data). When pregnancy is detected, consider alternative drug treatment and discontinue sacubitril and valsartan. However, if there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system, and if the drug is considered lifesaving for the mother, advise a pregnant woman of the potential risk to the fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Oligohydramnios in pregnant women who use drugs affecting the renin-angiotensin system in the second and third trimesters of pregnancy can result in the following: reduced fetal renal function leading to anuria and renal failure, fetal lung hypoplasia, skeletal deformations, including skull hypoplasia, hypotension, and death.
Perform serial ultrasound examinations to assess the intra-amniotic environment. Fetal testing may be appropriate, based on the week of gestation. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury. If oligohydramnios is observed, consider alternative drug treatment. Closely observe neonates with histories of in utero exposure to sacubitril and valsartan for hypotension, oliguria, and hyperkalemia. In neonates with a history of in utero exposure to sacubitril and valsartan, if oliguria or hypotension occurs, support blood pressure and renal perfusion. Exchange transfusions or dialysis may be required as a means of reversing hypotension and replacing renal function.
Data
Animal Data
Sacubitril and valsartan treatment during organogenesis resulted in increased embryo-fetal lethality in rats at doses greater than or equal to 49 mg sacubitril/51 mg valsartan/kg/day (less than or equal to 0.06 [LBQ657, the active metabolite] and 0.72 [valsartan]-fold the maximum recommended human dose [MRHD] of 97/103 mg twice-daily on the basis of the area under the plasma drug concentration-time curve [AUC]) and rabbits at doses greater than or equal to 5 mg sacubitril/5 mg valsartan/kg/day (2-fold and 0.03-fold the MRHD on the basis of valsartan and LBQ657 AUC, respectively). Sacubitril and valsartan is teratogenic based on a low incidence of fetal hydrocephaly, associated with maternally toxic doses, which was observed in rabbits at a sacubitril and valsartan dose of greater than or equal to 5 mg sacubitril/5 mg valsartan/kg/day. The adverse embryo-fetal effects of sacubitril and valsartan are attributed to the angiotensin receptor antagonist activity.
Pre- and postnatal development studies in rats at sacubitril doses up to 750 mg/kg/day (2.2-fold the MRHD on the basis of LBQ657 AUC) and valsartan at doses up to 600 mg/kg/day (0.86-fold the MRHD on the basis of AUC) indicate that treatment with sacubitril and valsartan during organogenesis, gestation and lactation may affect pup development and survival.
8.2 Lactation
Risk Summary
There is no information regarding the presence of sacubitril/valsartan in human milk, the effects on the breastfed infant, or the effects on milk production. Sacubitril/valsartan is present in rat milk (see Data). Because of the potential for serious adverse reactions in breastfed infants from exposure to sacubitril/valsartan, advise a nursing woman that breastfeeding is not recommended during treatment with sacubitril and valsartan.
Data
Following an oral dose (15 mg sacubitril/15 mg valsartan/kg) of [14C] sacubitril and valsartan to lactating rats, transfer of LBQ657 into milk was observed. After a single oral administration of 3 mg/kg [14C] valsartan to lactating rats, transfer of valsartan into milk was observed.
8.4 Pediatric Use
The safety and effectiveness of sacubitril and valsartan have been established for the treatment of heart failure in pediatric patients 1 year to less than 18 years. Use of sacubitril and valsartan was evaluated in a multinational, randomized, double-blind trial comparing sacubitril and valsartan and enalapril in 375 patients aged 1 month to less than 18 years (sacubitril and valsartan n = 187; Enalapril n = 188) (PANORAMA-HF) [see Clinical Studies (14.2)]. The safety profile in pediatric patients (1 year to less than 18 years) receiving sacubitril and valsartan was similar to that seen in adult patients.
Limited safety and efficacy data in patients aged 1 month to less than 1 year were inadequate to support conclusions on safety and efficacy in this age group.
Juvenile Animal Toxicity Data
Sacubitril given orally to juvenile rats from postnatal day (PND) 7 to PND 35 or PND 70 (an age approximately equivalent to neonatal through pre-pubertal development or adulthood in humans) at doses greater than or equal to 400 mg/kg/day (approximately 2-fold the AUC exposure to the active metabolite of sacubitril, LBQ657, at a sacubitril and valsartan pediatric clinical dose of 3.1 mg/kg twice daily) resulted in decreases in body weight, bone length, and bone mass. The decrease in body weight was transient from PND 10 to PND 20 and the effects for most bone parameters were reversible after treatment stopped. Exposure at the No-Observed-Adverse-Effect-Level (NOAEL) of 100 mg/kg/day was approximately 0.5-fold the AUC exposure to LBQ657 at the 3.1 mg/kg twice daily dose of sacubitril and valsartan. The mechanism underlying bone effects in rats and the translatability to pediatric patients are unknown.
Valsartan given orally to juvenile rats from PND 7 to PND 70 (an age approximately equivalent to neonatal through adulthood in humans) produced persistent, irreversible kidney damage at all dose levels. Exposure at the lowest tested dose of 1 mg/kg/day was approximately 0.2-fold the exposure at 3.1 mg/kg twice daily dose of sacubitril and valsartan based on AUC. These kidney effects in neonatal rats represent expected exaggerated pharmacological effects that are observed if rats are treated during the first 13 days of life. This period coincides with 36 weeks of gestation in humans, which could occasionally extend up to 44 weeks after conception in humans. In humans, nephrogenesis is thought to be complete around birth; however, maturation of other aspects of kidney function (such as glomerular filtration and tubular function) may continue until approximately 2 years of age. It is unknown whether post-natal use of valsartan before maturation of renal function is complete has long-term deleterious effects on the kidney.
8.5 Geriatric Use
There were 4,143 and 3,971 heart failure patients 65 years of age and older in PARADIGM-HF and PARAGON-HF, respectively [see Clinical Studies (14)]. Of the total number of sacubitril and valsartan-treated patients, 2,087 (49.6%) and 1,995 (82.9%) were 65 years of age and older, while 786 (18.7%) and 1,100 (45.7%) were 75 years of age and older in PARADIGM-HF and PARAGON-HF, respectively. No overall differences in safety or effectiveness of sacubitril and valsartan have been observed between patients 65 years of age and older and younger adult patients in either study.
No relevant pharmacokinetic differences have been observed in elderly (≥ 65 years) or very elderly (≥ 75 years) patients compared to the overall population [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
No dose adjustment is required when administering sacubitril and valsartan to patients with mild hepatic impairment (Child-Pugh A classification). Half of the starting dose is recommended in adult and pediatric patients with heart failure and with moderate hepatic impairment (Child-Pugh B classification). The use of sacubitril and valsartan in patients with severe hepatic impairment (Child-Pugh C classification) is not recommended, as no studies have been conducted in these patients [see Dosage and Administration (2.8) and Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dose adjustment is required in patients with mild (eGFR 60 to 90 mL/min/1.73 m2) to moderate (eGFR 30 to 60 mL/min/1.73 m2) renal impairment. Half of the starting dose is recommended in adult and pediatric patients with heart failure and with severe renal impairment (eGFR less than 30 mL/min/1.73 m2). [see Dosage and Administration (2.7), Warnings and Precautions (5.4) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Limited data are available with regard to overdosage in human subjects with sacubitril and valsartan. In healthy volunteers, a single dose of sacubitril and valsartan 583 mg sacubitril/617 mg valsartan, and multiple doses of 437 mg sacubitril/463 mg valsartan (14 days) have been studied and were well tolerated.
