DUAVEE- conjugated estrogens/bazedoxifene tablet, film coated
Duavee by
Drug Labeling and Warnings
Duavee by is a Prescription medication manufactured, distributed, or labeled by U.S. Pharmaceuticals, Wyeth Pharmaceuticals Inc., Wyeth Pharmaceuticals Inc., a subsidiary of Pfizer Inc., Pfizer Ireland Pharmaceuticals, Pfizer Pharmaceuticals LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DUAVEE safely and effectively. See full prescribing information for DUAVEE.
DUAVEE® (conjugated estrogens/bazedoxifene) tablets for oral use
Initial U.S. approval: 2013WARNING: ENDOMETRIAL CANCER, CARDIOVASCULAR DISORDERS, AND PROBABLE DEMENTIA
See full prescribing information for complete Boxed Warning.
- Women taking DUAVEE should not take additional estrogens (5.1)
- There is an increased risk of endometrial cancer in a woman with a uterus who uses unopposed estrogens (5.1, 5.3)
- Estrogen therapy should not be used for the prevention of cardiovascular disease or dementia (5.2, 5.4)
- The Women's Health Initiative (WHI) estrogen-alone substudy reported increased risks of stroke and deep vein thrombosis (DVT) (5.2)
- The WHI Memory Study (WHIMS) estrogen-alone ancillary study of WHI reported an increased risk of probable dementia in postmenopausal women 65 years of age and older (5.4)
INDICATIONS AND USAGE
DUAVEE is a combination of conjugated estrogens with an estrogen agonist/antagonist indicated for treatment of the following conditions in women with a uterus:
- Treatment of moderate to severe vasomotor symptoms associated with menopause (1.1)
- Prevention of postmenopausal osteoporosis (1.2)
Limitation of Use: DUAVEE should be used for the shortest duration consistent with treatment goals and risks for the individual woman (1.3)
DOSAGE AND ADMINISTRATION
- Take one tablet orally once daily (2)
DOSAGE FORMS AND STRENGTHS
Tablet containing conjugated estrogens 0.45 mg and bazedoxifene 20 mg (3)
CONTRAINDICATIONS
- Undiagnosed abnormal uterine bleeding (4, 5.3)
- Known, suspected, or past history of breast cancer (4, 5.3)
- Known or suspected estrogen-dependent neoplasia (4, 5.3)
- Active or past history of venous thromboembolism (4, 5.2)
- Active or past history of arterial thromboembolism (4, 5.2)
- Hypersensitivity (angioedema, anaphylaxis) to estrogens, bazedoxifene, or any ingredients (4)
- Known hepatic impairment or disease (4, 5.9, 8.7, 12.3)
- Known protein C, protein S, or antithrombin deficiency or other known thrombophilic disorders (4)
- Pregnancy, women who may become pregnant, and nursing mothers (4, 8.1, 8.3)
WARNINGS AND PRECAUTIONS
- Women taking DUAVEE should not take progestins, additional estrogens or additional estrogen agonist/antagonists (5.1)
- Cardiovascular disorders, including venous thromboembolism, pulmonary embolism, stroke, and retinal vascular thrombosis (5.2, 5.6)
- Malignant neoplasms, including endometrial cancer, breast cancer, and ovarian cancer (5.3)
- Estrogens increase the risk of gallbladder disease (5.5)
- Discontinue estrogen if loss of vision, severe hypertriglyceridemia or cholestatic jaundice occurs (5.6, 5.8, 5.9)
- Monitor thyroid function in women on thyroid replacement therapy (5.10, 5.17)
ADVERSE REACTIONS
In four prospective, randomized, placebo-controlled trials the common adverse reactions (incidence ≥ 5%) were muscle spasms, nausea, diarrhea, dyspepsia, abdominal pain upper, oropharyngeal pain, dizziness, and neck pain (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
No drug interaction studies were conducted with DUAVEE. Estrogens are metabolized partially by CYP3A4. Concomitant use of DUAVEE with CYP3A4 inhibitors may increase the exposure of conjugated estrogens and thereby may increase the risk of endometrial hyperplasia (7.1).
USE IN SPECIFIC POPULATIONS
- Geriatric Use: DUAVEE was not studied in women aged 75 or older (8.5, 12.3); use in this population is not recommended (2.7, 8.5)
- An increased risk of probable dementia in women over 65 years of age was reported in the Women's Health Initiative Memory ancillary studies of the Women's Health Initiative (5.4, 8.5, 14.6)
- Renal Impairment: DUAVEE was not studied in women with renal impairment; use in this population is not recommended (2.6, 8.6, 12.3)
- Body Mass Index: Women with BMI > 27 kg/m2 may have an increased risk of endometrial hyperplasia (8.8)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2013
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: ENDOMETRIAL CANCER, CARDIOVASCULAR DISORDERS, AND PROBABLE DEMENTIA
1 INDICATIONS AND USAGE
1.3 Important Limitations of Use
2 DOSAGE AND ADMINISTRATION
2.1 Treatment of Moderate to Severe Vasomotor Symptoms Associated with Menopause
2.2 Prevention of Postmenopausal Osteoporosis
2.3 General Dosing Information
2.4 Recommendations for Calcium and Vitamin D Supplementation
2.5 Administration Instructions for Missed Doses
2.6 Use in Patients with Renal Impairment
2.7 Use in the Elderly
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Drugs Containing Progestins, Estrogens or Estrogen Agonist/Antagonists
5.2 Cardiovascular Disorders
5.3 Malignant Neoplasms
5.4 Probable Dementia
5.5 Gallbladder Disease
5.6 Visual Abnormalities
5.7 Elevated Blood Pressure
5.8 Hypertriglyceridemia
5.9 Hepatic Impairment and Past History of Cholestatic Jaundice
5.10 Hypothyroidism
5.11 Fluid Retention
5.12 Hypocalcemia
5.13 Hereditary Angioedema
5.14 Exacerbation of Other Conditions
5.15 Premenopausal Women
5.16 Laboratory Tests
5.17 Drug-Laboratory Test Interactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Cytochrome P450 (CYP)
7.2 Uridine Diphosphate Glucuronosyltransferase (UGT)
7.3 Atorvastatin
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 Use in Women with Body Mass Index (BMI) > 27 kg/m2
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Treatment of Moderate to Severe Vasomotor Symptoms Associated with Menopause in Women with a Uterus
14.2 Prevention of Postmenopausal Osteoporosis in Women with a Uterus
14.3 Effects on the Endometrium
14.4 Effects on Uterine Bleeding and Spotting
14.5 Women's Health Initiative Studies
14.6 Women's Health Initiative Memory Study
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Instructions for Patients
17.2 Venous Thromboembolic Events
17.3 Abnormal Vaginal Bleeding
17.4 Possible Serious Adverse Reactions with Estrogen Therapy
17.5 Possible Less Serious Adverse Reactions with DUAVEE
17.6 Calcium and Vitamin D Intake
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: ENDOMETRIAL CANCER, CARDIOVASCULAR DISORDERS, AND PROBABLE DEMENTIA
- Women taking DUAVEE should not take additional estrogens [see Warnings and Precautions (5.1)]
- There is an increased risk of endometrial cancer in a woman with a uterus who uses unopposed estrogens. DUAVEE has been shown to reduce the risk of endometrial hyperplasia, which may be a precursor to endometrial cancer. Adequate diagnostic measures, including directed or random endometrial sampling when indicated, should be undertaken to rule out malignancy in postmenopausal women with undiagnosed persistent or recurring abnormal genital bleeding [see Warnings and Precautions (5.3)]
- Estrogen therapy should not be used for the prevention of cardiovascular disease or dementia [see Warnings and Precautions (5.2, 5.4)]
- The Women's Health Initiative (WHI) estrogen-alone substudy reported increased risks of stroke and deep vein thrombosis (DVT) in postmenopausal women (50 to 79 years of age) during 7.1 years of treatment with daily oral conjugated estrogens (0.625 mg)-alone, relative to placebo [see Warnings and Precautions (5.2)]
- The WHI Memory Study (WHIMS) estrogen-alone ancillary study of WHI reported an increased risk of probable dementia in postmenopausal women 65 years of age and older during 5.2 years of treatment with daily conjugated estrogens (0.625 mg)-alone, relative to placebo. It is unknown whether this finding applies to younger postmenopausal women [see Warnings and Precautions (5.4)]
In the absence of comparable data, these risks should be assumed to be similar for other doses of conjugated estrogens and other dosage forms of estrogens.
