XOSPATA- gilteritinib tablet
Xospata by
Drug Labeling and Warnings
Xospata by is a Prescription medication manufactured, distributed, or labeled by Astellas Pharma US, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use XOSPATA safely and effectively. See full prescribing information for XOSPATA.
XOSPATA® (gilteritinib) tablets, for oral use
Initial U.S. Approval: 2018WARNING: DIFFERENTIATION SYNDROME
See full prescribing information for complete boxed warning.
Patients treated with XOSPATA have experienced symptoms of differentiation syndrome, which can be fatal if not treated. If differentiation syndrome is suspected, initiate corticosteroid therapy and hemodynamic monitoring until symptom resolution. (5.1, 6.1)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
XOSPATA is a kinase inhibitor indicated for the treatment of adult patients who have relapsed or refractory acute myeloid leukemia (AML) with a FLT3 mutation as detected by an FDA-approved test. (1.1)
DOSAGE AND ADMINISTRATION
120 mg orally once daily. (2.2)
DOSAGE FORMS AND STRENGTHS
Tablets: 40 mg. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Posterior reversible encephalopathy syndrome (PRES): Discontinue XOSPATA in patients who develop PRES. (2.3, 5.2, 6.1)
- Prolonged QT Interval: Interrupt and reduce XOSPATA dosage in patients who have a QTcF >500 msec. Correct hypokalemia or hypomagnesemia prior to and during XOSPATA administration. (2.3, 5.3, 12.2, 6.1)
- Pancreatitis: Interrupt and reduce the dose in patients who develop pancreatitis. (2.3, 5.4)
- Embryo-Fetal Toxicity: XOSPATA can cause fetal harm when administered to a pregnant woman. Advise of the potential risk to a fetus and to use effective contraception. (5.5, 8.1, 8.3)
ADVERSE REACTIONS
The most common adverse reactions (≥20%) were transaminase increased, myalgia/arthralgia, fatigue/malaise, fever, mucositis, edema, rash, noninfectious diarrhea, dyspnea, nausea, cough, constipation, eye disorders, headache, dizziness, hypotension, vomiting, and renal impairment. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Astellas Pharma US, Inc. at 1-800-727-7003 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
Lactation: Advise women not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 5/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: DIFFERENTIATION SYNDROME
1 INDICATIONS AND USAGE
1.1 Relapsed or Refractory Acute Myeloid Leukemia
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage
2.3 Dose Modification
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Differentiation Syndrome
5.2 Posterior Reversible Encephalopathy Syndrome
5.3 Prolonged QT Interval
5.4 Pancreatitis
5.5 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on XOSPATA
7.2 Effect of XOSPATA on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Relapsed or Refractory Acute Myeloid Leukemia
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: DIFFERENTIATION SYNDROME
Patients treated with XOSPATA have experienced symptoms of differentiation syndrome, which can be fatal or life-threatening if not treated. Symptoms may include fever, dyspnea, hypoxia, pulmonary infiltrates, pleural or pericardial effusions, rapid weight gain or peripheral edema, hypotension, or renal dysfunction. If differentiation syndrome is suspected, initiate corticosteroid therapy and hemodynamic monitoring until symptom resolution [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for the treatment of AML with XOSPATA based on the presence of FLT3 mutations in the blood or bone marrow [see Clinical Studies (14)]. Information on FDA-approved tests for the detection of a FLT3 mutation in AML is available at http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage
The recommended starting dose of XOSPATA is 120 mg orally once daily with or without food. Response may be delayed. In the absence of disease progression or unacceptable toxicity, treatment for a minimum of 6 months is recommended to allow time for a clinical response.
Do not break or crush XOSPATA tablets. Administer XOSPATA tablets orally about the same time each day. If a dose of XOSPATA is missed or not taken at the usual time, administer the dose as soon as possible on the same day, and at least 12 hours prior to the next scheduled dose. Return to the normal schedule the following day. Do not administer 2 doses within 12 hours.
2.3 Dose Modification
Assess blood counts and blood chemistries, including creatine phosphokinase, prior to the initiation of XOSPATA, at least once weekly for the first month, once every other week for the second month, and once monthly for the duration of therapy. Perform electrocardiogram (ECG) prior to initiation of treatment with gilteritinib, on days 8 and 15 of cycle 1, and prior to the start of the next two subsequent cycles.
Interrupt dosing or reduce dose for toxicities as per Table 1.
Table 1: Dosage Modifications for XOSPATA-Related Toxicities* - * Grade 1 is mild, Grade 2 is moderate, Grade 3 is serious, Grade 4 is life-threatening.
Adverse Reaction
Recommended Action
Differentiation Syndrome
- If differentiation syndrome is suspected, administer systemic corticosteroids and initiate hemodynamic monitoring until symptom resolution and for a minimum of 3 days [see Warnings and Precautions (5.1)].
- Interrupt XOSPATA if severe signs and/or symptoms persist for more than 48 hours after initiation of corticosteroids [see Warnings and Precautions (5.1)].
- Resume XOSPATA when signs and symptoms improve to Grade 2* or lower.
Posterior Reversible Encephalopathy Syndrome
- Discontinue XOSPATA.
QTc interval greater than 500 msec
- Interrupt XOSPATA.
- Resume XOSPATA at 80 mg when QTc interval returns to within 30 msec of baseline or less than or equal to 480 msec.
QTc interval increased by >30 msec on ECG on day 8 of cycle 1
- Confirm with ECG on day 9.
- If confirmed, consider dose reduction to 80 mg.
Pancreatitis
- Interrupt XOSPATA until pancreatitis is resolved.
- Resume XOSPATA at 80 mg.
Other Grade 3* or higher toxicity considered related to treatment.
- Interrupt XOSPATA until toxicity resolves or improves to Grade 1*.
- Resume XOSPATA at 80 mg.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Differentiation Syndrome
Of 319 patients treated with XOSPATA in the clinical trials, 3% experienced differentiation syndrome. Differentiation syndrome is associated with rapid proliferation and differentiation of myeloid cells and may be life-threatening or fatal if not treated. Symptoms of differentiation syndrome in patients treated with XOSPATA included fever, dyspnea, pleural effusion, pericardial effusion, pulmonary edema, hypotension, rapid weight gain, peripheral edema, rash, and renal dysfunction. Some cases had concomitant acute febrile neutrophilic dermatosis. Differentiation syndrome occurred as early as 2 days and up to 75 days after XOSPATA initiation and has been observed with or without concomitant leukocytosis. Of the 11 patients who experienced differentiation syndrome, 9 (82%) recovered after treatment or after dose interruption of XOSPATA.