Hypotension is the most likely result of overdosage due to the blood pressure lowering effects of sacubitril and valsartan. Symptomatic treatment should be provided.
Sacubitril and valsartan is unlikely to be removed by hemodialysis because of high protein binding.
-
11 DESCRIPTION
Sacubitril and valsartan tablets are a combination of a neprilysin inhibitor and an angiotensin II receptor blocker.
Sacubitril and valsartan tablets contain a complex comprised of anionic forms of sacubitril and valsartan, and sodium cations in the molar ratio of 1:1:3, respectively. Following oral administration, the complex dissociates into sacubitril (which is further metabolized to LBQ657) and valsartan. The complex is chemically described as Tri sodium [3-((1S,3R)-1-biphenyl-4-ylmethyl-3-ethoxycarbonyl-1-butylcarbamoyl)propionate-(S)-3’-methyl-2’-(pentanoyl{2”-(tetrazol-5-ylate)biphenyl-4’-ylmethyl}amino)butyrate].
Its molecular formula is C48H55N6O8●3Na. Its molecular mass is 912.95 and its schematic structural formula is:
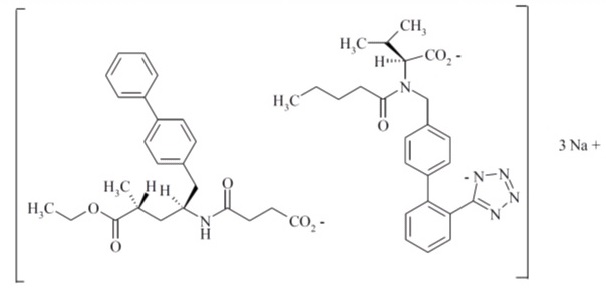
Sacubitril and valsartan tablets are available as film-coated tablets for oral administration, containing 24 mg of sacubitril and 26 mg of valsartan; 49 mg of sacubitril and 51 mg of valsartan; and 97 mg of sacubitril and 103 mg of valsartan. The tablet inactive ingredients are crospovidone, low-substituted hydroxypropylcellulose, magnesium stearate, microcrystalline cellulose, and silicon dioxide. The film-coat inactive ingredients are hypromellose, titanium dioxide, Macrogol 4000, talc, and iron oxide red. The film-coat for the 24 mg of sacubitril and 26 mg of valsartan tablet and the 97 mg of sacubitril and 103 mg of valsartan tablet also contains iron oxide black. The film-coat for the 49 mg of sacubitril and 51 mg of valsartan tablet contains iron oxide yellow.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Sacubitril and valsartan contains a neprilysin inhibitor, sacubitril, and an angiotensin receptor blocker, valsartan. Sacubitril and valsartan inhibits neprilysin (neutral endopeptidase; NEP) via LBQ657, the active metabolite of the prodrug sacubitril, and blocks the angiotensin II type-1 (AT1) receptor via valsartan. The cardiovascular and renal effects of sacubitril and valsartan in heart failure patients are attributed to the increased levels of peptides that are degraded by neprilysin, such as natriuretic peptides, by LBQ657, and the simultaneous inhibition of the effects of angiotensin II by valsartan. Valsartan inhibits the effects of angiotensin II by selectively blocking the AT1 receptor, and also inhibits angiotensin II-dependent aldosterone release.
12.2 Pharmacodynamics
The pharmacodynamic effects of sacubitril and valsartan were evaluated after single and multiple dose administrations in healthy subjects and in patients with heart failure, and are consistent with simultaneous neprilysin inhibition and renin-angiotensin system blockade.
In a 7-day valsartan-controlled study in patients with reduced ejection fraction (HFrEF), administration of sacubitril and valsartan resulted in a significant non-sustained increase in natriuresis, increased urine cGMP, and decreased plasma MR-proANP and NT-proBNP compared to valsartan.
In a 21-day study in HFrEF patients, sacubitril and valsartan significantly increased urine ANP and cGMP and plasma cGMP, and decreased plasma NT-proBNP, aldosterone and endothelin-1. Sacubitril and valsartan also blocked the AT1-receptor as evidenced by increased plasma renin activity and plasma renin concentrations. In PARADIGM-HF, sacubitril and valsartan decreased plasma NT-proBNP (not a neprilysin substrate) and increased plasma BNP (a neprilysin substrate) and urine cGMP compared with enalapril.
In PARAMOUNT, a randomized, double-blind, 36-week study in patients with heart failure with LVEF greater than or equal to 45% comparing 97/103 mg of sacubitril and valsartan (n=149) to 160 mg of valsartan (n =152) twice-daily, sacubitril and valsartan decreased NT-proBNP by 17% while valsartan increased NT-proBNP by 8% at Week 12 (p = 0.005).
In PARAGON-HF, sacubitril and valsartan decreased NT-proBNP by 24% (Week 16) and 19% (Week 48) compared to 6% and 3% reductions on valsartan, respectively.
In PANORAMA-HF, a reduction in NT-proBNP was observed at Weeks 4 and 12 for sacubitril and valsartan (40% and 50%) compared to baseline. The NT-proBNP levels continued to decrease over the duration of the study with a reduction of 65% for sacubitril and valsartan at Week 52 compared to baseline.
QT Prolongation: In a thorough QTc clinical study in healthy male subjects, single doses of sacubitril and valsartan 194 mg sacubitril/206 mg valsartan and 583 mg sacubitril/617 mg valsartan had no effect on cardiac repolarization.
Amyloid-β: Neprilysin is one of multiple enzymes involved in the clearance of amyloid-β (Aβ) from the brain and cerebrospinal fluid (CSF). Administration of sacubitril and valsartan 194 mg sacubitril/206 mg valsartan once-daily for 2 weeks to healthy subjects was associated with an increase in CSF Aβ1 to 38 compared to placebo; there were no changes in concentrations of CSF Aβ1 to 40 or CSF Aβ1 to 42. The clinical relevance of this finding is unknown [see Nonclinical Toxicology (13)].
Blood Pressure: Addition of a 50 mg single dose of sildenafil to sacubitril and valsartan at steady state (194 mg sacubitril/206 mg valsartan once daily for 5 days) in patients with hypertension was associated with additional blood pressure (BP) reduction (approximately 5/4 mmHg, systolic/diastolic BP) compared to administration of sacubitril and valsartan alone.
Co-administration of sacubitril and valsartan did not significantly alter the BP effect of intravenous nitroglycerin.12.3 Pharmacokinetics
Absorption
Following oral administration, sacubitril and valsartan tablets dissociates into sacubitril and valsartan. Sacubitril is further metabolized to LBQ657. The peak plasma concentrations of sacubitril, LBQ657, and valsartan are reached in 0.5 hours, 2 hours, and 1.5 hours, respectively. The oral absolute bioavailability of sacubitril is estimated to be greater than or equal to 60%. The valsartan in sacubitril and valsartan tablets is more bioavailable than the valsartan in other marketed tablet formulations; 26 mg, 51 mg, and 103 mg of valsartan in sacubitril and valsartan tablets is equivalent to 40 mg, 80 mg, and 160 mg of valsartan in other marketed tablet formulations, respectively.
