SUMANSETRON- sumatriptan succinate, ondansetron kit
Sumansetron by
Drug Labeling and Warnings
Sumansetron by is a Prescription medication manufactured, distributed, or labeled by PureTek Corporation, NATCO PHARMA LIMITED. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
Sumansetron
These highlights do not include all the information needed to use ONDANSETRON TABLETS, USP safely and effectively. See full prescribing information for ONDANSETRON TABLETS, USP.
ONDANSETRON tablets USP, for oral use
Initial U.S. Approval: 1991INDICATIONS AND USAGE
Ondansetron tablets are a 5-HT 3 receptor antagonist indicated for the prevention of:
nausea and vomiting associated with highly emetogenic cancer chemotherapy, including cisplatin ≥50 mg/m 2. ( 1)
nausea and vomiting associated with initial and repeat courses of moderately emetogenic cancer chemotherapy. ( 1)
nausea and vomiting associated with radiotherapy in patients receiving either total body irradiation, single high-dose fraction to the abdomen, or daily fractions to the abdomen. ( 1)
postoperative nausea and/or vomiting. ( 1)DOSAGE AND ADMINISTRATION
DOSAGE AND ADMINISTRATION
Tablets: 4 mg and 8 mg ( 3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Hypersensitivity reactions including anaphylaxis and bronchospasm: Discontinue ondansetron tablets if suspected. Monitor and treat promptly per standard of care until signs and symptoms resolve ( 5.1)
- QT interval prolongation and Torsade de Pointes: Avoid in patients with congenital long QT syndrome; monitor with electrocardiograms (ECGs) if concomitant electrolyte abnormalities, cardiac failure or arrhythmias, or use of other QT prolonging drugs. ( 5.2)
- Serotonin syndrome: Reported with 5-HT 3 receptor antagonists alone but particularly with concomitant use of serotonergic drugs. If such symptoms occur, discontinue ondansetron tablets and initiate supportive treatment. If concomitant use of ondansetron tablets with other serotonergic drugs is clinically warranted, patients should be made aware of a potential increased risk for serotonin syndrome. ( 5.3)
- Masking of progressive ileus and/or gastric distention following abdominal surgery or chemotherapy-induced nausea and vomiting: Monitor for decreased bowel activity, particularly in patients with risk factors for gastrointestinal obstruction. ( 5.4)
ADVERSE REACTIONS
The most common adverse reactions in adults for the:
prevention of chemotherapy-induced (greater than or equal to 5%) are: headache, malaise/fatigue, constipation, diarrhea. ( 6.1)
prevention of radiation-induced nausea and vomiting (greater than or equal to 2%) are: headache, constipation, and diarrhea. ( 6.1)
prevention of postoperative nausea and vomiting (greater than or equal to 9%) are: headache and hypoxia. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Actavis Pharma Inc., at 1-800-432-8534 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.See 17 for PATIENT COUNSELING INFORMATION, FDA-approved patient labeling and PATIENT COUNSELING INFORMATION.
Revised: 12/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
DESCRIPTION
CLINICAL PHARMACOLOGY
Mechanism of Action
Pharmacokinetics
Special Populations
Drug Interactions
CLINICAL STUDIES
INDICATIONS AND USAGE
CONTRAINDICATIONS
WARNINGS
Risk of Myocardial Ischemia and/or Infarction and Other Adverse Cardiac Events
Drug-Associated Cardiac Events and Fatalities
Drug-Associated Cerebrovascular Events and Fatalities
Other Vasospasm-Related Events
Serotonin Syndrome
Increase in Blood Pressure
Concomitant Drug Use
Hypersensitivity
PRECAUTIONS
General
Overuse:
Information for Patients
Laboratory Tests
Drug Interactions
Drug/Laboratory Test Interactions
Carcinogenesis, Mutagenesis, Impairment of Fertility
Pregnancy
Nursing Mothers
Pediatric Use
Geriatric Use
ADVERSE REACTIONS
Incidence in Controlled Clinical Trials
Other Events Observed in Association With the Administration of Sumatriptan Tablets
Other Events Observed in the Clinical Development of Sumatriptan
Postmarketing Experience (Reports for Subcutaneous or Oral Sumatriptan)
DRUG ABUSE AND DEPENDENCE
OVERDOSAGE
DOSAGE AND ADMINISTRATION
HOW SUPPLIED
ANIMAL TOXICOLOGY
Corneal Opacities
PATIENT PACKAGE INSERT
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
2.2 Dosage in Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 QT Prolongation
5.3 Serotonin Syndrome
5.4 Masking of Progressive Ileus and Gastric Distension
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Serotonergic Drugs
7.2 Drugs Affecting Cytochrome P-450 Enzymes
7.3 Tramadol
7.4 Chemotherapy
7.5 Alfentanil and Atracurium
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
9 DRUG ABUSE AND DEPENDENCE
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Prevention of Chemotherapy-Induced Nausea and Vomiting
14.2 Radiation-Induced Nausea and Vomiting
14.3 Postoperative Nausea and Vomiting
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
DESCRIPTION
Sumatriptan tablets USP contain sumatriptan (as the succinate), a selective 5-hydroxytryptamine 1 receptor subtype agonist. Sumatriptan succinate USP is chemically designated as 3-[2- (dimethylamino) ethyl]-N-methyl-indole-5-methanesulfonamide succinate (1:1), and it has the following structure:
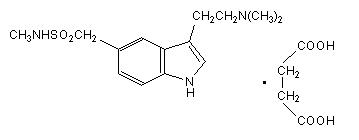
The molecular formula is C 14H 21N 3O 2SC 4H 6O 4, representing a molecular weight of 413.5. Sumatriptan succinate USP is a white to off-white powder that is readily soluble in water and in saline. Each sumatriptan tablet USP for oral administration contains 35, 70, or 140 mg of sumatriptan succinate USP equivalent to 25, 50, or 100 mg of sumatriptan, respectively. Each tablet also contains the inactive ingredients croscarmellose sodium, lactose anhydrous, lactose monohydrate, magnesium stearate, mannitol, microcrystalline cellulose, talc, titanium dioxide and triacetin.
- INDICATIONS & USAGE
-
CLINICAL PHARMACOLOGY
Mechanism of Action
Sumatriptan is an agonist for a vascular 5-hydroxytryptamine 1 receptor subtype (probably a member of the 5-HT 1D family) having only a weak affinity for 5-HT 1A, 5-HT 5A, and 5-HT 7 receptors and no significant affinity (as measured using standard radioligand binding assays) or pharmacological activity at 5-HT 2, 5-HT 3, or 5-HT 4 receptor subtypes or at alpha 1-, alpha 2-, or beta-adrenergic; dopamine 1; dopamine 2; muscarinic; or benzodiazepine receptors.
The vascular 5-HT 1 receptor subtype that sumatriptan activates is present on cranial arteries in both dog and primate, on the human basilar artery, and in the vasculature of human dura mater and mediates vasoconstriction. This action in humans correlates with the relief of migraine headache. In addition to causing vasoconstriction, experimental data from animal studies show that sumatriptan also activates 5-HT 1 receptors on peripheral terminals of the trigeminal nerve innervating cranial blood vessels. Such an action may also contribute to the antimigrainous effect of sumatriptan in humans.
In the anesthetized dog, sumatriptan selectively reduces the carotid arterial blood flow with little or no effect on arterial blood pressure or total peripheral resistance. In the cat, sumatriptan selectively constricts the carotid arteriovenous anastomoses while having little effect on blood flow or resistance in cerebral or extracerebral tissues.
Pharmacokinetics
The mean maximum concentration following oral dosing with 25 mg is 18 ng/mL (range: 7 to 47 ng/mL) and 51 ng/mL (range: 28 to 100 ng/mL) following oral dosing with 100 mg of sumatriptan. This compares with a C max of 5 and 16 ng/mL following dosing with a 5 and 20 mg intranasal dose, respectively. The mean C max following a 6 mg subcutaneous injection is 71 ng/mL (range: 49 to 110 ng/mL). The bioavailability is approximately 15%, primarily due to presystemic metabolism and partly due to incomplete absorption. The C max is similar during a migraine attack and during a migraine-free period, but the t max is slightly later during the attack, approximately 2.5 hours compared to 2.0 hours. When given as a single dose, sumatriptan displays dose proportionality in its extent of absorption (area under the curve [AUC]) over the dose range of 25 to 200 mg, but the C max after 100 mg is approximately 25% less than expected (based on the 25 mg dose).
A food effect study involving administration of sumatriptan tablets 100 mg to healthy volunteers under fasting conditions and with a high-fat meal indicated that the C max and AUC were increased by 15% and 12 %, respectively, when administered in the fed state.
Plasma protein binding is low (14% to 21%). The effect of sumatriptan on the protein binding of other drugs has not been evaluated, but would be expected to be minor, given the low rate of protein binding. The apparent volume of distribution is 2.4 L/kg.
The elimination half-life of sumatriptan is approximately 2.5 hours. Radiolabeled 14C-sumatriptan administered orally is largely renally excreted (about 60%) with about 40% found in the feces. Most of the radiolabeled compound excreted in the urine is the major metabolite, indole acetic acid (IAA), which is inactive, or the IAA glucuronide. Only 3% of the dose can be recovered as unchanged sumatriptan.
In vitro studies with human microsomes suggest that sumatriptan is metabolized by monoamine oxidase (MAO), predominantly the A isoenzyme, and inhibitors of that enzyme may alter sumatriptan pharmacokinetics to increase systemic exposure. No significant effect was seen with an MAO-B inhibitor (see CONTRAINDICATIONS,WARNINGS, and PRECAUTIONS: Drug Interactions).
Special Populations
Renal Impairment
Renal Impairment: The effect of renal impairment on the pharmacokinetics of sumatriptan has not been examined, but little clinical effect would be expected as sumatriptan is largely metabolized to an inactive substance.
Hepatic Impairment
The liver plays an important role in the presystemic clearance of orally administered sumatriptan. Accordingly, the bioavailability of sumatriptan following oral administration may be markedly increased in patients with liver disease. In 1 small study of hepatically impaired patients (N = 8) matched for sex, age, and weight with healthy subjects, the hepatically impaired patients had an approximately 70% increase in AUC and C max and a t max 40 minutes earlier compared to the healthy subjects (see DOSAGE AND ADMINISTRATION).
Age
The pharmacokinetics of oral sumatriptan in the elderly (mean age, 72 years; 2 males and 4 females) and in patients with migraine (mean age, 38 years; 25 males and 155 females) were similar to that in healthy male subjects (mean age, 30 years) (see PRECAUTIONS: Geriatric Use).
Drug Interactions
Monoamine Oxidase Inhibitors (MAO)
Treatment with MAO-A inhibitors generally leads to an increase of sumatriptan plasma levels (see CONTRAINDICATIONS and PRECAUTIONS).
Due to gut and hepatic metabolic first-pass effects, the increase of systemic exposure after coadministration of an MAO-A inhibitor with oral sumatriptan is greater than after coadministration of the monoamine oxidase inhibitors (MAOI) with subcutaneous sumatriptan. In a study of 14 healthy females, pretreatment with an MAO-A inhibitor decreased the clearance of subcutaneous sumatriptan. Under the conditions of this experiment, the result was a 2-fold increase in the area under the sumatriptan plasma concentration x time curve (AUC), corresponding to a 40% increase in elimination half-life. This interaction was not evident with an MAO-B inhibitor.
A small study evaluating the effect of pretreatment with an MAO-A inhibitor on the bioavailability from a 25 mg oral sumatriptan tablet resulted in an approximately 7-fold increase in systemic exposure.
-
CLINICAL STUDIES
The efficacy of sumatriptan tablets in the acute treatment of migraine headaches was demonstrated in 3, randomized, double-blind, placebo-controlled studies. Patients enrolled in these 3 studies were predominately female (87%) and Caucasian (97%), with a mean age of 40 years (range, 18 to 65 years). Patients were instructed to treat a moderate to severe headache. Headache response, defined as a reduction in headache severity from moderate or severe pain to mild or no pain, was assessed up to 4 hours after dosing. Associated symptoms such as nausea, photophobia, and phonophobia were also assessed. Maintenance of response was assessed for up to 24 hours postdose. A second dose of sumatriptane tablets or other medication was allowed 4 to 24 hours after the initial treatment for recurrent headache. Acetaminophen was offered to patients in Studies 2 and 3 beginning at 2 hours after initial treatment if the migraine pain had not improved or worsened. Additional medications were allowed 4 to 24 hours after the initial treatment for recurrent headache or as rescue in all 3 studies. The frequency and time to use of these additional treatments were also determined. In all studies, doses of 25, 50, and 100 mg were compared to placebo in the treatment of migraine attacks. In 1 study, doses of 25, 50, and 100 mg were also compared to each other.