Estrogens should be prescribed at the lowest effective doses and for the shortest duration consistent with treatment goals and risks for the individual woman.
-
1 INDICATIONS AND USAGE
DUAVEE is indicated in women with a uterus for:
1.3 Important Limitations of Use
- Use DUAVEE for the shortest duration consistent with treatment goals and risks for the individual woman. Postmenopausal women should be re-evaluated periodically as clinically appropriate to determine if treatment is still necessary.
- When prescribing solely for the prevention of postmenopausal osteoporosis, therapy should only be considered for women at significant risk of osteoporosis and non-estrogen medication should be carefully considered.
-
2 DOSAGE AND ADMINISTRATION
2.1 Treatment of Moderate to Severe Vasomotor Symptoms Associated with Menopause
The recommended dosage is one DUAVEE tablet daily.
2.3 General Dosing Information
Take DUAVEE once daily, without regard to meals. Tablets should be swallowed whole.
2.4 Recommendations for Calcium and Vitamin D Supplementation
Women taking DUAVEE for prevention of postmenopausal osteoporosis should add supplemental calcium and/or vitamin D to their diet if daily intake is inadequate.
2.5 Administration Instructions for Missed Doses
If a dose of DUAVEE is missed, instruct patients to take it as soon as remembered unless it is almost time for the next scheduled dose. They should not take two doses at the same time.
2.6 Use in Patients with Renal Impairment
The pharmacokinetics of DUAVEE have not been evaluated in patients with renal impairment. Use in patients with renal impairment is not recommended [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
DUAVEE is contraindicated in women with any of the following conditions:
- Undiagnosed abnormal uterine bleeding
- Known, suspected, or past history of breast cancer
- Known or suspected estrogen-dependent neoplasia
- Active deep venous thrombosis, pulmonary embolism, or history of these conditions
- Active arterial thromboembolic disease (for example, stroke, myocardial infarction) or history of these conditions
- Hypersensitivity (for example, anaphylaxis, angioedema) to estrogens, bazedoxifene, or any ingredients
- Known hepatic impairment or disease
- Known protein C, protein S, or antithrombin deficiency or other known thrombophilic disorders
- Pregnancy, women who may become pregnant, and nursing mothers. DUAVEE may cause fetal harm when administered to a pregnant woman. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus
-
5 WARNINGS AND PRECAUTIONS
5.1 Drugs Containing Progestins, Estrogens or Estrogen Agonist/Antagonists
DUAVEE contains conjugated estrogens and bazedoxifene, an estrogen agonist/antagonist. Women taking DUAVEE should not take progestins, additional estrogens or additional estrogen agonist/antagonists.
5.2 Cardiovascular Disorders
Estrogen agonist/antagonists (including bazedoxifene, a component of DUAVEE) and estrogens individually are known to increase the risk of VTE.
An increased risk of stroke and DVT has been reported with estrogen-alone therapy. Should any of these occur or be suspected, DUAVEE should be discontinued immediately.
Risk factors for arterial vascular disease (for example, hypertension, diabetes mellitus, tobacco use, hypercholesterolemia, and obesity) and/or VTE (for example, personal history or family history of VTE, obesity, and systemic lupus erythematosus) should be managed appropriately.
Stroke
In the WHI estrogen-alone substudy, a statistically significant increased risk of stroke was reported in women 50 to 79 years of age receiving daily conjugated estrogens (CE) (0.625 mg)-alone compared to women in the same age group receiving placebo (45 versus 33 per 10,000 women-years). The increase in risk was demonstrated in year 1 and persisted [see Clinical Studies (14.5)].
Subgroup analyses of women 50 to 59 years of age suggest no increased risk of stroke for those women receiving conjugated estrogens (0.625 mg)-alone versus those receiving placebo (18 versus 21 per 10,000 women-years).
Should a stroke occur or be suspected, DUAVEE should be discontinued immediately [see Contraindications (4)].
Coronary Heart Disease
In the WHI estrogen-alone substudy, no overall effect on coronary heart disease (CHD) events (defined as nonfatal myocardial infarction, silent myocardial infarction, or CHD death) was reported in women receiving estrogen-alone compared to placebo [see Clinical Studies (14.5)].
Subgroup analyses of women 50 to 59 years of age suggest a statistically non-significant reduction in CHD events (CE [0.625 mg]-alone compared to placebo) in women with less than 10 years since menopause (8 versus 16 per 10,000 women-years).
Venous Thromboembolism (VTE)
In the WHI estrogen-alone substudy, the risk of VTE [DVT and pulmonary embolism (PE)] was increased for women receiving daily conjugated estrogens (0.625 mg)-alone compared to placebo (30 versus 22 per 10,000 women-years), although only the increased risk of DVT reached statistical significance (23 versus 15 per 10,000 women-years). The increase in VTE risk was demonstrated during the first 2 years [see Clinical Studies (14.5)].
If feasible, DUAVEE should be discontinued at least 4 to 6 weeks before surgery of the type associated with an increased risk of thromboembolism, or during periods of prolonged immobilization. Because immobilization increases the risk for venous thromboembolic events independent of therapy, DUAVEE should be discontinued prior to and during prolonged immobilization (e.g., post-surgical recovery, prolonged bed rest) and DUAVEE therapy should be resumed only after the patient is fully ambulatory. In addition, women taking DUAVEE should be advised to move about periodically during travel involving prolonged immobilization.
5.3 Malignant Neoplasms
Endometrial Cancer
An increased risk of endometrial cancer has been reported with the use of unopposed estrogen therapy in women with a uterus. The reported endometrial cancer risk among unopposed estrogen users is about 2 to 12 times greater than in non-users, and appears dependent on duration of treatment and on estrogen dose. Most studies show no significant increased risk associated with use of estrogens for less than 1 year. The greatest risk appears associated with prolonged use, with increased risks of 15- to 24-fold for 5 to 10 years or more of treatment. This risk has been shown to persist for at least 8 to 15 years after estrogen therapy is discontinued.
DUAVEE contains an estrogen agonist/antagonist. This component reduces the risk of endometrial hyperplasia that can occur with the conjugated estrogens component. Endometrial hyperplasia may be a precursor to endometrial cancer. Women taking DUAVEE should not take additional estrogens as this may increase the risk of endometrial hyperplasia.
Clinical surveillance of all women taking DUAVEE is important. Adequate diagnostic measures, including directed or random endometrial sampling when indicated, should be undertaken to rule out malignancy in postmenopausal women with undiagnosed persistent or recurring abnormal genital bleeding.
Breast Cancer
The most important randomized clinical study providing information about breast cancer in estrogen-alone users is the WHI substudy of daily conjugated estrogens (0.625 mg)-alone. In the WHI estrogen-alone substudy, after an average follow-up of 7.1 years, daily conjugated estrogen (0.625 mg)-alone was not associated with an increased risk of invasive breast cancer (relative risk [RR] 0.80).
The use of estrogen-alone has been reported to result in an increase in abnormal mammograms requiring further evaluation. The effect of treatment with DUAVEE on the risk of breast cancer is unknown.
All women should receive yearly breast examinations by a healthcare provider and perform monthly breast self-examinations. In addition, mammography examinations should be scheduled based on patient age, risk factors, and prior mammogram results.
Ovarian Cancer
In some epidemiological studies, the use of estrogen-only products, in particular for 5 or more years, has been associated with an increased risk of ovarian cancer. However, the duration of exposure associated with increased risk is not consistent across all epidemiologic studies, and some report no association. The effect of treatment with DUAVEE on the risk of ovarian cancer is unknown.
5.4 Probable Dementia
In the WHIMS estrogen-alone ancillary study of WHI, a population of 2,947 hysterectomized women 65 to 79 years of age was randomized to daily CE (0.625 mg)-alone or placebo.
After an average follow-up of 5.2 years, 28 women in the estrogen-alone group and 19 women in the placebo group were diagnosed with probable dementia. The relative risk of probable dementia for CE-alone versus placebo was 1.49 (95 percent CI, 0.83–2.66). The absolute risk of probable dementia for CE-alone versus placebo was 37 versus 25 cases per 10,000 women-years [see Use in Specific Populations (8.5) and Clinical Studies (14.6)].