Following twice-daily dosing of sacubitril and valsartan tablets, steady-state levels of sacubitril, LBQ657, and valsartan are reached in 3 days. At steady state, sacubitril and valsartan do not accumulate significantly, whereas LBQ657 accumulates by 1.6-fold. Sacubitril and valsartan administration with food has no clinically significant effect on the systemic exposures of sacubitril, LBQ657, or valsartan. Although there is a decrease in exposure to valsartan when sacubitril and valsartan is administered with food, this decrease is not accompanied by a clinically significant reduction in the therapeutic effect. Sacubitril and valsartan tablets can therefore be administered with or without food.
Distribution
Sacubitril, LBQ657 and valsartan are highly bound to plasma proteins (94% to 97%). Based on the comparison of plasma and CSF exposures, LBQ657 crosses the blood brain barrier to a limited extent (0.28%). The average apparent volumes of distribution of valsartan and sacubitril are 75 and 103 L, respectively.
Metabolism
Sacubitril is readily converted to LBQ657 by esterases; LBQ657 is not further metabolized to a significant extent. Valsartan is minimally metabolized; only about 20% of the dose is recovered as metabolites. A hydroxyl metabolite has been identified in plasma at low concentrations (less than 10%).
Elimination
Following oral administration, 52% to 68% of sacubitril (primarily as LBQ657) and approximately 13% of valsartan and its metabolites are excreted in urine; 37% to 48% of sacubitril (primarily as LBQ657), and 86% of valsartan and its metabolites are excreted in feces. Sacubitril, LBQ657, and valsartan are eliminated from plasma with a mean elimination half-life (T1/2) of approximately 1.4 hours, 11.5 hours, and 9.9 hours, respectively.
Linearity/Nonlinearity
The pharmacokinetics of sacubitril, LBQ657, and valsartan were linear over a sacubitril and valsartan dose range of 24 mg sacubitril/26 mg valsartan to 194 mg sacubitril/206 mg valsartan.
Drug Interactions:
Effect of Co-administered Drugs on Sacubitril and Valsartan:
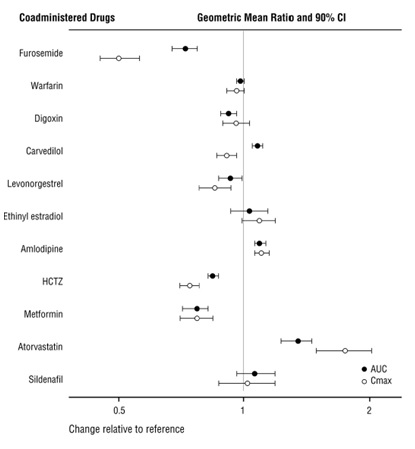
Because CYP450 enzyme-mediated metabolism of sacubitril and valsartan is minimal, coadministration with drugs that impact CYP450 enzymes is not expected to affect the pharmacokinetics of sacubitril and valsartan. Dedicated drug interaction studies demonstrated that coadministration of furosemide, warfarin, digoxin, carvedilol, a combination of levonorgestrel/ethinyl estradiol, amlodipine, omeprazole, hydrochlorothiazide (HCTZ), metformin, atorvastatin, and sildenafil, did not alter the systemic exposure to sacubitril, LBQ657 or valsartan.
Effect of Sacubitril and Valsartan on Co-administered Drugs:
In vitro data indicate that sacubitril inhibits OATP1B1 and OATP1B3 transporters. The effects of sacubitril and valsartan on the pharmacokinetics of coadministered drugs are summarized in Figure 1.
Figure 1: Effect of Sacubitril and Valsartan on Pharmacokinetics of Coadministered Drugs
Specific Populations
Effect of specific populations on the pharmacokinetics of LBQ657 and valsartan are shown in Figure 2.
Figure 2: Pharmacokinetics of Sacubitril and Valsartan in Specific Populations
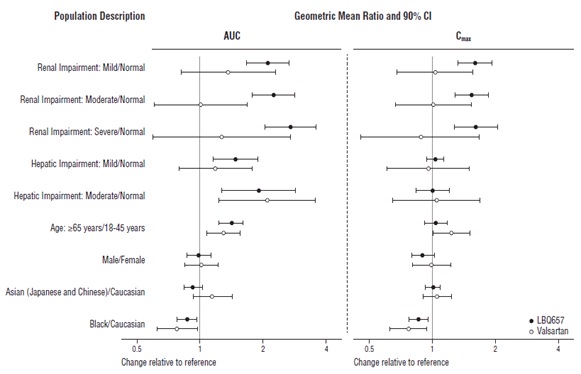
Note: Child-Pugh Classification was used for hepatic impairment.
Pediatric Patients: The pharmacokinetics of sacubitril and valsartan were evaluated in pediatric heart failure patients 1 to less than 18 years old administered oral doses of 0.8 mg/kg and 3.1 mg/kg of sacubitril and valsartan. Pharmacokinetic data indicated that exposure to sacubitril and valsartan in pediatric and adult patients is similar.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
Carcinogenicity studies conducted in mice and rats with sacubitril and valsartan did not identify any carcinogenic potential for sacubitril and valsartan. The LBQ657 Cmax at the high dose (HD) of 1,200 mg/kg/day in male and female mice was, respectively, 14 and 16 times that in humans at the MRHD. The LBQ657 Cmax in male and female rats at the HD of 400 mg/kg/day was, respectively, 1.7 and 3.5 times that at the MRHD. The doses of valsartan studied (high dose of 160 and 200 mg/kg/day in mice and rats, respectively) were about 4 and 10 times, respectively, the MRHD on a mg/m2 basis.
Mutagenicity and clastogenicity studies conducted with sacubitril and valsartan tablets, sacubitril, and valsartan did not reveal any effects at either the gene or chromosome level.
Impairment of Fertility
Sacubitril and valsartan did not show any effects on fertility in rats up to a dose of 73 mg sacubitril/77 mg valsartan/kg/day (≤ 1.0-fold and ≤ 0.18-fold the MRHD on the basis of the AUCs of valsartan and LBQ657, respectively).
13.2 Animal Toxicology and/or Pharmacology
The effects of sacubitril and valsartan on amyloid-β concentrations in CSF and brain tissue were assessed in young (2 to 4 years old) cynomolgus monkeys treated with sacubitril and valsartan (24 mg sacubitril/26 mg valsartan/kg/day) for 2 weeks. In this study, sacubitril and valsartan affected CSF Aβ clearance, increasing CSF Aβ 1 to 40, 1 to 42, and 1 to 38 levels in CSF; there was no corresponding increase in Aβ levels in the brain. In addition, in a toxicology study in cynomolgus monkeys treated with sacubitril and valsartan at 146 mg sacubitril/154 mg valsartan/kg/day for 39-weeks, there was no amyloid-β accumulation in the brain.
