YONSA- abiraterone acetate tablet
YONSA by
Drug Labeling and Warnings
YONSA by is a Prescription medication manufactured, distributed, or labeled by Sun Pharmaceutical Industries, Inc., Mayne Pharma Inc. , Sterling Chemical Malta Ltd . Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use YONSA safely and effectively. See full prescribing information for YONSA.
YONSA® (abiraterone acetate) tablets, for oral use
Initial U.S. Approval: 2011INDICATIONS AND USAGE
YONSA is a CYP17 inhibitor indicated in combination with methylprednisolone for the treatment of patients with metastatic castration-resistant prostate cancer (CRPC). (1) (1)
DOSAGE AND ADMINISTRATION
To avoid medication errors and overdose, be aware that YONSA tablets may have different dosing and food effects than other abiraterone acetate products. (2)
Recommended dose: YONSA 500 mg (four 125 mg tablets) administered orally once daily in combination with methylprednisolone 4 mg administered orally twice daily. (2)
Patients receiving YONSA should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy. (2.2) (2)
YONSA tablets can be taken with or without food. The tablets should be swallowed whole with water. Do not crush or chew tablets. (2.1) (2)
Dose Modification: (2)
- For patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the YONSA starting dose to 125 mg once daily. (2.2)
- For patients who develop hepatotoxicity during treatment, hold YONSA until recovery. Retreatment may be initiated at a reduced dose. YONSA should be discontinued if patients develop severe hepatotoxicity. (2.3)
DOSAGE FORMS AND STRENGTHS
- Tablets: 125 mg (3)
CONTRAINDICATIONS
- Pregnancy. (4.1, 8.1)
WARNINGS AND PRECAUTIONS
- Mineralocorticoid excess: Closely monitor patients with cardiovascular disease. Control hypertension and correct hypokalemia before treatment. Monitor blood pressure, serum potassium and symptoms of fluid retention at least monthly. (5.1)
- Adrenocortical insufficiency: Monitor for symptoms and signs of adrenocortical insufficiency. Increased dosage of corticosteroids may be indicated before, during and after stressful situations. (5.2)
- Hepatotoxicity: Can be severe and fatal. Monitor liver function and modify, interrupt, or discontinue YONSA dosing as recommended. (5.3)
ADVERSE REACTIONS
The most common adverse reactions (≥ 10%) are fatigue, joint swelling or discomfort, edema, hot flush, diarrhea, vomiting, cough, hypertension, dyspnea, urinary tract infection and contusion.
The most common laboratory abnormalities (> 20%) are anemia, elevated alkaline phosphatase, hypertriglyceridemia, lymphopenia, hypercholesterolemia, hyperglycemia, elevated AST, hypophosphatemia, elevated ALT and hypokalemia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Sun Pharmaceutical Industries, Inc. at 1-800-818-4555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- CYP3A4 Inducers: Avoid concomitant strong CYP3A4 inducers during YONSA treatment. If a strong CYP3A4 inducer must be co-administered, increase the YONSA dosing frequency (2.4, 7.1)
- CYP2D6 Substrates: Avoid co-administration of YONSA with CYP2D6 substrates that have a narrow therapeutic index. If an alternative treatment cannot be used, exercise caution and consider a dose reduction of the concomitant CYP2D6 substrate (7.2)
USE IN SPECIFIC POPULATIONS
- Females and Males of Reproductive Potential: Advise males with female partners of reproductive potential to use effective contraception. (8.3)
- Do not use YONSA in patients with baseline severe hepatic impairment (Child-Pugh Class C). (8.6)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Important Administration Instructions
2.3 Dose Modification Guidelines in Hepatic Impairment and Hepatotoxicity
2.4 Dose Modification Guidelines for Strong CYP3A4 Inducers
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypertension, Hypokalemia and Fluid Retention Due to Mineralocorticoid Excess
5.2 Adrenocortical Insufficiency
5.3 Hepatotoxicity
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Post Marketing Experience
7 DRUG INTERACTIONS
7.1 Drugs that Inhibit of Induce CYP3A4 Enzymes
7.2 Effects of Abiraterone on Drug Metabolizing Enzymes
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Hepatic Impairment
8.7 Patients with Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 QT Prolongation
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dose of YONSA is 500 mg (four 125 mg tablets) administered orally once daily in combination with methylprednisolone 4 mg administered orally twice daily.
2.2 Important Administration Instructions
To avoid medication errors and overdose, be aware that YONSA (abiraterone acetate) tablets may have different dosing and food effects than other abiraterone acetate products. Patients receiving YONSA should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy.
YONSA tablets can be taken with or without food [see Clinical Pharmacology (12.3)]. The tablets should be swallowed whole with water. Do not crush or chew tablets.
2.3 Dose Modification Guidelines in Hepatic Impairment and Hepatotoxicity
Hepatic Impairment
In patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the recommended dose of YONSA to 125 mg once daily. In patients with moderate hepatic impairment monitor ALT, AST, and bilirubin prior to the start of treatment, every week for the first month, every two weeks for the following two months of treatment and monthly thereafter. If elevations in ALT and/or AST greater than 5X upper limit of normal (ULN) or total bilirubin greater than 3X ULN occur in patients with baseline moderate hepatic impairment, discontinue YONSA and do not re-treat patients with abiraterone acetate [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Do not use YONSA in patients with baseline severe hepatic impairment (Child-Pugh Class C).
Hepatotoxicity
For patients who develop hepatotoxicity during treatment with YONSA (ALT and/or AST greater than 5X ULN or total bilirubin greater than 3X ULN), interrupt treatment with YONSA [see Warnings and Precautions (5.3)]. Treatment may be restarted at a reduced dose of 375 mg once daily following return of liver function tests to the patient’s baseline or to AST and ALT less than or equal to 2.5X ULN and total bilirubin less than or equal to 1.5X ULN. For patients who resume treatment, monitor serum transaminases and bilirubin at a minimum of every two weeks for three months and monthly thereafter.
If hepatotoxicity recurs at the dose of 375 mg once daily, re-treatment may be restarted at a reduced dose of 250 mg once daily following return of liver function tests to the patient’s baseline or to AST and ALT less than or equal to 2.5X ULN and total bilirubin less than or equal to 1.5X ULN.
If hepatotoxicity recurs at the reduced dose of 250 mg once daily, discontinue treatment with abiraterone acetate.
Permanently discontinue YONSA for patients who develop a concurrent elevation of ALT greater than 3 x ULN and total bilirubin greater than 2 x ULN in the absence of biliary obstruction or other causes responsible for the concurrent elevation [see Warnings and Precautions (5.3)].
2.4 Dose Modification Guidelines for Strong CYP3A4 Inducers
Avoid concomitant strong CYP3A4 inducers (e.g., phenytoin, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital) during YONSA treatment.
If a strong CYP3A4 inducer must be co-administered, increase the YONSA dosing frequency to twice a day only during the co-administration period (e.g., from 500 mg once daily to 500 mg twice a day). Reduce the dose back to the previous dose and frequency, if the concomitant strong CYP3A4 inducer is discontinued [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Pregnancy
YONSA can cause fetal harm and potential loss of pregnancy [see Use in Specific Populations (8.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypertension, Hypokalemia and Fluid Retention Due to Mineralocorticoid Excess
YONSA may cause hypertension, hypokalemia, and fluid retention as a consequence of increased mineralocorticoid levels resulting from CYP17 inhibition [see Clinical Pharmacology (12.1)]. Monitor patients for hypertension, hypokalemia, and fluid retention at least once a month. Control hypertension and correct hypokalemia before and during treatment with YONSA.
In the two randomized clinical trials, grade 3 to 4 hypertension occurred in 2% of patients, grade 3 to 4 hypokalemia in 4% of patients, and grade 3 to 4 edema in 1% of patients treated with abiraterone acetate [see Adverse Reactions (6)].