5.5 Gallbladder Disease
A 2- to 4-fold increase in the risk of gallbladder disease requiring surgery in postmenopausal women receiving estrogens has been reported.
5.6 Visual Abnormalities
Retinal vascular thrombosis has been reported in patients receiving estrogens. Discontinue medication pending examination if there is sudden partial or complete loss of vision, or a sudden onset of proptosis, diplopia, or migraine. If examination reveals papilledema or retinal vascular lesions, DUAVEE should be permanently discontinued.
5.7 Elevated Blood Pressure
In a small number of case reports in women receiving estrogens, substantial increases in blood pressure have been attributed to idiosyncratic reactions to estrogens. In a large, randomized, placebo-controlled clinical study, a generalized effect of estrogens on blood pressure was not seen.
5.8 Hypertriglyceridemia
In women with pre-existing hypertriglyceridemia, treatment with estrogens may be associated with elevations of plasma triglycerides leading to pancreatitis. Consider discontinuation of DUAVEE if pancreatitis occurs.
5.9 Hepatic Impairment and Past History of Cholestatic Jaundice
DUAVEE has not been studied in women with impaired liver function or past history of cholestatic jaundice.
Estrogens may be poorly metabolized in women with impaired liver function.
On average, women with hepatic impairment treated with bazedoxifene alone showed a 4.3-fold increase in overall exposures compared with controls [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
For women with a history of cholestatic jaundice associated with past estrogen use or with pregnancy, caution should be exercised; and in the case of recurrence, DUAVEE should be discontinued. Use of DUAVEE in patients with hepatic impairment is contraindicated [see Contraindications (4)].
5.10 Hypothyroidism
Estrogen administration leads to increased thyroid-binding globulin (TBG) levels. Women with normal thyroid function can compensate for the increased TBG by making more thyroid hormone, thus maintaining free T4 and T3 serum concentrations in the normal range. Women dependent on thyroid hormone replacement therapy who are also receiving estrogens may require increased doses of their thyroid replacement therapy. These women should have their thyroid function monitored in order to maintain their free thyroid hormone levels in an acceptable range.
5.11 Fluid Retention
Estrogens may cause some degree of fluid retention. Because of this, patients who have conditions that might be influenced by this factor, such as cardiac dysfunction or renal impairment, warrant careful observation when estrogens are prescribed. Use of DUAVEE in patients with renal impairment is not recommended [see Use in Specific Populations (8.6)].
5.12 Hypocalcemia
Estrogen therapy should be used with caution in women with hypoparathyroidism as estrogen-induced hypocalcemia may occur.
5.13 Hereditary Angioedema
Exogenous estrogens may exacerbate symptoms of angioedema in women with hereditary angioedema.
5.14 Exacerbation of Other Conditions
Estrogens may cause an exacerbation of asthma, diabetes mellitus, epilepsy, migraine or porphyria, systemic lupus erythematosus, and hepatic hemangiomas and should be used with caution in women with these conditions.
5.15 Premenopausal Women
There is no indication for premenopausal use of DUAVEE. The efficacy and safety of DUAVEE in premenopausal women have not been established, and its use is not recommended.
5.16 Laboratory Tests
Serum follicle stimulating hormone (FSH) and estradiol levels have not been shown to be useful in the management of moderate to severe vasomotor symptoms.
5.17 Drug-Laboratory Test Interactions
Accelerated prothrombin time, partial thromboplastin time, and platelet aggregation time; increased platelet count; increased factors II, VII antigen, VIII antigen, VIII coagulant activity, IX, X, XII, VII-X complex, II-VII-X complex, and beta-thromboglobulin; decreased levels of antifactor Xa and antithrombin III, decreased antithrombin III activity; increased levels of fibrinogen and fibrinogen activity; increased plasminogen antigen and activity.
Increased thyroid-binding globulin (TBG) leading to increased circulating total thyroid hormone, as measured by protein-bound iodine (PBI), T4 levels (by column or by radioimmunoassay), or T3 levels by radioimmunoassay. T3 resin uptake is decreased, reflecting the elevated TBG. Free T4 and free T3 concentrations are unaltered. Women on thyroid replacement therapy may require higher doses of thyroid hormone.
Other binding proteins may be elevated in serum, for example, corticosteroid binding globulin (CBG), sex hormone-binding globulin (SHBG), leading to increased total circulating corticosteroids and sex steroids, respectively. Free hormone concentrations, such as testosterone and estradiol, may be decreased. Other plasma proteins may be increased (angiotensinogen/renin substrate, alpha-1-antitrypsin, ceruloplasmin).
Increased plasma high-density lipoprotein (HDL) and HDL2 cholesterol subfraction concentrations, reduced low-density lipoprotein (LDL) cholesterol concentrations, increased triglyceride levels.
Impaired glucose tolerance.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Cardiovascular Disorders [see Warnings and Precautions (5.2)]
- Malignant Neoplasms [see Warnings and Precautions (5.3)]
- Gallbladder Disease [see Warnings and Precautions (5.5)]
- Hypertriglyceridemia [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of conjugated estrogens/bazedoxifene was evaluated in four Phase 3 clinical trials ranging from 12 weeks to 24 months in duration and enrolling 6,210 postmenopausal women age 40 to 75 years (mean age 55 years). A total of 1,224 patients were treated with DUAVEE and 1,069 patients received placebo. Women enrolled in Studies 1 and 2 received calcium (600–1200 mg) and vitamin D (200–400 IU) daily, while women in Studies 3 and 4 received no calcium and vitamin D supplementation as part of the protocol.
The incidence of all-cause mortality was 0.0% in the DUAVEE group and 0.2% in the placebo group. The incidence of serious adverse reactions was 3.5% in the DUAVEE group and 4.8% in the placebo group. The percentage of patients who withdrew from treatment due to adverse reactions was 7.5% in the DUAVEE group and 10.0% in the placebo group. The most common adverse reactions leading to discontinuation were hot flush, abdominal pain upper, and nausea.
The most commonly observed adverse reactions (incidence ≥ 5%) more frequently reported in women treated with DUAVEE than placebo are presented in Table 1.
Table 1: Adverse Reactions (Incidence ≥ 5%) More Common in the DUAVEE Treatment Group in Placebo-controlled Trials DUAVEE (N=1224)
n (%)Placebo (N=1069)
n (%)Gastrointestinal disorders Nausea 100 (8) 58 (5) Diarrhea 96 (8) 57 (5) Dyspepsia 84 (7) 59 (6) Abdominal pain upper 81 (7) 58 (5) Musculoskeletal and connective tissue disorders Muscle spasms 110 (9) 63 (6) Neck pain 62 (5) 46 (4) Nervous system disorders Dizziness 65 (5) 37 (3) Respiratory, thoracic, and mediastinal disorders Oropharyngeal pain 80 (7) 61 (6) Venous thromboembolism: In the clinical studies with DUAVEE, the reporting rates for venous thromboembolism (deep venous thrombosis, pulmonary embolism, and retinal vein thrombosis) were low in all treatment groups. Adverse reactions of venous thromboembolism were reported in 0.0% of patients treated with DUAVEE and 0.1% of patients treated with placebo. Due to the low rate of events in both groups, it is not possible to conclude that the risk of venous thromboembolism with DUAVEE is different from that seen with other estrogen therapies [see Warnings and Precautions (5.2)].
-
7 DRUG INTERACTIONS
No drug interaction studies were conducted with DUAVEE. Results from in vitro and in vivo studies and clinical studies conducted with the conjugated estrogens or bazedoxifene components of DUAVEE are noted below:
7.1 Cytochrome P450 (CYP)
In vitro and in vivo studies have shown that estrogens are metabolized partially by cytochrome P450 3A4 (CYP3A4). Therefore, inducers or inhibitors of CYP3A4 may affect estrogen drug metabolism. Inducers of CYP3A4, such as St. John's Wort (Hypericum perforatum) preparations, phenobarbital, carbamazepine, and rifampin, may reduce plasma concentrations of estrogens, possibly resulting in a decrease in therapeutic effects and/or changes in the uterine bleeding profile.