In all 3 trials, the percentage of patients achieving headache response 2 and 4 hours after treatment was significantly greater among patients receiving sumatriptan tablets at all doses compared to those who received placebo. In 1 of the 3 studies, there was a statistically significant greater percentage of patients with headache response at 2 and 4 hours in the 50 or 100 mg group when compared to the 25 mg dose groups. There were no statistically significant differences between the 50 and 100 mg dose groups in any study. The results from the 3 controlled clinical trials are summarized in Table 1.
Comparisons of drug performance based upon results obtained in different clinical trials are never reliable. Because studies are conducted at different times, with different samples of patients, by different investigators, employing different criteria and/or different interpretations of the same criteria, under different conditions (dose, dosing regimen, etc.), quantitative estimates of treatment response and the timing of response may be expected to vary considerably from study to study.
Table 1. Percentage of Patients With Headache Response (No or Mild Pain) 2 and 4 Hours Following Treatment Placebo
2 hr 4 hr
Sumatriptan Tablets
25 mg
2 hr 4 hr
Sumatriptan Tablets
50 mg
2 hr 4 hr
Sumatriptan Tablets
100 mg
2 hr 4 hr
Study 1
27% 38%
52% a 67% a
61% ab % ab
62% ab 79% ab
(N = 94)
(N = 298)
(N = 296)
(N = 296)
Study 2
26% 38%
52% a 70% a
50% a 68% a
56% a 71% a
(N = 65)
(N = 66)
(N = 62)
(N = 66)
Study 3
17% 19%
52% a 65% a
54% a 72% a
57% a 78% a
(N = 47)
(N = 48)
(N = 46)
(N = 46)
ap<0.05 in comparison with placebo.
bp<0.05 in comparison with 25 mg.
The estimated probability of achieving an initial headache response over the 4 hours following treatment is depicted in Figure 1.
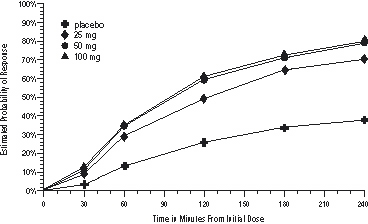
a The figure shows the probability over time of obtaining headache response (no or mild pain) following treatment with sumatriptan. The averages displayed are based on pooled data from the 3 clinical controlled trials providing evidence of efficacy. Kaplan-Meier plot with patients not achieving response and/or taking rescue within 240 minutes censored to 240 minutes.
For patients with migraine-associated nausea, photophobia, and/or phonophobia at baseline, there was a lower incidence of these symptoms at 2 hours (Study 1) and at 4 hours (Studies 1, 2, and 3) following administration of sumatriptan tablets compared to placebo.
As early as 2 hours in Studies 2 and 3 or 4 hours in Study 1, through 24 hours following the initial dose of study treatment, patients were allowed to use additional treatment for pain relief in the form of a second dose of study treatment or other medication. The estimated probability of patients taking a second dose or other medication for migraine over the 24 hours following the initial dose of study treatment is summarized in Figure 2.
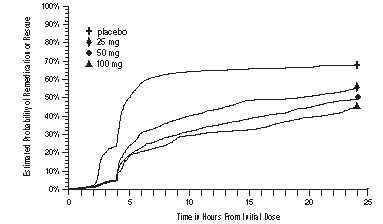
aKaplan-Meier plot based on data obtained in the 3 clinical controlled trials providing evidence of efficacy with patients not using additional treatments censored to 24 hours. Plot also includes patients who had no response to the initial dose. No remedication was allowed within 2 hours postdose.
There is evidence that doses above 50 mg do not provide a greater effect than 50 mg. There was no evidence to suggest that treatment with sumatriptan was associated with an increase in the severity of recurrent headaches. The efficacy of sumatriptan tablets was unaffected by presence of aura; duration of headache prior to treatment; gender, age, or weight of the patient; relationship to menses; or concomitant use of common migraine prophylactic drugs (e.g., beta-blockers, calcium channel blockers, tricyclic antidepressants). There were insufficient data to assess the impact of race on efficacy.
-
INDICATIONS AND USAGE
Sumatriptan tablets are indicated for the acute treatment of migraine attacks with or without aura in adults.
Sumatriptan tablets are not intended for the prophylactic therapy of migraine or for use in the management of hemiplegic or basilar migraine (see CONTRAINDICATIONS). Safety and effectiveness of sumatriptan tablets have not been established for cluster headache, which is present in an older, predominantly male population.
-
CONTRAINDICATIONS
Sumatriptan tablets should not be given to patients with history, symptoms, or signs of ischemic cardiac, cerebrovascular, or peripheral vascular syndromes. In addition, patients with other significant underlying cardiovascular diseases should not receive sumatriptane tablets. Ischemic cardiac syndromes include, but are not limited to, angina pectoris of any type (e.g., stable angina of effort, vasospastic forms of angina such as the Prinzmetal variant), all forms of myocardial infarction, and silent myocardial ischemia.
Cerebrovascular syndromes include, but are not limited to, strokes of any type as well as transient ischemic attacks. Peripheral vascular disease includes, but is not limited to, ischemic bowel disease (see WARNINGS).
Because sumatriptan tablets may increase blood pressure, they should not be given to patients with uncontrolled hypertension.
Concurrent administration of MAO-A inhibitors or use within 2 weeks of discontinuation of MAO-A inhibitor therapy is contraindicated (see CLINICAL PHARMACOLOGY: Drug Interactions and PRECAUTIONS: Drug Interactions).
Sumatriptan tablets should not be administered to patients with hemiplegic or basilar migraine.
Sumatriptan tablets and any ergotamine-containing or ergot-type medication (like dihydroergotamine or methysergide) should not be used within 24 hours of each other, nor should sumatriptan and another 5-HT 1 agonist.
Sumatriptan tablets are contraindicated in patients with hypersensitivity to sumatriptan or any of their components.
Sumatriptan tablets are contraindicated in patients with severe hepatic impairment.
-
WARNINGS
Sumatriptan tablets should only be used where a clear diagnosis of migraine headache has been established.
Risk of Myocardial Ischemia and/or Infarction and Other Adverse Cardiac Events
Sumatriptan should not be given to patients with documented ischemic or vasospastic coronary artery disease (CAD) (see CONTRAINDICATIONS). It is strongly recommended that sumatriptan not be given to patients in whom unrecognized CAD is predicted by the presence of risk factors (e.g., hypertension, hypercholesterolemia, smoker, obesity, diabetes, strong family history of CAD, female with surgical or physiological menopause, or male over 40 years of age) unless a cardiovascular evaluation provides satisfactory clinical evidence that the patient is reasonably free of coronary artery and ischemic myocardial disease or other significant underlying cardiovascular disease. The sensitivity of cardiac diagnostic procedures to detect cardiovascular disease or predisposition to coronary artery vasospasm is modest, at best. If, during the cardiovascular evaluation, the patient’s medical history or electrocardiographic investigations reveal findings indicative of, or consistent with, coronary artery vasospasm or myocardial ischemia, sumatriptan should not be administered (see CONTRAINDICATIONS).
For patients with risk factors predictive of CAD, who are determined to have a satisfactory cardiovascular evaluation, it is strongly recommended that administration of the first dose of sumatriptan tablets take place in the setting of a physician’s office or similar medically staffed and equipped facility unless the patient has previously received sumatriptan. Because cardiac ischemia can occur in the absence of clinical symptoms, consideration should be given to obtaining on the first occasion of use an electrocardiogram (ECG) during the interval immediately following sumatriptan tablets, in these patients with risk factors.
It is recommended that patients who are intermittent long-term users of sumatriptan and who have or acquire risk factors predictive of CAD, as described above, undergo periodic interval cardiovascular evaluation as they continue to use sumatriptan.
The systematic approach described above is intended to reduce the likelihood that patients with unrecognized cardiovascular disease will be inadvertently exposed to sumatriptan.
Drug-Associated Cardiac Events and Fatalities
Serious adverse cardiac events, including acute myocardial infarction, life-threatening disturbances of cardiac rhythm, and death have been reported within a few hours following the administration of sumatriptan succinate injection or sumatriptan tablets. Considering the extent of use of sumatriptan in patients with migraine, the incidence of these events is extremely low.
The fact that sumatriptan can cause coronary vasospasm, that some of these events have occurred in patients with no prior cardiac disease history and with documented absence of CAD, and the close proximity of the events to sumatriptan use support the conclusion that some of these cases were caused by the drug. In many cases, however, where there has been known underlying coronary artery disease, the relationship is uncertain.
Premarketing Experience With Sumatriptan
Of 6,348 patients with migraine who participated in premarketing controlled and uncontrolled clinical trials of oral sumatriptan, 2 experienced clinical adverse events shortly after receiving oral sumatriptan that may have reflected coronary vasospasm. Neither of these adverse events was associated with a serious clinical outcome.
Among the more than 1,900 patients with migraine who participated in premarketing controlled clinical trials of subcutaneous sumatriptan, there were 8 patients who sustained clinical events during or shortly after receiving sumatriptan that may have reflected coronary artery vasospasm. Six of these 8 patients had ECG changes consistent with transient ischemia, but without accompanying clinical symptoms or signs. Of these 8 patients, 4 had either findings suggestive of CAD or risk factors predictive of CAD prior to study enrollment.
Among approximately 4,000 patients with migraine who participated in premarketing controlled and uncontrolled clinical trials of sumatriptan nasal spray, 1 patient experienced an asymptomatic subendocardial infarction possibly subsequent to a coronary vasospastic event.
Postmarketing Experience With Sumatriptan
Serious cardiovascular events, some resulting in death, have been reported in association with the use of sumatriptan succinate injection or sumatriptan tablets. The uncontrolled nature of postmarketing surveillance, however, makes it impossible to determine definitively the proportion of the reported cases that were actually caused by sumatriptan or to reliably assess causation in individual cases. On clinical grounds, the longer the latency between the administration of sumatriptan and the onset of the clinical event, the less likely the association is to be causative. Accordingly, interest has focused on events beginning within 1 hour of the administration of sumatriptan.
Cardiac events that have been observed to have onset within 1 hour of sumatriptan administration include: coronary artery vasospasm, transient ischemia, myocardial infarction, ventricular tachycardia and ventricular fibrillation, cardiac arrest, and death.
Some of these events occurred in patients who had no findings of CAD and appear to represent consequences of coronary artery vasospasm. However, among domestic reports of serious cardiac events within 1 hour of sumatriptan administration, almost all of the patients had risk factors predictive of CAD and the presence of significant underlying
Drug-Associated Cerebrovascular Events and Fatalities
Cerebral hemorrhage, subarachnoid hemorrhage, stroke, and other cerebrovascular events have been reported in patients treated with oral or subcutaneous sumatriptan, and some have resulted in fatalities. The relationship of sumatriptan to these events is uncertain. In a number of cases, it appears possible that the cerebrovascular events were primary, sumatriptan having been administered in the incorrect belief that the symptoms experienced were a consequence of migraine when they were not. As with other acute migraine therapies, before treating headaches in patients not previously diagnosed as migraineurs, and in migraineurs who present with atypical symptoms, care should be taken to exclude other potentially serious neurological conditions. It should also be noted that patients with migraine may be at increased risk of certain cerebrovascular events (e.g., cerebrovascular accident, transient ischemic attack).
Other Vasospasm-Related Events
Sumatriptan may cause vasospastic reactions other than coronary artery vasospasm. Both peripheral vascular ischemia and colonic ischemia with abdominal pain and bloody diarrhea have been reported. Very rare reports of transient and permanent blindness and significant partial vision loss have been reported with the use of sumatriptan. Visual disorders may also be part of a migraine attack.
Serotonin Syndrome
Serotonin syndrome may occur with triptans, including Sumatriptan, particularly during combined use with selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs).Serotonin synd rome symptoms may include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g ., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperr eflexia, incoordination), and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). The on set of symptoms can occur within minutes to hours of receiving a new or a greater dose of a serotonergic medication. Treatment with Sumatriptan treatment should be discontinued if serotonin syndrome is suspected.
Increase in Blood Pressure
Significant elevation in blood pressure, including hypertensive crisis, has been reported on rare occasions in patients with and without a history of hypertension. Sumatriptan is contraindicated in patients with uncontrolled hypertension (see CONTRAINDICATIONS). Sumatriptan should be administered with caution to patients with controlled hypertension as transient increases in blood pressure and peripheral vascular resistance have been observed in a small proportion of patients.
Concomitant Drug Use
In patients taking MAO-A inhibitors, sumatriptan plasma levels attained after treatment with recommended doses are 7-fold higher following oral administration than those obtained under other conditions. Accordingly, the coadministration of sumatriptan tablets and an MAO-A inhibitor is contraindicated (see CLINICAL PHARMACOLOGY and CONTRAINDICATIONS).