If differentiation syndrome is suspected, initiate dexamethasone 10 mg IV every 12 hours (or an equivalent dose of an alternative oral or IV corticosteroid) and hemodynamic monitoring until improvement. Taper corticosteroids after resolution of symptoms and administer corticosteroids for a minimum of 3 days. Symptoms of differentiation syndrome may recur with premature discontinuation of corticosteroid treatment. If severe signs and/or symptoms persist for more than 48 hours after initiation of corticosteroids, interrupt XOSPATA until signs and symptoms are no longer severe [see Dosage and Administration (2.3)].
5.2 Posterior Reversible Encephalopathy Syndrome
Of 319 patients treated with XOSPATA in the clinical trials, 1% experienced posterior reversible encephalopathy syndrome (PRES) with symptoms including seizure and altered mental status. Symptoms have resolved after discontinuation of XOSPATA. A diagnosis of PRES requires confirmation by brain imaging, preferably magnetic resonance imaging (MRI). Discontinue XOSPATA in patients who develop PRES [see Dosage and Administration (2.3) and Adverse Reactions (6.1)].
5.3 Prolonged QT Interval
XOSPATA has been associated with prolonged cardiac ventricular repolarization (QT interval). Of the 317 patients with a post-baseline QTc measurement on treatment with XOSPATA in the clinical trial, 1% were found to have a QTc interval greater than 500 msec and 7% of patients had an increase from baseline QTc greater than 60 msec. Perform electrocardiogram (ECG) prior to initiation of treatment with gilteritinib, on days 8 and 15 of cycle 1, and prior to the start of the next two subsequent cycles. Interrupt and reduce XOSPATA dosage in patients who have a QTcF >500 msec [see Dosage and Administration (2.3), Adverse Reactions (6.1) and Clinical Pharmacology (12.2)].
Hypokalemia or hypomagnesemia may increase the QT prolongation risk. Correct hypokalemia or hypomagnesemia prior to and during XOSPATA administration.
5.4 Pancreatitis
Of 319 patients treated with XOSPATA in the clinical trials, 4% experienced pancreatitis. Evaluate patients who develop signs and symptoms of pancreatitis. Interrupt and reduce the dose of XOSPATA in patients who develop pancreatitis [see Dosage and Administration (2.3)].
5.5 Embryo-Fetal Toxicity
Based on findings in animals and its mechanism of action, XOSPATA can cause embryo-fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of gilteritinib to pregnant rats during organogenesis caused embryo-fetal lethality, suppressed fetal growth and teratogenicity at maternal exposures (AUC24) approximately 0.4 times the AUC24 in patients receiving the recommended dose. Advise females of reproductive potential to use effective contraception during treatment with XOSPATA and for at least 6 months after the last dose of XOSPATA. Advise males with female partners of reproductive potential to use effective contraception during treatment with XOSPATA and for at least 4 months after the last dose of XOSPATA. Pregnant women, patients becoming pregnant while receiving XOSPATA or male patients with pregnant female partners should be apprised of the potential risk to the fetus [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Differentiation syndrome [see Boxed Warning and Warnings and Precautions (5.1)]
- Posterior reversible encephalopathy syndrome [see Warnings and Precautions (5.2)]
- Prolonged QT interval [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety profile of XOSPATA is based on 319 patients with relapsed or refractory AML treated with gilteritinib 120 mg daily in three clinical trials. The median duration of exposure to XOSPATA was 3.6 months (range 0.1 to 43.4 months).
Fatal adverse reactions occurred in 2% of patients receiving XOSPATA. These included cardiac arrest (1%) and one case each of differentiation syndrome and pancreatitis. The most frequent (≥5%) nonhematological serious adverse reactions reported in patients were fever (13%), dyspnea (9%), renal impairment (8%), transaminase increased (6%) and noninfectious diarrhea (5%).
Of the 319 patients, 91 (29%) required a dose interruption due to an adverse reaction; the most common adverse reactions leading to dose interruption were aspartate aminotransferase increased (6%), alanine aminotransferase increased (6%) and fever (4%). Twenty patients (6%) required a dose reduction due to an adverse reaction. Twenty-two (7%) discontinued XOSPATA treatment permanently due to an adverse reaction. The most common (>1%) adverse reactions leading to discontinuation were aspartate aminotransferase increased (2%) and alanine aminotransferase increased (2%).
Overall, for the 319 patients, the most frequent (≥10%) all-grade nonhematological adverse reactions reported in patients were transaminase increased (51%), myalgia/arthralgia (50%), fatigue/malaise (44%), fever (41%), mucositis (41%), edema (40%), rash (36%), noninfectious diarrhea (35%), dyspnea (35%), nausea (30%), cough (28%), constipation (28%), eye disorders (25%), headache (24%), dizziness (22%), hypotension (22%), vomiting (21%), renal impairment (21%), abdominal pain (18%), neuropathy (18%), insomnia (15%) and dysgeusia (11%). The most frequent (≥5%) grade ≥3 nonhematological adverse reactions reported in patients were transaminase increased (21%), dyspnea (12%), hypotension (7%), mucositis (7%), myalgia/arthralgia (7%), and fatigue/malaise (6%). Shifts to grades 3-4 nonhematologic laboratory abnormalities included phosphate decreased 42/309 (14%), alanine aminotransferase increased 41/317 (13%), sodium decreased 37/314 (12%), aspartate aminotransferase increased 33/317 (10%), calcium decreased 19/312 (6%), creatine kinase increased 20/317 (6%), triglycerides increased 18/310 (6%), creatinine increased 10/316 (3%), and alkaline phosphatase increased 5/317 (2%).
Adverse reactions reported in the first 30 days of therapy on the ADMIRAL Study [see Clinical Studies (14)] are shown in Tables 2 and 3, according to whether patients were preselected for high intensity or low intensity chemotherapy.