Inhibitors of CYP3A4, such as erythromycin, clarithromycin, ketoconazole, itraconazole, ritonavir and grapefruit juice, may increase the exposure of conjugated estrogens resulting in an increased risk of endometrial hyperplasia. Therefore, for chronically administered CYP3A4 inhibitors (>30 days) concurrently administered with DUAVEE, adequate diagnostic measures, including directed or random endometrial sampling when indicated by signs and symptoms of endometrial hyperplasia, should be undertaken to rule out malignancy in postmenopausal women with undiagnosed persistent or recurring abnormal genital bleeding.
Bazedoxifene undergoes little or no cytochrome P450 (CYP)-mediated metabolism. Bazedoxifene does not induce or inhibit the activities of major CYP isoenzymes. In vitro data suggest that bazedoxifene is unlikely to interact with co-administered drugs via CYP-mediated metabolism.
7.2 Uridine Diphosphate Glucuronosyltransferase (UGT)
Bazedoxifene undergoes metabolism by UGT enzymes in the intestinal tract and liver. The metabolism of bazedoxifene may be increased by concomitant use of substances known to induce UGTs, such as rifampin, phenobarbital, carbamazepine, and phenytoin. A reduction in bazedoxifene exposure may be associated with an increase risk of endometrial hyperplasia. Adequate diagnostic measures, including directed or random endometrial sampling when indicated, should be undertaken to rule out malignancy in postmenopausal women with undiagnosed persistent or recurring abnormal genital bleeding.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category X [see Contraindications (4)]
DUAVEE must not be used in women who are or may become pregnant.
No studies were performed on animals to evaluate the effects on reproduction with conjugated estrogens/bazedoxifene.
Administration of bazedoxifene to rats at maternally toxic dosages ≥ 1 mg/kg/day (≥ 0.3 times the human area under the curve (AUC) at the 20 mg dose) resulted in reduced numbers of live fetuses and/or reductions in fetal body weights. No fetal developmental anomalies were observed. In studies conducted with pregnant rabbits treated with bazedoxifene, abortion and an increased incidence of heart (ventricular septal defect) and skeletal system (ossification delays, misshapen or misaligned bones, primarily of the spine and skull) anomalies in the fetuses were present at maternally toxic dosages of ≥ 0.5 mg/kg/day (2 times the human AUC at the 20 mg dose).
8.3 Nursing Mothers
DUAVEE should not be used by lactating women [see Contraindications (4)]. It is not known whether this drug is excreted in human milk. Detectable amounts of estrogens have been identified in the milk of mothers receiving conjugated estrogens. Estrogen administration to nursing mothers has been shown to decrease the quantity and quality of the milk.
8.5 Geriatric Use
DUAVEE is not recommended for use in women greater than 75 years of age [see Dosage and Administration (2.7) and Clinical Pharmacology 12.3)].
Of the total number of women in phase 3 clinical studies who received DUAVEE, 4.60% (n=224) were 65 years and over. DUAVEE was not studied in women aged 75 and over. No overall differences in safety or effectiveness were observed between women 65–74 years of age and younger women, and other reported clinical experience has not identified differences in responses between the elderly and younger women, but greater sensitivity of some older women cannot be ruled out.
An increased risk of probable dementia in women over 65 years of age was reported in the Women's Health Initiative Memory ancillary studies of the Women's Health Initiative using daily conjugated estrogens (0.625 mg) [see Clinical Studies (14.6)].
8.6 Renal Impairment
DUAVEE is not recommended for use in patients with renal impairment [see Dosage and Administration (2.6) and Clinical Pharmacology (12.3)].
The pharmacokinetics, safety, and efficacy of DUAVEE have not been evaluated in women with renal impairment.
8.7 Hepatic Impairment
DUAVEE is contraindicated in patients with hepatic impairment [see Contraindications (4) and Clinical Pharmacology (12.3)].
The pharmacokinetics, safety, and efficacy of DUAVEE have not been evaluated in women with hepatic impairment. In a pharmacokinetics study of bazedoxifene 20 mg alone, the Cmax and AUC of bazedoxifene increased 67% and 143%, respectively, in women with mild hepatic impairment (Child Pugh Class A), compared to healthy women. The Cmax and AUC of bazedoxifene increased 32% and 109%, respectively, in women with moderate hepatic impairment (Child Pugh Class B). The Cmax and AUC of bazedoxifene increased 20% and 268%, respectively, in women with severe hepatic impairment (Child Pugh Class C).
No pharmacokinetic studies with conjugated estrogens were conducted in women with hepatic impairment.
8.8 Use in Women with Body Mass Index (BMI) > 27 kg/m2
A 17% reduction in bazedoxifene exposure was predicted in women with BMI > 27 kg/m2 (N=144) compared to those with BMI ≤ 27 kg/m2 (N=93) after administration of DUAVEE, based on a population pharmacokinetic model using data from four Phase 1 studies. A reduction in bazedoxifene exposure may be associated with an increased risk of endometrial hyperplasia. Regardless of BMI, adequate diagnostic measures, including directed or random endometrial sampling when indicated, should be undertaken to rule out malignancy in postmenopausal women with undiagnosed persistent or recurring abnormal genital bleeding.
- 10 OVERDOSAGE
-
11 DESCRIPTION
DUAVEE (conjugated estrogens/bazedoxifene), contains conjugated estrogens with bazedoxifene, an estrogen agonist/antagonist.
Conjugated estrogens are purified from pregnant mares' urine and consist of the sodium salts of water-soluble estrogen sulfates blended to represent the average composition of material derived from pregnant mares' urine. Conjugated estrogens are a mixture of sodium estrone sulfate and sodium equilin sulfate, and also contain as concomitant components, sodium sulfate conjugates, 17α-dihydroequilin, 17α-estradiol, and 17β-dihydroequilin.
Bazedoxifene is supplied as the acetate salt (bazedoxifene acetate) and has the chemical name 1H-Indol-5-ol, 1-[[4-[2-(hexahydro-1H-azepin-1-yl) ethoxy]phenyl]methyl]-2-(4-hydroxyphenyl)-3-methyl-, monoacetate. The empirical formula is C30H34N2O3 ∙ C2H4O2, and the molecular weight is 530.65.
Bazedoxifene acetate is a white to tan powder. The aqueous solubility of bazedoxifene is pH-dependent. Solubility is higher at lower pH. The solubility of bazedoxifene acetate in unbuffered sterile water was measured to be 923 μgA/mL at pH 5.4. The following represents the chemical structure of bazedoxifene acetate:
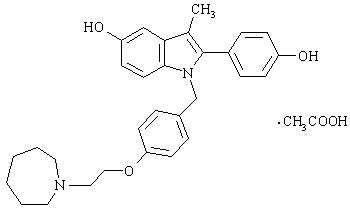
DUAVEE is available for oral administration as tablets containing 0.45 mg of conjugated estrogens with 20 mg of bazedoxifene (equivalent to 22.6 mg of bazedoxifene acetate). Each tablet of DUAVEE contains the following inactive ingredients: calcium phosphate tribasic, hydroxypropyl cellulose, microcrystalline cellulose, powdered cellulose, hypromellose, lactose monohydrate, magnesium stearate, polyethylene glycol, sucrose, ascorbic acid, sucrose palmitic acid ester, hydroxyethylcellulose, titanium dioxide, red iron oxide, yellow iron oxide, black iron oxide, povidone, polydextrose, maltitol, poloxamer 188, propylene glycol, and isopropyl alcohol.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
DUAVEE pairs conjugated estrogens with bazedoxifene. Conjugated estrogens and bazedoxifene function by binding to and activating estrogen receptors (ER) α and β, which vary in proportion from tissue to tissue. Conjugated estrogens are composed of multiple estrogens and are agonists of ER- α and β. Bazedoxifene is an estrogen agonist/antagonist that acts as an agonist in some estrogen-sensitive tissues and an antagonist in others (e.g., uterus). The pairing of conjugated estrogens with bazedoxifene produces a composite effect that is specific to each target tissue. The bazedoxifene component reduces the risk of endometrial hyperplasia that can occur with the conjugated estrogens component.
Pharmacodynamic studies have not been conducted with DUAVEE.
12.3 Pharmacokinetics
Absorption
Following administration of multiple doses of conjugated estrogens 0.45 mg/bazedoxifene 20 mg to healthy women who were naturally postmenopausal or who had undergone bilateral oophorectomy, the mean steady state pharmacokinetic parameters at Day 10 for conjugated estrogens (baseline adjusted for total estrone) and bazedoxifene are summarized in Table 2.