Co-administration of a corticosteroid suppresses adrenocorticotropic hormone (ACTH) drive, resulting in a reduction in the incidence and severity of these adverse reactions. Closely monitor patients whose underlying medical conditions might be compromised by increases in blood pressure, hypokalemia or fluid retention, such as those with heart failure, recent myocardial infarction, cardiovascular disease, or ventricular arrhythmia. The safety of YONSA in patients with left ventricular ejection fraction < 50% or New York Heart Association (NYHA) Class III or IV heart failure (in Study 1) or NYHA Class II to IV heart failure (in Study 2) was not established because these patients were excluded from these randomized clinical trials [see Clinical Studies (14)].
5.2 Adrenocortical Insufficiency
Adrenal insufficiency occurred in the two randomized clinical studies in 0.5% of patients taking abiraterone acetate and in 0.2% of patients taking placebo. Adrenocortical insufficiency was reported in patients receiving abiraterone acetate in combination with corticosteroid, following interruption of daily steroids and/or with concurrent infection or stress.
Monitor patients for symptoms and signs of adrenocortical insufficiency, particularly if patients are withdrawn from corticosteroids, have corticosteroid dose reductions, or experience unusual stress. Symptoms and signs of adrenocortical insufficiency may be masked by adverse reactions associated with mineralocorticoid excess seen in patients treated with YONSA. If clinically indicated, perform appropriate tests to confirm the diagnosis of adrenocortical insufficiency. Increased dosage of corticosteroids may be indicated before, during and after stressful situations [see Warnings and Precautions (5.1)].
5.3 Hepatotoxicity
In postmarketing experience, there have been abiraterone acetate-associated severe hepatic toxicity, including fulminant hepatitis, acute liver failure and deaths [see Adverse Reactions (6.2)].
In the two randomized clinical trials, grade 3 or 4 ALT or AST increases (at least 5X ULN) were reported in 4% of patients who received abiraterone acetate, typically during the first 3 months after starting treatment. Patients whose baseline ALT or AST were elevated were more likely to experience liver test elevation than those beginning with normal values. Treatment discontinuation due to liver enzyme increases occurred in 1% of patients taking abiraterone acetate. No deaths clearly related to abiraterone acetate were reported due to hepatotoxicity events.
Measure serum transaminases (ALT and AST) and bilirubin levels prior to starting treatment with YONSA, every two weeks for the first three months of treatment and monthly thereafter. In patients with baseline moderate hepatic impairment receiving a reduced YONSA dose of 125 mg, measure ALT, AST, and bilirubin prior to the start of treatment, every week for the first month, every two weeks for the following two months of treatment and monthly thereafter. Promptly measure serum total bilirubin, AST, and ALT if clinical symptoms or signs suggestive of hepatotoxicity develop. Elevations of AST, ALT, or bilirubin from the patient's baseline should prompt more frequent monitoring. If at any time AST or ALT rise above five times the ULN, or the bilirubin rises above three times the ULN, interrupt YONSA treatment and closely monitor liver function.
Re-treatment with YONSA at a reduced dose level may take place only after return of liver function tests to the patient’s baseline or to AST and ALT less than or equal to 2.5X ULN and total bilirubin less than or equal to 1.5X ULN [see Dosage and Administration (2.2)].
Permanently discontinue treatment with abiraterone acetate for patients who develop a concurrent elevation of ALT greater than 3 x ULN and total bilirubin greater than 2 x ULN in the absence of biliary obstruction or other causes responsible for the concurrent elevation [see Dosage and Administration (2.2)].
The safety of YONSA re-treatment of patients who develop AST or ALT greater than or equal to 20X ULN and/or bilirubin greater than or equal to 10X ULN is unknown.
-
6 ADVERSE REACTIONS
The following are discussed in more detail in other sections of the labeling:
- Hypertension, Hypokalemia, and Fluid Retention due to Mineralocorticoid Excess [see Warnings and Precautions (5.1)].
- Adrenocortical Insufficiency [see Warnings and Precautions (5.2)].
- Hepatotoxicity [see Warnings and Precautions (5.3)].
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Two randomized placebo-controlled, multicenter clinical trials enrolled patients who had metastatic castration-resistant prostate cancer who were using a gonadotropin-releasing hormone (GnRH) agonist or were previously treated with orchiectomy. In both Study 1 and Study 2 abiraterone acetate was administered at a dose equivalent to 500 mg of YONSA daily in combination with a different corticosteroid twice daily in the active treatment arms. Placebo plus corticosteroid was given to control patients.
The most common adverse drug reactions (>10%) reported in the two randomized clinical trials that occurred more commonly (>2%) in the abiraterone acetate arm were fatigue, joint swelling or discomfort, edema, hot flush, diarrhea, vomiting, cough, hypertension, dyspnea, urinary tract infection and contusion.
The most common laboratory abnormalities (>20%) reported in the two randomized clinical trials that occurred more commonly (≥2%) in the abiraterone acetate arm were anemia, elevated alkaline phosphatase, hypertriglyceridemia, lymphopenia, hypercholesterolemia, hyperglycemia, elevated AST, hypophosphatemia, elevated ALT and hypokalemia.
Study 1: Metastatic CRPC Following Chemotherapy
Study 1 enrolled 1195 patients with metastatic CRPC who had received prior docetaxel chemotherapy. Patients were not eligible if AST and/or ALT ≥ 2.5 XULN in the absence of liver metastases. Patients with liver metastases were excluded if AST and/or ALT > 5X ULN.
Table 1 shows adverse reactions on the abiraterone acetate arm in Study 1 that occurred with a ≥2% absolute increase in frequency compared to placebo or were events of special interest. The median duration of treatment with abiraterone acetate was 8 months.
Table 1 Adverse Reactions due to Abiraterone Acetate in Study 1 Abiraterone Acetate with Corticosteroid
(N=791)
Placebo with
Corticosteroid (N=394)
System Organ Class
Adverse Reaction
All Grades1
%
Grade 3-4
%
All Grades
%
Grade 3-4
%
Musculoskeletal and connective tissue disorders
Joint swelling/ discomfort2
30
4.2
23
4.1
Muscle discomfort3
26
3.0
23
2.3
General Disorders
Edema4
27
1.9
18
0.8
Vascular Disorders
Hot Flush
19
0.3
17
0.3
Hypertension
8.5
1.3
6.9
0.3
Gastrointestinal Disorders
Diarrhea
18
0.6
14
1.3
Dyspepsia
6.1
0
3.3
0
Infections and infestations
Urinary tract infection
12
2.1
7.1
0.5
Upper respiratory tract infection
5.4
0
2.5
0
Respiratory, thoracic and mediastinal disorders
Cough
11
0
7.6
0
Renal and urinary disorders
Urinary frequency
7.2
0.3
5.1
0.3
Nocturia
6.2
0
4.1
0
Injury, poisoning and procedural complications
Fractures5
5.9
1.4
2.3
0
Cardiac disorders
Arrhythmia6
7.2
1.1
4.6
1.0
Chest pain or chest discomfort7
3.8
0.5
2.8
0
Cardiac failure8
2.3
1.9
1.0
0.3
1 Adverse events graded according to CTCAE version 3.0
2 Includes terms Arthritis, Arthralgia, Joint swelling, and Joint stiffness
3 Includes terms Muscle spasms, Musculoskeletal pain, Myalgia, Musculoskeletal discomfort, and Musculoskeletal stiffness
4 Includes terms Edema, Edema peripheral, Pitting edema, and Generalized edema
5 Includes all fractures with the exception of pathological fracture
6 Includes terms Arrhythmia, Tachycardia, Atrial fibrillation, Supraventricular tachycardia, Atrial tachycardia, Ventricular tachycardia, Atrial flutter, Bradycardia, Atrioventricular block complete, Conduction disorder, and Bradyarrhythmia
7 Includes terms Angina pectoris, Chest pain, and Angina unstable. Myocardial infarction or ischemia occurred more commonly in the placebo arm than in the abiraterone acetate arm (1.3% vs. 1.1% respectively).
