These highlights do not include all the information needed to use PURINETHOL safely and effectively. See full prescribing information for PURINETHOL. PURINETHOL ® (mercaptopurine) tablets, for oral use Initial U.S. Approval: 1953
PURINETHOL by
Drug Labeling and Warnings
PURINETHOL by is a Prescription medication manufactured, distributed, or labeled by Stason Pharmaceuticals, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
PURINETHOL- mercaptopurine tablet
Stason Pharmaceuticals, Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use PURINETHOL safely and effectively. See full prescribing information for PURINETHOL.
PURINETHOL ® (mercaptopurine) tablets, for oral use Initial U.S. Approval: 1953 RECENT MAJOR CHANGES
Warnings and Precautions, Hepatotoxicity (5.2) 7/2024 INDICATIONS AND USAGEPURINETHOL is a nucleoside metabolic inhibitor indicated for treatment of adult and pediatric patients with acute lymphoblastic leukemia (ALL) as part of a combination chemotherapy maintenance regimen. ( 1.1) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSTablets: 50 mg ( 3) CONTRAINDICATIONSNone . WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSThe most common adverse reaction (> 20%) is myelosuppression, including anemia, leukopenia and thrombocytopenia. Adverse reactions occurring in 5% to 20% of patients include anorexia, nausea, vomiting, diarrhea, malaise and rash. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Stason Pharmaceuticals at (888) 598-7707 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSUSE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 7/2024 |
FULL PRESCRIBING INFORMATION
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended starting dosage of PURINETHOL is 1.5mg/kg to 2.5 mg/kg orally once daily as part of combination chemotherapy maintenance regimen. A recommended dosage for patients less than 17 kg is not achievable, because the only available strength is 50 mg. Take PURINETHOL either consistently with or without food.
After initiating PURINETHOL, monitor complete blood count (CBC) and adjust the dose to maintain absolute neutrophil count (ANC) at a desirable level and for excessive myelosuppression. Evaluate the bone marrow in patients with prolonged myelosuppression or repeated episodes of myelosuppression to assess leukemia status and marrow cellularity.
Evaluate thiopurine S-methyltransferase (TPMT) and nucleotide diphosphatase (NUDT15) status in patients with severe myelosuppression or repeated episodes or myelosuppression [see Dosage and Administration (2.2)] .
Do not administer to patients who are unable to swallow tablets.
If a patient misses a dose, instruct the patient to continue with the next scheduled dose.
PURINETHOL is a cytotoxic drug. Follow special handling and disposal procedures.
2.2 Dosage Modifications in Patients with TPMT and NUDT15 Deficiency
Consider testing for TPMT and NUDT15 deficiency in patients who experience severe myelosuppression or repeated episodes of myelosuppression [see Warnings and Precautions (5.1), Clinical Pharmacology (12.5)] .
Homozygous Deficiency in either TPMT or NUDT15
Patients with homozygous deficiency of either enzyme typically require 10% or less of the recommended dosage. Reduce the recommended starting dosage of PURINETHOL in patients who are known to have homozygous TPMT or NUDT15 deficiency.
Heterozygous Deficiency in TPMT and/or NUDT15
Reduce the PURINETHOL dose based on tolerability. Most patients with heterozygous TPMT or NUDT15 deficiency tolerate the recommended dosage, but some require a dose reduction based on adverse reactions. Patients who are heterozygous for both TPMT and NUDT15 may require more substantial dose reductions.
2.3 Dosage Modifications in Renal and Hepatic Impairment
Renal Impairment
Use the lowest recommended starting dosage for PURINETHOL in patients with renal impairment (CLcr less than 50 mL/min). Adjust the dosage to maintain absolute neutrophil count (ANC) at a desirable level and for adverse reactions [see Uses in Specific Populations (8.6)] .