Table 2: Mean ± SD Steady-State Pharmacokinetic Parameters (n=24) Cmax
(ng/mL)Tmax
(hr)AUCss
(ng∙hr/mL)Baseline-Adjusted Total Estrone 2.6 ± 0.8 6.5 ± 1.6 35 ± 12 Bazedoxifene 6.9 ± 3.9 2.5 ± 2.1 71 ± 34 Results from monotherapy studies with conjugated estrogens or bazedoxifene components of DUAVEE, are noted below:
Conjugated estrogens are soluble in water and are well-absorbed from the gastrointestinal tract after release from the drug formulation.
Bazedoxifene exhibits a linear increase in plasma concentrations for single doses from 0.5 mg up to 120 mg and multiple daily doses from 1 mg to 80 mg. The absolute bioavailability of bazedoxifene is approximately 6%.
Food Effect
In a single-dose, crossover study in 23 postmenopausal women given conjugated estrogens 0.625 mg/bazedoxifene 20 mg with a high fat/high calorie meal, food increased AUC0–inf of bazedoxifene by 25%. The Cmax of bazedoxifene was unchanged.
Distribution
The distribution of conjugated estrogens and bazedoxifene after administration of DUAVEE has not been studied.
Results from monotherapy studies with conjugated estrogens or bazedoxifene, components of DUAVEE, are noted below:
The distribution of exogenous estrogens is similar to that of endogenous estrogens. Estrogens are widely distributed in the body and are generally found in higher concentrations in the sex hormone target organs. Estrogens circulate in the blood largely bound to sex hormone binding globulin (SHBG) and albumin.
Following intravenous (IV) administration of a 3 mg dose of bazedoxifene, the volume of distribution is 14.7 ± 3.9 L/kg. Bazedoxifene is highly bound (98%–99%) to plasma proteins in vitro, but does not bind to SHBG.
Metabolism
The metabolic disposition of conjugated estrogens and bazedoxifene, after administration of DUAVEE, has not been studied.
Results from monotherapy studies with conjugated estrogens or bazedoxifene, components of DUAVEE, are noted below:
Exogenous estrogens are metabolized in the same manner as endogenous estrogens. Circulating estrogens exist in a dynamic equilibrium of metabolic interconversions. 17-β estradiol is converted reversibly to estrone, and both can be converted to estriol, which is the major urinary metabolite. In postmenopausal women, a significant proportion of the circulating estrogens exists as sulfate conjugates, especially estrone sulfate, which serves as a circulating reservoir for the formation of more active estrogens.
The metabolic disposition of bazedoxifene has been determined following oral administration of 20 mg of radiolabeled bazedoxifene. Bazedoxifene is extensively metabolized in women. Glucuronidation is the major metabolic pathway. Little or no cytochrome P450-mediated metabolism is evident. Bazedoxifene-5-glucuronide is the major circulating metabolite. The concentrations of this glucuronide are approximately 10-fold higher than those of unchanged drug in plasma.
Excretion
After administration of a single dose of conjugated estrogens/bazedoxifene, baseline-adjusted total estrone (representing conjugated estrogens) is eliminated with a half-life of approximately 17 hours. Bazedoxifene is eliminated with a half-life of approximately 30 hours. Steady-state concentrations are achieved by the second week of once-daily administration.
Results from monotherapy studies with conjugated estrogens or bazedoxifene, components of DUAVEE, are noted below:
The conjugated estrogens components, 17β-estradiol, estrone, and estriol are excreted in the urine, along with glucuronide and sulfate conjugates.
The clearance of bazedoxifene is 0.4 ± 0.1 L/h/kg based on intravenous administration. The major route of excretion after oral administration of 20 mg of radiolabeled bazedoxifene is via biliary excretion, followed by elimination in the feces (~85%), with < 1% of the radioactive dose eliminated in the urine. Based on these results, it is expected that bazedoxifene undergoes entero-hepatic recycling from the gut back to the systemic circulation, therefore, some drugs may potentially interfere with bazedoxifene recycling process in the gut by various mechanisms resulting in a decrease in its systemic exposure.
Use in Specific Populations
Pediatric
The pharmacokinetics of conjugated estrogens/bazedoxifene tablets have not been evaluated in a pediatric population [see Use in Specific Populations (8.4)].
Geriatric
The effect of age on the pharmacokinetics of conjugated estrogens/bazedoxifene tablets have not been evaluated [see Use in Specific Populations (8.5)].
No pharmacokinetic studies with conjugated estrogens were conducted in specific populations, including women over 75 years of age.
The pharmacokinetics of a 20 mg single-dose of bazedoxifene, were evaluated in postmenopausal women. On average, compared to women 51 to 64 years of age (n=8), women 65 to 74 years of age (n=8) showed a 1.5-fold increase in AUC, and women ≥ 75 years of age (n=8) showed a 2.6-fold increase in AUC.
Renal Impairment
The pharmacokinetics of conjugated estrogens/bazedoxifene tablets have not been evaluated in women with renal impairment [see Dosage and Administration (2.6) and Use in Specific Populations (8.6)].
Hepatic Impairment
The pharmacokinetics of conjugated estrogens/bazedoxifene tablets have not been evaluated in women with hepatic impairment [see Contraindications (4), Warnings and Precautions (5.5), and Use in Specific Populations (8.7)].
No pharmacokinetic studies with conjugated estrogens were conducted in specific populations, including women with hepatic impairment.
A single dose of bazedoxifene 20 mg was given to fasted, healthy (N=18) and hepatically impaired postmenopausal women. In six mild hepatic impairment patients (Child Pugh Class A), Cmax and AUC of bazedoxifene increased 67% and 143%, respectively, compared to healthy subjects. In six moderate hepatic impairment patients (Child Pugh Class B), Cmax and AUC of bazedoxifene increased 32% and 109%, respectively, compared to healthy subjects. In six severe hepatic impairment patients (Child Pugh Class C), Cmax and AUC of bazedoxifene increased 20% and 268%, respectively, compared to healthy subjects. Half-life was prolonged from 32 to 50 hrs in patients with severe hepatic impairment, compared to healthy subjects.
Drug Interactions
No drug-drug interaction studies were conducted with conjugated estrogens/bazedoxifene tablets.
Effect of Co-Administered Drugs on the Pharmacokinetics of Bazedoxifene
Conjugated Estrogens
Conjugated estrogens 0.625 mg were administered alone for 6 consecutive days prior to the co-administration of a single dose of 20 mg bazedoxifene and conjugated estrogens 0.625 mg in thirty postmenopausal women. Conjugated estrogens 0.625 mg were continued for 2 additional days after the co-administration of bazedoxifene and conjugated estrogens. The Cmax of bazedoxifene increased by 3% and AUC of bazedoxifene decreased by 6%.
Ibuprofen
A single dose of ibuprofen 600 mg was given with a bazedoxifene 20 mg capsule in twelve postmenopausal women after an overnight fast. Co-administration of ibuprofen and bazedoxifene increased Cmax and AUC of bazedoxifene by 18% and 7%, respectively.
Atorvastatin
Atorvastatin 20 mg was given once with bazedoxifene 40 mg in thirty postmenopausal women. Co-administration of atorvastatin and bazedoxifene decreased Cmax of bazedoxifene by 3% and increased AUC of bazedoxifene by 6%.
Azithromycin
Azithromycin 500 mg was given once daily for 8 consecutive days in thirty postmenopausal women. Azithromycin 500 mg and a bazedoxifene 40 mg tablet were co-administered on Day 9. Azithromycin 250 mg administration once daily continued on Days 10 to 13. Co-administration of azithromycin and bazedoxifene increased Cmax of bazedoxifene by 6% and decreased AUC of bazedoxifene by 15%.
Aluminum and Magnesium Hydroxide
A single dose of 460 mg aluminum hydroxide and 400 mg magnesium hydroxide was given with a bazedoxifene 40 mg tablet in thirty postmenopausal women after an overnight fast. Co-administration of aluminum/magnesium hydroxide and bazedoxifene decreased Cmax of bazedoxifene by 8% and increased AUC of bazedoxifene by 7%.