Table 2: Adverse Reactions Reported in ≥10% (Any Grade) or ≥5% (Grade 3-5)* of Patients with Relapsed or Refractory AML in the Pre-selected High Intensity Chemotherapy Subgroup in the First 30 Days of the ADMIRAL Trial - * Grade 3-5 includes serious, life-threatening and fatal adverse reactions
- † Grouped terms: arthralgia, back pain, bone pain, flank pain, limb discomfort, medial tibial stress syndrome, myalgia, muscle twitching, musculoskeletal discomfort, musculoskeletal pain, muscle spasms, neck pain, non-cardiac chest pain, pain and pain in extremity
- ‡ Grouped terms: aspartate aminotransferase increased, alanine aminotransferase increased, blood alkaline phosphatase increased, gamma-glutamyltransferase increased, hepatic enzyme increased, hepatic function abnormal, hepatoxicity, liver function test increased and transaminases increased
- § Grouped terms: asthenia, fatigue, lethargy and malaise
- ¶ Grouped terms: edema, edema peripheral, face edema, fluid overload, generalized edema, hypervolemia, localized edema, periorbital edema and swelling face
- # Grouped terms: aphthous ulcer, colitis, enteritis, esophageal pain, gingival pain, large intestinal ulcer, laryngeal inflammation, lip blister, lip ulceration, mouth hemorrhage, mouth ulceration, mucosal inflammation, oral discomfort, oral pain, oropharyngeal pain, proctalgia, stomatitis, swollen tongue, tongue discomfort and tongue ulceration
- Þ Grouped terms: abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper and gastrointestinal pain
- ß Grouped terms: acne, dermatitis bullous, dermatitis contact, drug eruption, eczema asteatotic, erythema, hyperkeratosis, lichenoid keratosis, palmar-plantar erythrodysesthesia syndrome, rash, rash maculo-papular, rash papular, skin exfoliation, skin lesion and skin hyperpigmentation
- à Grouped terms: acute respiratory distress syndrome, dyspnea, dyspnea exertional, hypoxia, pulmonary edema, respiratory failure, tachypnea and wheezing
- è Grouped terms: hyperesthesia, hypoesthesia, neuralgia, neuropathy peripheral, peripheral sensory neuropathy and paresthesia
- ð Grouped terms: coordination abnormal and dizziness
Adverse Reaction
Any Grade
n (%)
Grade ≥3
n (%)
XOSPATA
(120 mg daily)
n=149
Chemotherapy
n=68XOSPATA
(120 mg daily)
n=149
Chemotherapy
n=68Musculoskeletal and connective tissue disorders
Myalgia/arthralgia†
56 (38)
20 (29)
1 (1)
3 (4)
Investigations
Transaminase increased‡
46 (31)
11 (16)
15 (10)
5 (7)
General disorders and administration site conditions
Fatigue/malaise§
36 (24)
9 (13)
1 (1)
2 (3)
Fever
25 (17)
21 (31)
2 (1)
4 (6)
Edema¶
20 (13)
13 (19)
0
0
Gastrointestinal disorders
Constipation
29 (20)
10 (15)
0
0
Mucositis#
18 (12)
30 (44)
0
5 (7)
Nausea
23 (15)
26 (38)
0
0
Abdominal painÞ
16 (11)
16 (24)
0
0
Blood and lymphatic system disorder
Febrile neutropenia
26 (17)
30 (44)
26 (17)
30 (44)
Skin and subcutaneous tissue disorders
Rashß
23 (15)
21 (31)
1 (1)
2 (3)
Respiratory, thoracic and mediastinal disorders
Dyspneaà
20 (13)
9 (13)
1 (1)
6 (9)
Cough
18 (12)
5 (7)
1 (1)
0
Nervous system disorders
Neuropathyè
19 (13)
0
0
0
Dizzinessð
17 (11)
2 (3)
0
0
Headache
17 (11)
12 (18)
0
0
Table 3: Adverse Reactions Reported in ≥10% (Any Grade) or ≥5% (Grade 3-5)* of Patients with Relapsed or Refractory AML in the Pre-selected Low Intensity Chemotherapy Subgroup in the First 30 Days of the ADMIRAL Trial - * Grade 3-5 includes serious, life-threatening and fatal adverse reactions
- † Grouped terms: aspartate aminotransferase increased, alanine aminotransferase increased, blood alkaline phosphatase increased and transaminases increased
- ‡ Grouped terms: arthralgia, arthritis, back pain, limb discomfort, myalgia, muscle contracture, muscle spasms, myositis, non-cardiac chest pain, pain, pain in extremity and polyarthritis
- § Grouped terms: asthenia, fatigue and malaise
- ¶ Grouped terms: edema, face edema, localized edema, edema peripheral, peripheral swelling, periorbital edema, scrotal edema and swelling face
- # Grouped terms: colitis, mouth hemorrhage, mouth ulceration, mucosal inflammation, oropharyngeal pain, proctalgia, stomatitis, tongue discomfort and tongue ulceration
- Þ Grouped terms: acute respiratory failure, dyspnea, hypoxia and respiratory failure
- ß Grouped terms: dermatitis acneiform, dermatitis bullous, dermatitis exfoliative, erythema, rash, rash maculo-papular, rash papular, rosacea and skin ulcer
Adverse Reaction
Any Grade
n (%)
Grade ≥3
n (%)
XOSPATA
(120 mg daily)
n=97
Chemotherapy
n=41XOSPATA
(120 mg daily)
n=97
Chemotherapy
n=41Investigations
Transaminase increased†
35 (36)
6 (15)
9 (9)
1 (2)
Blood and lymphatic system disorder
Febrile neutropenia
26 (27)
5 (12)
25 (26)
5 (12)
Musculoskeletal and connective tissue disorders
Myalgia/arthralgia‡
21 (22)
7 (17)
2 (2)
0
General disorders and administration site conditions
Fatigue/malaise§
20 (21)
9 (22)
4 (4)
1 (2)
Edema¶
19 (20)
5 (12)
1 (1)
0
Fever
11 (11)
7 (17)
0
0
Gastrointestinal disorders
Mucositis#
19 (20)
7 (17)
1 (1)
1 (2)
Constipation
13 (13)
5 (12)
1 (1)
0
Diarrhea
12 (12)
2 (5)
0
0
Nausea
10 (10)
7 (17)
0
0
Respiratory, thoracic and mediastinal disorders
DyspneaÞ
11 (11)
2 (5)
3 (3)
2 (5)
Skin and subcutaneous tissue disorders
Rashß
10 (10)
2 (5)
2 (2)
0
Other clinically significant adverse reactions occurring in ≤10% of patients included: electrocardiogram QT prolonged (9%), hypersensitivity* (8%), pancreatitis* (5%), cardiac failure* (4%), pericardial effusion (4%), acute febrile neutrophilic dermatosis (3%), differentiation syndrome (3%), pericarditis/myocarditis* (2%), large intestine perforation (1%), and posterior reversible encephalopathy syndrome (1%).
*Grouped terms: cardiac failure (cardiac failure, cardiac failure congestive, cardiomegaly, cardiomyopathy, chronic left ventricular failure, and ejection fraction decreased), hypersensitivity (anaphylactic reaction, angioedema, dermatitis allergic, drug hypersensitivity, erythema multiforme, hypersensitivity, and urticaria), pancreatitis (amylase increased, lipase increased, pancreatitis, pancreatitis acute), pericarditis/myocarditis (myocarditis, pericardial hemorrhage, pericardial rub, and pericarditis).
Selected post-baseline laboratory values that were observed in patients with relapsed or refractory AML are shown in Table 4.