Hypersensitivity
Hypersensitivity (anaphylaxis/anaphylactoid) reactions have occurred on rare occasions in patients receiving sumatriptan. Such reactions can be life threatening or fatal. In general, hypersensitivity reactions to drugs are more likely to occur in individuals with a history of sensitivity to multiple allergens (see CONTRAINDICATIONS).
-
PRECAUTIONS
General
Chest discomfort and jaw or neck tightness have been reported following use of sumatriptan tablets and have also been reported infrequently following administration of sumatriptan succinate Nasal Spray. Chest, jaw, or neck tightness is relatively common after administration of sumatriptan succinate injection. Only rarely have these symptoms been associated with ischemic ECG changes. However, because sumatriptan may cause coronary artery vasospasm, patients who experience signs or symptoms suggestive of angina following sumatriptan should be evaluated for the presence of CAD or a predisposition to Prinzmetal variant angina before receiving additional doses of sumatriptan, and should be monitored electrocardiographically if dosing is resumed and similar symptoms recur. Similarly, patients who experience other symptoms or signs suggestive of decreased arterial flow, such as ischemic bowel syndrome or Raynaud syndrome following sumatriptan should be evaluated for atherosclerosis or predisposition to vasospasm (see WARNINGS).
Sumatriptan should also be administered with caution to patients with diseases that may alter the absorption, metabolism, or excretion of drugs, such as impaired hepatic or renal function.
There have been rare reports of seizure following administration of sumatriptan. Sumatriptan should be used with caution in patients with a history of epilepsy or conditions associated with a lowered seizure threshold.
Care should be taken to exclude other potentially serious neurologic conditions before treating headache in patients not previously diagnosed with migraine headache or who experience a headache that is atypical for them. There have been rare reports where patients received sumatriptan for severe headaches that were subsequently shown to have been secondary to an evolving neurologic lesion (see WARNINGS).
For a given attack, if a patient does not respond to the first dose of sumatriptan, the diagnosis of migraine should be reconsidered before administration of a second dose.
Overuse:
Overuse of acute migraine drugs (e.g., ergotamine, triptans, opioids, or a combination of drugs for 10 or more days per month) may lead to exacerbation of headache (medication overuse headache). Medication overuse headache may present as migraine-like daily headaches, or as a marked increase in frequency of migraine attacks. Detoxification of patients, including withdrawal of the overused drugs, and treatment of withdrawal symptoms (which often includes a transient worsening of headache) may be necessary. Migraine patients should be informed about the risks of medication overuse, and encouraged to record headache frequency and drug use.
Information for Patients
See PATIENTINFORMATION at the end of this labeling for the text of the separate leaflet provided for patients.
Patients should be cautioned about the risk of serotonin syndrome with the use of sumatriptan or other triptans, especially during combined use with SSRIs or SNRIs.
Laboratory Tests
No specific laboratory tests are recommended for monitoring patients prior to and/or after treatment with sumatriptan.
Drug Interactions
Selective Serotonin Reuptake Inhibitors/Serotonin Norepinephrine Reuptake Inhibitors and Serotonin Syndrome:
Cases of life‑threatening serotonin syndrome have been reported during combined use of SSRIs or SNRIs and triptans (see WARNINGS).
Ergot-Containing Drugs
Ergot-containing drugs have been reported to cause prolonged vasospastic reactions. Because there is a theoretical basis that these effects may be additive, use of ergotamine-containing or ergot-type medications (like dihydroergotamine or methysergide) and sumatriptan within 24 hours of each other should be avoided (see CONTRAINDICATIONS).
Monoamine Oxidase-A Inhibitors
MAO-A inhibitors reduce sumatriptan clearance, significantly increasing systemic exposure. Therefore, the use of sumatriptan tablets in patients receiving MAO-A inhibitors is contraindicated (see CLINICALPHARMACOLOGY and CONTRAINDICATIONS).
Drug/Laboratory Test Interactions
Sumatriptan tablets are not known to interfere with commonly employed clinical laboratory tests.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis:
In carcinogenicity studies, rats and mice were given sumatriptan by oral gavage (rats: 104 weeks) or drinking water (mice: 78 weeks). Average exposures achieved in mice receiving the highest dose (target dose of 160 mg/kg/day) were approximately 40 times the exposure attained in humans after the maximum recommended single oral dose of 100 mg. The highest dose administered to rats (160 mg/kg/day, reduced from 360 mg/kg/day during week 21) was approximately 15 times the maximum recommended single human oral dose of 100 mg on a mg/m 2 basis. There was no evidence of an increase in tumors in either species related to sumatriptan administration.
Mutagenesis
Sumatriptan was not mutagenic in the presence or absence of metabolic activation when tested in 2 gene mutation assays (the Ames test and the in vitro mammalian Chinese hamster V79/HGPRT assay). In 2 cytogenetics assays (the in vitro human lymphocyte assay and the in vivo rat micronucleus assay) sumatriptan was not associated with clastogenic activity.
Impairment of Fertility
In a study in which male and female rats were dosed daily with oral sumatriptan prior to and throughout the mating period, there was a treatment-related decrease in fertility secondary to a decrease in mating in animals treated with 50 and 500 mg/kg/day. The highest no-effect dose for this finding was 5 mg/kg/day, or approximately one half of the maximum recommended single human oral dose of 100 mg on a mg/m 2 basis. It is not clear whether the problem is associated with treatment of the males or females or both combined. In a similar study by the subcutaneous route there was no evidence of impaired fertility at 60 mg/kg/day, the maximum dose tested, which is equivalent to approximately 6 times the maximum recommended single human oral dose of 100 mg on a mg/m 2 basis.
Pregnancy
Teratogenic Effects: Pregnancy Category C. In reproductive toxicity studies in rats and rabbits, oral treatment with sumatriptan was associated with embryolethality, fetal abnormalities, and pup mortality. When administered by the intravenous route to rabbits, sumatriptan has been shown to be embryolethal. There are no adequate and well-controlled studies in pregnant women. Therefore, sumatriptan should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. In assessing this information, the following findings should be considered.
Embryolethality
When given orally or intravenously to pregnant rabbits daily throughout the period of organogenesis, sumatriptan caused embryolethality at doses at or close to those producing maternal toxicity. In the oral studies this dose was 100 mg/kg/day, and in the intravenous studies this dose was 2 mg/kg/day. The mechanism of the embryolethality is not known. The highest no-effect dose for embryolethality by the oral route was 50 mg/kg/day, which is approximately 9 times the maximum single recommended human oral dose of 100 mg on a mg/m 2 basis. By the intravenous route, the highest no-effect dose was 0.75 mg/kg/day, or approximately one tenth of the maximum single recommended human oral dose of 100 mg on a mg/m 2 basis.
The intravenous administration of sumatriptan to pregnant rats throughout organogenesis at 12.5 mg/kg/day, the maximum dose tested, did not cause embryolethality. This dose is equivalent to the maximum single recommended human oral dose of 100 mg on a mg/m 2 basis. Additionally, in a study in rats given subcutaneous sumatriptan daily prior to and throughout pregnancy at 60 mg/kg/day, the maximum dose tested, there was no evidence of increased embryo/fetal lethality. This dose is equivalent to approximately 6 times the maximum recommended single human oral dose of 100 mg on a mg/m 2 basis.
Teratogenicity
Oral treatment of pregnant rats with sumatriptan during the period of organogenesis resulted in an increased incidence of blood vessel abnormalities (cervicothoracic and umbilical) at doses of approximately 250 mg/kg/day or higher. The highest no-effect dose was approximately 60 mg/kg/day, which is approximately 6 times the maximum single recommended human oral dose of 100 mg on a mg/m 2 basis. Oral treatment of pregnant rabbits with sumatriptan during the period of organogenesis resulted in an increased incidence of cervicothoracic vascular and skeletal abnormalities. The highest no-effect dose for these effects was 15 mg/kg/day, or approximately 3 times the maximum single recommended human oral dose of 100 mg on a mg/m 2 basis.
A study in which rats were dosed daily with oral sumatriptan prior to and throughout gestation demonstrated embryo/fetal toxicity (decreased body weight, decreased ossification, increased incidence of rib variations) and an increased incidence of a syndrome of malformations (short tail/short body and vertebral disorganization) at 500 mg/kg/day. The highest no-effect dose was 50 mg/kg/day, or approximately 5 times the maximum single recommended human oral dose of 100 mg on a mg/m 2 basis. In a study in rats dosed daily with subcutaneous sumatriptan prior to and throughout pregnancy, at a dose of 60 mg/kg/day, the maximum dose tested, there was no evidence of teratogenicity. This dose is equivalent to approximately 6 times the maximum recommended single human oral dose of 100 mg on a mg/m 2 basis.
Pup Deaths
Oral treatment of pregnant rats with sumatriptan during the period of organogenesis resulted in a decrease in pup survival between birth and postnatal day 4 at doses of approximately 250 mg/kg/day or higher. The highest no-effect dose for this effect was approximately 60 mg/kg/day, or 6 times the maximum single recommended human oral dose of 100 mg on a mg/m 2 basis.
Oral treatment of pregnant rats with sumatriptan from gestational day 17 through postnatal day 21 demonstrated a decrease in pup survival measured at postnatal days 2, 4, and 20 at the dose of 1,000 mg/kg/day. The highest no-effect dose for this finding was 100 mg/kg/day, approximately 10 times the maximum single recommended human oral dose of 100 mg on a mg/m 2 basis. In a similar study in rats by the subcutaneous route there was no increase in pup death at 81 mg/kg/day, the highest dose tested, which is equivalent to 8 times the maximum single recommended human oral dose of 100 mg on a mg/m 2 basis.
Nursing Mothers
Sumatriptan is excreted in human breast milk following subcutaneous administration. Infant exposure to sumatriptan can be minimized by avoiding breastfeeding for 12 hours after treatment with sumatriptan tablets.
Pediatric Use
Safety and effectiveness of sumatriptan tablets in pediatric patients have not been established.
Completed placebo-controlled clinical trials evaluating oral sumatriptan (25 to 100 mg) in pediatric patients aged 12 to 17 years enrolled a total of 701 adolescent migraineurs. These studies did not establish the efficacy of oral sumatriptan compared to placebo in the treatment of migraine in adolescents. Adverse events observed in these clinical trials were similar in nature to those reported in clinical trials in adults. The frequency of all adverse events in theses patients appeared to be both dose- and age-dependent, with younger patients reporting events more commonly than older adolescents. Post-marketing experience includes a limited number of reports that describe pediatric patients who have experienced adverse events, some clinically serious, after use of subcutaneous sumatriptan and/or oral sumatriptan. These reports include events similar in nature to those reported rarely in adults. A myocardial infarct has been reported in a 14-year-old male following the use of oral sumatriptan; clinical signs occurred within 1 day of drug administration. Since clinical data to determine the frequency of serious adverse events in pediatric patients who might receive injectable, oral, or intranasal sumatriptan are not presently available, the use of sumatriptan in patients aged younger than 18 years is not recommended.
Geriatric Use
The use of sumatriptan in elderly patients is not recommended because elderly patients are more likely to have decreased hepatic function, they are at higher risk for CAD, and blood pressure increases may be more pronounced in the elderly (see WARNINGS).
-
ADVERSE REACTIONS
Serious cardiac events, including some that have been fatal, have occurred following the use of sumatriptan succinate injection or tablets. These events are extremely rare and most have been reported in patients with risk factors predictive of CAD. Events reported have included coronary artery vasospasm, transient myocardial ischemia, myocardial infarction, ventricular tachycardia, and ventricular fibrillation (see CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS) .
Significant hypertensive episodes, including hypertensive crises, have been reported on rare occasions in patients with or without a history of hypertension (see WARNINGS).
Incidence in Controlled Clinical Trials
Table 2 lists adverse events that occurred in placebo-controlled clinical trials in patients who took at least 1 dose of study drug. Only events that occurred at a frequency of 2% or more in any group treated with sumatriptan tablets and were more frequent in that group than in the placebo group are included in Table 2. The events cited reflect experience gained under closely monitored conditions of clinical trials in a highly selected patient population. In actual clinical practice or in other clinical trials, these frequency estimates may not apply, as the conditions of use, reporting behavior, and the kinds of patients treated may differ.
Table 2. Treatment Emergent Adverse Events Reported by at Least 2% of Patients in Controlled Migraine Trials a Adverse Event Type
Percent of Patients Reporting
Placebo
(N = 309)
Sumatriptan
25 mg
(N = 417)
Sumatriptan
50 mg
(N = 771)
Sumatriptan
100 mg
(N = 437)
Atypical sensations
4%
5%
6%
6%
Paresthesia (all types)
2%
3%
5%
3%
Sensation warm/cold
2%
3%
2%
3%
Pain and other pressure sensations
4%
6%
6%
8%
Chest - pain/tightness/pressure and/or heaviness
1%
1%
2%
2%
Neck/throat/jaw - pain/ tightness/pressure
<1%
<1%
2%
3%
Pain - location specified
1%
2%
1%
1%
Other - pressure/tightness/ heaviness
2%
1%
1%
3%
Neurological
Vertigo
<1%
<1%
<1%
2%
Other
Malaise/fatigue
<1%
2%
2%
3%
aEvents that occurred at a frequency of 2% or more in the group treated with sumatriptan tablets and that occurred more frequently in that group than the placebo group.