Effect of Bazedoxifene on the Pharmacokinetics of Co-Administered Drugs
Conjugated Estrogens
Bazedoxifene 20 mg was administered alone for 8 consecutive days prior to co-administration of a single dose of conjugated estrogens 0.625 mg and bazedoxifene 20 mg in twenty-six postmenopausal women. Bazedoxifene 20 mg was continued for 2 additional days after co-administration of bazedoxifene and conjugated estrogens. The Cmax and AUC of unconjugated estrone increased by 11% and 3%, respectively. The Cmax and AUC of unconjugated equilin increased by 17% and 14%, respectively.
Ibuprofen
A single dose of bazedoxifene 20 mg capsule was given with a single dose of ibuprofen 600 mg in twelve fasted, postmenopausal women. Co-administration of bazedoxifene and ibuprofen increased the Cmax of ibuprofen by 6%. The AUC of ibuprofen was unchanged.
Atorvastatin
Bazedoxifene 40 mg was given for 8 consecutive days prior to co-administration of bazedoxifene 40 mg and atorvastatin 20 mg. Co-administration of bazedoxifene and atorvastatin decreased Cmax of atorvastatin by 14%. The AUC of atorvastatin was unchanged. The Cmax and AUC of 2-OH atorvastatin were decreased by 18% and 8%, respectively.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies with conjugated estrogens/bazedoxifene have not been conducted.
Long-term continuous administration of natural and synthetic estrogens in certain animal species increases the frequency of carcinomas of the breast, uterus, cervix, vagina, testis, and liver.
In 6-month oral gavage carcinogenicity studies of bazedoxifene in transgenic Tg.RasH2 mice, there was a drug-related increased incidence of benign, ovarian granulosa-cell tumors in female mice given 150 or 500 mg/kg/day. In a two-year dietary carcinogenicity study of bazedoxifene in rats (administered at 0.003%, 0.01%, 0.03%, or 0.1%) a drug-related marked increased incidence of benign, ovarian granulosa-cell tumors was observed in female rats at concentrations of 0.03% and 0.1%. Systemic exposure (AUC) of bazedoxifene in these groups was 3 and 8 times that observed in postmenopausal women administered 20 mg/day. In male rats, drug-related renal tumors (adenomas and carcinomas), in the presence of renal toxicity, were observed at all doses tested, which corresponded to exposure ratios of 0.06 to 5 times the clinical AUC at a dose of 20 mg.
Mutagenesis
Mutagenicity studies with conjugated estrogens/bazedoxifene have not been conducted.
Bazedoxifene was not genotoxic or mutagenic in a battery of tests, including in vitro bacterial reverse mutation assay, in vitro mammalian cell forward mutation assay at the thymidine kinase (TK+/-) locus in L5178Y mouse lymphoma cells, in vitro chromosome aberration assay in Chinese hamster ovary (CHO) cells, and in vivo mouse micronucleus assay.
Impairment of Fertility
Impairment of fertility studies with conjugated estrogens/bazedoxifene have not been conducted.
Female rats were administered daily dosages of 0.3 to 30 mg/kg bazedoxifene (0.03 to 10 times human AUC at the 20 mg dose) prior to and during mating with untreated males. Estrous cycles and fertility were adversely affected in all bazedoxifene-treated female groups.
13.2 Animal Toxicology and/or Pharmacology
In a 12-month study in ovariectomized rats, co-administration of conjugated estrogens (2.5 mg/kg/day) and bazedoxifene (0.1, 0.3, or 1 mg/kg/day) prevented the loss of bone mass at the spine, femur, and tibia with concomitant maintenance of biomechanical strength parameters.
-
14 CLINICAL STUDIES
14.1 Treatment of Moderate to Severe Vasomotor Symptoms Associated with Menopause in Women with a Uterus
The safety and efficacy of DUAVEE as a treatment for moderate to severe vasomotor symptoms associated with menopause was established in a 12-week randomized, double-blind, placebo-controlled study (Study 3). Study 3 enrolled a total of 318 women, age 42–64 (mean age of 53 years), who had at least 7 moderate to severe hot flushes per day or at least 50 per week at baseline. The mean number of years since menopause was 4.5 years with all women undergoing natural menopause. A total of 127 women were assigned to DUAVEE and 63 women were assigned to placebo.
In Study 3, DUAVEE significantly reduced the number and severity of moderate to severe hot flushes, as measured by the daily severity score, compared with placebo at Weeks 4 and 12. The change from baseline in the number and severity of moderate to severe hot flushes observed and the difference from placebo in Study 3 are shown in Table 3.
Table 3: Adjusted Mean Change from Baseline in the Average Daily Frequency and Severity of Hot Flushes (Study 3) Frequency Severity DUAVEE Placebo DUAVEE Placebo - * Change from baseline using raw data
- † Based on raw data analysis using ANCOVA model: Difference= Treatment + Baseline + Site
- ‡ p<0.001
N 122 63 122 63 Baseline 10.3 10.5 2.3 2.3 Week 4 Mean Change* -5.9 -2.8 -0.6 -0.1 Treatment Difference† -3.1 (-4.4, -1.7)‡ -- -0.5 (-0.7, -0.3)‡ -- Week 12 Mean Change* -7.6 -4.9 -0.9 -0.3 Treatment Difference† -2.7 (-3.8, -1.6)‡ -- -0.6 (-0.9, -0.4)‡ -- 14.2 Prevention of Postmenopausal Osteoporosis in Women with a Uterus
The safety and efficacy of DUAVEE for the prevention of postmenopausal osteoporosis was demonstrated in Study 1 and Study 2.
Study 1 was a 24-month, double-blind, randomized, placebo- and active-controlled study evaluating the safety and efficacy of multiple combinations of conjugated estrogen/bazedoxifene (including conjugated estrogens 0.45 mg/bazedoxifene 20 mg) compared to placebo. The primary endpoint of the study was the incidence of endometrial hyperplasia at Year 1. Bone mineral density change at the lumbar spine at Year 2 was the key secondary endpoint, assessed in two subsets of patients (Substudy I and Substudy II). Patients enrolled into Substudy I had to be more than 5 years postmenopausal, have a lumbar spine or total hip T-score of -1 to -2.5, and have at least one additional risk factor for osteoporosis (e.g., Caucasian race, family history of osteoporosis, early menopause, thin/small frame, inactive lifestyle, tobacco abuse). Those enrolled into Substudy II had to be 1–5 years postmenopausal with at least one additional risk factor for osteoporosis. A total of 3,397 women age 40–75 (mean age of 56 years) were enrolled in the overall study. Substudy I enrolled a total of 1,454 women (182 women receiving DUAVEE) with mean baseline T-scores of -1.43 and -1.52 in the DUAVEE and placebo groups, respectively. Substudy II enrolled a total of 861 women (with 111 women receiving DUAVEE) with mean baseline T-scores of -0.81 and -0.94 in the DUAVEE and placebo groups, respectively. Women also took calcium (600–1200 mg) and vitamin D (200–400 IU) daily.
In these substudies, treatment with DUAVEE significantly increased lumbar spine bone mineral density (BMD) at 24 months compared to placebo in both groups of postmenopausal women (Table 4).
Table 4: Lumbar Spine Bone Mineral Density Results at 24 Months (Study 1) DUAVEE Placebo ** Adjusted mean changes, confidence intervals, and p-values based on an ANCOVA model with treatment and region (U.S. or non-U.S.) as factors and baseline BMD value and years since menopause as covariates using the Modified Intention to Treat population with Last Observation Carried Forward. Study 1 excludes those subjects with missing source documentation. - * p-value < 0.001
Between 1 and 5 Years Postmenopausal N 95 95 % Mean Change 1.72 -1.90 Difference from Placebo (95% C.I.) 3.62 (2.64, 4.60)* More Than 5 Years Postmenopausal N 155 151 % Mean Change 1.64 -1.47 Difference from Placebo (95% C.I.) 3.11 (2.29, 3.93)* ─ In Study 1, treatment with DUAVEE also significantly increased total hip BMD. The treatment difference (or difference from placebo) in total hip BMD at 24 months was 1.96% (DUAVEE minus placebo) in women who had been postmenopausal between 1 and 5 years and 1.73% (DUAVEE minus placebo) in women who had been postmenopausal for more than 5 years.