8 Includes terms Cardiac failure, Cardiac failure congestive, Left ventricular dysfunction, Cardiogenic shock, Cardiomegaly, Cardiomyopathy, and Ejection fraction decreased
Table 2 shows laboratory abnormalities of interest from Study 1. Grade 3-4 low serum phosphorus (7%) and low potassium (5%) occurred at a greater than or equal to 5% rate in the abiraterone acetate arm.
Table 2 Laboratory Abnormalities of Interest in Study 1 Abiraterone Acetate with Corticosteroid
(N=791)
Placebo with
Corticosteroid
(N=394)
Laboratory Abnormality
All Grades
%
Grade 3-4
%
All Grades
%
Grade 3-4
%
Hypertriglyceridemia
63
0.4
53
0
High AST
31
2.1
36
1.5
Hypokalemia
28
5.3
20
1.0
Hypophosphatemia
24
7.2
16
5.8
High ALT
11
1.4
10
0.8
High Total Bilirubin
6.6
0.1
4.6
0
Study 2: Metastatic CRPC Prior to Chemotherapy
Study 2 enrolled 1088 patients with metastatic CRPC who had not received prior cytotoxic chemotherapy. Patients were ineligible if AST and/or ALT ≥ 2.5X ULN and patients were excluded if they had liver metastases.
Table 3 shows adverse reactions on the abiraterone acetate arm in Study 2 that occurred with a ≥ 2% absolute increase in frequency compared to placebo. The median duration of treatment with abiraterone acetate was 13.8 months.
Table 3 Adverse Reactions in ≥5% of Patients on the Abiraterone Acetate Arm in Study 2 Abiraterone Acetate with Corticosteroid
(N=542)
Placebo with
Corticosteroid (N=540)
System Organ Class
Adverse Reaction
All Grades1
%
Grade 3-4
%
All Grades
%
Grade 3-4
%
General Disorders
Fatigue
39
2.2
34
1.7
Edema2
25
0.4
21
1.1
Pyrexia
8.7
0.6
5.9
0.2
Musculoskeletal and connective tissue disorders
Joint swelling/ discomfort3
30
2.0
25
2.0
Groin Pain
6.6
0.4
4.1
0.7
Gastrointestinal Disorders
Constipation
23
0.4
19
0.6
Diarrhea
22
0.9
18
0.9
Dyspepsia
11
0.0
5.0
0.2
Vascular Disorders
Hot Flush
22
0.2
18
0.0
Hypertension
22
3.9
13
3.0
Respiratory, thoracic and mediastinal disorders
Cough
17
0.0
14
0.2
Dyspnea
12
2.4
9.6
0.9
Psychiatric Disorders
Insomnia
14
0.2
11
0.0
Injury, poisoning and procedural complications
Contusion
13
0.0
9.1
0.0
Falls
5.9
0.0
3.3
0.0
Infections and infestations
Upper respiratory tract infection
13
0.0
8.0
0.0
Nasopharyngitis
11
0.0
8.1
0.0
Renal and urinary disorders
Hematuria
10
1.3
5.6
0.6
Skin and subcutaneous tissue disorders
Rash
8.1
0.0
3.7
0.0
1 Adverse events graded according to CTCAE version 3.0
2 Includes terms Edema peripheral, Pitting edema, and Generalized edema
3 Includes terms Arthritis, Arthralgia, Joint swelling, and Joint stiffness
Table 4 shows laboratory abnormalities that occurred in greater than 15% of patients, and more frequently (>5%) in the abiraterone acetate arm compared to placebo in Study 2.
Table 4 Laboratory Abnormalities in > 15% of Patients in the Abiraterone Acetate Arm of Study 2 Abiraterone Acetate with Corticosteroid
(N=542)
Placebo with
Corticosteroid
(N=540)
Laboratory Abnormality
Grade 1-4
%
Grade 3-4
%
Grade 1-4
%
Grade 3-4
%
Hematology
Lymphopenia
38
8.7
32
7.4
Chemistry
Hyperglycemia1
57
6.5
51
5.2
High ALT
42
6.1
29
0.7
High AST
37
3.1
29
1.1
Hypernatremia
33
0.4
25
0.2
Hypokalemia
17
2.8
10
1.7
1 Based on non-fasting blood draws
Cardiovascular Adverse Reactions:
In the combined data for studies 1 and 2, cardiac failure occurred more commonly in patients treated with abiraterone acetate compared to patients on the placebo arm (2.1% versus 0.7%). Grade 3-4 cardiac failure occurred in 1.6% of patients taking abiraterone acetate and led to 5 treatment discontinuations and 2 deaths. Grade 3-4 cardiac failure occurred in 0.2% of patients taking placebo. There were no treatment discontinuations and one death due to cardiac failure in the placebo group.
In Study 1 and 2, the majority of arrhythmias were grade 1 or 2. There was one death associated with arrhythmia and one patient with sudden death in the abiraterone acetate arms and no deaths in the placebo arms. There were 7 (0.5 %) deaths due to cardiorespiratory arrest in the abiraterone acetate arms and 3 (0.3 %) deaths in the placebo arms. Myocardial ischemia or myocardial infarction led to death in 3 patients in the placebo arms and 2 deaths in the abiraterone acetate arms.
6.2 Post Marketing Experience
The following additional adverse reactions have been identified during post approval use of abiraterone acetate with a different corticosteroid. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Respiratory, Thoracic and Mediastinal Disorders: non-infectious pneumonitis.
Musculoskeletal and Connective Tissue Disorders: myopathy, including rhabdomyolysis.
Hepatobiliary Disorders: fulminant hepatitis, including acute hepatic failure and death.
-
7 DRUG INTERACTIONS
7.1 Drugs that Inhibit of Induce CYP3A4 Enzymes
Based on in vitro data, YONSA is a substrate of CYP3A4.
In a dedicated drug interaction trial, co-administration of rifampin, a strong CYP3A4 inducer, decreased exposure of abiraterone by 55%. Avoid concomitant strong CYP3A4 inducers during YONSA treatment. If a strong CYP3A4 inducer must be co-administered, increase the YONSA dosing frequency [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
In a dedicated drug interaction trial, co-administration of ketoconazole, a strong inhibitor of CYP3A4, had no clinically meaningful effect on the pharmacokinetics of abiraterone [see Clinical Pharmacology (12.3)].
7.2 Effects of Abiraterone on Drug Metabolizing Enzymes
Abiraterone is an inhibitor of the hepatic drug-metabolizing enzymes CYP2D6 and CYP2C8. In a CYP2D6 drug-drug interaction trial, the Cmax and AUC of dextromethorphan (CYP2D6 substrate) were increased 2.8- and 2.9-fold, respectively, when dextromethorphan was given with an abiraterone acetate dose equivalent to YONSA 500 mg daily and a different corticosteroid twice daily. Avoid coadministration of abiraterone acetate with substrates of CYP2D6 with a narrow therapeutic index (e.g., thioridazine). If alternative treatments cannot be used, exercise caution and consider a dose reduction of the concomitant CYP2D6 substrate drug [see Clinical Pharmacology (12.3)].
In a CYP2C8 drug-drug interaction trial in healthy subjects, the AUC of pioglitazone (CYP2C8 substrate) was increased by 46% when pioglitazone was given together with an abiraterone acetate single dose equivalent to YONSA 500 mg. Therefore, patients should be monitored closely for signs of toxicity related to a CYP2C8 substrate with a narrow therapeutic index if used concomitantly with abiraterone acetate [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and the mechanism of action, YONSA is contraindicated for use in pregnant women because the drug can cause fetal harm and potential loss of pregnancy. YONSA is not indicated for use in females.
There are no human data on the use of YONSA in pregnant women. In animal reproduction studies, oral administration of abiraterone acetate in pregnant mice during organogenesis, caused adverse developmental effects at maternal exposures of approximately ≥ 0.03 times the human exposure (AUC) at the recommended dose [see Data].