Hepatic Impairment
Use the lowest recommended starting dosage for PURINETHOL in patients with hepatic impairment. Adjust the dosage to maintain absolute neutrophil count (ANC) at a desirable level and for adverse reactions [see Uses in Specific Populations (8.7)] .
2.4 Dosage Modification with Concomitant Use of Allopurinol
Reduce the dose of PURINETHOL to one-third to one-quarter of the current dosage when coadministered with allopurinol [see Drug Interactions (7.1)] .
3 DOSAGE FORMS AND STRENGTHS
Tablets: 50 mg, biconvex, round, pale yellow to buff, scored tablets imprinted with “9|3”
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
The most consistent, dose-related adverse reaction is myelosuppression, manifested by anemia, leukopenia, thrombocytopenia, or any combination of these. Monitor CBC and adjust the dosage of PURINETHOL for excessive myelosuppression [see Dosage and Administration (2.1)] .
Consider testing for TPMT or NUDT15 deficiency in patients with severe myelosuppression or repeated episodes of myelosuppression. TPMT genotyping or phenotyping (red blood cell TPMT activity) and NUDT15 genotyping can identify patients who have reduced activity of these enzymes. Patients with heterozygous or homozygous TPMT or NUDT15 deficiency may require a dose reduction [see Dosage and Administration (2.2),Clinical Pharmacology (12.5)] .
Myelosuppression can be exacerbated by coadministration with allopurinol, aminosalicylates or other products that cause myelosuppression [see Drug Interactions (7.1, 7.3, 7.4)] . Reduce the dose of PURINETHOL when coadministered with allopurinol [see Dosage and Administration (2.4)] .
5.2 Hepatoxicity
Mercaptopurine is hepatotoxic. There are reports of deaths attributed to hepatic necrosis associated with the administration of mercaptopurine. Hepatic injury can occur with any dosage but seems to occur with greater frequency when the recommended dosage is exceeded. In some patients, jaundice has cleared following withdrawal of mercaptopurine and reappeared with rechallenge.
Usually, clinically detectable jaundice appears early in the course of treatment (1 to 2 months); however, jaundice has been reported as early as 1 week and as late as 8 years after the starting mercaptopurine. The hepatotoxicity has been associated in some cases with anorexia, diarrhea, jaundice, ascites, and pruritus. Hepatic encephalopathy has occurred.
Monitor serum transaminase levels, alkaline phosphatase, and bilirubin levels at weekly intervals when first beginning therapy and at monthly intervals thereafter. Monitor liver tests more frequently in patients who are receiving PURINETHOL with other hepatotoxic products [see Drug Interactions (7.5)] or with known pre-existing liver disease. Withhold PURINETHOL at onset of hepatotoxicity.
Intrahepatic Cholestasis of Pregnancy
Postmarketing cases of intrahepatic cholestasis of pregnancy (ICP) have been reported in patients with inflammatory bowel disease who received mercaptopurine during pregnancy. PURINETHOL is not indicated for use in inflammatory bowel disease [see Indications and Usage (1.1)].
Discontinue PURINETHOL if ICP develops in a pregnant woman.
5.3 Immunosuppression
Mercaptopurine is immunosuppressive and may impair the immune response to infectious agents or vaccines. Due to the immunosuppression associated with maintenance chemotherapy for ALL, response to all vaccines may be diminished and there is a risk of infection with live virus vaccines. Consult immunization guidelines for immunocompromised patients.
5.4 Treatment Related Malignancies
Hepatosplenic T-cell lymphoma has been reported in patients treated with mercaptopurine for inflammatory bowel disease (IBD), an unapproved use. Mercaptopurine is mutagenic in animals and humans, carcinogenic in animals, and may increase the risk of secondary malignancies.
Patients receiving immunosuppressive therapy, including mercaptopurine, are at an increased risk of developing lymphoproliferative disorders and other malignancies, notably skin cancers (melanoma and non-melanoma), sarcomas (Kaposi's and non-Kaposi's) and uterine cervical cancer in situ. The increased risk appears to be related to the degree and duration of immunosuppression. It has been reported that discontinuation of immunosuppression may provide partial regression of the lymphoproliferative disorder.