Study 2 was a 12-month, double-blind, randomized, placebo- and active-controlled study. The primary endpoint was the incidence of endometrial hyperplasia at 12 months. The prevention of osteoporosis was assessed in a substudy that enrolled women (n=590) who were less than 5 years postmenopausal (mean 2.5 years). The mean baseline T-score in the substudy was -0.91 in the DUAVEE group and -0.95 in the placebo group. The mean age of women (n=135) taking DUAVEE was 53 years (range 46–60 years). Women also took calcium (600 mg) and vitamin D (400 IU) daily.
In Study 2, treatment with DUAVEE significantly increased mean lumbar spine BMD (treatment difference, 1.51%), at 12 months compared to placebo in women who had been postmenopausal between 1 and 5 years. Treatment with DUAVEE also increased total hip BMD. The treatment difference in total hip BMD at 12 months was 1.21%.
14.3 Effects on the Endometrium
Effects of DUAVEE on endometrial hyperplasia and endometrial malignancy were assessed in Study 1 and Study 2. The Efficacy Evaluable population included patients who had taken at least one dose of DUAVEE, had baseline and post baseline endometrial biopsies, or had been diagnosed with hyperplasia. By endometrial biopsy, the incidence of endometrial hyperplasia or malignancy for DUAVEE was below 1% in both studies (see Table 5).
14.4 Effects on Uterine Bleeding and Spotting
Uterine bleeding or spotting were evaluated in two clinical studies (Studies 1 and 2) by daily diary. In Study 1, cumulative amenorrhea at Year 1 was 83% in women treated with DUAVEE and 85% in women who received placebo. In Study 2, cumulative amenorrhea at Year 1 was 88% in women treated with DUAVEE and 84% in women who received placebo.
14.5 Women's Health Initiative Studies
The WHI enrolled approximately 11,000 predominantly healthy postmenopausal women to assess the risks and benefits of daily oral conjugated estrogens 0.625 mg compared to placebo in the prevention of certain chronic diseases. The primary endpoint was the incidence of CHD (defined as nonfatal MI, silent MI and CHD death), with invasive breast cancer as the primary adverse outcome. A "global index" included the earliest occurrence of CHD, invasive breast cancer, stroke, PE, colorectal cancer, hip fracture, or death due to other cause. These substudies did not evaluate the effects of conjugated estrogens on menopausal symptoms.
The WHI estrogen-alone substudy was stopped early because an increased risk of stroke was observed, and it was deemed that no further information would be obtained regarding the risks and benefits of estrogen-alone in predetermined primary endpoints.
Results of the estrogen-alone substudy, which included 10,739 women (average 63 years of age, range 50 to 79; 75.3 percent White, 15.1 percent Black, 6.1 percent Hispanic, 3.6 percent Other), after an average follow- up of 7.1 years are presented in Table 6.
Table 6: Relative and Absolute Risk Seen in the Estrogen Alone Substudy of WHI Event Relative Risk
CE vs. Placebo
(95% nCI*)CE
n = 5,310Placebo
N = 5,429Absolute Risk per 10,000
Women-Yearsa Adapted from numerous WHI publications. WHI publications can be viewed at www.nhlbi.nih.gov/whi. For those outcomes included in the WHI "global index" that reached statistical significance, the absolute excess risk per 10,000 women-years in the group treated with CE-alone was 12 more strokes, while the absolute risk reduction per 10,000 women-years was 7 fewer hip fractures. The absolute excess risk of events included in the "global index" was a non-significant 5 events per 10,000 women-years. There was no difference between the groups in terms of all-cause mortality. - * Nominal confidence intervals unadjusted for multiple looks and multiple comparisons.
- † Results are based on centrally adjudicated data for an average follow-up of 7.1 years.
- ‡ Not included in "global index".
- § Results are based on an average follow-up of 6.8 years.
- ¶ All deaths, except from breast or colorectal cancer, definite or probable CHD, PE, or cerebrovascular disease.
- # A subset of the events was combined in a "global index", defined as the earliest occurrence of CHD events, invasive breast cancer, stroke, PE, endometrial cancer, colorectal cancer, hip fracture, or death due to other causes.
CHD events† 0.95 (0.78–1.16) 54 57 Non-fatal MI† 0.91 (0.73–1.14) 40 43 CHD death† 1.01 (0.71–1.43) 16 16 All strokes† 1.33 (1.15–1.68) 45 33 Ischemic stroke† 1.55 (1.19–2.01) 38 25 Deep vein thrombosis†,‡ 1.47 (1.06–2.06) 23 15 Pulmonary embolism† 1.37 (0.90–2.07) 14 10 Invasive breast cancer† 0.80 (0.62–1.04) 28 34 Colorectal cancer§ 1.08 (0.75–1.55) 17 16 Hip fracture† 0.65 (0.45–0.94) 12 19 Vertebral fractures†,‡ 0.64 (0.44–0.93) 11 18 Lower arm/wrist fractures†,‡ 0.58 (0.47–0.72) 35 59 Total fractures†,‡ 0.71 (0.64–0.80) 144 197 Death due to other causes§,¶ 1.08 (0.88–1.32) 53 50 Overall mortality†,‡ 1.04 (0.88–1.22) 79 75 Global Index# 1.02 (0.92–1.13) 206 201 No overall difference for primary CHD events (nonfatal MI, silent MI and CHD death) and invasive breast cancer incidence in women receiving conjugated estrogens-alone compared to placebo was reported in final centrally adjudicated results from the estrogen-alone substudy, after an average follow-up of 7.1 years.
Centrally adjudicated results for stroke events from the estrogen-alone substudy, after an average follow-up of 7.1 years, reported no significant differences in distribution of stroke subtype or severity, including fatal strokes, in women receiving conjugated estrogens-alone compared to placebo. Estrogen-alone increased the risk for ischemic stroke, and this excess risk was present in all subgroups of women examined.
Timing of the initiation of estrogen-alone therapy relative to the start of menopause may affect the overall risk benefit profile. The WHI estrogen-alone substudy, stratified by age, showed in women 50 to 59 years of age a non-significant trend toward reduced risk for CHD [hazard ratio (HR) 0.63 (95 percent CI, 0.36–1.09)] and overall mortality [HR 0.71 (95 percent CI, 0.46–1.11)].
14.6 Women's Health Initiative Memory Study
The WHIMS estrogen-alone ancillary study of WHI enrolled 2,947 predominantly healthy hysterectomized postmenopausal women 65 to 79 years of age (45 percent were 65 to 69 years of age, 36 percent were 70 to 74 years of age, and 19 percent were 75 years of age and older) to evaluate the effects of daily conjugated estrogens (0.625 mg)-alone on the incidence of probable dementia (primary outcome) compared to placebo.
After an average follow-up of 5.2 years, the relative risk of probable dementia for conjugated estrogens-alone versus placebo was 1.49 (95 percent CI, 0.83–2.66). The absolute risk of probable dementia for conjugated estrogens-alone versus placebo was 37 versus 25 cases per 10,000 women-years. Probable dementia as defined in this study included Alzheimer's disease (AD), vascular dementia (VaD) and mixed type (having features of both AD and VaD). The most common classification of probable dementia in the treatment group and the placebo group was AD. Since the ancillary study was conducted in women 65 to 79 years of age, it is unknown whether these findings apply to younger postmenopausal women [see Warnings and Precautions (5.4) and Use in Specific Populations (8.5)].
-
16 HOW SUPPLIED/STORAGE AND HANDLING
DUAVEE tablets contain 0.45 mg conjugated estrogens and 20 mg bazedoxifene. The tablets are oval, biconvex, and pink, branded with "0.45/20" in black ink on one side.
DUAVEE® tablets are supplied as follows:
Package NDC number Conjugated estrogens 0.45 mg/bazedoxifene 20 mg 2 blisters of 15 tablets each NDC: 0008-1123-12 Storage
Blisters
DUAVEE tablets should be stored at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F). See USP Controlled Room Temperature. Dispense product in the original package. Tablets should not be removed from blisters until immediately before use. Protect from moisture. After opening foil pouch, product must be used within 60 days.
-
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Patient Information).
17.1 Instructions for Patients
- Keep DUAVEE in the original container to protect from moisture. Do not place DUAVEE in pill boxes or pill organizers.
- If more than one blister package is dispensed to the patient, instruct them to open one foil pouch at a time.
- Instruct patient to record the date the blister package is opened in the space provided on the blister package label. Do not use if the blister package has been open more than 60 days.
- Instruct patient to remove only one tablet from the blister package at the time of use.