Other events that occurred in more than 1% of patients receiving sumatriptan tablets and at least as often on placebo included nausea and/or vomiting, migraine, headache, hyposalivation, dizziness, and drowsiness/sleepiness.
Sumatriptan tablets are generally well tolerated. Across all doses, most adverse reactions were mild and transient and did not lead to long-lasting effects. The incidence of adverse events in controlled clinical trials was not affected by gender or age of the patients. There were insufficient data to assess the impact of race on the incidence of adverse events.
Other Events Observed in Association With the Administration of Sumatriptan Tablets
In the paragraphs that follow, the frequencies of less commonly reported adverse clinical events are presented. Because the reports include events observed in open and uncontrolled studies, the role of sumatriptan tablets in their causation cannot be reliably determined. Furthermore, variability associated with adverse event reporting, the terminology used to describe adverse events, etc., limit the value of quantitative frequency estimates provided. Event frequencies are calculated as the number of patients who used sumatriptane tablets (25, 50, or 100 mg) and reported an event divided by the total number of patients (N = 6,348) exposed to sumatriptan tablets. All reported events are included except those already listed in the previous table, those too general to be informative, and those not reasonably associated with the use of the drug. Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are defined as those occurring in at least 1/100 patients, infrequent adverse events are those occurring in 1/100 to 1/1,000 patients, and rare adverse events are those occurring in fewer than 1/1,000 patients.
Atypical Sensations
Frequent were burning sensation and numbness. Infrequent was tight feeling in head. Rare were dysesthesia.
Cardiovascular
Frequent were palpitations, syncope, decreased blood pressure, and increased blood pressure. Infrequent were arrhythmia, changes in ECG, hypertension, hypotension, pallor, pulsating sensations, and tachycardia. Rare were angina, atherosclerosis, bradycardia, cerebral ischemia, cerebrovascular lesion, heart block, peripheral cyanosis, thrombosis, transient myocardial ischemia, and vasodilation.
Ear, Nose, and Throat
Frequent were sinusitis, tinnitus; allergic rhinitis; upper respiratory inflammation; ear, nose, and throat hemorrhage; external otitis; hearing loss; nasal inflammation; and sensitivity to noise. Infrequent were hearing disturbances and otalgia. Rare was feeling of fullness in the ear(s).
Endocrine and Metabolic
Infrequent was thirst. Rare were elevated thyrotropin stimulating hormone (TSH) levels; galactorrhea; hyperglycemia; hypoglycemia; hypothyroidism; polydipsia; weight gain; weight loss; endocrine cysts, lumps, and masses; and fluid disturbances.
Eye
Rare were disorders of sclera, mydriasis, blindness and low vision, visual disturbances, eye edema and swelling, eye irritation and itching, accommodation disorders, external ocular muscle disorders, eye hemorrhage, eye pain, and keratitis and conjunctivitis.
Gastrointestinal
Frequent were diarrhea and gastric symptoms. Infrequent were constipation, dysphagia, and gastroesophageal reflux. Rare were gastrointestinal bleeding, hematemesis, melena, peptic ulcer, gastrointestinal pain, dyspeptic symptoms, dental pain, feelings of gastrointestinal pressure, gastritis, gastroenteritis, hypersalivation, abdominal distention, oral itching and irritation, salivary gland swelling, and swallowing disorders.
Musculoskeletal
Frequent was myalgia. Infrequent was muscle cramps. Rare were tetany; muscle atrophy, weakness, and tiredness; arthralgia and articular rheumatitis; acquired musculoskeletal deformity; muscle stiffness, tightness, and rigidity; and musculoskeletal inflammation.
Neurological
Frequent were phonophobia and photophobia. Infrequent were confusion, depression, difficulty concentrating, disturbance of smell, dysarthria, euphoria, facial pain, heat sensitivity, incoordination, lacrimation, monoplegia, sleep disturbance, shivering, syncope, and tremor. Rare were aggressiveness, apathy, bradylogia, cluster headache, convulsions, decreased appetite, drug abuse, dystonic reaction, facial paralysis, hallucinations, hunger, hyperesthesia, hysteria, increased alertness, memory disturbance, neuralgia, paralysis, personality change, phobia, radiculopathy, rigidity, suicide, twitching, agitation, anxiety, depressive disorders, detachment, motor dysfunction, neurotic disorders, psychomotor disorders, taste disturbances, and raised intracranial pressure.
Respiratory
Frequent was dyspnea. Infrequent was asthma. Rare were hiccoughs, breathing disorders, cough, and bronchitis.
Skin
Frequent was sweating. Infrequent were erythema, pruritus, rash, and skin tenderness. Rare were dry/scaly skin, tightness of skin, wrinkling of skin, eczema, seborrheic dermatitis, and skin nodules.
Breasts
Infrequent was tenderness. Rare were nipple discharge; breast swelling; cysts, lumps, and masses of breasts; and primary malignant breast neoplasm.
Urogenital
Infrequent were dysmenorrhea, increased urination, and intermenstrual bleeding. Rare were abortion and hematuria, urinary frequency, bladder inflammation, micturition disorders, urethritis, urinary infections, menstruation symptoms, abnormal menstrual cycle, inflammation of fallopian tubes, and menstrual cycle symptoms.
Other Events Observed in the Clinical Development of Sumatriptan
The following adverse events occurred in clinical trials with sumatriptan succinate injection and sumatriptan succinate nasal spray. Because the reports include events observed in open and uncontrolled studies, the role of sumatriptan in their causation cannot be reliably determined. All reported events are included except those already listed, those too general to be informative, and those not reasonably associated with the use of the drug.
Cardiovascular
Abdominal aortic aneurysm, abnormal pulse, flushing, phlebitis, Raynaud syndrome, and various transient ECG changes (nonspecific ST or T wave changes, prolongation of PR or QTc intervals, sinus arrhythmia, nonsustained ventricular premature beats, isolated junctional ectopic beats, atrial ectopic beats, delayed activation of the right ventricle).
Ear, Nose, and Throat
Disorder/discomfort nasal cavity and sinuses, ear infection, Meniere disease, and throat discomfort.
Gastrointestinal
Abdominal discomfort, colitis, disturbance of liver function tests, flatulence/eructation, gallstones, intestinal obstruction, pancreatitis, and retching.
Miscellaneous
Difficulty in walking, hypersensitivity to various agents, jaw discomfort, miscellaneous laboratory abnormalities, “serotonin agonist effect,” swelling of the extremities, and swelling of the face.
Mouth and Teeth
Disorder of mouth and tongue (e.g., burning of tongue, numbness of tongue, dry mouth).
Musculoskeletal
Arthritis, backache, intervertebral disc disorder, neck pain/stiffness, need to flex calf muscles, and various joint disturbances (pain, stiffness, swelling, ache).
Neurological
Bad/unusual taste, chills, diplegia, disturbance of emotions, sedation, globus hystericus, intoxication, myoclonia, neoplasm of pituitary, relaxation, sensation of lightness, simultaneous hot and cold sensations, stinging sensations, stress, tickling sensations, transient hemiplegia, and yawning.
Postmarketing Experience (Reports for Subcutaneous or Oral Sumatriptan)
The following section enumerates potentially important adverse events that have occurred in clinical practice and that have been reported spontaneously to various surveillance systems. The events enumerated represent reports arising from both domestic and nondomestic use of oral or subcutaneous dosage forms of sumatriptan. The events enumerated include all except those already listed in the ADVERSE REACTIONS section above or those too general to be informative. Because the reports cite events reported spontaneously from worldwide postmarketing experience, frequency of events and the role of sumatriptan in their causation cannot be reliably determined. It is assumed, however, that systemic reactions following sumatriptan use are likely to be similar regardless of route of administration.
Cardiovascular
Atrial fibrillation, cardiomyopathy, colonic ischemia (see WARNINGS), Prinzmetal variant angina, pulmonary embolism, shock, thrombophlebitis.
Neurological
Central nervous system vasculitis, cerebrovascular accident, dysphasia, serotonin syndrome, subarachnoid hemorrhage.
Skin
Exacerbation of sunburn, hypersensitivity reactions (allergic vasculitis, erythema, pruritus, rash, shortness of breath, urticaria; in addition, severe anaphylaxis/anaphylactoid reactions have been reported [see WARNINGS]), photosensitivity.
- DRUG ABUSE AND DEPENDENCE
-
OVERDOSAGE
Patients (N = 670) have received single oral doses of 140 to 300 mg without significant adverse effects. Volunteers (N = 174) have received single oral doses of 140 to 400 mg without serious adverse events.
Overdose in animals has been fatal and has been heralded by convulsions, tremor, paralysis, inactivity, ptosis, erythema of the extremities, abnormal respiration, cyanosis, ataxia, mydriasis, salivation, and lacrimation. The elimination half-life of sumatriptan is approximately 2.5 hours (see CLINICALPHARMACOLOGY), and therefore monitoring of patients after overdose with sumatriptan tablets should continue for at least 12 hours or while symptoms or signs persist.
It is unknown what effect hemodialysis or peritoneal dialysis has on the serum concentrations of sumatriptan.
-
DOSAGE AND ADMINISTRATION
In controlled clinical trials, single doses of 25, 50, or 100 mg of sumatriptan tablets were effective for the acute treatment of migraine in adults. There is evidence that doses of 50 and 100 mg may provide a greater effect than 25 mg (see CLINICALTRIALS). There is also evidence that doses of 100 mg do not provide a greater effect than 50 mg. Individuals may vary in response to doses of sumatriptan tablets. The choice of dose should therefore be made on an individual basis, weighing the possible benefit of a higher dose with the potential for a greater risk of adverse events.
If the headache returns or the patient has a partial response to the initial dose, the dose may be repeated after 2 hours, not to exceed a total daily dose of 200 mg. If a headache returns following an initial treatment with sumatriptan succinate injection, additional single sumatriptan tablets (up to 100 mg/day) may be given with an interval of at least 2 hours between tablet doses. The safety of treating an average of more than 4 headaches in a 30-day period has not been established.
Because of the potential of MAO-A inhibitors to cause unpredictable elevations in the bioavailability of oral sumatriptan, their combined use is contraindicated (see CONTRAINDICATIONS).
Hepatic disease/functional impairment may also cause unpredictable elevations in the bioavailability of orally administered sumatriptan. Consequently, if treatment is deemed advisable in the presence of liver disease, the maximum single dose should in general not exceed 50 mg (see CLINICALPHARMACOLOGYfor the basis of this recommendation).
-
HOW SUPPLIED
Sumatriptan tablets USP, 25, 50, and 100 mg of sumatriptan (base) as the succinate.
Sumatriptan tablets USP, 25 mg are white, round, biconvex film-coated tablets debossed with “RDY” on one side and “291” on the other side. The tablets are supplied in bottles of 30, 36, 90 100, 500, unit of use blister pack of 9's and unit dose package of 100's.
Bottles of 30 NDC: 55111-291-30
Bottles of 36 NDC: 55111-291-36
Bottles of 90 NDC: 55111-291-90
Bottles of 100 NDC: 55111-291-01
Bottles of 500 NDC: 55111-291-05
Unit of use blister pack of 9 (1 x 9) NDC: 55111-291-09
Unit dose package of 100 (10 x 10) NDC: 55111-291-78
Sumatriptan tablets USP, 50 mg are white, round, biconvex film-coated tablets debossed with “RDY” on one side and “292” on the other side. The tablets are supplied in bottles of 30, 36, 90 100, 500, unit of use blister pack of 9's and unit dose package of 100's.
Bottles of 30 NDC: 55111-292-30
Bottles of 36 NDC: 55111-292-36
Bottles of 90 NDC: 55111-292-90
Bottles of 100 NDC: 55111-292-01
Bottles of 500 NDC: 55111-292-05
Unit of use blister pack of 9 (1 x 9) NDC: 55111-292-09
Unit dose package of 100 (10 x 10) NDC: 55111-292-78
Sumatriptan tablets USP, 100 mg are white, capsule shaped, biconvex film-coated tablets debossed with “RDY” on one side and “293” on the other side. The tablets are supplied in bottles of 30, 36, 90 100, 500, unit of use blister pack of 9's and unit dose package of 100's.
Bottles of 30 NDC: 55111-293-30
Bottles of 36 NDC: 55111-293-36
Bottles of 90 NDC: 55111-293-90
Bottles of 100 NDC: 55111-293-01
Bottles of 500 NDC: 55111-293-05
Unit of use blister pack of 9 (1 x 9) NDC: 55111-293-09
Unit dose package of 100 (10 x 10) NDC: 55111-293-78
Store at 20to 25°C (68 – 77°F) [See USP Controlled Room Temperature].
Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).
-
ANIMAL TOXICOLOGY
Corneal Opacities
Dogs receiving oral sumatriptan developed corneal opacities and defects in the corneal epithelium. Corneal opacities were seen at the lowest dosage tested, 2 mg/kg/day, and were present after 1 month of treatment. Defects in the corneal epithelium were noted in a 60-week study. Earlier examinations for these toxicities were not conducted and no-effect doses were not established; however, the relative exposure at the lowest dose tested was approximately 5 times the human exposure after a 100 mg oral dose. There is evidence of alterations in corneal appearance on the first day of intranasal dosing to dogs. Changes were noted at the lowest dose tested, which was approximately one half the maximum single human oral dose of 100 mg on a mg/m 2 basis.
R X Only
Manufactured by:
Dr. Reddy’s Laboratories Limited
Bachepalli – 502 325 INDIA
Revised: 0312
-
PATIENT PACKAGE INSERT
Patient Information
Sumatriptan Tablets USP
Read this Patient Information before you start taking sumatriptan tablets and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or treatment.
What is the most important information I should know about sumatriptan tablets?
Sumatriptan tablets can cause serious side effects, including: Heart attack and other heart problems. Heart problems may lead to death.
Stop taking sumatriptan tablets and get emergency medical help right away if you have any of the following symptoms of a heart attack:
- discomfort in the center of your chest that lasts for more than a few minutes, or that goes away and comes back
- chest pain or chest discomfort that feels like uncomfortable heavy pressure, squeezing, or fullness, or pain
- pain or discomfort in your arms, back, neck, jaw, or stomach
- shortness of breath with or without chest discomfort
- breaking out in a cold sweat
- nausea or vomiting
- feeling lightheaded
Sumatriptan tablets are not for people with risk factors for heart disease unless a heart exam is done and shows no problem. You have a higher risk for heart disease if you:
- have high blood pressure
- have high cholesterol levels
- smoke
- are overweight
- have diabetes
- have a family history of heart disease
- are a female who has gone through menopause
- are a male over age 40
Serotonin syndrome. Serotonin syndrome is a serious and life-threatening problem that can happen in people taking sumatriptan tablets, especially if sumatriptan tablets are used with antidepressant medicines called selective serotonin reuptake inhibitors (SSRIs) or selective norepinephrine reuptake inhibitors (SNRIs).
Ask your healthcare provider or pharmacist for a list of these medicines if you are not sure.
Call your healthcare provider right away if you have any of the following symptoms of serotonin syndrome:
- mental changes such as seeing things that are not there (hallucinations), agitation, or coma
- fast heartbeat
- changes in blood pressure
- high body temperature
- tight muscles
- trouble walking
- nausea, vomiting, or diarrhea
What are sumatriptan tablets?
Sumatriptan tablets are a prescription medicine used to treat acute migraine headaches with or without aura in adults.
Sumatriptan tablets are not used to prevent or decrease the number of migraine headaches you have.
Sumatriptan tablets are not used to treat other types of headaches such as hemiplegic (that make you unable to move on one side of your body) or basilar migraines (rare form of migraine with aura).
It is not known if sumatriptan tablets are safe and effective to treat cluster headaches.
It is not known if sumatriptan tablets are safe and effective in children under 18 years of age.
Who should not take sumatriptan tablets?
Do not take sumatriptan tablets if you have:
- heart problems or a history of heart problems
- narrowing of blood vessels to your legs, arms, stomach, or kidney (peripheral vascular disease)
- uncontrolled high blood pressure
- severe liver problems
- hemiplegic migraines or basilar migraines. If you are not sure if you have these types of migraines, ask your healthcare provider.
- had a stroke, transient ischemic attacks (TIAs), or problems with your blood circulation
- taken any of the following medicines in the last 24 hours:
- almotriptan (AXERT ®)
- eletriptan (RELPAX ®)
- frovatriptan (FROVA ®)
- naratriptan (AMERGE ®)
- rizatriptan (MAXALT ®, MAXALT-MLT ®)
- sumatriptan and naproxen (TREXIMET ®)
- ergotamines (CAFERGOT ®, ERGOMAR ®, MIGERGOT ®)
- dihydroergotamine (D.H.E. 45 ®, MIGRANAL ®)
Ask your doctor if you are not sure if your medicine is listed above.
- an allergy to sumatriptan or any of the ingredients in sumatriptan tablets. See the end of this leaflet for a complete list of ingredients in sumatriptan tablets.
What should I tell my healthcare provider before taking sumatriptan tablets?
Before you take sumatriptan tablets, tell your healthcare provider about all of your medical conditions, including if you:
- have high blood pressure
- have high cholesterol
- have diabetes
- smoke
- are overweight
- are a female who has gone through menopause
- have heart disease or a family history of heart disease or stroke
- have kidney problems
- have liver problems
- have had epilepsy or seizures
- are not using effective birth control
- are pregnant or plan to become pregnant. It is not known if sumatriptan tablets will harm your unborn baby.
- are breastfeeding or plan to breastfeed. Sumatriptan tablets passes into your breast milk and may harm your baby. Talk with your healthcare provider about the best way to feed your baby if you take sumatriptan tablets.
Tell your healthcare provider about all the medicines you take, including prescription and nonprescription medicines, vitamins, and herbal supplements.
Sumatriptan tablets and other medicines may affect each other, causing side effects.
Especially tell your healthcare provider if you take anti-depressant medicines called:
- selective serotonin reuptake inhibitors (SSRIs)
- serotonin norepinephrine reuptake inhibitors (SNRIs)
- monoamine oxidase inhibitors (MAOIs)
Ask your healthcare provider or pharmacist for a list of these medicines if you are not sure.
Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine.
How should I take sumatriptan tablets?
- Certain people should take their first dose of sumatriptan tablets in their healthcare provider’s office or in another medical setting. Ask your healthcare provider if you should take your first dose in a medical setting.
- Take sumatriptan tablets exactly as your healthcare provider tells you to take it.
- Your healthcare provider may change your dose. Do not change your dose without first talking to your healthcare provider.
- Take sumatriptan tablets with water or other liquids.
- If you do not get any relief after your first sumatriptan tablet, do not take a second tablet without first talking with your healthcare provider.
- If your headache comes back or you only get some relief from your headache, you can take a second tablet 2 hours after the first tablet
- Do not take more than a total of 200 mg of sumatriptan tablets in a 24-hour period.
- Some people who take too many sumatriptan tablets may have worse headaches (medication overuse headache). If your headaches get worse, your healthcare provider may decide to stop your treatment with sumatriptan tablets.
- If you take too much sumatriptan tablets, call your healthcare provider or go to the nearest hospital emergency room right away.
- You should write down when you have headaches and when you take sumatriptan tablets so you can talk with your healthcare provider about how sumatriptan tablets are working for you.
What should I avoid while taking sumatriptan tablets?
Sumatriptan tablets can cause dizziness, weakness, or drowsiness. If you have these symptoms, do not drive a car, use machinery, or do anything where you need to be alert.
What are the possible side effects of sumatriptan tablets? Sumatriptan tablets may cause serious side effects. See “What is the most important information I should know about sumatriptan tablets?”
These serious side effects include:
- changes in color or sensation in your fingers and toes (Raynaud’s syndrome)
- stomach and intestinal problems (gastrointestinal and colonic ischemic events). Symptoms of gastrointestinal and colonic ischemic events include:
- sudden or severe stomach pain
- stomach pain after meals
- weight loss
- nausea or vomiting
- constipation or diarrhea
- bloody diarrhea
- fever
- problems with blood circulation to your legs and feet (peripheral vascular ischemia). Symptoms of peripheral vascular ischemia include:
- cramping and pain in your legs or hips
- feeling of heaviness or tightness in your leg muscles
- burning or aching pain in your feet or toes while resting
- numbness, tingling, or weakness in your legs
- cold feeling or color changes in 1 or both legs or feet
- shortness of breath or wheezing
- hives (itchy bumps); swelling of your tongue, mouth, or throat
The most common side effects of sumatriptan tablets include:
- tingling or numbness in your fingers or toes
- dizziness
- warm, hot, burning feeling to your face (flushing)
- feeling weak, drowsy, or tired
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of sumatriptan tablets. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Sumatriptan Tablets?
Store at 20to 25°C (68 – 77°F) [See USP Controlled Room Temperature].
Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).
Keep sumatriptan tablets and all medicines out of the reach of children.
General information about the safe and effective use of sumatriptan tablets.
Medicines are sometimes prescribed for purposes other than those listed in Patient Information leaflets. Do not use sumatriptan tablets for a condition for which it was not prescribed. Do not give sumatriptan tablets to other people, even if they have the same symptoms you have. It may harm them.
This Patient Information leaflet summarizes the most important information about sumatriptan tablets. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about sumatriptan tablets that is written for healthcare professionals.
For more information, call 1-888-375-3784.
What are the ingredients in Sumatriptan Tablets?
Active ingredients : Sumatriptan succinate
Inactive ingredients: croscarmellose sodium, lactose anhydrous, lactose monohydrate, magnesium stearate, mannitol, microcrystalline cellulose, talc, titanium dioxide and triacetin.
To reorder additional Patient Information Leaflets, please contact Dr. Reddy’s Customer Service at 1-866-733-3952.
IMITREX, AMERGE, and TREXIMET are registered trademarks of GlaxoSmithKline. The other brands listed are trademarks of their respective owners.
This Patient Information has been approved by the U.S. Food and Drug Administration.
RX Only
Manufactured by:
Dr. Reddy’s Laboratories Limited
Bachepalli – 502 325 INDIA
Revised : 0312
-
1 INDICATIONS AND USAGE
Ondansetron tablets are indicated for the prevention of nausea and vomiting associated with:
highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m 2.
initial and repeat courses of moderately emetogenic cancer chemotherapy.
radiotherapy in patients receiving either total body irradiation, single high-dose fraction to the abdomen, or daily fractions to the abdomen.Ondansetron tablets are also indicated for the prevention of postoperative nausea and/or vomiting.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
The recommended dosage regimens for adult and pediatric patients are described in Table 1 and Table 2, respectively.
Table 1: Adult Recommended Dosage Regimen for Prevention of Nausea and VomitingIndication
Dosage Regimen
Highly Emetogenic Cancer Chemotherapy
A single 24 mg dose administered 30 minutes before the start of single-day highly emetogenic chemotherapy, including cisplatin greater than or equal to 50 mg/m 2
Moderately Emetogenic Cancer Chemotherapy
8 mg administered 30 minutes before the start of chemotherapy, with a subsequent 8 mg dose 8 hours after the first dose.
Then administer 8 mg twice a day (every 12 hours) for 1 to 2 days after completion of chemotherapy
Radiotherapy
For total body irradiation: 8 mg administered 1 to 2 hours before each fraction of radiotherapy each day.
For single high-dose fraction radiotherapy to the abdomen: 8 mg administered 1 to 2 hours before radiotherapy, with subsequent 8 mg doses every 8 hours after the first dose for 1 to 2 days after completion of radiotherapy.
For daily fractionated radiotherapy to the abdomen: 8 mg administered 1 to 2 hours before radiotherapy, with subsequent 8 mg doses every 8 hours after the first dose for each day radiotherapy is given.
Postoperative
16 mg administered 1 hour before induction of anesthesia
Table 2: Pediatric Recommended Dosage Regimen for Prevention of Nausea and Vomiting
Indication
Dosage Regimen
Moderately Emetogenic Cancer Chemotherapy
12 to 17 years of age: 8 mg administered 30 minutes before the start of chemotherapy, with a subsequent 8 mg dose 4 and 8 hours after the first dose.
Then administer 8 mg three times a day for 1 to 2 days after completion of chemotherapy.
4 to 11 years of age: 4 mg administered 30 minutes before the start of chemotherapy, with a subsequent 4 mg dose 4 and 8 hours after the first dose.
Then administer 4 mg three times a day for 1 to 2 days after completion of chemotherapy.
2.2 Dosage in Hepatic Impairment
In patients with severe hepatic impairment (Child-Pugh score of 10 or greater), do not exceed a total daily dose of 8 mg [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Ondansetron tablets are contraindicated in patients:
known to have hypersensitivity (e.g., anaphylaxis) to ondansetron or any of the components of the formulation [see Adverse Reactions (6.2)].
receiving concomitant apomorphine due to the risk of profound hypotension and loss of consciousness. -
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis and bronchospasm, have been reported in patients who have exhibited hypersensitivity to other selective 5-HT 3 receptor antagonists. If hypersensitivity reactions occur, discontinue use of ondansetron tablets; treat promptly per standard of care and monitor until signs and symptoms resolve [see Contraindications (4)].