-
14 CLINICAL STUDIES
Dosing in clinical trials was based on the total amount of both components of sacubitril and valsartan tablets, i.e., 24/26 mg, 49/51 mg, and 97/103 mg were referred to as 50 mg, 100 mg, and 200 mg, respectively.
14.1 Adult Heart Failure
PARADIGM-HF
PARADIGM-HF was a multinational, randomized, double-blind trial comparing sacubitril and valsartan and enalapril in 8,442 adult patients with symptomatic chronic heart failure (NYHA class II to IV) and systolic dysfunction (left ventricular ejection fraction ≤ 40%). Patients had to have been on an ACE inhibitor or ARB for at least four weeks and on maximally tolerated doses of beta-blockers. Patients with a systolic blood pressure of less than 100 mmHg at screening were excluded.
The primary objective of PARADIGM-HF was to determine whether sacubitril and valsartan tablets, a combination of sacubitril and an RAS inhibitor (valsartan), was superior to an RAS inhibitor (enalapril) alone in reducing the risk of the combined endpoint of cardiovascular (CV) death or hospitalization for heart failure (HF).
After discontinuing their existing ACE inhibitor or ARB therapy, patients entered sequential single-blind run-in periods during which they received enalapril 10 mg twice-daily, followed by sacubitril and valsartan 100 mg twice-daily, increasing to 200 mg twice-daily. Patients who successfully completed the sequential run-in periods were randomized to receive either sacubitril and valsartan 200 mg (N = 4,209) twice-daily or enalapril 10 mg (N = 4,233) twice-daily. The primary endpoint was the first event in the composite of CV death or hospitalization for HF. The median follow-up duration was 27 months and patients were treated for up to 4.3 years.
The population was 66% Caucasian, 18% Asian, and 5% Black; the mean age was 64 years and 78% were male. At randomization, 70% of patients were NYHA Class II, 24% were NYHA Class III, and 0.7% were NYHA Class IV. The mean left ventricular ejection fraction was 29%. The underlying cause of heart failure was coronary artery disease in 60% of patients; 71% had a history of hypertension, 43% had a history of myocardial infarction, 37% had an eGFR less than 60 mL/min/1.73 m2, and 35% had diabetes mellitus. Most patients were taking beta-blockers (94%), mineralocorticoid antagonists (58%), and diuretics (82%). Few patients had an implantable cardioverter-defibrillator (ICD) or cardiac resynchronization therapy-defibrillator (CRT-D) (15%).
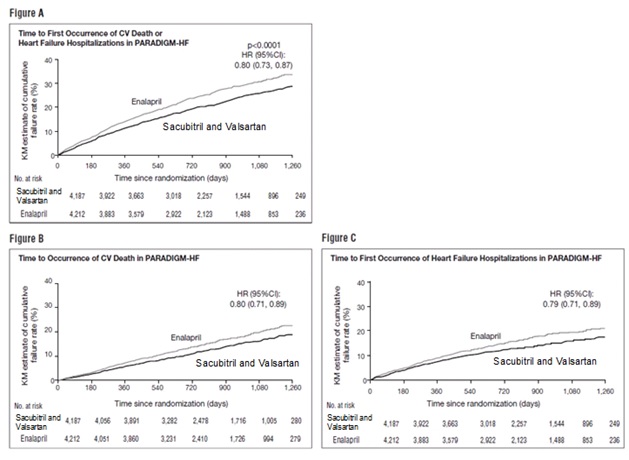
PARADIGM-HF demonstrated that sacubitril and valsartan tablets, a combination of sacubitril and an RAS inhibitor (valsartan), was superior to a RAS inhibitor (enalapril), in reducing the risk of the combined endpoint of cardiovascular death or hospitalization for heart failure, based on a time-to-event analysis (hazard ratio [HR] 0.80; 95% confidence interval [CI], 0.73, 0.87, p < 0.0001). The treatment effect reflected a reduction in both cardiovascular death and heart failure hospitalization; see Table 4 and Figure 3. Sudden death accounted for 45% of cardiovascular deaths, followed by pump failure, which accounted for 26%.
Sacubitril and valsartan also improved overall survival (HR 0.84; 95% CI [0.76, 0.93], p = 0.0009) (Table 4). This finding was driven entirely by a lower incidence of cardiovascular mortality on sacubitril and valsartan.
Table 4: Treatment Effect for the Primary Composite Endpoint, Its Components, and All-cause Mortality in PARADIGM-HF *Analyses of the components of the primary composite endpoint were not prospectively planned to be adjusted for multiplicity.
**Includes patients who had heart failure hospitalization prior to death.
Sacubitril and Valsartan
N = 4,187
n (%)
Enalapril
N = 4,212
n (%)
Hazard Ratio
(95% CI)
p-value
Primary composite endpoint of cardiovascular
death or heart failure hospitalization
Cardiovascular death as first event
Heart failure hospitalization as first event
914 (21.8)
377 (9.0)
537 (12.8)
1,117 (26.5)
459 (10.9)
658 (15.6)
0.80 (0.73, 0.87)
< 0.0001
Number of patients with events: *
Cardiovascular death**
Heart failure hospitalizations
558 (13.3)
537 (12.8)
693 (16.5)
658 (15.6)
0.80 (0.71, 0.89)
0.79 (0.71, 0.89)
All-cause mortality
711 (17.0)
835 (19.8)
0.84 (0.76, 0.93)
0.0009
The Kaplan-Meier curves presented below (Figure 3) show time to first occurrence of the primary composite endpoint (3A), and time to occurrence of cardiovascular death at any time (3B) and first heart failure hospitalization (3C).
Figure 3: Kaplan-Meier Curves for the Primary Composite Endpoint (A), Cardiovascular Death (B), and Heart Failure Hospitalization (C)
A wide range of demographic characteristics, baseline disease characteristics, and baseline concomitant medications were examined for their influence on outcomes. The results of the primary composite endpoint were consistent across the subgroups examined (Figure 4).