Table 4: Shifts to Grade 3-4 Laboratory Abnormalities in Patients with Relapsed or Refractory AML by Pre-selected High Intensity and Low Intensity Chemotherapy in the First 30 Days of the ADMIRAL Trial Pre-selected High Intensity Chemotherapy Subgroup
Pre-selected Low Intensity Chemotherapy Subgroup
XOSPATA
(120 mg daily)
Chemotherapy
XOSPATA
(120 mg daily)
Chemotherapy
Alanine aminotransferase increased
7/149 (5%)
1/66 (2%)
7/95 (7%)
1/41 (2%)
Alkaline phosphatase increased
1/149 (1%)
0
0
0
Aspartate aminotransferase increased
8/149 (5%)
2/65 (3%)
5/95 (5%)
0
Calcium decreased
2/149 (1%)
3/65 (5%)
3/94 (3%)
0
Creatine kinase increased
1/149 (1%)
0
1/95 (1%)
0
Phosphatase decreased
4/144 (3%)
6/65 (9%)
4/93 (4%)
3/38 (8%)
Sodium decreased
7/148 (5%)
5/65 (8%)
6/93 (6%)
2/41 (5%)
Triglycerides increased
1/146 (1%)
0
2/94 (2%)
0
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on XOSPATA
Combined P-gp and Strong CYP3A Inducers
Concomitant use of XOSPATA with a combined P-gp and strong CYP3A inducer decreases gilteritinib exposure which may decrease XOSPATA efficacy [see Clinical Pharmacology (12.3)]. Avoid concomitant use of XOSPATA with combined P-gp and strong CYP3A inducers.
Strong CYP3A Inhibitors
Concomitant use of XOSPATA with a strong CYP3A inhibitor increases gilteritinib exposure [see Clinical Pharmacology (12.3)]. Consider alternative therapies that are not strong CYP3A inhibitors. If the concomitant use of these inhibitors is considered essential for the care of the patient, monitor patient more frequently for XOSPATA adverse reactions. Interrupt and reduce XOSPATA dosage in patients with serious or life-threatening toxicity [see Dosage and Administration (2.3)].
7.2 Effect of XOSPATA on Other Drugs
Drugs that Target 5HT2B Receptor or Sigma Nonspecific Receptor
Concomitant use of gilteritinib may reduce the effects of drugs that target the 5HT2B receptor or the sigma nonspecific receptor (e.g., escitalopram, fluoxetine, sertraline). Avoid concomitant use of these drugs with XOSPATA unless their use is considered essential for the care of the patient [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies (see Data) and its mechanism of action, XOSPATA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)].
There are no available data on XOSPATA use in pregnant women to inform a drug-associated risk of adverse developmental outcomes. In animal reproduction studies, administration of gilteritinib to pregnant rats during organogenesis caused adverse developmental outcomes including embryo-fetal lethality, suppressed fetal growth, and teratogenicity at maternal exposures (AUC24) approximately 0.4 times the AUC24 in patients receiving the recommended dose (see Data). Advise pregnant women of the potential risk to a fetus.
Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Data
Animal Data
In an embryo-fetal development study in rats, pregnant animals received oral doses of gilteritinib of 0, 0.3, 3, 10, and 30 mg/kg/day during the period of organogenesis. Maternal findings at 30 mg/kg/day (resulting in exposures approximately 0.4 times the AUC24 in patients receiving the recommended dose) included decreased body weight and food consumption. Administration of gilteritinib at the dose of 30 mg/kg/day also resulted in embryo-fetal death (postimplantation loss), decreased fetal body and placental weight, and decreased numbers of ossified sternebrae and sacral and caudal vertebrae, and increased incidence of fetal gross external (anasarca, local edema, exencephaly, cleft lip, cleft palate, short tail, and umbilical hernia), visceral (microphthalmia; atrial and/or ventricular defects; and malformed/absent kidney, and malpositioned adrenal, and ovary), and skeletal (sternoschisis, absent rib, fused rib, fused cervical arch, misaligned cervical vertebra, and absent thoracic vertebra) abnormalities.
Single oral administration of [14C] gilteritinib to pregnant rats resulted in transfer of radioactivity to the fetus similar to that observed in maternal plasma on day 14 of gestation. In addition, distribution profiles of radioactivity in most maternal tissues and the fetus on day 18 of gestation were similar to that on day 14 of gestation.
8.2 Lactation
Risk Summary
There are no data on the presence of gilteritinib and/or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Following administration of radiolabeled gilteritinib to lactating rats, milk concentrations of radioactivity were higher than radioactivity in maternal plasma at 4 and 24 hours post-dose. In animal studies, gilteritinib and/or its metabolite(s) were distributed to the tissues in infant rats via the milk. Because of the potential for serious adverse reactions in a breastfed child, advise a lactating woman not to breastfeed during treatment with XOSPATA and for at least 2 months after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy testing
Pregnancy testing is recommended for females of reproductive potential within seven days prior to initiating XOSPATA treatment [see Use in Specific Populations (8.1)].
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment and for at least 6 months after the last dose of XOSPATA.
Males
Advise males of reproductive potential to use effective contraception during treatment and for at least 4 months after the last dose of XOSPATA.
-
11 DESCRIPTION
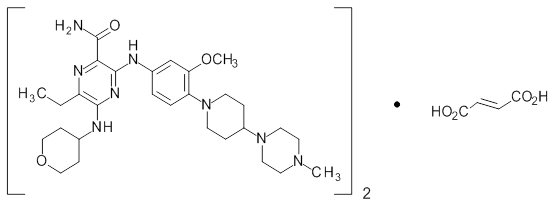
Gilteritinib is a tyrosine kinase inhibitor. The chemical name is 2-Pyrazinecarboxamide, 6-ethyl-3-[[3-methoxy-4-[4-(4-methyl-1-piperazinyl)-1-piperidinyl] phenyl] amino]-5-[(tetrahydro-2H-pyran-4-yl) amino]-, (2E)-2-butenedioate (2:1). The molecular weight is 1221.50 and the molecular formula is (C29H44N8O3)2·C4H4O4. The structural formula is:
Gilteritinib fumarate is a light yellow to yellow powder or crystals that is sparingly soluble in water and very slightly soluble in anhydrous ethanol.
XOSPATA (gilteritinib) is provided as a tablet for oral administration. Each tablet contains 40 mg of gilteritinib active ingredient as free base (corresponding to 44.2 mg gilteritinib fumarate). The inactive ingredients are ferric oxide, hydroxypropyl cellulose, hypromellose, low-substituted hydroxypropyl cellulose, mannitol, magnesium stearate, talc, polyethylene glycol and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Gilteritinib is a small molecule that inhibits multiple receptor tyrosine kinases, including FMS-like tyrosine kinase 3 (FLT3). Gilteritinib demonstrated the ability to inhibit FLT3 receptor signaling and proliferation in cells exogenously expressing FLT3 including FLT3-ITD, tyrosine kinase domain mutations (TKD) FLT3-D835Y and FLT3-ITD-D835Y, and it induced apoptosis in leukemic cells expressing FLT3-ITD.