Data
Animal Data
In an embryo-fetal developmental toxicity study in rats, abiraterone acetate caused developmental toxicity when administered at oral doses of 10, 30 or 100 mg/kg/day during the period of organogenesis (gestational days 6-17). Findings included embryo-fetal lethality (increased post implantation loss and resorptions and decreased number of live fetuses), fetal developmental delay (skeletal effects) and urogenital effects (bilateral ureter dilation) at doses >10 mg/kg/day, decreased fetal ano-genital distance at >30 mg/kg/day, and decreased fetal body weight at 100 mg/kg/day. Doses >10 mg/kg/day caused maternal toxicity. These doses tested in rats resulted in systemic exposures (AUC) approximately 0.03, 0.1 and 0.3 times, respectively, of the AUC achieved in patients.
8.2 Lactation
Risk Summary
YONSA is not indicated for use in women. There is no information available on the presence of YONSA in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production.
8.3 Females and Males of Reproductive Potential
Contraception
Males
Based on findings in animal reproduction studies, advise male patients with female partners of reproductive potential to use effective contraception during treatment and for at least 3 weeks after the final dose of YONSA [see Use in Specific Populations (8.1)].
Infertility
Based on animal studies, abiraterone acetate may impair reproductive function and fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of abiraterone acetate in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of patients receiving abiraterone acetate in the two randomized trials, 73% of patients were 65 years and over and 30% were 75 years and over. No overall differences in safety or effectiveness were observed between these elderly patients and younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Patients with Hepatic Impairment
The pharmacokinetics of abiraterone were examined in subjects with baseline mild (N=8) or moderate (N=8) hepatic impairment (Child-Pugh Class A and B, respectively) and in 8 healthy control subjects with normal hepatic function. The systemic exposure (AUC) of abiraterone after a single oral dose equivalent to 500 mg of YONSA increased by approximately 1.1-fold and 3.6-fold in subjects with mild and moderate baseline hepatic impairment, respectively compared to subjects with normal hepatic function.
In another trial, the pharmacokinetics of abiraterone were examined in subjects with baseline severe (N=8) hepatic impairment (Child-Pugh Class C) and in 8 healthy control subjects with normal hepatic function. The systemic exposure (AUC) of abiraterone increased by approximately 7-fold and the fraction of free drug increased 2-fold in subjects with severe baseline hepatic impairment compared to subjects with normal hepatic function.
No dosage adjustment is necessary for patients with baseline mild hepatic impairment. In patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the recommended dose of YONSA to 125 mg once daily. Do not use YONSA in patients with baseline severe hepatic impairment (Child-Pugh Class C). If elevations in ALT or AST >5X ULN or total bilirubin >3X ULN occur in patients with baseline moderate hepatic impairment, discontinue abiraterone acetate treatment [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
For patients who develop hepatotoxicity during treatment, interruption of treatment and dosage adjustment may be required [see Dosage and Administration (2.3), Warnings and Precautions (5.3), and Clinical Pharmacology (12.3)].
8.7 Patients with Renal Impairment
No dosage adjustment is necessary for patients with renal impairment [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
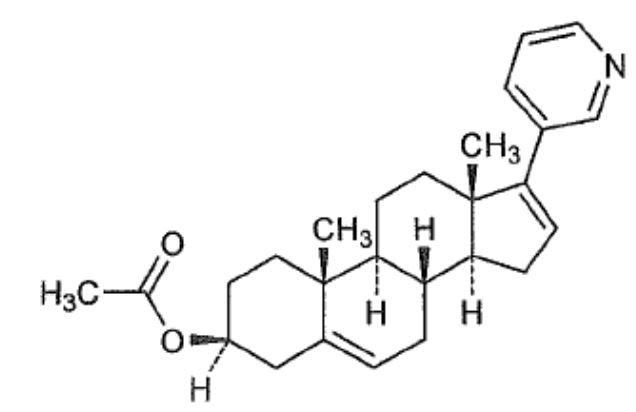
Abiraterone acetate, the active ingredient of YONSA tablet is the acetyl ester of abiraterone. Abiraterone is an inhibitor of CYP17 (17α-hydroxylase/C17,20-lyase). Each YONSA Tablet contains 125 mg of abiraterone acetate. Abiraterone acetate is designated chemically as (3β)17-(3-pyridinyl) androsta-5,16-dien-3-yl acetate and its structure is:
Abiraterone acetate is micronized (smaller particle size) white to off-white, non-hygroscopic, crystalline powder. Its molecular formula is C26H33NO2 and it has a molecular weight of 391.55. Abiraterone acetate is a lipophilic compound with an octanol-water partition coefficient of 5.12 (Log P) and is practically insoluble in water. The pKa of the aromatic nitrogen is 5.19.
Inactive ingredients in the tablets are lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, sodium lauryl sulfate, sodium stearyl fumarate, butylated hydroxyanisole, butylated hydroxytoluene.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Abiraterone acetate (YONSA) is converted in vivo to abiraterone, an androgen biosynthesis inhibitor, that inhibits 17 α-hydroxylase/C17, 20-lyase (CYP17). This enzyme is expressed in testicular, adrenal, and prostatic tumor tissues and is required for androgen biosynthesis.
CYP17 catalyzes two sequential reactions: 1) the conversion of pregnenolone and progesterone to their 17α-hydroxy derivatives by 17α-hydroxylase activity and 2) the subsequent formation of dehydroepiandrosterone (DHEA) and androstenedione, respectively, by C17, 20 lyase activity. DHEA and androstenedione are androgens and are precursors of testosterone. Inhibition of CYP17 by abiraterone can also result in increased mineralocorticoid production by the adrenals [see Warnings and Precautions (5.1)].
Androgen sensitive prostatic carcinoma responds to treatment that decreases androgen levels. Androgen deprivation therapies, such as treatment with GnRH agonists or orchiectomy, decrease androgen production in the testes but do not affect androgen production by the adrenals or in the tumor.
Abiraterone acetate decreased serum testosterone and other androgens in patients in the placebo-controlled phase 3 clinical trial. It is not necessary to monitor the effect of YONSA on serum testosterone levels.
Changes in serum prostate specific antigen (PSA) levels may be observed but have not been shown to correlate with clinical benefit in individual patients.
12.2 Pharmacodynamics
In a clinical study in patients with metastatic CRPC who were treated with YONSA 500 mg once daily and methylprednisolone 4 mg twice daily for 84 days, the average serum testosterone level ± standard deviation (SD) on days 9 and 10 of treatment was 0.33 ± 0.09 ng/dL.
12.3 Pharmacokinetics
Following administration of abiraterone acetate, the pharmacokinetics of abiraterone and abiraterone acetate have been studied in healthy volunteers and in patients with metastatic CRPC. In vivo, abiraterone acetate is converted to abiraterone. In clinical studies of other abiraterone acetate formulations, abiraterone acetate plasma concentrations were below detectable levels (< 0.2 ng/mL) in > 99% of the analyzed samples.
Geometric mean ±SD abiraterone Cmax was 73 ± 44 ng/mL and AUCINF was 373 ± 249 ng·hr/mL following a single dose of YONSA 500 mg in overnight fasted healthy volunteers. Dose proportionality was observed in single doses of YONSA in a range of 125 mg to 625 mg.
Absorption
Following oral administration of YONSA to healthy volunteers and patients with metastatic CRPC, the mean time to reach maximum plasma abiraterone concentrations is approximately 2 hours.
Effect of Food
Abiraterone Cmax was approximately 6.5-fold higher and AUCINF was 4.4-fold higher when a single dose of YONSA 500 mg was administered with a high-fat meal (56-60% fat, 900-1,000 calories) compared to overnight fasting in healthy volunteers.
Other formulations of abiraterone acetate may differ in their food effects and dose. This may impact the ability to take other abiraterone formulations with food. YONSA can be taken with or without food [see Dosage and Administration (2.2)].
Distribution and Protein Binding
Abiraterone is highly bound (>99%) to the human plasma proteins, albumin and alpha-1 acid glycoprotein. The apparent steady-state volume of distribution (mean ± SD) is 19,669 ± 13,358 L. In vitro studies show that at clinically relevant concentrations, abiraterone acetate and abiraterone are not substrates of P-glycoprotein (P-gp) and that abiraterone acetate is an inhibitor of P-gp.