Figure 4: Primary Composite Endpoint (CV Death or HF Hospitalization) - Subgroup Analysis (PARADIGM-HF)
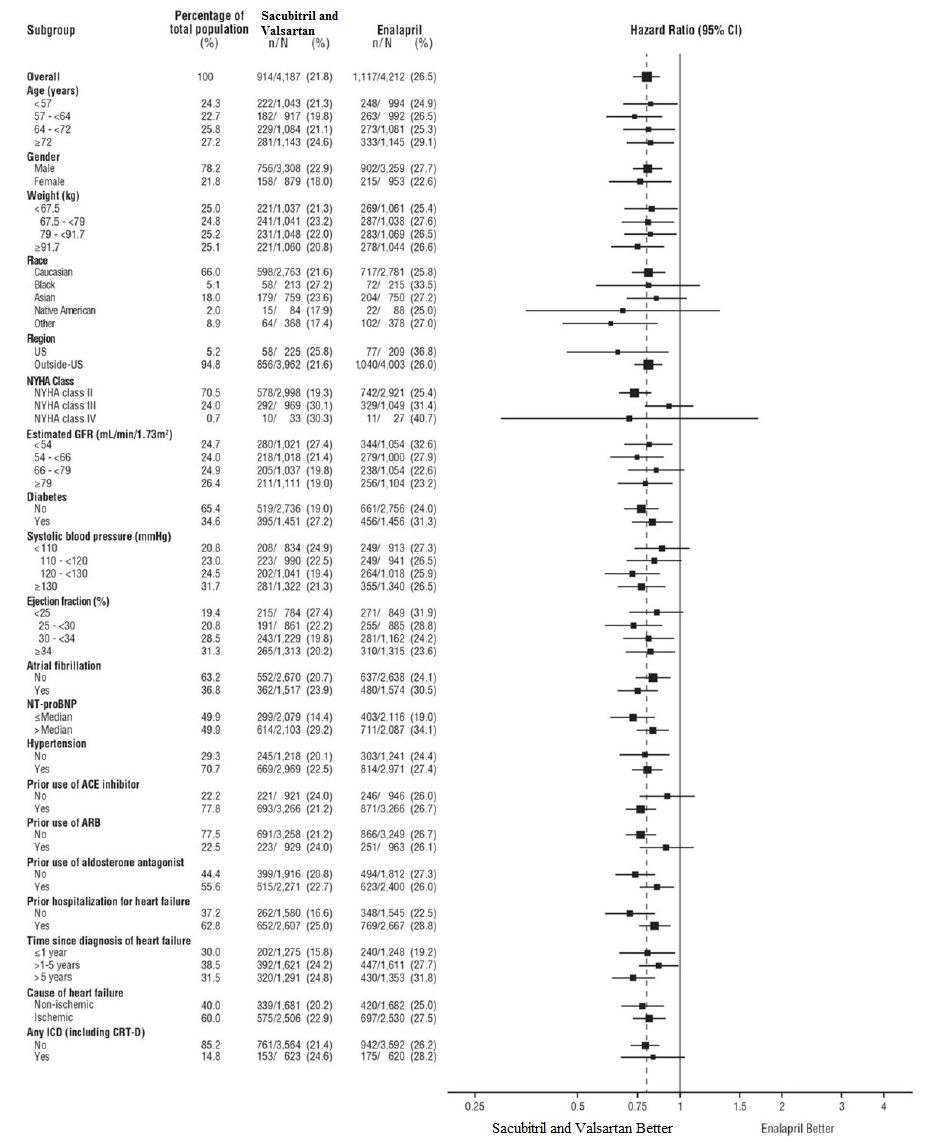
Note: The figure above presents effects in various subgroups, all of which are baseline characteristics. The 95% confidence limits that are shown do not take into account the number of comparisons made, and may not reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
PARAGON-HF
PARAGON-HF, was a multicenter, randomized, double-blind trial comparing sacubitril and valsartan and valsartan in 4,796 adult patients with symptomatic heart failure with left ventricular ejection fraction greater than or equal to 45%, and structural heart disease [either left atrial enlargement (LAE) or left ventricular hypertrophy (LVH)]. Patients with a systolic blood pressure of less than 110 mmHg and patients with any prior echocardiographic LVEF less than 40% at screening were excluded.
The primary objective of PARAGON-HF was to determine whether sacubitril and valsartan reduced the rate of the composite endpoint of total (first and recurrent) heart failure (HF) hospitalizations and cardiovascular (CV) death.
After discontinuing their existing ACE inhibitor or ARB therapy, patients entered sequential single-blind run-in periods during which they received valsartan 80 mg twice-daily, followed by sacubitril and valsartan 100 mg twice-daily. Patients on prior low doses of an ACEi or ARB began the run-in period receiving valsartan 40 mg twice-daily for 1 to 2 weeks. Patients who successfully completed the sequential run-in periods were randomized to receive either sacubitril and valsartan 200 mg (N = 2,419) twice-daily or valsartan 160 mg (N = 2,403) twice-daily. The median follow-up duration was 35 months and patients were treated for up to 4.7 years.
The population was 81% Caucasian, 13% Asian, and 2% Black; the mean age was 73 years and 52% were female. At randomization, 77% of patients were NYHA Class II, 19% were NYHA Class III, and 0.4% were NYHA Class IV. The median left ventricular ejection fraction was 57%. The underlying cause of heart failure was of ischemic etiology in 36% of patients. Furthermore, 96% had a history of hypertension, 23% had a history of myocardial infarction, 46% had an eGFR less than 60 mL/min/1.73 m2, and 43% had diabetes mellitus. Most patients were taking beta-blockers (80%) and diuretics (95%).
PARAGON-HF demonstrated that sacubitril and valsartan had a numerical reduction in the rate of the composite endpoint of total (first and recurrent) HF hospitalizations and CV death, based on an analysis using a proportional rates model (rate ratio [RR] 0.87; 95% CI [0.75, 1.01], p = 0.06); see Table 5. The treatment effect was primarily driven by the reduction in total HF hospitalizations in patients randomized to sacubitril and valsartan (RR 0.85; 95% CI [0.72, 1.00]).
Table 5: Treatment Effect for the Primary Composite Endpoint and Its Components in PARAGON-HF Abbreviations: RR = rate ratio, HR = hazard ratio.
a Event rate per 100 patient-years.
b Includes patients who had CV death following HF hospitalization event.
Sacubitril and valsartan
N = 2,407
Valsartan
N = 2,389
Effect Size
(95% CI)
Efficacy Endpoints
n
Event Ratea
n
Event Ratea
Composite of total (first and recurrent)
HF hospitalizations and CV death
894
12.8
1,009
14.6
RR = 0.87 (0.75, 1.01)
p-value 0.06
Total HF Hospitalizations
690
9.9
797
11.6
RR = 0.85 (0.72, 1.00)
CV Deathb
204
2.9
212
3.1
HR = 0.95 (0.79, 1.16)
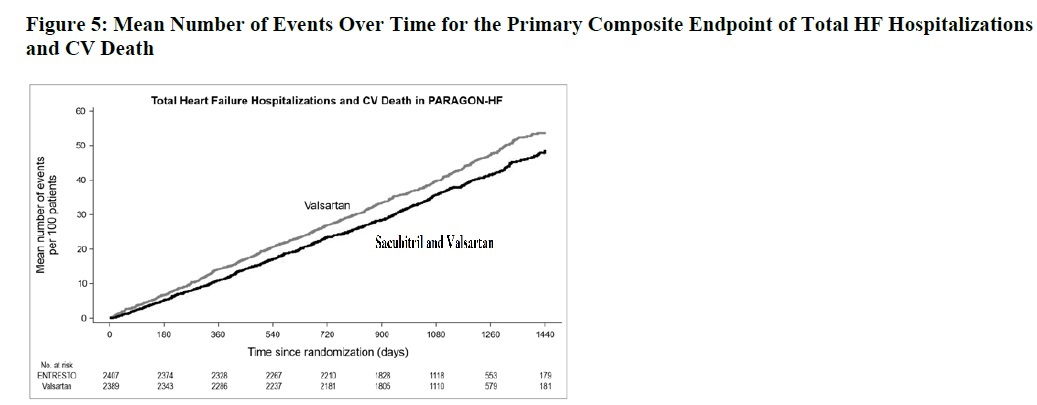
Figure 5 shows the mean number of composite endpoint events of total HF hospitalizations and CV death over time.