5.2 QT Prolongation
Electrocardiogram (ECG) changes including QT interval prolongation have been seen in patients receiving ondansetron. In addition, postmarketing cases of Torsade de Pointes have been reported in patients using ondansetron tablets. Avoid ondansetron tablets in patients with congenital long QT syndrome. ECG monitoring is recommended in patients with electrolyte abnormalities (e.g., hypokalemia or hypomagnesemia), congestive heart failure, bradyarrhythmias, or patients taking other medicinal products that lead to QT prolongation [see Clinical Pharmacology (12.2)].
5.3 Serotonin Syndrome
The development of serotonin syndrome has been reported with 5-HT 3 receptor antagonists alone. Most reports have been associated with concomitant use of serotonergic drugs (e.g., selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), monoamine oxidase inhibitors, mirtazapine, fentanyl, lithium, tramadol, and intravenous methylene blue). Some of the reported cases were fatal. Serotonin syndrome occurring with overdose of ondansetron tablets alone has also been reported. The majority of reports of serotonin syndrome related to 5-HT 3 receptor antagonist use occurred in a post-anesthesia care unit or an infusion center.
Symptoms associated with serotonin syndrome may include the following combination of signs and symptoms: mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, with or without gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Patients should be monitored for the emergence of serotonin syndrome, especially with concomitant use of ondansetron tablets and other serotonergic drugs. If symptoms of serotonin syndrome occur, discontinue ondansetron tablets and initiate supportive treatment. Patients should be informed of the increased risk of serotonin syndrome, especially if ondansetron tablets is used concomitantly with other serotonergic drugs [see Drug Interactions (7.1), Overdosage (10)].5.4 Masking of Progressive Ileus and Gastric Distension
The use of ondansetron tablets in patients following abdominal surgery or in patients with chemotherapy-induced nausea and vomiting may mask a progressive ileus and/or gastric distension. Monitor for decreased bowel activity, particularly in patients with risk factors for gastrointestinal obstruction.
Ondansetron tablets are not a drug that stimulates gastric or intestinal peristalsis. It should not be used instead of nasogastric suction. -
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The following adverse reactions have been reported in clinical trials of patients treated with ondansetron, the active ingredient of ondansetron tablets. A causal relationship to therapy with ondansetron tablets was unclear in many cases.
Prevention of Chemotherapy-induced Nausea and Vomiting
The most common adverse reactions reported in greater than or equal to 4% of 300 adults receiving a single 24 mg dose of ondansetron tablets orally in 2 trials for the prevention of nausea and vomiting associated with highly emetogenic chemotherapy (cisplatin greater than or equal to 50 mg/m 2) were: headache (11%) and diarrhea (4%).
The most common adverse reactions reported in 4 trials in adults for the prevention of nausea and vomiting associated with moderately emetogenic chemotherapy (primarily cyclophosphamide-based regimens) are shown in Table 3.
Table 3. Most Common Adverse Reactions in Adults a for the Prevention of Nausea and Vomiting Associated with Moderately Emetogenic Chemotherapy [Primarily Cyclophosphamide-based Regimens]a Reported in greater than or equal to 5% of patients treated with ondansetron tablets and at a rate that exceeded placebo.
Adverse Reaction
Ondansetron Tablets 8 mg Twice Daily (n = 242)
Placebo
(n = 262)
Headache
58 (24%)
34 (13%)
Malaise/fatigue
32 (13%)
6 (2%)
Constipation
22 (9%)
1 (<1%)
Diarrhea
15 (6%)
10 (4%)Less Common Adverse Reactions
Central Nervous System: Extrapyramidal reactions (less than 1% of patients).
Hepatic: Aspartate transaminase (AST) and/or alanine transaminase (ALT) values exceeded twice the upper limit of normal in approximately 1% to 2% of 723 patients receiving ondansetron tablets and cyclophosphamide-based chemotherapy in US clinical trials. The increases were transient and did not appear to be related to dose or duration of therapy. On repeat exposure, similar transient elevations in transaminase values occurred in some courses, but symptomatic hepatic disease did not occur. The role of cancer chemotherapy in these biochemical changes is unclear.
Liver failure and death has been reported in cancer patients receiving concurrent medications, including potentially hepatotoxic cytotoxic chemotherapy and antibiotics. The etiology of the liver failure is unclear.
Integumentary: Rash (approximately 1% of patients).
Other (less than 2%): Anaphylaxis, bronchospasm, tachycardia, angina, hypokalemia, electrocardiographic alterations, vascular occlusive events, and grand mal seizures. Except for bronchospasm and anaphylaxis, the relationship to ondansetron tablets is unclear.
Prevention of Radiation-induced Nausea and Vomiting
The most common adverse reactions (greater than or equal to 2%) reported in patients receiving ondansetron tablets and concurrent radiotherapy were similar to those reported in patients receiving ondansetron tablets and concurrent chemotherapy and were headache, constipation, and diarrhea.
Prevention of Postoperative Nausea and Vomiting
The most common adverse reactions reported in adults in trial(s) of prevention of postoperative nausea and vomiting are shown in Table 4. In these trial(s) patients were receiving multiple concomitant perioperative and postoperative medications in both treatment groups.
Table 4. Most Common Adverse Reactions in Adults a for the Prevention of Postoperative Nausea and Vomitinga Reported in greater than or equal to 5% of patients treated with ondansetron tablets and at a rate that exceeded placebo.
Adverse Reaction
Ondansetron Tablets 16 mg as
a Single Dose
(n = 550)
Placebo
(n = 531)
Headache
49 (9%)
27 (5%)
Hypoxia
49 (9%)
35 (7%)
Pyrexia
45 (8%)
34 (6%)
Dizziness
36 (7%)
34 (6%)
Gynecological disorder
36 (7%)
33 (6%)
Anxiety/agitation
33 (6%)
29 (5%)
Urinary retention
28 (5%)
18 (3%)
Pruritus
27 (5%)
20 (4%)6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ondansetron. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiovascular
Arrhythmias (including ventricular and supraventricular tachycardia, premature ventricular contractions, and atrial fibrillation), bradycardia, electrocardiographic alterations (including second-degree heart block, QT/QTc interval prolongation, and ST segment depression), palpitations, and syncope. Rarely and predominantly with intravenous ondansetron, transient ECG changes including QT interval prolongation have been reported.
General
Flushing. Rare cases of hypersensitivity reactions, sometimes severe (e.g., anaphylactic reactions, angioedema, bronchospasm, shortness of breath, hypotension, laryngeal edema, stridor) have also been reported.
Laryngospasm, shock, and cardiopulmonary arrest have occurred during allergic reactions in patients receiving injectable ondansetron.
Hepatobiliary
Liver enzyme abnormalities.
Lower Respiratory
Hiccups.
Neurology
Oculogyric crisis, appearing alone, as well as with other dystonic reactions.
Skin
Urticaria, Stevens-Johnson syndrome, and toxic epidermal necrolysis.
Eye Disorders
Cases of transient blindness, predominantly during intravenous administration, have been reported. These cases of transient blindness were reported to resolve within a few minutes up to 48 hours. -
7 DRUG INTERACTIONS
7.1 Serotonergic Drugs
Serotonin syndrome (including altered mental status, autonomic instability, and neuromuscular symptoms) has been described following the concomitant use of 5-HT 3 receptor antagonists and other serotonergic drugs, including selective serotonin reuptake inhibitors (SSRIs) and serotonin and noradrenaline reuptake inhibitors (SNRIs). Monitor for the emergence of serotonin syndrome. If symptoms occur, discontinue ondansetron tablets and initiate supportive treatment [see Warnings and Precautions (5.3)].
7.2 Drugs Affecting Cytochrome P-450 Enzymes
Ondansetron does not itself appear to induce or inhibit the cytochrome P-450 drug-metabolizing enzyme system of the liver [see Clinical Pharmacology (12.3)]. Because ondansetron is metabolized by hepatic cytochrome P-450 drug-metabolizing enzymes (CYP3A4, CYP2D6, CYP1A2), inducers or inhibitors of these enzymes may change the clearance and, hence, the half-life of ondansetron. In patients treated with potent inducers of CYP3A4 (i.e., phenytoin, carbamazepine, and rifampin), the clearance of ondansetron was significantly increased and ondansetron blood concentrations were decreased. However, on the basis of available data, no dosage adjustment for ondansetron tablets is recommended for patients on these drugs [see Clinical Pharmacology (12.3)].
7.3 Tramadol
Although no pharmacokinetic drug interaction between ondansetron and tramadol has been observed, data from 2 small trials indicate that when used together, ondansetron tablets may increase patient-controlled administration of tramadol. Monitor patients to ensure adequate pain control when ondansetron is administered with tramadol.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data do not reliably inform the association of ondansetron tablets and adverse fetal outcomes. Published epidemiological studies on the association between ondansetron and fetal outcomes have reported inconsistent findings and have important methodological limitations hindering interpretation [see Data]. Reproductive studies in rats and rabbits did not show evidence of harm to the fetus when ondansetron was administered during organogenesis at approximately 6 and 24 times the maximum recommended human oral dose of 24 mg/day, based on body surface area, respectively [see Data].
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the US general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
Methodological limitations of the epidemiology studies preclude a reliable evaluation of the potential risk of adverse fetal outcomes with the use of ondansetron in pregnancy.
Two large retrospective cohort studies of ondansetron use in pregnancy have been published. In one study with 1,349 infants born to women who reported the use of ondansetron or received an ondansetron prescription in the first trimester, no increased risk for major congenital malformations was seen in aggregate analysis. In this same study, however, a sub-analysis for specific malformations reported an association between ondansetron exposure and cardiovascular defect (odds ratio (OR) 1.62 [95% CI (1.04, 2.14)]) and cardiac septal defect (OR) 2.05 [95% CI (1.19, 3.28)]). The second study examined 1970 women who received ondansetron prescription during pregnancy and reported no association between ondansetron exposure and major congenital malformations, miscarriage or stillbirth, and infants of low birth weight or small for gestational age. Important methodological limitations with these studies include the uncertainty of whether women who filled a prescription actually took the medication, the concomitant use of other medications or treatments, and other unadjusted confounders that may account for the study findings.
A case-control study evaluating associations between several common non-cardiac malformations and multiple antiemetic drugs reported an association between maternal use of ondansetron and isolated cleft palate (reported adjusted OR = 2.37 [95% CI (1.18, 4.76)]). However, this association could be a chance finding, given the large number of drugs-birth defect comparisons in this study. It is unknown whether ondansetron exposure in utero in the cases of cleft palate occurred during the time of palate formation (the palate is formed between the 6th and 9th weeks of pregnancy) or whether mothers of infants with cleft palate used other medications or had other risk factors for cleft palate in the offspring. In addition, no cases of isolated cleft palate were identified in the aforementioned two large retrospective cohort studies. At this time, there is no clear evidence that ondansetron exposure in early pregnancy can cause cleft palate.
Animal Data
In embryo-fetal development studies in rats and rabbits, pregnant animals received oral doses of ondansetron up to 15 mg/kg/day and 30 mg/kg/day, respectively, during the period of organogenesis. With the exception of a slight decrease in maternal body weight gain in the rabbits, there were no significant effects of ondansetron on the maternal animals or the development of the offspring. At doses of 15 mg/kg/day in rats and 30 mg/kg/day in rabbits, the maternal exposure margin was approximately 6 and 24 times the maximum recommended human oral dose of 24 mg/day, respectively, based on body surface area.
In a pre-and postnatal developmental toxicity study, pregnant rats received oral doses of ondansetron up to 15 mg/kg/day from Day 17 of pregnancy to litter Day 21. With the exception of a slight reduction in maternal body weight gain, there were no effects upon the pregnant rats and the pre-and postnatal development of their offspring, including reproductive performance of the mated F1 generation. At a dose of 15 mg/kg/day in rats, the maternal exposure margin was approximately 6 times the maximum recommended human oral dose of 24 mg/day, based on body surface area.8.2 Lactation
Risk Summary
It is not known whether ondansetron is present in human milk. There are no data on the effects of ondansetron tablets on the breastfed infant or the effects on milk production. However, it has been demonstrated that ondansetron is present in the milk of rats.The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ondansetron tablets and any potential adverse effects on the breast-fed infant from ondansetron tablets or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of orally administered ondansetron tablets have been established in pediatric patients 4 years and older for the prevention of nausea and vomiting associated with moderately emetogenic cancer chemotherapy. Use of ondansetron tablets in these age-groups is supported by evidence from adequate and well-controlled studies of ondansetron tablets in adults with additional data from 3 open-label, uncontrolled, non-US trials in 182 pediatric patients aged 4 to 18 years with cancer who were given a variety of cisplatin or noncisplatin regimens [see Dosage and Administration (2.2),Clinical Studies (14.1)].
Additional information on the use of ondansetron in pediatric patients may be found in ondansetron injection prescribing information.