Metabolism
Following oral administration of 14C-abiraterone acetate as capsules, abiraterone acetate is hydrolyzed to abiraterone (active metabolite). The conversion is likely through esterase activity (the esterases have not been identified) and is not CYP mediated. The two main circulating metabolites of abiraterone in human plasma are abiraterone sulphate (inactive) and N-oxide abiraterone sulphate (inactive), which account for about 43% of exposure each. CYP3A4 and SULT2A1 are the enzymes involved in the formation of N-oxide abiraterone sulphate and SULT2A1 is involved in the formation of abiraterone sulphate.
Excretion
In patients with metastatic CRPC, the mean terminal half-life of abiraterone in plasma (mean ± SD) is 12 ± 5 hours. Following oral administration of 14C-abiraterone acetate, approximately 88% of the radioactive dose is recovered in feces and approximately 5% in urine. The major compounds present in feces are unchanged abiraterone acetate and abiraterone (approximately 55% and 22% of the administered dose, respectively).
Patients with Hepatic Impairment
The pharmacokinetics of abiraterone was examined in subjects with baseline mild (N=8) or moderate (N=8) hepatic impairment (Child-Pugh Class A and B, respectively) and in 8 healthy control subjects with normal hepatic function. Systemic exposure to abiraterone after a single oral 1,000 mg dose of another abiraterone acetate product given under fasting conditions increased approximately 1.1-fold and 3.6-fold in subjects with mild and moderate baseline hepatic impairment, respectively. The mean half-life of abiraterone is prolonged to approximately 18 hours in subjects with mild hepatic impairment and to approximately 19 hours in subjects with moderate hepatic impairment.
In another trial, the pharmacokinetics of abiraterone were examined in subjects with baseline severe (N=8) hepatic impairment (Child-Pugh Class C) and in 8 healthy control subjects with normal hepatic function. The systemic exposure (AUC) of abiraterone increased by approximately 7-fold in subjects with severe baseline hepatic impairment compared to subjects with normal hepatic function. In addition, the mean protein binding was found to be lower in the severe hepatic impairment group compared to the normal hepatic function group, which resulted in a two-fold increase in the fraction of free drug in patients with severe hepatic impairment [see Dosage and Administration (2.3) and Use in Specific Populations (8.6)].
Patients with Renal Impairment
The pharmacokinetics of abiraterone were examined in patients with end-stage renal disease (ESRD) on a stable hemodialysis schedule (N=8) and in matched control subjects with normal renal function (N=8). In the ESRD cohort of the trial, a single 1,000 mg dose of another abiraterone acetate product was given under fasting conditions 1 hour after dialysis, and samples for pharmacokinetic analysis were collected up to 96 hours post dose. Systemic exposure to abiraterone after a single oral 1,000 mg dose of another abiraterone acetate product did not increase in subjects with end-stage renal disease on dialysis, compared to subjects with normal renal function [see Use in Specific Populations (8.7)].
Drug Interactions
In vitro studies with human hepatic microsomes showed that abiraterone has the potential to inhibit CYP1A2, CYP2D6 and CYP2C8 and to a lesser extent CYP2C9, CYP2C19 and CYP3A4/5.
In an in vivo drug-drug interaction trial, the Cmax and AUC of dextromethorphan (CYP2D6 substrate) were increased 2.8- and 2.9-fold, respectively when dextromethorphan 30 mg was given with 1,000 mg daily of another abiraterone acetate product (plus a different corticosteroid twice daily). The AUC for dextrorphan, the active metabolite of dextromethorphan, increased approximately 1.3 fold [see Drug Interactions (7.2)].
In a clinical study to determine the effects of 1,000 mg daily of another abiraterone acetate product (plus a different corticosteroid twice daily) on a single 100 mg dose of the CYP1A2 substrate theophylline, no increase in systemic exposure of theophylline was observed.
Abiraterone is a substrate of CYP3A4, in vitro. In a clinical pharmacokinetic interaction study of healthy subjects, pretreated with a strong CYP3A4 inducer (rifampin, 600 mg daily for 6 days) followed by a single dose of 1,000 mg of another abiraterone acetate product, the mean plasma AUCINF of abiraterone was decreased by 55% [see Drug Interactions (7.1)].
In a separate clinical pharmacokinetic interaction study of healthy subjects, co-administration of ketoconazole, a strong inhibitor of CYP3A4, had no clinically meaningful effect on the pharmacokinetics of abiraterone [see Drug Interactions (7.1)].
In a CYP2C8 drug-drug interaction trial in healthy subjects, the AUC of pioglitazone was increased by 46% when pioglitazone was given together with a single dose of 1,000 mg of another abiraterone acetate product [see Drug Interactions (7.2)].
In vitro, abiraterone and its major metabolites were shown to inhibit the hepatic uptake transporter OATP1B1. There are no clinical data available to confirm transporter based interaction.
12.6 QT Prolongation
In a multi-center, open-label, single-arm trial, 33 patients with metastatic CRPC received a dose of 1,000 mg once daily orally of another abiraterone acetate product at least 1 hour before or 2 hours after a meal in combination with a different corticosteroid orally twice daily. Assessments up to Cycle 2 Day 2 showed no large changes in the QTc interval (i.e., >20 ms) from baseline. However, small increases in the QTc interval (i.e., <10 ms) due to abiraterone acetate cannot be excluded due to study design limitations.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A two-year carcinogenicity study was conducted in rats at abiraterone acetate oral doses of 5, 15, and 50 mg/kg/day for males and 15, 50, and 150 mg/kg/day for females. Abiraterone acetate increased the combined incidence of interstitial cell adenomas and carcinomas in the testes at all dose levels tested. This finding is considered to be related to the pharmacological activity of abiraterone. Rats are regarded as more sensitive than humans to developing interstitial cell tumors in the testes. Abiraterone acetate was not carcinogenic in female rats at exposure levels up to 0.8 times the human clinical exposure based on AUC. Abiraterone acetate was not carcinogenic in a 6-month study in the transgenic (Tg.rasH2) mouse.
Abiraterone acetate and abiraterone was not mutagenic in the microbial mutagenesis (Ames) assay or clastogenic in an in vitro cytogenetic assay using primary human lymphocytes or an in vivo rat micronucleus assay.
In repeat-dose toxicity studies in male rats (13- and 26-weeks) and monkeys (39-weeks), atrophy, aspermia/hypospermia, and hyperplasia in the reproductive system were observed at ≥ 50 mg/kg/day in rats and ≥ 250 mg/kg/day in monkeys and were consistent with the antiandrogenic pharmacological activity of abiraterone [see Nonclinical Toxicology (13.2)]. These effects were observed in rats at systemic exposures similar to humans and in monkeys at exposures approximately 0.6 times the AUC in humans.
In fertility studies in male rats, reduced organ weights of the reproductive system, sperm counts, sperm motility, altered sperm morphology and decreased fertility were observed in males dosed for 4 weeks at ≥ 30 mg/kg/day. Mating of untreated females with males that received 30 mg/kg/day abiraterone acetate resulted in a reduced number of corpora lutea, implantations and live embryos and an increased incidence of pre-implantation loss. Effects on male rats were reversible after 16 weeks from the last abiraterone acetate administration.
In a fertility study in female rats, animals dosed orally for 2 weeks until day 7 of pregnancy at > 30 mg/kg/day had an increased incidence of irregular or extended estrous cycles and pre-implantation loss (300 mg/kg/day). There were no differences in mating, fertility, and litter parameters in female rats that received abiraterone acetate. Effects on female rats were reversible after 4 weeks from the last abiraterone acetate administration.
The dose of 30 mg/kg/day in rats is approximately 0.6 times the recommended dose of 500 mg of YONSA/day based on body surface area.