12.2 Pharmacodynamics
In patients with relapsed or refractory AML administered gilteritinib 120 mg, substantial (>90%) inhibition of FLT3 phosphorylation was rapid (within 24 hours after first dose) and sustained, as characterized by an ex vivo plasma inhibitory activity (PIA) assay.
Cardiac Electrophysiology
The effect of XOSPATA 120 mg once a day on the QTc interval has been evaluated in patients, which showed an absence of large mean increases (i.e., 20 msec) in the QTc interval.
Of 317 patients with a post-baseline QTc measurement on treatment with gilteritinib at 120 mg in clinical trials, 4 patients (1.3%) experienced a QTcF >500 msec. Additionally, across all doses 2.3% of patients with relapse/refractory AML had a maximum post-baseline QTcF interval >500 msec [see Warnings and Precautions (5.3)].
12.3 Pharmacokinetics
The following pharmacokinetic parameters were observed following administration of gilteritinib 120 mg once daily, unless otherwise specified.
Gilteritinib exposure (Cmax and AUC24) increases proportionally with once daily doses ranging from 20 mg to 450 mg (0.17 to 3.75 times the recommended dosage) in patients with relapsed or refractory AML. Gilteritinib mean (±SD) steady-state Cmax is 374 ng/mL (±190) and AUC24 is 6943 nghr/mL (±3221). Steady-state plasma levels are reached within 15 days of dosing with an approximate 10-fold accumulation.
Absorption
The time to maximum gilteritinib concentration (tmax) observed is approximately between 4 and 6 hours postdose in the fasted state.
Effect of Food
In healthy adults administered a single gilteritinib 40 mg dose (0.3 times the recommended dosage), gilteritinib Cmax decreased by 26% and AUC decreased by less than 10% when co-administered with a high-fat meal (approximately 800 to 1,000 total calories with 500 to 600 fat calories, 250 carbohydrate calories, 150 protein calories) compared to a fasted state. Median tmax was delayed 2 hours when gilteritinib was administered with a high-fat meal.
Distribution
The population mean (%CV) estimates of apparent central and peripheral volume of distribution were 1092 L (9.22%) and 1100 L (4.99%), respectively, which may indicate extensive tissue distribution. In vivo, gilteritinib is approximately 94% bound to human plasma proteins. In vitro, gilteritinib is primarily bound to human serum albumin.
Elimination
The estimated half-life of gilteritinib is 113 hours, and the estimated apparent clearance is 14.85 L/h.
Metabolism
Gilteritinib is primarily metabolized via CYP3A4 in vitro. At steady state, the primary metabolites in humans include M17 (formed via N-dealkylation and oxidation), M16 and M10 (both formed via N‑dealkylation). None of these 3 metabolites exceeded 10% of overall parent exposure.
Excretion
After a single radiolabeled dose, gilteritinib is excreted in feces with 64.5% of the total administered dose recovered in feces. Of the total radiolabeled dose of gilteritinib, 16.4% was recovered in urine as unchanged drug and metabolites.
Specific Populations
Age (20-87 years), sex, race, mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment and mild (creatinine clearance (CLCr) 50-80 mL/min) or moderate (CLCr 30-50 mL/min) renal impairment do not have clinically meaningful effects on the pharmacokinetics of gilteritinib.
The effect of severe hepatic (Child-Pugh Class C) or severe renal impairment (CLCr ≤ 29 mL/min) on gilteritinib pharmacokinetics is unknown.
Drug Interaction Studies
Clinical Studies
Combined P-gp and Strong CYP3A Inducers:
Gilteritinib Cmax decreased approximately 30% and AUC decreased approximately 70% when co-administered with rifampin (a combined P-gp and strong CYP3A inducer).
Strong CYP3A Inhibitors:
Gilteritinib Cmax increased approximately 20% and AUC increased approximately 120% when co-administered with itraconazole (a strong CYP3A inhibitor).
Moderate CYP3A Inhibitors:
Gilteritinib Cmax increased approximately 16% and AUC increased approximately 40% when co-administered with fluconazole (a moderate CYP3A inhibitor).
CYP3A Substrates:
Midazolam (a CYP3A substrate) Cmax and AUC increased approximately 10% when co-administered with gilteritinib.
MATE1 Substrates:
Cephalexin (a MATE1 substrate) Cmax and AUC decreased by less than 10% when co-administered with gilteritinib.
In Vitro Studies
Gilteritinib inhibits human 5HT2B receptor or sigma nonspecific receptors, which may reduce the effects of drugs that target these receptors such as escitalopram, fluoxetine and sertraline.
Gilteritinib is a substrate of P-gp transporter and has the potential to inhibit breast cancer resistance protein (BCRP) and organic cation transporter 1 (OCT1) transporters.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with gilteritinib.
Gilteritinib was not mutagenic in a bacterial mutagenesis (Ames) assay and was not clastogenic in a chromosome aberration test assay in Chinese hamster lung cells. Gilteritinib was positive for the induction of micronuclei in mouse bone marrow cells from 65 mg/kg (195 mg/m2) the mid dose tested (approximately 2.6 times the recommended human dose of 120 mg).
The effect of XOSPATA on human fertility is unknown. Administration of 10 mg/kg/day gilteritinib in the 4-week study in dogs (12 days of dosing) resulted in degeneration and necrosis of germ cells and spermatid giant cell formation in the testis as well as single cell necrosis of the epididymal duct epithelia of the epididymal head.
-
14 CLINICAL STUDIES
14.1 Relapsed or Refractory Acute Myeloid Leukemia
The efficacy of XOSPATA was assessed in the ADMIRAL Trial (NCT02421939), which included adult patients with relapsed or refractory AML having a FLT3 ITD, D835, or I836 mutation by the LeukoStrat® CDx FLT3 Mutation Assay. XOSPATA was given orally at a starting dose of 120 mg daily until unacceptable toxicity or lack of clinical benefit.
First Interim Analysis
The efficacy of XOSPATA was established on the basis of the rate of complete remission (CR)/CR with partial hematological recovery (CRh), the duration of CR/CRh (DOR), and the rate of conversion from transfusion dependence to transfusion independence at the first interim analysis in the ADMIRAL trial (n=138). The median follow-up was 4.6 months (95% CI: 2.8, 15.8). Fourteen patients were still in remission at the time of the first interim DOR analysis. The efficacy results are shown in Table 5. For patients who achieved a CR/CRh, the median time to first response was 3.6 months (range, 0.9 to 9.6 months). The CR/CRh rate was 29 of 126 in patients with FLT3-ITD or FLT3-ITD/TKD and 0 of 12 in patients with FLT3-TKD only.