A wide range of demographic characteristics, baseline disease characteristics, and baseline concomitant medications were examined for their influence on outcomes (Figure 6).
Figure 6: Primary Composite Endpoint of Total HF Hospitalizations and CV Death – Subgroup Analysis (PARAGON – HF)
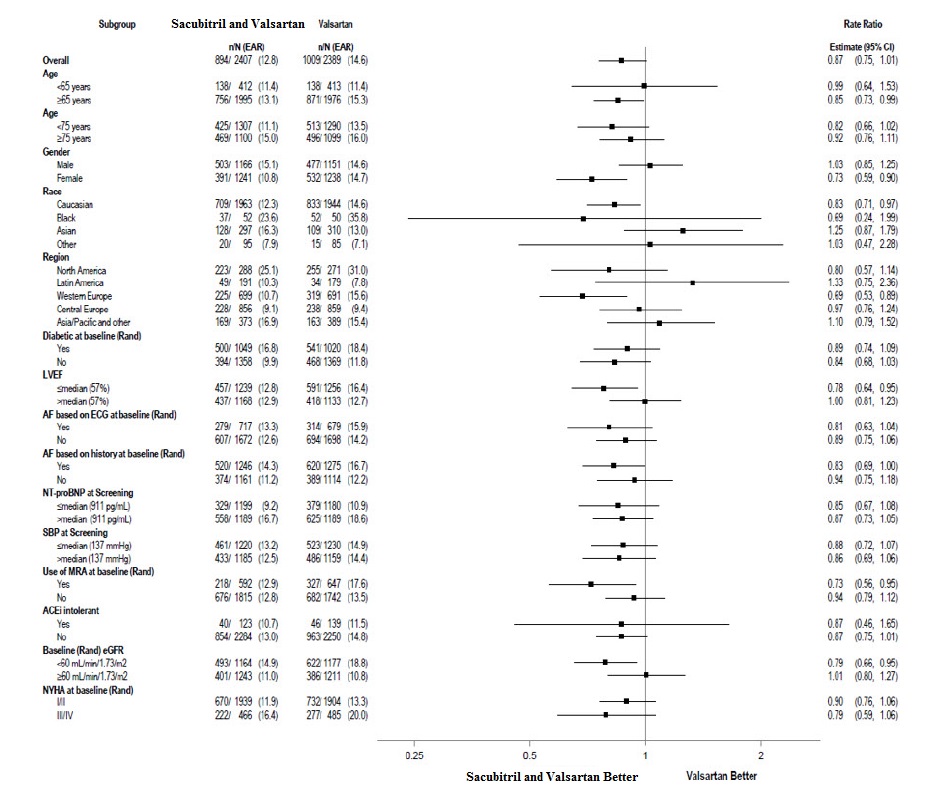
Note: The figure above presents effects in various subgroups, all of which are baseline characteristics. The 95% confidence limits that are shown do not take into account the number of comparisons made, and may not reflect the effect of a particular factor after adjustment for all other factors.
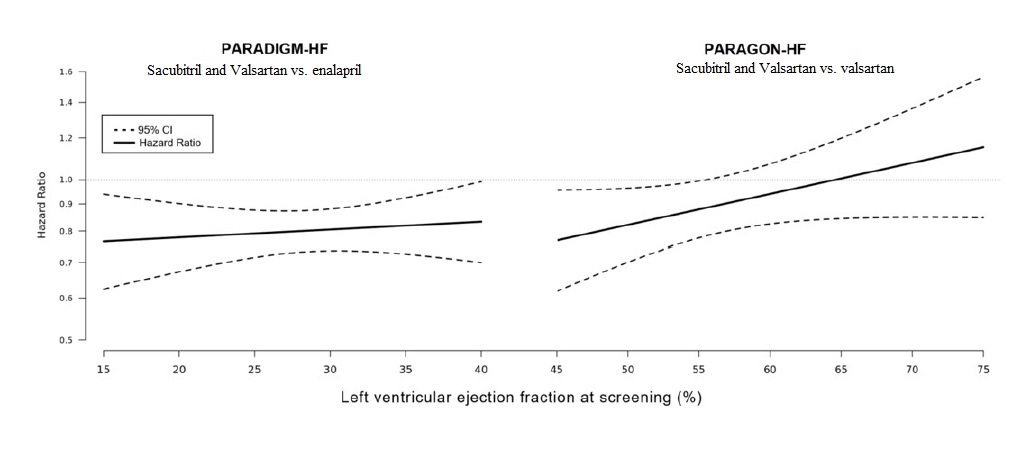
In an analysis of the relationship between LVEF and outcome in PARADIGM-HF and PARAGON-HF, patients with LVEF below normal treated with sacubitril and valsartan experienced greater risk reduction (Figure 7).
Figure 7: Treatment Effect for the Composite Endpoint of Time to First HF Hospitalization or CV Death by LVEF in PARADIGM-HF and PARAGON – HF
14.2 Pediatric Heart Failure
The efficacy of sacubitril and valsartan was evaluated in a multinational, randomized, double-blind trial PANORAMA-HF comparing sacubitril and valsartan (n = 187) and enalapril (n = 188) in pediatric patients aged 1 month to less than 18 years old due to systemic left ventricular systolic dysfunction (LVEF ≤ 45% or fractional shortening ≤ 22.5%). Patients with systemic right ventricle, single ventricle, restrictive cardiomyopathy or hypertrophic cardiomyopathy were excluded from the trial. Efficacy of sacubitril and valsartan in patients less than 1 year old was not established. At Week 52, there were 144 sacubitril and valsartan and 133 enalapril patients with a post-baseline assessment of NT-proBNP. The estimated least squares mean percent reduction from baseline in NT-proBNP was 65% and 62% in the sacubitril and valsartan and enalapril groups, respectively. While the between-group difference was not nominally statistically significant, the reductions for sacubitril and valsartan and enalapril were larger than what was seen in adults; these reductions did not appear to be attributable to post-baseline changes in background therapy.
Because sacubitril and valsartan improved outcomes and reduced NT-proBNP in adults in PARADIGM-HF, the effect on NT-proBNP was the basis to infer improved cardiovascular outcomes in pediatric patients.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Sacubitril and valsartan tablets are available as described below:
Sacubitril and valsartan tablets 24/26 mg, (sacubitril 24 mg and valsartan 26 mg) are violet white color, oval shaped, biconvex film-coated tablets debossed with “S5” on one side and plain on the other side.
Bottles of 30 NDC: 42385-930-30
Bottles of 60 NDC: 42385-930-60
Bottles of 180 NDC: 42385-930-18
Carton with 100 (10 x 10) Unit-Dose Tablets
containing cold form with desiccant blisters NDC: 42385-930-68
Carton with 100 (10 x 10) Unit-Dose Tablets
containing PVC/ACLAR blisters NDC: 42385-930-72
Sacubitril and valsartan tablets 49/51 mg, (sacubitril 49 mg and valsartan 51 mg) are pale yellow color, oval shaped, biconvex film-coated tablets debossed with “S1” on one side and plain on the other side.