In 13- and 26-week studies in rats and 13- and 39-week studies in monkeys, a reduction in circulating testosterone levels occurred with abiraterone acetate at approximately one half the human clinical exposure based on AUC. As a result, decreases in organ weights and toxicities were observed in the male and female reproductive system, adrenal glands, liver, pituitary (rats only), and male mammary glands. The changes in the reproductive organs are consistent with the antiandrogenic pharmacological activity of abiraterone acetate.
13.2 Animal Toxicology and/or Pharmacology
A dose-dependent increase in cataracts was observed in rats at 26 weeks starting at > 50 mg/kg/day (similar to the human clinical exposure based on AUC). In the 39-week monkey study, no cataracts were observed at higher doses (2 times greater than the clinical exposure based on AUC).
-
14 CLINICAL STUDIES
The efficacy and safety of abiraterone acetate in patients with metastatic castration-resistant prostate cancer (CRPC) that has progressed on androgen deprivation therapy was demonstrated in two randomized, placebo-controlled, multicenter phase 3 clinical trials. Patients with prior ketoconazole treatment for prostate cancer and a history of adrenal gland or pituitary disorders were excluded from these trials. Concurrent use of spironolactone was not allowed during the study period.
Study 1
Patients with metastatic CRPC who had received prior docetaxel chemotherapy:
A total of 1195 patients were randomized 2:1 to receive either abiraterone acetate orally at a dose equivalent to 500 mg of YONSA once daily in combination with a different corticosteroid orally twice daily (N=797) or placebo once daily plus a different corticosteroid orally twice daily (N=398). Patients randomized to either arm were to continue treatment until disease progression (defined as a 25% increase in PSA over the patient’s baseline/nadir together with protocol-defined radiographic progression and symptomatic or clinical progression), initiation of new treatment, unacceptable toxicity or withdrawal.
The following patient demographics and baseline disease characteristics were balanced between the treatment arms. The median age was 69 years (range 39-95) and the racial distribution was 93.3% Caucasian, 3.6% Black, 1.7% Asian, and 1.6% Other. Eighty-nine percent of patients enrolled had an ECOG performance status score of 0-1 and 45% had a Brief Pain Inventory-Short Form score of ≥ 4 (patient’s reported worst pain over the previous 24 hours). Ninety percent of patients had metastases in bone and 30% had visceral involvement. Seventy percent of patients had radiographic evidence of disease progression and 30% had PSA-only progression. Seventy percent of patients had previously received one cytotoxic chemotherapy regimen and 30% received two regimens.
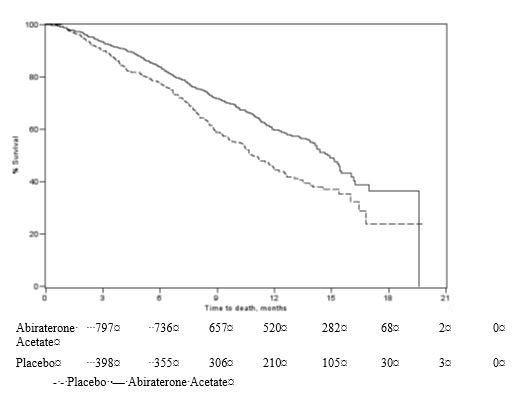
The protocol pre-specified interim analysis was conducted after 552 deaths and showed a statistically significant improvement in overall survival in patients treated with abiraterone acetate compared to patients in the placebo arm (Table 5 and Figure 1). An updated survival analysis was conducted when 775 deaths (97% of the planned number of deaths for final analysis) were observed. Results from this analysis were consistent with those from the interim analysis (Table 5).
Table 5 Overall Survival of Patients Treated with Either Abiraterone Acetate or Placebo in Combination with Corticosteroid in Study 1 (Intent-to-Treat Analysis) Abiraterone Acetate with Corticosteroid
(N=797)
Placebo with
Corticosteroid
(N=398)
Primary Survival Analysis
Deaths (%)
333 (42%)
219 (55%)
Median survival (months)
14.8 (14.1, 15.4)
10.9 (10.2, 12.0)
(95% CI)
p-value1
<0.0001
Hazard ratio (95% CI)2
0.646 (0.543, 0.768)
Updated Survival Analysis
Deaths (%)
501 (63%)
274 (69%)
Median survival (months)
15.8 (14.8, 17.0)
11.2 (10.4, 13.1)
(95% CI)
Hazard ratio (95% CI)2
0.740 (0.638, 0.859)
1 p-value is derived from a log-rank test stratified by ECOG performance status score (0-1 vs. 2), pain score (absent vs. present), number of prior chemotherapy regimens (1 vs. 2), and type of disease progression (PSA only vs. radiographic)
2 Hazard Ratio is derived from a stratified proportional hazards model. Hazard ratio <1 favors abiraterone acetate
Figure 1 Kaplan-Meier Overall Survival Curves in Study 1 (Intent-to-Treat Analysis)
Study 2
Patients with metastatic CRPC who had not received prior cytotoxic chemotherapy
In Study 2, 1088 patients were randomized 1:1 to receive either abiraterone acetate at a dose equivalent to 500 mg of YONSA once daily (N=546) or placebo once daily (N=542). Both arms were also given a different corticosteroid twice daily. Patients continued treatment until radiographic or clinical (cytotoxic chemotherapy, radiation or surgical treatment for cancer, pain requiring chronic opioids, or ECOG performance status decline to 3 or more) disease progression, unacceptable toxicity or withdrawal. Patients with moderate or severe pain, opiate use for cancer pain, or visceral organ metastases were excluded.
Patient demographics were balanced between the treatment arms. The median age was 70 years. The racial distribution of patients treated with abiraterone acetate was 95% Caucasian, 2.8% Black, 0.7% Asian and 1.1% Other. The ECOG performance status was 0 for 76% of patients, and 1 for 24% of patients. Co-primary efficacy endpoints were overall survival and radiographic progression-free survival (rPFS). Baseline pain assessment was 0-1 (asymptomatic) in 66% of patients and 2-3 (mildly symptomatic) in 26% of patients as defined by the Brief Pain Inventory-Short Form (worst pain over the last 24 hours).
Radiographic progression-free survival was assessed with the use of sequential imaging studies and was defined by bone scan identification of 2 or more new bone lesions with confirmation (Prostate Cancer Working Group 2 criteria) and/or modified Response Evaluation Criteria In Solid Tumors (RECIST) criteria for progression of soft tissue lesions. Analysis of rPFS utilized centrally-reviewed radiographic assessment of progression.