Among the 106 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 33 (31.1%) became independent of RBC and platelet transfusions during any 56-day post-baseline period. For the 32 patients who were independent of both RBC and platelet transfusions at baseline, 17 (53.1%) remained transfusion-independent during any 56-day post-baseline period.
Table 5: Efficacy Results in Patients with Relapsed or Refractory AML Treated with XOSPATA in the First Interim Analysis (ADMIRAL Trial) CI: confidence interval; NE: not estimable; NR: not reached; Only responses prior to HSCT were included in response rate. - * CR was defined as an absolute neutrophil count ≥1.0 x 109/L, platelets ≥100 x 109/L, normal marrow differential with <5% blasts, must have been red blood cells, platelet transfusion independent and no evidence of extramedullary leukemia.
- † CRh was defined as marrow blasts <5%, partial hematologic recovery absolute neutrophil count ≥0.5 x 109/L and platelets ≥50 x 109/L, no evidence of extramedullary leukemia and could not have been classified as CR.
- ‡ The 95% CI rate was calculated using the exact method based on binomial distribution.
- § DOR was defined as the time from the date of either first CR or CRh until the date of a documented relapse of any type. Deaths were counted as events.
- ¶ Response was ongoing.
Remission Rate
XOSPATA
N=138
29/138 (21)
95% CI‡
14.5, 28.8
Median DOR§ (months)
4.6
Range (months)
0.1 to 15.8¶
CR* n/N (%)
16/138 (11.6)
95% CI‡
6.8, 18.1
Median DOR§ (months)
8.6
Range (months)
1 to 13.8
CRh† n/N (%)
13/138 (9.4)
95% CI‡
5.1, 15.6
Median DOR§ (months)
2.9
Range (months)
0.1 to 15.8¶
Final Analysis
The final analysis of the ADMIRAL trial included 371 adult patients randomized 2:1 to receive XOSPATA 120 mg once daily (n=247) over continuous 28-day cycles or a prespecified chemotherapy regimen (n=124). Randomization was stratified by response to first-line AML therapy and prespecified chemotherapy. The prespecified chemotherapy regimens included high intensity combinations (MEC and FLAG-IDA) and low intensity regimens (LDAC and AZA).
The demographic and disease characteristics of the randomized patients are shown in Table 6.
Table 6: Baseline Demographic and Disease Characteristics in Patients with Relapsed or Refractory AML in the Final Analysis (ADMIRAL Trial) AML: acute myeloid leukemia; FLT3: FMS-related tyrosine kinase 3; ITD: internal tandem duplication; TKD: D835/I836 tyrosine kinase domain point mutation; ECOG PS: Eastern Cooperative Oncology Group performance status; CRc: Composite complete remission (complete remission [CR] + complete remission with incomplete hematologic recovery [CRi] + complete remission with incomplete platelet recovery [CRp]); HSCT: Hematopoietic stem cell transplantation - * Patients were defined as transfusion dependent at baseline if they were dosed and received any red blood cell or platelet transfusions within the 56-day baseline period.
- † Prior use of FLT3 inhibitor is defined as “Yes” if patients received prior AML therapy of midostaurin, sorafenib or quizartinib; otherwise, prior use of FLT3 inhibitor was assigned as “No.”
- ‡ MEC: mitoxantrone 8 mg/m2, etoposide 100 mg/m2 and cytarabine 1000 mg/m2 once daily by IV for 5 days
- § FLAG-IDA: granulocyte colony-stimulating factor 300 mcg/m2 once daily by SC days 1 to 5, fludarabine 30 mg/m2 once daily by IV days 2 through 6, cytarabine 2000 mg/m2 once daily by IV for days 2 through 6, idarubicin 10 mg/m2 once daily by IV days 2 through 4
- ¶ LDAC: cytarabine 20 mg twice daily by subcutaneous (SC) or intravenous (IV) for 10 days
- # AZA: azacitidine 75 mg/m2 once daily by SC or IV for 7 days
Demographic and Disease Characteristics
Xospata
(120 mg daily)
N=247
Chemotherapy
N=124
Demographics
Median Age (Years) (Range)
62 (20, 84)
62 (19, 85)
Age Categories, n (%)
<65 years
141 (57)
75 (60)
≥65 years
106 (43)
49 (40)
Sex, n (%)
Male
116 (47)
54 (44)
Female
131 (53)
70 (57)
Race, n (%)
White
145 (59)
75 (60)
Asian
69 (28)
33 (27)
Black or African American
14 (6)
7 (6)
Native Hawaiian or Other Pacific Islander
1 (0.4)
0
Other
5 (2)
1 (0.8)
Unknown/Missing
13 (5)
8 (6)
Baseline ECOG, n (%)
0-1
206 (83)
105 (85)
≥2
41 (17)
19 (15)
Disease Characteristics
Untreated relapse AML, n (%)
151 (61)
74 (60)
Primary refractory AML, n (%)
96 (39)
49 (40)
Refractory relapse AML, n (%)
0
1 (0.8)
Number of Relapses, n (%)
0
96 (39)
49 (40)
1
149 (60)
74 (60)
2 or more
2 (0.8)
1 (0.8)
Median number of relapses (Range)
1 (0, 2)
1 (0, 2)
Transfusion Dependent at Baseline, n (%)*
197 (80)
97 (89)
FLT3 Mutation Status, n (%)
ITD alone
215 (87)
113 (91)
TKD alone
21 (9)
10 (8)
ITD and TKD
7 (3)
0
Prior Use of FLT3 Inhibitor†, n (%)
No
215 (87)
110 (89)
Yes
32 (13)
14 (11)
Prespecified Chemotherapy
High Intensity
149 (60)
75 (60)
MEC‡
-
33 (27)
FLAG-IDA§
-
42 (34)
Low Intensity
98 (40)
49 (40)
LDAC¶
-
17 (14)
AZA#
-
32 (26)
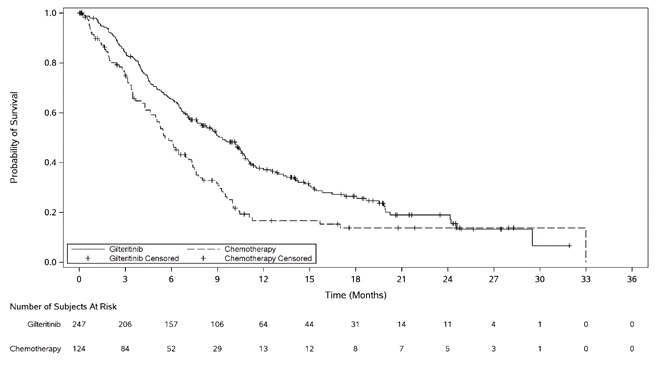
The final analysis included an assessment of OS, measured from the date of randomization until death by any cause. At the time of analysis, median follow-up was 17.8 months (range, 14.9 to 19.1). Patients randomized to the XOSPATA arm had significantly longer survival compared to the chemotherapy arm (HR 0.64; 95% CI: 0.49 – 0.83; 1-sided p-value: 0.0004). Figure 1 and Table 7 show the results of the OS analysis.