The safety and effectiveness of orally administered ondansetron tablets have not been established in pediatric patients for:- prevention of nausea and vomiting associated with highly emetogenic cancer chemotherapy.
- prevention of nausea and vomiting associated with radiotherapy.
- prevention of postoperative nausea and/or vomiting.
8.5 Geriatric Use
Of the total number of subjects enrolled in cancer chemotherapy-induced and postoperative nausea and vomiting in US-and foreign-controlled clinical trials, for which there were subgroup analyses, 938 (19%) were aged 65 years and older.
No overall differences in safety or effectiveness were observed between subjects 65 years of age and older and younger subjects. A reduction in clearance and increase in elimination half-life were seen in patients older than 75 years compared with younger subjects [see Clinical Pharmacology (12.3)].There were an insufficient number of patients older than 75 years of age and older in the clinical trials to permit safety or efficacy conclusions in this age-group. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. No dosage adjustment is needed in elderly patients.
8.6 Hepatic Impairment
No dosage adjustment is needed in patients with mild or moderate hepatic impairment.
In patients with severe hepatic impairment, clearance is reduced and the apparent volume of distribution is increased, resulting in a significant increase in the half-life of ondansetron. Therefore, do not exceed a total daily dose of 8 mg in patients with severe hepatic impairment (Child-Pugh score of 10 or greater) [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].8.7 Renal Impairment
No dosage adjustment is recommended for patients with any degree of renal impairment (mild, moderate, or severe). There is no experience beyond first-day administration of ondansetron [see Clinical Pharmacology (12.3)].
- 9 DRUG ABUSE AND DEPENDENCE
-
10 OVERDOSAGE
There is no specific antidote for ondansetron overdose. Patients should be managed with appropriate supportive therapy.
In addition to the adverse reactions listed above, the following adverse reactions have been described in the setting of ondansetron overdose: “Sudden blindness” (amaurosis) of 2 to 3 minutes’ duration plus severe constipation occurred in one patient that was administered 72 mg of ondansetron intravenously as a single dose. Hypotension (and faintness) occurred in a patient that took 48 mg of ondansetron tablets. Following infusion of 32 mg over only a 4-minute period, a vasovagal episode with transient second-degree heart block was observed. In all instances, the adverse reactions resolved completely.
Pediatric cases consistent with serotonin syndrome have been reported after inadvertent oral overdoses of ondansetron (exceeding estimated ingestion of 5 mg per kg) in young children. Reported symptoms included somnolence, agitation, tachycardia, tachypnea, hypertension, flushing, mydriasis, diaphoresis, myoclonic movements, horizontal nystagmus, hyperreflexia, and seizure. Patients required supportive care, including intubation in some cases, with complete recovery without sequelae within 1 to 2 days.
-
11 DESCRIPTION
The active ingredient in ondansetron tablets is ondansetron hydrochloride as the dihydrate, the racemic form of ondansetron and a selective blocking agent of the serotonin 5-HT3 receptor type. Chemically it is (±) 1, 2, 3, 9-tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl) methyl]-4H-carbazol-4-one, monohydrochloride, dihydrate. It has the following structural formula:
The molecular formula is C18H19N3OHCl2H2O, representing a molecular weight of 365.5.Ondansetron hydrochloride dihydrate is a white to off-white powder that is soluble in water and normal saline.
Each 4 mg ondansetron tablet, USP for oral administration contains ondansetron hydrochloride dihydrate equivalent to 4 mg of ondansetron. Each 8 mg ondansetron tablet, USP for oral administration contains ondansetron hydrochloride dihydrate equivalent to 8 mg of ondansetron. Each tablet also contains the inactive ingredients croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, pregelatinized starch, titanium dioxide, triacetin, and iron oxide yellow (8 mg tablet only).
This product meets USP Dissolution Test 3.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ondansetron is a selective 5-HT3 receptor antagonist. While its mechanism of action has not been fully characterized, ondansetron is not a dopamine-receptor antagonist. Serotonin receptors of the 5-HT3 type are present both peripherally on vagal nerve terminals and centrally in the chemoreceptor trigger zone of the area postrema. It is not certain whether ondansetron’s antiemetic action is mediated centrally, peripherally, or in both sites. However, cytotoxic chemotherapy appears to be associated with release of serotonin from the enterochromaffin cells of the small intestine. In humans, urinary 5-hydroxyindoleacetic acid (5-HIAA) excretion increases after cisplatin administration in parallel with the onset of emesis. The released serotonin may stimulate the vagal afferents through the 5-HT3 receptors and initiate the vomiting reflex.
12.2 Pharmacodynamics
In healthy subjects, single intravenous doses of 0.15 mg/kg of ondansetron had no effect on esophageal motility, gastric motility, lower esophageal sphincter pressure, or small intestinal transit time. Multiday administration of ondansetron has been shown to slow colonic transit in healthy subjects. Ondansetron has no effect on plasma prolactin concentrations.
Cardiac Electrophysiology
QTc interval prolongation was studied in a double-blind, single intravenous dose, placebo-and positive-controlled, crossover trial in 58 healthy subjects. The maximum mean (95% upper confidence bound) difference in QTcF from placebo after baseline correction was 19.5 (21.8) milliseconds and 5.6 (7.4) milliseconds after 15minute intravenous infusions of 32 mg and 8 mg of ondansetron injection, respectively. A significant exposure-response relationship was identified between ondansetron concentration and ΔΔQTcF. Using the established exposure-response relationship, 24 mg infused intravenously over 15 minutes had a mean predicted (95% upper prediction interval) ΔΔQTcF of 14.0 (16.3) milliseconds. In contrast, 16 mg infused intravenously over 15 minutes using the same model had a mean predicted (95% upper prediction interval) ΔΔQTcF of 9.1 (11.2) milliseconds. In this study, the 8 mg dose infused over 15 minutes did not prolong the QT interval to any clinically relevant extent.12.3 Pharmacokinetics
Absorption
Ondansetron is absorbed from the gastrointestinal tract and undergoes some first-pass metabolism. Mean bioavailability in healthy subjects, following administration of a single 8 mg tablet, is approximately 56%.
Ondansetron systemic exposure does not increase proportionately to dose. The AUC from a 16 mg tablet was 24% greater than predicted from an 8 mg tablet dose. This may reflect some reduction of first-pass metabolism at higher oral doses.
Food Effects: Bioavailability is also slightly enhanced by the presence of food.
Distribution
Plasma protein binding of ondansetron as measured in vitro was 70% to 76% over the concentration range of 10 to 500 ng/mL. Circulating drug also distributes into erythrocytes.
Elimination
Metabolism and Excretion: Ondansetron is extensively metabolized in humans, with approximately 5% of a radiolabeled dose recovered as the parent compound from the urine. The metabolites are observed in the urine. The primary metabolic pathway is hydroxylation on the indole ring followed by subsequent glucuronide or sulphate conjugation.
In vitro metabolism studies have shown that ondansetron is a substrate for human hepatic cytochrome P-450 enzymes, including CYP1A2, CYP2D6, and CYP3A4. In terms of overall ondansetron turnover, CYP3A4 played the predominant role. Because of the multiplicity of metabolic enzymes capable of metabolizing ondansetron, it is likely that inhibition or loss of one enzyme (e.g., CYP2D6 genetic deficiency) will be compensated by others and may result in little change in overall rates of ondansetron elimination.
Although some nonconjugated metabolites have pharmacologic activity, these are not found in plasma at concentrations likely to significantly contribute to the biological activity of ondansetron.
Specific Populations
Age:Geriatric Population: A reduction in clearance and increase in elimination half-life are seen in patients older than 75 years compared to younger subjects [see Use in Specific Populations (8.5)].
Sex: Gender differences were shown in the disposition of ondansetron given as a single dose. The extent and rate of absorption are greater in women than men. Slower clearance in women, a smaller apparent volume of distribution (adjusted for weight), and higher absolute bioavailability resulted in higher plasma ondansetron concentrations. These higher plasma concentrations may in part be explained by differences in body weight between men and women. It is not known whether these sex-related differences were clinically important. More detailed pharmacokinetic information is contained in Tables 5 and 6.
Table 5. Pharmacokinetics in Male and Female Healthy Subjects after a Single Dose of a Ondansetron 8 mg Tablet
Age- group (years)
Sex (M/F)
Mean Weight (kg)
N
Peak Plasma Concentration (ng/mL)
Time of Peak Plasma Concentration(h)
Mean Elimination Half- life
(h)
Systemic Plasma Clearance L/h/kg
Absolute Bioavailability
18-40 M
F
69.0
62.7
6
5
26.2
42.7
2.0
1.7
3.1
3.5
0.403
0.354
0.483
0.663
61-74 M
F
77.5
60.2
6
6
24.1
52.4
2.1
1.9
4.1
4.9
0.384
0.255
0.585
0.643
≥75 M
F
78.0
67.6
5
6
37.0
46.1
2.2
2.1
4.5
6.2
0.277
0.249
0.619
0.747
Table 6. Pharmacokinetics in Male and Female Healthy Subjects after a Single Dose of a Ondansetron 24 mg Tablet
Age- group (years)
Sex (M/F)
Mean Weight (kg)
N
Peak Plasma Concentration (ng/mL)
Time of Peak Plasma Concentration(h)
Mean Elimination Half- life
(h)
18-43 M
F
84.1
71.8
8
8
125.8
194.4
1.9
1.6
4.7
5.8
Renal Impairment: Renal impairment is not expected to significantly influence the total clearance of ondansetron as renal clearance represents only 5% of the overall clearance. However, the mean plasma clearance of ondansetron was reduced by about 50% in patients with severe renal impairment (creatinine clearance less than 30 mL/min). The reduction in clearance was variable and not consistent with an increase in half-life [see Use in Specific Populations (8.7)].
Hepatic Impairment: In patients with mild-to-moderate hepatic impairment, clearance is reduced 2-fold and mean half-life is increased to 11.6 hours compared with 5.7 hours in healthy subjects. In patients with severe hepatic impairment (Child-Pugh score of 10 or greater), clearance is reduced 2-fold to 3-fold and apparent volume of distribution is increased with a resultant increase in half-life to 20 hours [see Dosage and Administration (2.2),Use in Specific Populations (8.6)].
Drug Interaction Studies
CYP3A4 Inducers: Ondansetron elimination may be affected by cytochrome P-450 inducers. In a pharmacokinetic trial of 16 epileptic patients maintained chronically on CYP3A4 inducers, carbamazepine, or phenytoin, a reduction in AUC, C max, and t ½ of ondansetron was observed. This resulted in a significant increase in the clearance of ondansetron. However, this increase is not thought to be clinically relevant [see Drug Interactions (7.2)].Chemotherapeutic Agents: Carmustine, etoposide, and cisplatin do not affect the pharmacokinetics of ondansetron [see Drug Interactions (7.4)].
Antacids: Concomitant administration of antacids does not alter the absorption of ondansetron.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenic effects were not seen in 2-year studies in rats and mice with oral ondansetron doses up to 10 mg/kg per day and 30 mg/kg per day, respectively (approximately 4 and 6 times the maximum recommended human oral dose of 24 mg per day, based on body surface area).
Ondansetron was not mutagenic in standard tests for mutagenicity.
Oral administration of ondansetron up to 15 mg/kg per day (approximately 6 times the maximum recommended human oral dose of 24 mg per day, based on body surface area) did not affect fertility or general reproductive performance of male and female rats. -
14 CLINICAL STUDIES
Highly Emetogenic Chemotherapy
In two randomized, double-blind, monotherapy trials, a single 24 mg oral dose of ondansetron tablets was superior to a relevant historical placebo control in the prevention of nausea and vomiting associated with highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m 2. Steroid administration was excluded from these clinical trials. More than 90% of patients receiving a cisplatin dose ≥50 mg/m 2 in the historical placebo comparator experienced vomiting in the absence of antiemetic therapy.
The first trial compared oral doses of ondansetron 24 mg as a single dose, 8 mg every 8 hours for 2 doses, and 32 mg as a single dose in 357 adult cancer patients receiving chemotherapy regimens containing cisplatin greater than or equal to 50 mg/m 2. The first or single dose was administered 30 minutes prior to chemotherapy. A total of 66% of patients in the ondansetron 24 mg once-a-day group, 55% in the ondansetron 8 mg twice-a-day group, and 55% in the ondansetron 32 mg once-a-day group completed the 24-hour trial period with 0 emetic episodes and no rescue antiemetic medications, the primary endpoint of efficacy. Each of the 3 treatment groups was shown to be statistically significantly superior to a historical placebo control.
In the same trial, 56% of patients receiving a single 24 mg oral dose of ondansetron experienced no nausea during the 24-hour trial period, compared with 36% of patients in the oral ondansetron 8 mg twice-a-day group ( P = 0.001) and 50% in the oral ondansetron 32 mg once-a-day group. Dosage regimens of ondansetron tablets 8 mg twice daily and 32 mg once daily are not recommended for the prevention of nausea and vomiting associated with highly emetogenic chemotherapy [see Dosage and Administration (2.1)].