A treatment regimen containing multiple immunosuppressants (including thiopurines) should therefore be used with caution as this could lead to lymphoproliferative disorders, some with reported fatalities. A combination of multiple immunosuppressants, given concomitantly increases the risk of Epstein-Barr virus (EBV)-associated lymphoproliferative disorders.
5.5 Macrophage Activation Syndrome
Macrophage activation syndrome (MAS) (hemophagocytic lymphohistiocytosis) is a known, life-threatening disorder that may develop in patients with autoimmune conditions, in particular with inflammatory bowel disease (IBD), and there could potentially be an increased susceptibility for developing the condition with the use of mercaptopurine (an unapproved use). If MAS occurs, or is suspected, discontinue PURINETHOL. Monitor for and promptly treat infections such as EBV and cytomegalovirus (CMV), as these are known triggers for MAS.
5.6 Embryo-Fetal Toxicity
PURINETHOL can cause fetal harm when administered to a pregnant woman. An increased incidence of miscarriage has been reported in women who received mercaptopurine in the first trimester of pregnancy. Adverse embryo-fetal findings, including miscarriage and stillbirth, have been reported in women who received mercaptopurine after the first trimester of pregnancy. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with PURINETHOL and for 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with PURINETHOL and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)] .
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Myelosuppression [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Immunosuppression [see Warnings and Precautions (5.3)]
- Treatment related malignancies [see Warnings and Precautions (5.4)]
- Macrophage activation syndrome [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Based on multicenter cooperative group ALL trials, the most common adverse reaction occurring in > 20% of patients was myelosuppression, including anemia, neutropenia, lymphopenia and thrombocytopenia. Adverse reactions occurring in 5% to 20% of patients included anorexia, nausea, vomiting, diarrhea, malaise and rash. Adverse reactions occurring in < 5 % of patients included urticaria, hyperuricemia, oral lesions, increased transaminases, hyperbilirubinemia, hyperpigmentation, infections, and pancreatitis. Oral lesions resemble thrush rather than antifolic ulcerations. Delayed or late adverse reactions include hepatic fibrosis, hyperbilirubinemia, alopecia, pulmonary fibrosis, oligospermia and secondary malignancies [see Warnings and Precautions (5.1, 5.2)] .
Drug fever has been reported with mercaptopurine.
Additional adverse reactions that have been reported in patients who have received mercaptopurine include photosensitivity, hypoglycemia, and portal hypertension.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of PURINETHOL. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These reactions include: intrahepatic cholestasis of pregnancy (ICP).
7 DRUG INTERACTIONS
7.1 Allopurinol
Allopurinol can inhibit the first-pass oxidative metabolism of mercaptopurine by xanthine oxidase, which can lead to an increased risk of mercaptopurine adverse reactions (i.e., myelosuppression, nausea, and vomiting) [see Warnings and Precautions (5.1), Adverse Reactions (6.1)] . Reduce the dose of PURINETHOL when coadministered with allopurinol [see Dosage and Administration (2.4)] .
7.2 Warfarin
The concomitant administration of PURINETHOL and warfarin may decrease the anticoagulant effectiveness of warfarin. Monitor the international normalized ratio (INR) in patients receiving warfarin and adjust the warfarin dosage as appropriate.
7.3 Myelosuppressive Products
PURINETHOL can cause myelosuppression. Myelosuppression may be increased when PURINETHOL is coadministered with other products that cause myelosuppression. Enhanced myelosuppression has been noted in some patients also receiving trimethoprim-sulfamethoxazole. Monitor the CBC and adjust the dose of PURINETHOL for excessive myelosuppression [see Dosage and Administration (2.1), Warnings and Precautions (5.1)] .