Exploratory subgroup analyses demonstrated that the hazard ratio for survival was 0.66 (95% CI: 0.47 – 0.93) for patients in the high intensity chemotherapy stratum and 0.56 (95% CI: 0.38 – 0.84) for patients in the low intensity chemotherapy stratum. The CR rates are shown in Table 7. For patients on XOSPATA and chemotherapy arms, the CR rates were 15.4% (95% CI: 10% – 22.3%) and 16% (95% CI: 8.6% – 26.3%), respectively, for patients in the high intensity chemotherapy stratum, and 12.2% (95% CI: 6.5% – 20.4%) and 2% (95% CI 0.1% – 10.9%), respectively, for patients in the low intensity chemotherapy stratum.
Table 7: OS and CR* in Patients with Relapsed or Refractory AML in the Final Analysis (ADMIRAL Trial) CI: confidence interval; Only responses prior to HSCT were included in response rate. - * CR was defined as an absolute neutrophil count ≥1.0 x 109/L, platelets ≥100 x 109/L, normal marrow differential with <5% blasts, must have been red blood cells, platelet transfusion independent and no evidence of extramedullary leukemia.
- † The 95% CI rate was calculated using the exact method based on binomial distribution.
- ‡ DOR was defined as the time from the date of first remission until the date of a documented relapse.
XOSPATA
N=247
Chemotherapy
N=124
Overall Survival
Deaths, n (%)
171 (69.2%)
90 (72.6%)
Median in months (95% CI)
9.3 (7.7, 10.7)
5.6 (4.7, 7.3)
Hazard Ratio (95% CI)
0.64 (0.49, 0.83)
p-value (1-sided)
0.0004
Complete Remission
CR, n (%)
35 (14.2%)
13 (10.5%)
(95% CI† )
(10.1, 19.2)
(5.7, 17.3)
Median DOR‡ (range) (months)
14.8 (0.6 to 23.1+)
1.8 (<0.1+ to 1.8)
Figure 1: Kaplan-Meier Plot of Overall Survival in ADMIRAL Trial
In the final analysis, the CR/CRh rate in the gilteritinib arm was 22.6% (55/243) and the DOR was 7.4 months (range, <0.1+ to 23.1+). For patients who achieved a CR/CRh, the median time to first response was 2 months (range, 0.9 to 9.6 months). The CR/CRh rate was 49 of 215 in patients with FLT3-ITD only, 3 of 7 in patients with FLT3-ITD/TKD and 3 of 21 in patients with FLT3-TKD only.
Among the 197 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 68 (34.5%) became independent of RBC and platelet transfusions during any 56-day post-baseline period. For the 49 patients who were independent of both RBC and platelet transfusions at baseline, 29 (59.2%) remained transfusion-independent during any 56-day post-baseline period.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
XOSPATA (gilteritinib) 40 mg tablets are supplied as light yellow, round-shaped, film-coated tablets debossed with the Astellas logo and ‘235’ on the same side. XOSPATA tablets are available in the following package size:
- Bottles of 90 tablets with Child Resistant Closure, (NDC: 0469-1425-90)
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Differentiation Syndrome
Advise patients of the risks of developing differentiation syndrome as early as 2 days after the start of therapy and during the first 3 months on treatment. Ask patients to immediately report any symptoms suggestive of differentiation syndrome, such as fever, cough or difficulty breathing, rash, low blood pressure, rapid weight gain, swelling of their arms or legs, or decreased urinary output, to their healthcare provider for further evaluation [see Boxed Warning and Warnings and Precautions (5.1)].
Posterior Reversible Encephalopathy Syndrome
Advise patients of the risk of developing posterior reversible encephalopathy syndrome (PRES). Ask patients to immediately report any symptoms suggestive of PRES, such as seizure and altered mental status, to their healthcare provider for further evaluation [see Warnings and Precautions (5.2)].
Prolonged QT Interval
Advise patients to consult their healthcare provider immediately if they feel faint, lose consciousness, or have signs or symptoms suggestive of arrhythmia. Advise patients with a history of hypokalemia or hypomagnesemia of the importance of monitoring their electrolytes [see Warnings and Precautions (5.3)].
Pancreatitis
Advise patients of the risk of pancreatitis and to contact their healthcare provider for signs or symptoms of pancreatitis, which include severe and persistent stomach pain, with or without nausea and vomiting [see Warnings and Precautions (5.4)].
Use of Contraceptives
- Advise female patients with reproductive potential to use effective contraceptive methods while receiving XOSPATA and to avoid pregnancy while on treatment and for 6 months after completion of treatment.
- Advise patients to notify their healthcare provider immediately in the event of a pregnancy or if pregnancy is suspected during XOSPATA treatment.
- Advise males with female partners of reproductive potential to use effective contraception during treatment with XOSPATA and for at least 4 months after the last dose of XOSPATA [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with XOSPATA for at least 2 months after the final dose [see Use in Specific Populations (8.2)].
Dosing Instructions
- Advise patients not to break, crush or chew the tablets but to swallow them whole with a cup of water.
-
Instruct patients that, if they miss a dose of XOSPATA, to take it as soon as possible on the same day, and at least 12 hours prior to the next scheduled dose, and return to the normal schedule the following day. Instruct patients to not take 2 doses within 12 hours [see Dosage and Administration (2.2)].
Distributed by:
Astellas Pharma US, Inc.
Northbrook, Illinois 60062XOSPATA® is a registered trademark of Astellas Pharma Inc.
LeukoStrat® is a registered trademark of Invivoscribe, Inc.
©2019 Astellas Pharma US, Inc.
222317-GLT -
MEDICATION GUIDE
MEDICATION GUIDE
XOSPATA® (Zoh spah' tah)
(gilteritinib)
tabletsWhat is the most important information I should know about XOSPATA?
XOSPATA may cause serious side effects, including:
Differentiation Syndrome. Differentiation syndrome is a condition that affects your blood cells and may be life-threatening or lead to death if not treated. Differentiation syndrome can happen as early as 2 days after starting XOSPATA and during the first 3 months of treatment. Call your healthcare provider or go to the nearest hospital emergency room right away if you develop any of the following symptoms of differentiation syndrome while taking XOSPATA:
- o fever
- o cough
- o trouble breathing
- o rash
- o dizziness or lightheadedness
- o rapid weight gain
- o swelling of your arms or legs
- o decreased urination
If you develop any of these symptoms of differentiation syndrome, your healthcare provider may treat you with a corticosteroid medicine and may monitor you in the hospital.