In a second trial, efficacy of a single 24 mg oral dose of ondansetron tablets for the prevention of nausea and vomiting associated with highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m 2, was confirmed.
Moderately Emetogenic Chemotherapy
A randomized, placebo-controlled, double-blind trial was conducted in the US in 67 patients receiving a cyclophosphamide-based chemotherapy regimen containing doxorubicin. The first 8 mg dose of ondansetron tablets was administered 30 minutes before the start of chemotherapy, with a subsequent dose 8 hours after the first dose, followed by 8 mg of ondansetron tablets twice a day for 2 days after the completion of chemotherapy.
Ondansetron tablets were significantly more effective than placebo in preventing vomiting. Treatment response was based on the total number of emetic episodes over the 3-day trial period. The results of this trial are summarized in Table 7:
Table 7. Emetic Episodes: Treatment Response in Patients Receiving Moderately Emetogenic Chemotherapy (Cyclophosphamide-based Regimen Containing Doxorubicin)a Median undefined since at least 50% of the patients were withdrawn or had more than 2 emetic episodes.
b Median undefined since at least 50% of patients did not have any emetic episodes.Ondansetron Tablets
(n = 33)
Placebo
(n = 34)
P Value
Treatment response
0 Emetic episodes
1 to 2 Emetic episodes
More than 2 emetic episodes/withdrawn
20 (61%)
6 (18%)
7 (21%)
2 (6%)
8 (24%)
24 (71%)
<0.001
<0.001
Median number of emetic episodes
0.0
Undefined a
Median time to first emetic episode (hours)
Undefined b
6.5
In a double-blind US trial in 336 patients receiving a cyclophosphamide-based chemotherapy regimen containing either methotrexate or doxorubicin, ondansetron tablets 8 mg administered twice a day was as effective as ondansetron tablets 8 mg administered 3 times a day in preventing nausea and vomiting. Ondansetron Tablets 8 mg three times daily is not a recommended regimen for the treatment of moderately emetogenic chemotherapy [see Dosage and Administration (2.1)].
Treatment response was based on the total number of emetic episodes over the 3-day trial period. See Table 8 for the details of the dosage regimens studied and results of this trial.
Table 8. Emetic Episodes: Treatment Response after Ondansetron Tablets Administered Twice a Day and Three Times a Day14.1 Prevention of Chemotherapy-Induced Nausea and Vomiting
Highly Emetogenic Chemotherapy
In two randomized, double-blind, monotherapy trials, a single 24 mg oral dose of ondansetron tablets was superior to a relevant historical placebo control in the prevention of nausea and vomiting associated with highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m 2. Steroid administration was excluded from these clinical trials. More than 90% of patients receiving a cisplatin dose ≥50 mg/m 2 in the historical placebo comparator experienced vomiting in the absence of antiemetic therapy.
The first trial compared oral doses of ondansetron 24 mg as a single dose, 8 mg every 8 hours for 2 doses, and 32 mg as a single dose in 357 adult cancer patients receiving chemotherapy regimens containing cisplatin greater than or equal to 50 mg/m 2. The first or single dose was administered 30 minutes prior to chemotherapy. A total of 66% of patients in the ondansetron 24 mg once-a-day group, 55% in the ondansetron 8 mg twice-a-day group, and 55% in the ondansetron 32 mg once-a-day group completed the 24-hour trial period with 0 emetic episodes and no rescue antiemetic medications, the primary endpoint of efficacy. Each of the 3 treatment groups was shown to be statistically significantly superior to a historical placebo control.
In the same trial, 56% of patients receiving a single 24 mg oral dose of ondansetron experienced no nausea during the 24-hour trial period, compared with 36% of patients in the oral ondansetron 8 mg twice-a-day group ( P = 0.001) and 50% in the oral ondansetron 32 mg once-a-day group. Dosage regimens of ondansetron tablets 8 mg twice daily and 32 mg once daily are not recommended for the prevention of nausea and vomiting associated with highly emetogenic chemotherapy [see Dosage and Administration (2.1)].
In a second trial, efficacy of a single 24 mg oral dose of ondansetron tablets for the prevention of nausea and vomiting associated with highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m 2, was confirmed.
Moderately Emetogenic Chemotherapy
A randomized, placebo-controlled, double-blind trial was conducted in the US in 67 patients receiving a cyclophosphamide-based chemotherapy regimen containing doxorubicin. The first 8 mg dose of ondansetron tablets was administered 30 minutes before the start of chemotherapy, with a subsequent dose 8 hours after the first dose, followed by 8 mg of ondansetron tablets twice a day for 2 days after the completion of chemotherapy.
Ondansetron tablets were significantly more effective than placebo in preventing vomiting. Treatment response was based on the total number of emetic episodes over the 3-day trial period. The results of this trial are summarized in Table 7:
Table 7. Emetic Episodes: Treatment Response in Patients Receiving Moderately Emetogenic Chemotherapy (Cyclophosphamide-based Regimen Containing Doxorubicin)a Median undefined since at least 50% of the patients were withdrawn or had more than 2 emetic episodes.
b Median undefined since at least 50% of patients did not have any emetic episodes.Ondansetron Tablets
(n = 33)Placebo
(n = 34)P Value Treatment response
0 Emetic episodes
1 to 2 Emetic episodes
More than 2 emetic episodes/withdrawn20 (61%)
6 (18%)
7 (21%)2 (6%)
8 (24%)
24 (71%)<0.001
<0.001Median number of emetic episodes 0.0 Undefined a Median time to first emetic episode (hours) Undefined b 6.5 In a double-blind US trial in 336 patients receiving a cyclophosphamide-based chemotherapy regimen containing either methotrexate or doxorubicin, ondansetron tablets 8 mg administered twice a day was as effective as ondansetron tablets 8 mg administered 3 times a day in preventing nausea and vomiting. Ondansetron Tablets 8 mg three times daily is not a recommended regimen for the treatment of moderately emetogenic chemotherapy [see Dosage and Administration (2.1)].
Treatment response was based on the total number of emetic episodes over the 3-day trial period. See Table 8 for the details of the dosage regimens studied and results of this trial.
Table 8. Emetic Episodes: Treatment Response after Ondansetron Tablets Administered Twice a Day and Three Times a Day14.2 Radiation-Induced Nausea and Vomiting
Total Body Irradiation
In a randomized, placebo-controlled, double-blind trial in 20 patients, 8 mg of ondansetron tablets administered 1.5 hours before each fraction of radiotherapy for 4 days was significantly more effective than placebo in preventing vomiting induced by total body irradiation. Total body irradiation consisted of 11 fractions (120 cGy per fraction) over 4 days for a total of 1,320 cGy. Patients received 3 fractions for 3 days, then 2 fractions on Day 4.Single High-dose Fraction Radiotherapy
In an active-controlled, double-blind trial in 105 patients receiving single high-dose radiotherapy (800 to 1,000 cGy) over an anterior or posterior field size of greater than or equal to 80 cm2 to the abdomen, ondansetron tablets was significantly more effective than metoclopramide with respect to complete control of emesis (0 emetic episodes). Patients received the first dose of ondansetron tablets (8 mg) or metoclopramide (10 mg) 1 to 2 hours before radiotherapy. If radiotherapy was given in the morning, 8 mg of ondansetron tablets or 10 mg of metoclopramide was administered in the late afternoon and repeated again before bedtime. If radiotherapy was given in the afternoon, patients took 8 mg of ondansetron tablets or 10 mg of metoclopramide only once before bedtime. Patients continued the doses of oral medication three times daily for 3 days.Daily Fractionated Radiotherapy
In an active-controlled, double-blind trial in 135 patients receiving a 1-to 4-week course of fractionated radiotherapy (180 cGy doses) over a field size of greater than or equal to 100 cm 2 to the abdomen, ondansetron tablets was significantly more effective than prochlorperazine with respect to complete control of emesis (0 emetic episodes). Patients received the first dose of ondansetron tablets (8 mg) or prochlorperazine (10 mg) 1 to 2 hours before the first daily radiotherapy fraction, with subsequent 8 mg doses approximately every 8 hours on each day of radiotherapy.14.3 Postoperative Nausea and Vomiting
In 2 placebo-controlled, double-blind trials (one conducted in the US and the other outside the US) in 865 females undergoing inpatient surgical procedures, ondansetron tablets 16 mg as a single dose or placebo was administered one hour before the induction of general balanced anesthesia (barbiturate, opioid, nitrous oxide, neuromuscular blockade, and supplemental isoflurane or enflurane), ondansetron tablets was significantly more effective than placebo in preventing postoperative nausea and vomiting.
No trials have been performed in males. -
16 HOW SUPPLIED/STORAGE AND HANDLING
Ondansetron Tablets
Ondansetron Tablets USP, 4 mg (ondansetron hydrochloride dihydrate equivalent to 4 mg of ondansetron), are white, oval, film-coated tablets engraved with “4” on one side and “NO” on another side. They are supplied as follows:
NDC: 45963-538-30 Bottles of 30
Ondansetron Tablets USP, 8 mg (ondansetron hydrochloride dihydrate equivalent to 8 mg of ondansetron), are yellow, oval, film-coated tablets engraved with “8” on one side and “NO” on the other side. They are supplied as follows:
NDC: 45963-539-30 Bottles of 30
Store at 20° to 25°C (68° to 77°F). [See USP Controlled Room Temperature]. -
17 PATIENT COUNSELING INFORMATION
QT Prolongation
Inform patients that ondansetron tablets may cause serious cardiac arrhythmias such as QT prolongation. Instruct patients to tell their healthcare provider right away if they perceive a change in their heart rate, if they feel lightheaded, or if they have a syncopal episode.Hypersensitivity Reactions
Inform patients that ondansetron tablets may cause hypersensitivity reactions, some as severe as anaphylaxis and bronchospasm. Instruct patients to immediately report any signs and symptoms of hypersensitivity reactions, including fever, chills, rash, or breathing problems to their healthcare provider.
Masking of Progressive Ileus and Gastric Distension
Inform patients following abdominal surgery or those with chemotherapy-induced nausea and vomiting that ondansetron tablets may mask signs and symptoms of bowel obstruction. Instruct patients to immediately report any signs or symptoms consistent with a potential bowel obstruction to their healthcare provider.Drug Interactions
- Instruct the patient to report the use of all medications, especially apomorphine, to their healthcare provider. Concomitant use of apomorphine and ondansetron tablets may cause a significant drop in blood pressure and loss of consciousness.
- Advise patients of the possibility of serotonin syndrome with concomitant use of ondansetron tablets and another serotonergic agent such as medications to treat depression and migraines. Advise patients to seek immediate medical attention if the following symptoms occur: changes in mental status, autonomic instability, neuromuscular symptoms with or without gastrointestinal symptoms.
Distributed by:
Actavis Pharma Inc.,
Parsippany, NJ 07054 USA
Manufactured by:
Natco Pharma Limited
Kothur- 509 228, India
- Sumansetron
-
INGREDIENTS AND APPEARANCE
SUMANSETRON
sumatriptan succinate, ondansetron kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59088-772 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59088-772-00 1 in 1 PACKAGE; Type 0: Not a Combination Product 01/01/2021 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BLISTER PACK 9 Part 2 1 BOTTLE 30 Part 1 of 2 SUMATRIPTAN SUCCINATE
sumatriptan succinate tabletProduct Information Item Code (Source) NDC: 55111-292 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SUMATRIPTAN SUCCINATE (UNII: J8BDZ68989) (SUMATRIPTAN - UNII:8R78F6L9VO) SUMATRIPTAN 50 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) Product Characteristics Color white Score no score Shape ROUND Size 10mm Flavor Imprint Code RDY;292 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55111-292-09 9 in 1 CARTON 1 9 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA076847 11/17/2009 Part 2 of 2 ONDANSETRON
ondansetron tablet, film coatedProduct Information Item Code (Source) NDC: 45963-538 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ONDANSETRON HYDROCHLORIDE (UNII: NMH84OZK2B) (ONDANSETRON - UNII:4AF302ESOS) ONDANSETRON 4 mg Inactive Ingredients Ingredient Name Strength MAGNESIUM STEARATE (UNII: 70097M6I30) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) STARCH, CORN (UNII: O8232NY3SJ) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYPROMELLOSES (UNII: 3NXW29V3WO) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) Product Characteristics Color white Score no score Shape OVAL Size 10mm Flavor Imprint Code 4;NO Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 45963-538-30 30 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077851 07/27/2007 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date unapproved drug other 01/01/2021 Labeler - PureTek Corporation (785961046) Establishment Name Address ID/FEI Business Operations NATCO PHARMA LIMITED 918588174 manufacture(45963-538)

© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.