Bottles of 30 NDC: 42385-931-30
Bottles of 60 NDC: 42385-931-60
Bottles of 180 NDC: 42385-931-18
Carton with 100 (10 x 10) Unit-Dose Tablets
containing cold form with desiccant blisters NDC: 42385-931-68
Carton with 100 (10 x 10) Unit-Dose Tablets
containing PVC/ACLAR blisters NDC: 42385-931-72
Sacubitril and valsartan tablets 97/103 mg, (sacubitril 97 mg and valsartan 103 mg) are light pink color, oval shaped, biconvex film-coated tablets debossed with “S2” on one side and plain on the other side.
Bottles of 30 NDC: 42385-932-30
Bottles of 60 NDC: 42385-932-60
Bottles of 180 NDC: 42385-932-18
Carton with 100 (10 x 10) Unit-Dose Tablets
containing cold form with desiccant blisters NDC: 42385-932-68
Carton with 100 (10 x 10) Unit-Dose Tablets
containing PVC/ACLAR blisters NDC: 42385-932-72
Store at 20° to 25°C (68° to 77°F); excursions permitted between 15° to 30°C (59° to 86°F) [See USP Controlled Room Temperature]. Protect from moisture. -
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information).
Pregnancy: Advise female patients of childbearing age about the consequences of exposure to sacubitril and valsartan tablets during pregnancy. Discuss treatment options with women planning to become pregnant. Ask patients to report pregnancies to their physicians as soon as possible [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1)].
Lactation: Advise patients that breastfeeding is not recommended during treatment with sacubitril and valsartan tablets [see Use in Specific Populations (8.2)].
Angioedema: Advise patients to discontinue use of their previous ACE inhibitor or ARB. Advise patients to allow a 36-hour wash-out period if switching from or to an ACE inhibitor [see Contraindications (4) and Warnings and Precautions (5.2)].
Manufactured for:
Laurus Generics Inc.
400 Connell Drive
Suite 5200
Berkeley Heights, NJ 07922
Manufactured by:
Laurus Labs Limited
Anakapalli-531011
India
Revised: 5/2024 -
PATIENT PACKAGE INSERT
PATIENT INFORMATION
Sacubitril and Valsartan
(sak-UE-bi-tril and val-SAR-tan)
Tablets, for oral use
What is the most important information I should know about sacubitril and valsartan tablets?
Sacubitril and valsartan tablets can harm or cause death to your unborn baby. Talk to your doctor about other ways to treat heart failure if you plan to become pregnant. Tell your doctor right away if you become pregnant during treatment with sacubitril and valsartan tablets.
What are sacubitril and valsartan tablets?
Sacubitril and valsartan tablets are a prescription medicine used to treat:
- adults with long-lasting (chronic) heart failure to help reduce the risk of death and hospitalization. Sacubitril and valsartan tablet works better when the heart cannot pump a normal amount of blood to the body.
- certain children 1 year of age and older who have symptomatic heart failure.
Do not take sacubitril and valsartan tablets if you:
- are allergic to any of the ingredients in sacubitril and valsartan tablets. See the end of this Patient Information leaflet for a complete list of ingredients in sacubitril and valsartan tablets.
- have had an allergic reaction, including swelling of your face, lips, tongue, throat, or trouble breathing while taking a type of medicine called an angiotensin-converting enzyme (ACE) inhibitor or angiotensin II receptor blocker (ARB).
- take an ACE inhibitor medicine. Do not take sacubitril and valsartan tablets for at least 36 hours before or after you take an ACE inhibitor medicine. Talk with your doctor or pharmacist before taking sacubitril and valsartan tablets if you are not sure if you take an ACE inhibitor medicine.
- have diabetes and take a medicine that contains aliskiren.
Before taking sacubitril and valsartan tablets, tell your doctor about all of your medical conditions, including if you:
- have a history of hereditary angioedema
- have kidney or liver problems
- have diabetes
- are pregnant or plan to become pregnant. See “What is the most important information I should know about sacubitril and valsartan tablets?”
- are breastfeeding or plan to breastfeed. It is not known if sacubitril and valsartan passes into your breast milk. You should not breastfeed during treatment with sacubitril and valsartan tablets. You and your doctor should decide if you will take sacubitril and valsartan tablets or breastfeed.
- potassium supplements or a salt substitute
- nonsteroidal anti-inflammatory drugs (NSAIDs)
- lithium
- other medicines for high blood pressure or heart problems, such as an ACE inhibitor, ARB, or aliskiren
How should I take sacubitril and valsartan tablets?
Take sacubitril and valsartan tablets exactly as your doctor tells you to take it.
Take sacubitril and valsartan tablets 2 times each day. Your doctor may change your dose of sacubitril and valsartan tablets during treatment.
If you or your child cannot swallow tablets, or if tablets are not available in the prescribed strength, you or your child may take sacubitril and valsartan tablets prepared as a liquid (oral) suspension.
If you or your child switches between taking sacubitril and valsartan tablets and the liquid suspension prepared from sacubitril and valsartan tablets, your doctor will adjust the dose as needed.
If you or your child are prescribed sacubitril and valsartan tablets to be prepared as a liquid suspension:
o Your pharmacist will prepare sacubitril and valsartan tablets for you or your child to take as a liquid suspension.
o Shake the bottle of suspension well before measuring the dose of medicine and before taking or giving the dose.
If you miss a dose, take it as soon as you remember. If it is close to your next dose, do not take the missed dose. Take the next dose at your regular time.
If you take too many sacubitril and valsartan tablets, call your doctor right away.What are the possible side effects of sacubitril and valsartan tablets?
Sacubitril and valsartan tablets may cause serious side effects, including:
- See “What is the most important information I should know about sacubitril and valsartan tablets?”
- Serious allergic reactions causing swelling of your face, lips, tongue, and throat (angioedema) that may cause trouble breathing and death. Get emergency medical help right away if you have symptoms of angioedema or trouble breathing. Do not take sacubitril and valsartan tablets again if you have had angioedema during treatment with sacubitril and valsartan tablets.
People who have had angioedema before taking sacubitril and valsartan tablets may have a higher risk of having angioedema than people who have not had angioedema before takingsacubitril and valsartan tablets. See “Who should not take sacubitril and valsartan tablets?”- Low blood pressure (hypotension). Low blood pressure is common during treatment with sacubitril and valsartan tablets. Your risk of low blood pressure is greater if you also take water pills (diuretics). Call your doctor if you become dizzy or lightheaded, or you develop extreme tiredness (fatigue).
- Kidney problems. Kidney problems are common during treatment with sacubitril and valsartan tablets and can be serious and can lead to kidney failure. Your doctor will check your kidney function during your treatment with sacubitril and valsartan tablets.
- Increased amount of potassium in your blood (hyperkalemia). Increased blood potassium levels are common during treatment with sacubitril and valsartan tablets. Your doctor will check your potassium blood level during your treatment with sacubitril and valsartan tablets.
Your doctor may need to lower your dose, temporarily stop treatment, or permanently stop treatment if you develop certain side effects or if you have changes in your kidney function or increased blood levels of potassium during treatment with sacubitril and valsartan tablets.
These are not all of the possible side effects of sacubitril and valsartan tablets. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store sacubitril and valsartan tablets?
- Store sacubitril and valsartan tablets at room temperature between 68°F to 77°F (20°C to 25°C).