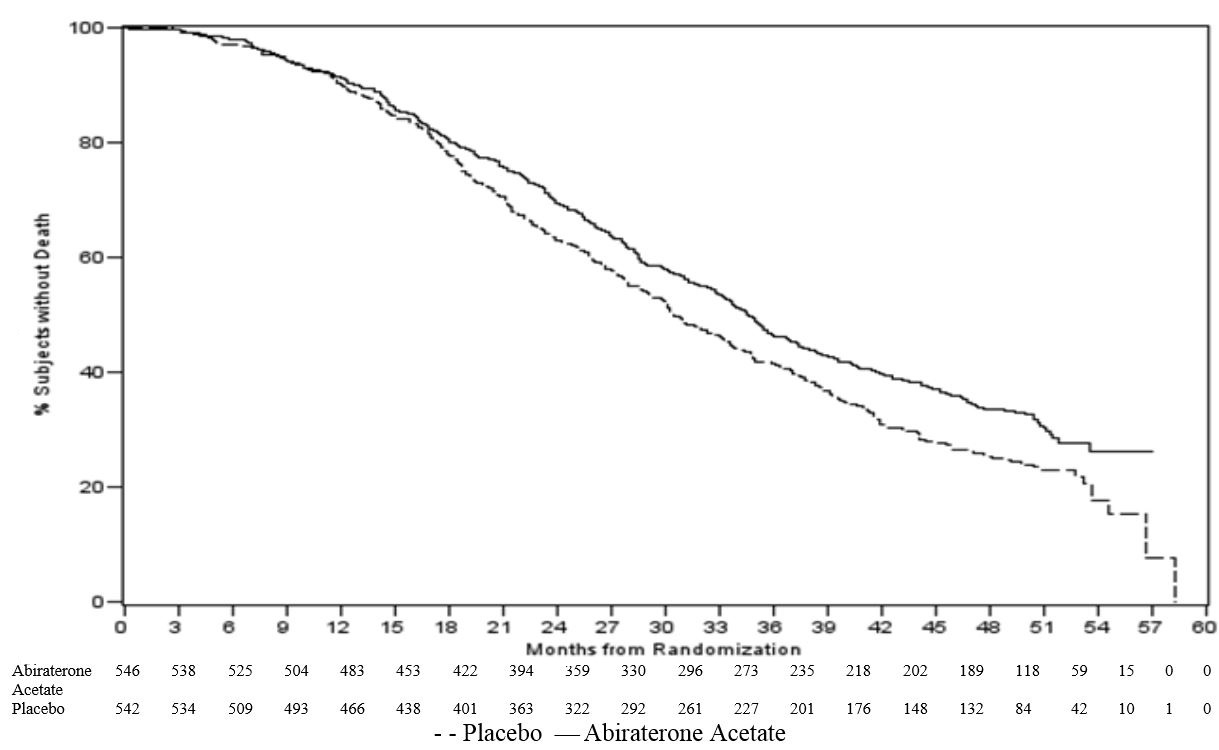
The planned final analysis for OS, conducted after 741 deaths (median follow up of 49 months) demonstrated a statistically significant OS improvement in patients treated with abiraterone acetate compared to those treated with placebo (Table 6 and Figure 2). Sixty-five percent of patients on the abiraterone acetate arm and 78% of patients on the placebo arm used subsequent therapies that may prolong OS in metastatic CRPC. Abiraterone acetate was used as a subsequent therapy in 13% of patients on the abiraterone acetate arm and 44% of patients on the placebo arm.
Table 6: Overall Survival of Patients Treated with Either Abiraterone Acetate or Placebo in Combination with Corticosteroid in Study 2 (Intent-to-Treat Analysis)
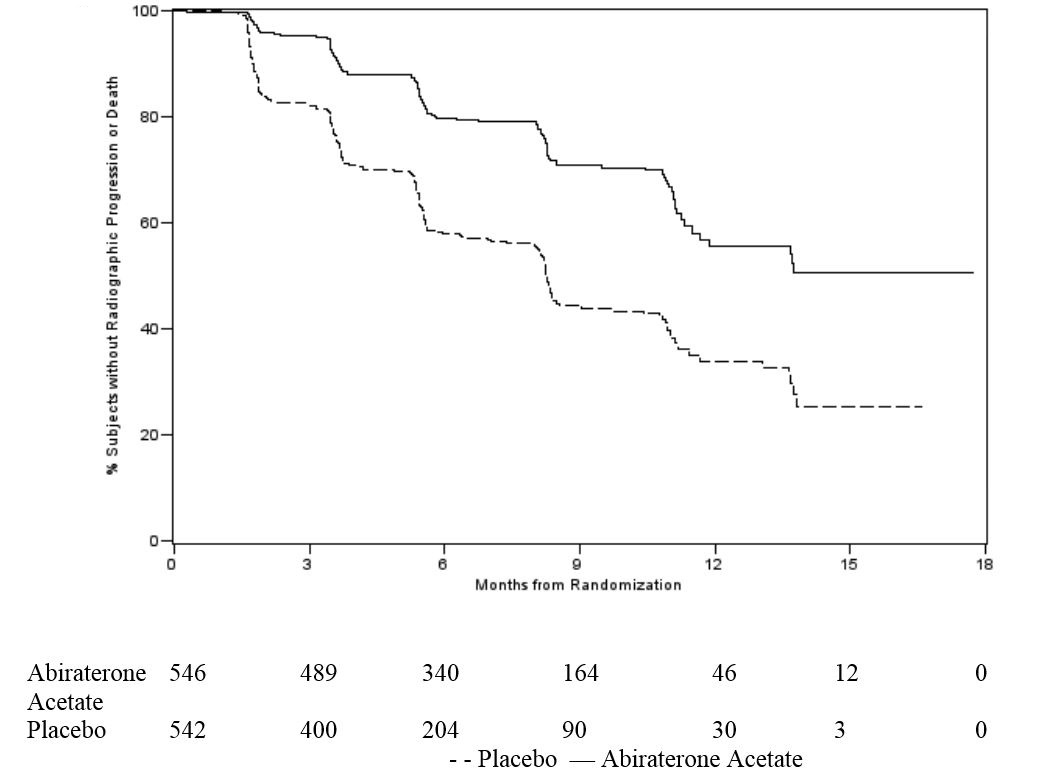
At the pre-specified rPFS analysis, 150 (28%) patients treated with abiraterone acetate and 251 (46%) patients treated with placebo had radiographic progression. A significant difference in rPFS between treatment groups was observed (Table 7 and Figure 3).
Table 7: Radiographic Progression-free Survival of Patients Treated with Either Abiraterone Acetate or Placebo in Combination with Corticosteroid in Study 2 (Intent-to-Treat Analysis)
Radiographic Progression-free Survival
Abiraterone Acetate with Corticosteroid
(N=546)
Placebo with Corticosteroid
(N=542)
Progression or death
150 (28%)
251 (46%)
Median rPFS (months)
(95% CI)
NR
(11.66, NR)
8.28
(8.12, 8.54)
p-value1
<0.0001
Hazard ratio2 (95% CI)
0.425 (0.347, 0.522)
NR= Not Reached
1 p-value is derived from a log-rank test stratified by ECOG performance status score (0 vs. 1).
2 Hazard Ratio is derived from a stratified proportional hazards model. Hazard ratio <1 favors abiraterone acetate
Figure 3 Kaplan Meier Curves of Radiographic Progression-free Survival in Study 2 (Intent-to-Treat Analysis)
The primary efficacy analyses are supported by the following prospectively defined endpoints. The median time to initiation of cytotoxic chemotherapy was 25.2 months for patients in the abiraterone acetate arm and 16.8 months for patients in the placebo arm (HR=0.580; 95% CI: [0.487, 0.691], p<0.0001).
The median time to opiate use for prostate cancer pain was not reached for patients receiving abiraterone acetate and was 23.7 months for patients receiving placebo (HR=0.686; 95% CI: [0.566, 0.833], p=0.0001). The time to opiate use result was supported by a delay in patient reported pain progression favoring the abiraterone acetate arm.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
YONSA (abiraterone acetate) tablets, 125 mg
White to off-white, oval-shaped tablets debossed with “125 FP” on one side
120 tablets available in high-density polyethylene bottles with child resistant closure
NDC Number 47335-401-81
Storage and Handling
Store at 20oC to 25oC (68oF to 77oF); excursions permitted in the range from 15oC to 30oC (59oF to 86°F) [see USP controlled room temperature].
Women who are pregnant or women who may be pregnant should not handle YONSA without protection, e.g., gloves [see Use in Specific Populations (8.1)].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information)
Hypertension, Hypokalemia, and Fluid Retention
- Inform patients that YONSA is associated with hypertension, hypokalemia, and peripheral edema. Advise patients to report symptoms of hypertension, hypokalemia, or edema to their healthcare provider [see Warnings and Precautions (5.1)].
Adrenocortical Insufficiency
- Inform patients that YONSA with methylprednisolone is associated with adrenal insufficiency. Advise patients to report symptoms of adrenocortical insufficiency to their healthcare provider [see Warnings and Precautions (5.2)].
Hepatotoxicity
- Inform patients that YONSA is associated with severe hepatotoxicity. Inform patients that their liver function will be monitored using blood tests. Advise patients to immediately report symptoms of hepatotoxicity to their healthcare provider [see Warnings and Precautions (5.3)].
Dosing and Administration
- Inform patients that YONSA tablets may not be interchangeable with other abiraterone acetate products due to different dosing and food effects [see Dosage and Administration (2.2)].