See “What are the possible side effects of XOSPATA?” for more information about side effects.
What is XOSPATA?
XOSPATA is a prescription medicine used to treat adults with acute myeloid leukemia (AML) who have a FMS-like tyrosine kinase 3 (FLT3) mutation:- when the disease has come back, or
- has not improved after previous treatment(s).
Your healthcare provider will perform a test to make sure that XOSPATA is right for you.
It is not known if XOSPATA is safe and effective in children.
Who should not take XOSPATA?
Do not take XOSPATA if you are allergic to gilteritinib or any of the ingredients in XOSPATA. See the end of this Medication Guide for a complete list of ingredients in XOSPATA.
Before taking XOSPATA, tell your healthcare provider about all of your medical conditions, including if you:
- have any heart problems, including a condition called long QT syndrome.
- have problems with abnormal electrolytes such as sodium, potassium, or magnesium levels.
-
are pregnant or plan to become pregnant. XOSPATA can cause harm to your unborn baby. You should avoid becoming pregnant during treatment with XOSPATA. Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with XOSPATA.
- o If you are able to become pregnant, your healthcare provider may perform a pregnancy test 7 days before you start treatment with XOSPATA.
- o Females who are able to become pregnant should use effective birth control (contraception) during treatment with XOSPATA and for at least 6 months after the last dose of XOSPATA.
- o Males who have female partners that are able to become pregnant should use effective birth control (contraception) during treatment with XOSPATA and for at least 4 months after the last dose of XOSPATA.
- are breastfeeding or plan to breastfeed. It is not known if XOSPATA passes into your breast milk. Do not breastfeed during treatment with XOSPATA and for at least 2 months after the last dose of XOSPATA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How should I take XOSPATA?
- Take XOSPATA exactly as your healthcare provider tells you to. Do not change your dose or stop taking XOSPATA without talking to your healthcare provider.
- Take XOSPATA 1 time a day at about the same time each day.
- Swallow XOSPATA tablets whole. Do not break, crush, or chew the tablet.
- XOSPATA can be taken with or without food.
- If you miss a dose of XOSPATA, or did not take it at the usual time, take your dose as soon as possible and at least 12 hours before your next dose. Return to your normal schedule the following day. Do not take 2 doses of XOSPATA within 12 hours.
What are the possible side effects of XOSPATA?
XOSPATA may cause serious side effects, including:- See “What is the most important information I should know about XOSPATA?”
- Posterior Reversible Encephalopathy Syndrome (PRES). If you take XOSPATA, you may be at risk of developing a condition involving the brain called PRES. Tell your healthcare provider right away if you have a seizure or quickly worsening symptoms such as headache, decreased alertness, confusion, reduced eyesight, blurred vision, or other visual problems. Your healthcare provider will do a test to check for PRES. Your healthcare provider will stop XOSPATA if you develop PRES.
- Changes in the electrical activity of your heart called QTc prolongation. QTc prolongation can cause irregular heartbeats that can be life-threatening. Your healthcare provider will check the electrical activity of your heart with a test called an electrocardiogram (ECG) before you start taking XOSPATA and during your treatment with XOSPATA. Tell your healthcare provider right away if you feel dizzy, lightheaded, or faint. The risk of QT prolongation is higher in people with low blood magnesium or low blood potassium levels. Your healthcare provider will do blood tests to check your potassium and magnesium levels before and during your treatment with XOSPATA.
- Inflammation of the pancreas (pancreatitis). Tell your healthcare provider right away if you have severe stomach (abdomen) pain that does not go away. This pain may happen with or without nausea and vomiting.
The most common side effects of XOSPATA include:
- o changes in liver function tests
- o joint or muscle pain
- o tiredness
- o fever
- o pain or sores in mouth or throat
- o swelling of arms or legs
- o rash
- o diarrhea
- o shortness of breath
- o nausea
- o cough
- o constipation
- o eye problems
- o headache
- o dizziness
- o low blood pressure
- o vomiting
- o decreased urination
Your healthcare provider may tell you to decrease your dose, temporarily stop, or completely stop taking XOSPATA if you develop certain side effects during treatment with XOSPATA.
These are not all of the possible side effects of XOSPATA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store XOSPATA?
- XOSPATA comes in a child-resistant package.
- Store XOSPATA at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep XOSPATA in the original container provided by your pharmacist to protect it from light, moisture and humidity.
- Keep XOSPATA and all medicines out of the reach of children.
General information about the safe and effective use of XOSPATA.
Medicines are sometimes prescribed for conditions not listed in a Medication Guide. Do not use XOSPATA for a condition for which it was not prescribed. Do not give XOSPATA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about XOSPATA that is written for healthcare professionals.What are the ingredients in XOSPATA?
Active ingredient: gilteritinib
Inactive ingredients: ferric oxide, hydroxypropyl cellulose, hypromellose, low-substituted hydroxypropyl cellulose, mannitol, magnesium stearate, talc, polyethylene glycol and titanium dioxide.Distributed by: Astellas Pharma US, Inc., Northbrook, Illinois 60062
XOSPATA® is a registered trademark of Astellas Pharma Inc.
©2019 Astellas Pharma US, Inc.
For more information about XOSPATA, call 1-800-727-7003, or visit www.XOSPATA.com.
222317-GLTThis Medication Guide has been approved by the U.S. Food and Drug Administration. Issued: May 2019
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL – 40 mg Tablet Bottle Carton
NDC: 0469-1425-90
XOSPATA®
(gilteritinib) tablets
40 mg
Do not break, crush or chew tablets.
90 tablets
Rx only -
INGREDIENTS AND APPEARANCE
XOSPATA
gilteritinib tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0469-1425 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength GILTERITINIB FUMARATE (UNII: 5RZZ0Z1GJT) (GILTERITINIB - UNII:66D92MGC8M) GILTERITINIB 40 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TALC (UNII: 7SEV7J4R1U) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color YELLOW (Light yellow) Score no score Shape ROUND Size 7mm Flavor Imprint Code Logo;235 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0469-1425-90 1 in 1 CARTON 11/29/2018 1 90 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211349 11/29/2018 Labeler - Astellas Pharma US, Inc. (605764828)
Trademark Results [Xospata]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 XOSPATA 88806053 not registered Live/Pending |
Astellas Pharma Inc. 2020-02-21 |
 XOSPATA 88806044 not registered Live/Pending |
Astellas Pharma Inc. 2020-02-21 |
 XOSPATA 79199845 5211443 Live/Registered |
Astellas Pharma Inc. 2016-10-14 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.