7.4 Aminosalicylates
Aminosalicylates (e.g., mesalamine, olsalazine or sulfasalazine) may inhibit the TPMT enzyme, which may increase the risk of myelosuppression when coadministered with PURINETHOL. When aminosalicylates and PURINETHOL are coadministered, use the lowest possible doses for each drug and monitor more frequently for myelosuppression [see Warnings and Precautions (5.1)] .
7.5 Hepatotoxic Products
PURINETHOL can cause hepatotoxicity. Hepatotoxicity may be increased when PURINETHOL is coadministered with other products that cause hepatotoxicity. Monitor liver tests more frequently in patients who are receiving PURINETHOL with other hepatotoxic products [see Warnings and Precautions (5.2)] .
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
PURINETHOL can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)] . Pregnant women who receive mercaptopurine have an increased incidence of miscarriage and stillbirth (see Data) . Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population(s) is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
Women receiving mercaptopurine in the first trimester of pregnancy have an increased incidence of miscarriage; the risk of malformation in offspring surviving first trimester exposure is not known. In a series of 28 women receiving mercaptopurine after the first trimester of pregnancy, 3 mothers died prior to delivery, 1 delivered a stillborn child, and 1 aborted; there were no cases of macroscopically abnormal fetuses.
8.2 Lactation
Risk Summary
There are no data on the presence of mercaptopurine or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with PURINETHOL and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
PURINETHOL can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)] .
Pregnancy Testing
Verify the pregnancy status in females of reproductive potential prior to initiating PURINETHOL [see Use in Specific Populations (8.1)] .
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with PURINETHOL and for 6 months after the last dose.
Males
Based on genotoxicity findings, advise males with female partners of reproductive potential to use effective contraception during treatment with PURINETHOL and for 3 months after the last dose [see Nonclinical Toxicology (13.1)] .
Infertility
Females and Males
Based on findings from animal studies, PURINETHOL can impair female and male fertility [see Nonclinical Toxicology (13.1)] . The long-term effects of mercaptopurine on female and male fertility, including the reversibility have not been studied.
8.4 Pediatric Use
Safety and effectiveness of PURINETHOL has been established in pediatric patients. Use of PURINETHOL in pediatrics is supported by evidence from the published literature and clinical experience. Symptomatic hypoglycemia has been reported in pediatric patients with ALL receiving mercaptopurine. Reported cases were in pediatrics less than 6 years of age or with a low body mass index.
8.5 Geriatric Use
Clinical studies of mercaptopurine did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or another drug therapy.
8.6 Renal Impairment
Use the lowest recommended starting dosage for PURINETHOL or increase the dosing interval to every 36-48 hours in patients with renal impairment (CLcr less than 50 mL/min). Adjust the dose to maintain absolute neutrophil count (ANC) at a desirable level and for adverse reactions [see Dosage and Administration (2.3)] .
8.7 Hepatic Impairment
Use the lowest recommended starting dosage for PURINETHOL in patients with hepatic impairment. Adjust the dose to maintain absolute neutrophil count (ANC) at a desirable level and for adverse reactions [see Dosage and Administration (2.3)] .
10 OVERDOSAGE
Signs and symptoms of mercaptopurine overdosage may be immediate (anorexia, nausea, vomiting, and diarrhea); or delayed (myelosuppression, liver dysfunction, and gastroenteritis). Dialysis cannot be expected to clear mercaptopurine. Hemodialysis is thought to be of marginal use due to the rapid intracellular incorporation of mercaptopurine into active metabolites with long persistence.
Withhold PURINETHOL immediately if severe or life-threatening adverse reactions occur during treatment. If a patient is seen immediately following an accidental overdosage, it may be useful to induce emesis.