- Protect sacubitril and valsartan tablets from moisture.
- Store bottles of sacubitril and valsartan tablets prepared as an oral suspension at room temperature less than 77°F (25°C) for up to 15 days. Do not refrigerate sacubitril and valsartan tablets prepared as an oral suspension.
General information about the safe and effective use of sacubitril and valsartan tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use sacubitril and valsartan tablets for a condition for which it was not prescribed. Do not give sacubitril and valsartan tablets to other people, even if they have the same symptoms that you have. It may harm them.You can ask your pharmacist or doctor for information about sacubitril and valsartan tablets that is written for health professionals.
What are the ingredients in sacubitril and valsartan tablets?
Active ingredients: sacubitril and valsartan
Inactive ingredients: crospovidone, low-substituted hydroxypropylcellulose, magnesium stearate, microcrystalline cellulose, and silicon dioxide. The film-coat inactive ingredients are hypromellose, titanium dioxide, Macrogol 4000, talc, and iron oxide red. The film-coat for the 24 mg of sacubitril and 26 mg of valsartan tablet and the 97 mg of sacubitril and 103 mg of valsartan tablet also contains iron oxide black. The film-coat for the 49 mg of sacubitril and 51 mg of valsartan tablet contains iron oxide yellow.
Prepared sacubitril and valsartan oral suspension also contains Ora-Sweet SF and Ora-Plus.
For more information, call Laurus Generics Inc. at 1-833-3-LAURUS (1-833-352-8787).
This Patient Information has been approved by the U.S. Food and Drug Administration.
Manufactured for:
Laurus Generics Inc.
400 Connell Drive
Suite 5200
Berkeley Heights, NJ 07922
Manufactured by:
Laurus Labs Limited
Anakapalli-531011
India
Revised: 5/2024
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL - 24 mg/26 mg - Container Label (30's count)
-
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL -24 mg/26 mg - cold form Blister Carton - 100 (10 x 10) unit-dose tablets
Laurus Labs NDC: 42385-930-68
Sacubitril and Valsartan Tablets 24 mg/26 mg
For Institutional Use Only.
100 (10 x 10) Unit-Dose Tablets Rx Only

- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL - 24 mg/26 mg - cold form Blister (1 x 10's count)
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL - 49 mg/51 mg - Container Label (30's count)
-
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL -49 mg/51 mg - cold form Blister Carton - 100 (10 x 10) unit-dose tablets
Laurus Labs NDC: 42385-931-68
Sacubitril and Valsartan Tablets 49 mg/51 mg
For Institutional Use Only.
100 (10 x 10) Unit-Dose Tablets Rx Only

- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL - 49 mg/51 mg - cold form Blister (1 x 10's count)
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL - 97 mg/103 mg - Container Label (30's count)
-
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL -97 mg/103 mg - cold form Blister Carton - 100 (10 x 10) unit-dose tablets
Laurus Labs NDC: 42385-932-68
Sacubitril and Valsartan Tablets 97 mg/103 mg
For Institutional Use Only.
100 (10 x 10) Unit-Dose Tablets Rx Only

- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL - 97 mg/103 mg - cold form Blister (1 x 10's count)
-
INGREDIENTS AND APPEARANCE
SACUBITRIL AND VALSARTAN
sacubitril and valsartan tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42385-930 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SACUBITRIL (UNII: 17ERJ0MKGI) (SACUBITRILAT - UNII:SPI5PBF81S) SACUBITRIL 24 mg VALSARTAN (UNII: 80M03YXJ7I) (VALSARTAN - UNII:80M03YXJ7I) VALSARTAN 26 mg Inactive Ingredients Ingredient Name Strength CROSPOVIDONE (120 .MU.M) (UNII: 68401960MK) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 4000 (UNII: 4R4HFI6D95) TALC (UNII: 7SEV7J4R1U) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color WHITE (Violet white color) Score no score Shape OVAL (biconvex) Size 9mm Flavor Imprint Code S5 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42385-930-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/11/2024 2 NDC: 42385-930-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 11/11/2024 3 NDC: 42385-930-18 180 in 1 BOTTLE; Type 0: Not a Combination Product 11/11/2024 4 NDC: 42385-930-68 100 in 1 CARTON 11/11/2024 4 NDC: 42385-930-10 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 5 NDC: 42385-930-72 100 in 1 CARTON 11/11/2024 5 NDC: 42385-930-17 10 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA213676 11/11/2024 SACUBITRIL AND VALSARTAN
sacubitril and valsartan tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42385-931 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SACUBITRIL (UNII: 17ERJ0MKGI) (SACUBITRILAT - UNII:SPI5PBF81S) SACUBITRIL 49 mg VALSARTAN (UNII: 80M03YXJ7I) (VALSARTAN - UNII:80M03YXJ7I) VALSARTAN 51 mg Inactive Ingredients Ingredient Name Strength CROSPOVIDONE (120 .MU.M) (UNII: 68401960MK) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 4000 (UNII: 4R4HFI6D95) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color YELLOW (Pale yellow color) Score no score Shape OVAL (biconvex) Size 12mm Flavor Imprint Code S1 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42385-931-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/11/2024 2 NDC: 42385-931-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 11/11/2024 3 NDC: 42385-931-18 180 in 1 BOTTLE; Type 0: Not a Combination Product 11/11/2024 4 NDC: 42385-931-68 100 in 1 CARTON 11/11/2024 4 NDC: 42385-931-10 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 5 NDC: 42385-931-72 100 in 1 CARTON 11/11/2024 5 NDC: 42385-931-17 10 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA213676 11/11/2024 SACUBITRIL AND VALSARTAN
sacubitril and valsartan tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42385-932 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SACUBITRIL (UNII: 17ERJ0MKGI) (SACUBITRILAT - UNII:SPI5PBF81S) SACUBITRIL 97 mg VALSARTAN (UNII: 80M03YXJ7I) (VALSARTAN - UNII:80M03YXJ7I) VALSARTAN 103 mg Inactive Ingredients Ingredient Name Strength CROSPOVIDONE (120 .MU.M) (UNII: 68401960MK) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 4000 (UNII: 4R4HFI6D95) TALC (UNII: 7SEV7J4R1U) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color PINK (Light pink color) Score no score Shape OVAL (biconvex) Size 15mm Flavor Imprint Code S2 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42385-932-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/11/2024 2 NDC: 42385-932-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 11/11/2024 3 NDC: 42385-932-18 180 in 1 BOTTLE; Type 0: Not a Combination Product 11/11/2024 4 NDC: 42385-932-68 100 in 1 CARTON 11/11/2024 4 NDC: 42385-932-10 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 5 NDC: 42385-932-72 100 in 1 CARTON 11/11/2024 5 NDC: 42385-932-17 10 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA213676 11/11/2024 Labeler - Laurus Labs Limited (915075687) Establishment Name Address ID/FEI Business Operations Laurus Labs Limited (VSP2) 650885309 ANALYSIS(42385-930, 42385-931, 42385-932) , API MANUFACTURE(42385-930, 42385-931, 42385-932) , MANUFACTURE(42385-930, 42385-931, 42385-932)






© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.