- Inform patients that YONSA is taken once daily with methylprednisolone twice daily and to not interrupt or stop either of these medications without consulting their healthcare provider [see Dosage and Administration (2.2)].
- Inform patients receiving GnRH therapy that they need to maintain this treatment during the course of treatment with YONSA [see Dosage and Administration (2.2)].
- Instruct patients that YONSA can be taken with or without food. Instruct patients to swallow tablets whole with water and not to crush or chew the tablets [see Dosage and Administration (2.2)].
- Inform patients that if they miss a dose of YONSA or methylprednisolone, they should take their normal dose the following day. If more than one daily dose is skipped, inform patients to contact their healthcare provider [see Dosage and Administration (2.2)].
Fetal Toxicity
- Inform patients that YONSA may harm a developing fetus. Advise males with female partners of reproductive potential to use effective contraception during treatment and for 3 weeks after the final dose of YONSA [see Use in Specific Populations (8.1)].
- Women who are pregnant or women who may be pregnant should not handle YONSA tablets without protection, e.g., gloves [see Use in Specific Populations (8.1) and How Supplied/Storage and Handling (16)].
Infertility
- Advise male patients that YONSA may impair fertility [see Use in Specific Populations (8.3)].
Manufactured for:
Sun Pharma Global FZE
Distributed by:
Sun Pharmaceutical Industries, Inc.
Cranbury, NJ 08512
YONSA is a registered trademark of Sun Pharma Global FZE
Copyright © 2018 Sun Pharma Global, FZE
All rights reserved.
Revised: May 2018
uspi-YONSA-tab-00001
-
Patient Medication Information
PATIENT INFORMATION
YONSA® (Yon-suh)
(abiraterone acetate)
tablets
What is YONSA?
YONSA is a prescription medicine used with methylprednisolone to treat prostate cancer that has spread to other parts of the body and no longer responds to medical or surgical treatment that lowers testosterone.
It is not known if YONSA is safe and effective in children.
Do not take YONSA if you:
- are pregnant or may become pregnant. YONSA may harm your unborn baby.
- are female.
YONSA is not for use in women.
Women who are pregnant or who may become pregnant should not touch YONSA tablets if broken, crushed, or damaged without protection, such as gloves.
Before you take YONSA, tell your healthcare provider about all of your medical conditions, including if you:
- have heart problems
- have liver problems
- have a history of adrenal problems
- have a history of pituitary problems
- have a partner who is pregnant or may become pregnant.
- Men who are sexually active with a woman who may become pregnant must use effective birth control during treatment and for at least 3 weeks after the final dose of YONSA.
- Men who are sexually active with a woman who is pregnant must use a condom.
- Talk with your healthcare provider if you have questions about birth control.
Tell your healthcare provider about all the medicines you take, including prescription and over-the counter medicines, vitamins, and herbal supplements. YONSA can interact with many other medicines.
You should not start or stop any medicine before you talk with the healthcare provider that prescribed YONSA.
Know the medicines you take. Keep a list of them with you to show to your healthcare provider and pharmacist when you get a new medicine.
How should I take YONSA?
- Take YONSA and methylprednisolone exactly as your healthcare provider tells you.
- Take your prescribed dose of YONSA 1 time a day. Take your prescribed dose of methylprednisolone 2 times a day.
- YONSA contains abiraterone acetate. YONSA and other medicines that contain abiraterone acetate may not be the same.
- Do not switch between YONSA and other medicines that contain abiraterone acetate unless your healthcare provider tells you to.
- Follow your healthcare provider’s instructions carefully if you are switching between YONSA and another medicine that contains abiraterone acetate.
- Do not take YONSA and other medicines that contain abiraterone acetate on the same day.
- If you do not have enough YONSA to take your full dose or if you have any other questions about YONSA, talk with your healthcare provider or pharmacist.
- Your healthcare provider may change your dose of YONSA if needed.
- Do not stop taking your prescribed dose of YONSA or methylprednisolone without talking with your healthcare provider first.
- Take YONSA with or without food.
- Swallow YONSA tablets whole with water. Do not crush or chew tablets.
- If you miss a dose of YONSA or methylprednisolone, take your prescribed dose the following day. If you miss more than 1 dose, tell your healthcare provider right away.
- If you are receiving gonadotropin-releasing hormone (GnRH) therapy, you should continue your therapy during treatment with YONSA and methylprednisolone.
- If you take too much YONSA, call your healthcare provider or go to the nearest hospital emergency room right away.
Your healthcare provider will do blood tests to check for side effects.
What are the possible side effects of YONSA?
YONSA may cause serious side effects, including:
- High blood pressure (hypertension), low blood potassium levels (hypokalemia) and fluid retention
- (edema). Tell your healthcare provider if you get any of the following symptoms:
- dizziness
- confusion
- fast heartbeats
- muscle weakness
- feel faint or lightheaded
- pain in your legs
- headache
- swelling in your legs or feet
- Adrenal problems may happen if you stop taking methylprednisolone, get an infection, or are under stress.
- Liver problems. You may develop changes in liver function blood tests. Your healthcare provider will do blood tests to check your liver before treatment with YONSA and during treatment with YONSA. Liver failure may occur, which can lead to death. Tell your healthcare provider if you notice any of the following changes:
- yellowing of the skin or eyes
- darkening of the urine
- severe nausea or vomiting
The most common side effects of YONSA include:
- weakness
- joint swelling or pain
- muscle pain
- swelling in your legs or feet
- hot flushes
- diarrhea
- vomiting
- cough
- high blood pressure
- shortness of breath
- trouble sleeping
- urinary tract infection
- bruising
- indigestion
- blood in the urine
- constipation
- upper respiratory tract infection
- low red blood cells (anemia)
- low blood potassium levels
- high blood sugars
- high blood cholesterol and triglycerides
- changes in liver function blood tests
- certain other abnormal blood tests
YONSA may affect fertility in males and may affect your ability to father a child. Talk to your healthcare provider if this is a concern for you.
These are not all the possible side effects of YONSA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store YONSA?
- Store YONSA at room temperature between 68°F to 77°F (20°C to 25°C).
- Women who are pregnant or who may become pregnant should not touch YONSA without protection, such as gloves.
Keep YONSA and all medicines out of the reach of children.
General information about the safe and effective use of YONSA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use YONSA for a condition for which it was not prescribed. Do not give YONSA to other people, even if they have the same symptoms that you have. It may harm them. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about YONSA that is written for health professionals.
What are the ingredients in YONSA?
Active ingredient: abiraterone acetate.
Inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, sodium lauryl sulfate, sodium stearyl fumarate, butylated hydroxyanisole, butylated hydroxytoluene.
Manufactured for Sun Pharma Global FZE
Distributed by Sun Pharmaceutical Industries, Inc.
Cranbury, NJ 08512
YONSA is a registered trademark of Sun Pharma Global FZE
Copyright © 2018 Sun Pharma Global, FZE
All rights reserved.
For more information, call Sun Pharmaceuticals Industries Inc at 1-800-818-4555 or go to www.YONSA.net
- Package/Label Display Panel
-
INGREDIENTS AND APPEARANCE
YONSA
abiraterone acetate tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 47335-401 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ABIRATERONE ACETATE (UNII: EM5OCB9YJ6) (ABIRATERONE - UNII:G819A456D0) ABIRATERONE ACETATE 125 mg Product Characteristics Color WHITE (to off-white) Score no score Shape OVAL Size 12mm Flavor Imprint Code 125;FP Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 47335-401-81 120 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 05/22/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210308 05/22/2018 Labeler - Sun Pharmaceutical Industries, Inc. (146974886) Registrant - Sun Pharmaceutical Industries, Inc. (146974886) Establishment Name Address ID/FEI Business Operations Sun Pharmaceutical Industries, Inc. 146974886 MANUFACTURE(47335-401)

Trademark Results [YONSA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 YONSA 86757216 5317612 Live/Registered |
SUN PHARMA GLOBAL FZE 2015-09-15 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.