11 DESCRIPTION
Mercaptopurine is a nucleoside metabolic inhibitor. The chemical name is 6 H-purine-6-thione, 1,7-dihydro-, monohydrate. The molecular formula is C 5H 4N 4SH 2O and the molecular weight is 170.20. Its structural formula is:
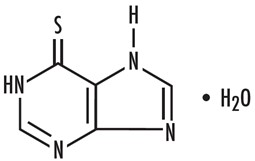
Mercaptopurine is a yellow, crystalline powder. Mercaptopurine is practically insoluble in water and in ether. It has a pKa of 7.8, an average tapped density of 1.0 g/mL and average bulk density of 0.85 g/mL. It dissolves in solutions of alkali hydroxides.
PURINETHOL is available for oral use. Each scored tablet contains 50 mg mercaptopurine and the following inactive ingredients: corn starch, pregelatinized, potato starch, lactose, magnesium stearate and stearic acid.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Mercaptopurine is a purine analog that undergoes intracellular transport and activation to form metabolites including thioguanine nucleotides (TGNs). Incorporation of TGNs into DNA or RNA results in cell-cycle arrest and cell death. TGNs and other mercaptopurine metabolites are also inhibitors of de novo purine synthesis and purine nucleotide interconversions. Mercaptopurine was cytotoxic to proliferating cancer cells in vitro and had antitumor activity in mouse tumor models. It is not known which of the biochemical effects of mercaptopurine and its metabolites are directly or predominantly responsible for cell death.
12.3 Pharmacokinetics
Following a single oral dose of mercaptopurine 50 mg under fasted conditions to adult healthy subjects, the mean AUC 0-INF was 129 hng/mL and C max was 69 ng/mL.
Distribution
The volume of distribution usually exceeded that of the total body water. There is negligible entry of mercaptopurine into cerebrospinal fluid.
Plasma protein binding averages 19% over the concentration range 10 to 50 mcg/mL (a concentration only achieved by intravenous administration of mercaptopurine at doses exceeding 5 to 10 mg/kg).
Elimination
The elimination half-life is less than 2 hours following a single oral dose.
Metabolism
Mercaptopurine is inactivated via two major pathways. One is thiol methylation, which is catalyzed by the polymorphic enzyme thiopurine S-methyltransferase (TPMT), to form the inactive metabolite methyl-mercaptopurine. The second inactivation pathway is oxidation, which is catalyzed by xanthine oxidase. The product of oxidation is the inactive metabolite 6-thiouric acid.
12.5 Pharmacogenomics
Several published studies indicate that patients with reduced TPMT or NUDT15 activity receiving usual doses of mercaptopurine, accumulate excessive cellular concentrations of active 6-TGNs, and are at higher risk for severe myelosuppression. In a study of 1028 children with ALL, the approximate tolerated mercaptopurine dosage for patients with TPMT and/or NUDT15 deficiency on mercaptopurine maintenance therapy (as a percentage of the planned dosage) was as follows: heterozygous for either TPMT or NUDT15, 50-90%; heterozygous for both TPMT and NUDT15, 30-50%; homozygous for either TPMT or NUDT15, 5-10%.
Approximately 0.3% (1:300) of patients of European or African ancestry have two loss-of-function alleles of the TPMT gene and have little or no TPMT activity (homozygous deficient or poor metabolizers), and approximately 10% of patients have one loss-of-function TPMT allele leading to intermediate TPMT activity (heterozygous deficient or intermediate metabolizers). The TPMT *2, TPMT *3A, and TPMT *3C alleles account for about 95% of individuals with reduced levels of TPMT activity.
NUDT15 deficiency is detected in < 1% of patients of European or African ancestry. Among patients of East Asian ancestry (i.e., Chinese, Japanese, Vietnamese), 2% have two loss-of-function alleles of the NUDT15 gene, and approximately 21% have one loss-of-function allele. The p.R139C variant of NUDT15 (present on the *2 and *3 alleles) is the most commonly observed, but other less common loss-of-function NUDT15 alleles have been observed.
Consider all clinical information when interpreting results from phenotypic testing used to determine the level of thiopurine nucleotides or TPMT activity in erythrocytes, since some coadministered drugs can influence measurement of TPMT activity in blood and blood from recent transfusions will misrepresent a patient’s actual TPMT activity [see Dosage and Administration (2.2), Warnings and Precautions (5.1)] .
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Mercaptopurine is carcinogenic in animals.
Mercaptopurine causes chromosomal aberrations in cells derived from animals and humans and induces dominant-lethal mutations in the germ cells of male mice.
Mercaptopurine can impair fertility. In mice, surviving female offspring of mothers who received chronic low doses of mercaptopurine during pregnancy were found sterile, or if they became pregnant, had smaller litters and more dead fetuses as compared to control animals.
16 HOW SUPPLIED/STORAGE AND HANDLING
PURINETHOL is supplied as biconvex, round, pale yellow to buff, scored tablets containing 50 mg mercaptopurine, imprinted with “9|3” available in:
- bottles of 25 NDC: 60763-601-13
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Store in a dry place. Dispense in tight container as defined in the USP.
PURINETHOL is a cytotoxic drug. Follow special handling and disposal procedures 1.
17 PATIENT COUNSELING INFORMATION
Major Adverse Reactions
Advise patients and caregivers that PURINETHOL can cause myelosuppression, hepatotoxicity, and gastrointestinal toxicity. Advise patients to contact their healthcare provider if they experience fever, sore throat, jaundice, nausea, vomiting, signs of local infection, bleeding from any site, or symptoms suggestive of anemia [see Warnings and Precautions (5.1, 5.2, 5.3)] .
Embryo-Fetal Toxicity
- Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.2, 5.6), Use in Specific Populations (8.1)] .
- Advise females of reproductive potential to use effective contraception during treatment with PURINETHOL and for 6 months after the last dose [see Use in Specific Populations (8.3)] .
- Advise males with female partners of reproductive potential to use effective contraception during treatment with PURINETHOL and for 3 months after the last dose [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)] .
Lactation
Advise women not to breastfeed during treatment with PURINETHOL and for 1 week after the last dose [see Use in Specific Populations (8.2)] .
Infertility
Advise males and females of reproductive potential that PURINETHOL can impair fertility [see Use in Specific Populations (8.3)] .
Other Adverse Reactions
Instruct patients to minimize sun exposure due to risk of photosensitivity [see Adverse Reactions (6.1)] .
Distributed by:
Stason Pharmaceuticals, Inc.
Irvine, CA 92618
Made in U.S.A.
PRINCIPAL DISPLAY PANEL - 50 mg
NDC: 60763-601-13
PURINETHOL ®
(Mercaptopurine)
Tablets, USP
50 mg
Each tablet contains 50 mg
mercaptopurine
Cytotoxic Agent
Rx only 25 Tablets
Dosage: See prescribing information.
WARNING: This drug is
only to be
taken under close medical supervi-
sion. Do not take in larger doses or
more frequently or for a longer time
than specifically directed by the
physician. Periodic blood counts are
necessary to determine proper dose
and to avoid ill effects.
Store at 20° to 25°C (68° to 77°F).
[See USP Controlled Room
Temperature.] Store in a dry place.
Dispense in a tight container as defined in the USP.
Distributed by: Stason Pharmaceuticals, Inc.
Irvine, CA 92618
Made in U.S.A. Rev. 5/21
| PURINETHOL
mercaptopurine tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Stason Pharmaceuticals, Inc. (807437553) |
| Registrant - Stason Pharmaceuticals, Inc. (807437553) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Stason Pharmaceuticals, Inc. | 807437553 | manufacture(60763-601) | |
Trademark Results [PURINETHOL]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 PURINETHOL 77140307 3391060 Dead/Cancelled |
STASON PHARMACEUTICALS, INC. 2007-03-26 |
 PURINETHOL 71647991 0587330 Live/Registered |
BURROUGHS WELLCOME & CO. (U. S. A.) INC. 1953-06-01 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.