17.2 Venous Thromboembolic Events
Advise patients to immediately report to their physician any signs or symptoms related to venous thrombosis and thromboembolic events [see Warnings and Precautions (5.2)].
17.3 Abnormal Vaginal Bleeding
Inform postmenopausal women of the importance of reporting abnormal vaginal bleeding to their healthcare provider as soon as possible [see Warnings and Precautions (5.3)].
17.4 Possible Serious Adverse Reactions with Estrogen Therapy
Inform postmenopausal women of possible serious adverse reactions of estrogen therapy including Cardiovascular Disorders, Malignant Neoplasms, and Probable Dementia [see Warnings and Precautions (5.2, 5.3, 5.4)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
Patient Information
DUAVEE® (DEW' ah-vee)
(conjugated estrogens/bazedoxifene)
TabletsRead this Patient Information before you start taking DUAVEE and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about DUAVEE?
- Do not take additional estrogen products while you are taking DUAVEE.
- Using estrogens may increase your chance of getting cancer of the uterus (womb).
- Report any unusual vaginal bleeding right away while you are taking DUAVEE. Vaginal bleeding after menopause may be a warning sign of cancer of the uterus (womb). Your healthcare provider should check any unusual vaginal bleeding to find out the cause.
- Do not use estrogens to prevent heart disease, heart attacks, strokes or dementia (decline in brain function).
- Using estrogens may increase your chances of getting strokes or blood clots.
- Using estrogens may increase your chance of getting dementia, based on a study of women 65 years of age or older.
- You and your healthcare provider should talk regularly about whether you still need treatment with DUAVEE.
What is DUAVEE?
DUAVEE is a prescription medicine that contains a mixture of estrogens and bazedoxifene.
What is DUAVEE used for?
DUAVEE is used after menopause for women with a uterus to:
-
reduce moderate to severe hot flushes
Estrogens are hormones made by a woman's ovaries. The ovaries normally stop making estrogens when a woman is between 45 and 55 years old. This drop in body estrogen levels causes the "change of life" or menopause (the end of monthly menstrual periods). Sometimes both ovaries are removed during an operation before natural menopause takes place. The sudden drop in estrogen levels causes "surgical menopause."
When the estrogen levels begin dropping, some women get very uncomfortable symptoms, such as feelings of warmth in the face, neck, and chest, or sudden intense episodes of heat and sweating ("hot flashes" or "hot flushes"). In some women, the symptoms are mild, and they will not need to take medicines. In other women, symptoms can be more severe. -
help reduce your chances of developing osteoporosis (thin, weak bones)
If you use DUAVEE only to prevent osteoporosis due to menopause, talk with your healthcare provider about whether a different treatment or medicine without estrogens might be better for you.
DUAVEE should be taken for the shortest time possible and only for as long as treatment is needed.
You and your healthcare provider should talk regularly about whether you still need treatment with DUAVEE.
DUAVEE is not for use in children.
It is not known if DUAVEE is safe and effective in people with kidney problems.
Who should not take DUAVEE?
Do not take DUAVEE if you:
- currently have or have had blood clots
- are allergic to estrogens or bazedoxifene, the active ingredients in DUAVEE, or any of its ingredients. See the list of ingredients in DUAVEE at the end of this leaflet.
- have unusual vaginal bleeding. Vaginal bleeding after menopause may be a warning sign of cancer of the uterus (womb). Your healthcare provider should check any unusual vaginal bleeding to find out the cause.
- currently have or have had certain cancers. Estrogens may increase the chances of getting certain types of cancers, including cancer of the breast or uterus. If you have or have had cancer, talk with your healthcare provider about whether you should use DUAVEE.
- currently have or have had liver problems
- have been diagnosed with a bleeding disorder
- think you may be pregnant. DUAVEE is not for pregnant women. If you think you may be pregnant, you should have a pregnancy test and know the results. Do not take DUAVEE if the test is positive and talk to your healthcare provider.
- are breastfeeding or plan to breastfeed. It is not known if DUAVEE passes into your breast milk. You and your healthcare provider should decide if you will take DUAVEE or breastfeed. You should not do both.
What should I tell my healthcare provider before taking DUAVEE?
Before you take DUAVEE, tell your healthcare provider if you:
- have any unusual vaginal bleeding.
- have any other medical conditions. Your healthcare provider may need to check you more carefully if you have certain conditions, such as asthma (wheezing), epilepsy (seizures), diabetes, migraine, endometriosis, lupus, or problems with your heart, liver, thyroid, kidneys, or have high calcium levels in your blood.
- are going to have surgery or will be on bed rest. Your healthcare provider will let you know if you need to stop taking DUAVEE.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Especially tell your healthcare provider if you take other hormonal medicines, including progestins or other medicines like DUAVEE. Ask your healthcare provider if you do not know if you take any of these medicines.
Some medicines may affect how DUAVEE works. DUAVEE may also affect how your other medicines work. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take DUAVEE?
- DUAVEE comes in a blister package.
- Record the date you open the foil pouch in the space provided on the blister package label. Do not use if the blister package has been open for more than 60 days.
- Take DUAVEE exactly as your healthcare provider tells you to take it.
- Take 1 DUAVEE tablet at the same time each day.
- DUAVEE should be swallowed whole.
- Take DUAVEE with or without food.
- You should not remove DUAVEE from the blister until right before you are ready to take it. Remove 1 tablet at a time from the blister package. Do not place DUAVEE in pill boxes or pill organizers.
- If you miss a dose of DUAVEE, take it as soon as you remember. If it is almost time for your next dose, skip the missed dose. Take the next dose at your regular time. Do not take 2 doses at the same time unless your healthcare provider tells you to. If you are not sure about your dosing, call your healthcare provider.
- If you take a calcium or vitamin D supplement, you may take it at the same time you take DUAVEE.
- If you take too much DUAVEE, call your healthcare provider. Symptoms of taking too much DUAVEE include:
- nausea
- vomiting
- breast tenderness
- dizziness
- abdominal pain
- feeling tired
- vaginal bleeding
What are the possible side effects of DUAVEE?
Side effects are grouped by how serious they are and how often they happen when you are treated.
Serious side effects include:
- blood clots
- stroke
- heart attack
- cancer of the lining of the uterus
- breast cancer
- cancer of the ovary
- dementia
- gallbladder problems
- loss of vision
- high blood pressure
- increased fats in your blood
- liver problems
- thyroid problems
- fluid retention
- low calcium
- swelling of your mouth or tongue
- worsening of other medical problems such as asthma, diabetes, epilepsy, migraines, a genetic problem called porphyria, lupus and liver problems
Call your healthcare provider right away if you get any of the following warning signs, or any other unusual symptoms that concern you:
- new breast lumps
- unusual vaginal bleeding
- changes in vision or speech
- sudden new severe headaches
- severe pains in your chest or legs with or without shortness of breath, weakness and fatigue
Less serious, but common side effects include:
- muscle spasms
- nausea
- diarrhea
- upset stomach
- abdominal pain
- throat pain
- dizziness
- neck pain
These are not all the possible side effects of DUAVEE. For more information, ask your healthcare provider or pharmacist. Tell your healthcare provider if you have any side effects that bother you or do not go away.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
What can I do to lower my chances of a serious side effect with DUAVEE?
- Talk with your healthcare provider regularly about whether you should continue taking DUAVEE.
- See you healthcare provider right away if you get vaginal bleeding while taking DUAVEE.
- Have a pelvic exam, breast exam and mammogram (breast X-ray) every year unless your healthcare provider tells you something else.
- If members of your family have had breast cancer or if you have ever had breast lumps or an abnormal mammogram, you may need to have breast exams more often.
- If you have high blood pressure, high cholesterol (fat in the blood), diabetes, are overweight, or if you use tobacco, you may have higher chances of getting heart disease.
Ask your healthcare provider for ways to lower your chances of getting heart disease.
How do I store DUAVEE?
- Store DUAVEE at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep DUAVEE in the blister until you are ready to take it to protect the tablet from moisture.
- Do not place DUAVEE in pill boxes or pill organizers.
- After opening the foil pouch the DUAVEE blisters come in, DUAVEE must be used within 60 days.
Keep DUAVEE and all other medicines out of the reach of children.
General information about the safe and effective use of DUAVEE
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use DUAVEE for a condition for which it was not prescribed. Do not give DUAVEE to other people, even if they have the same symptoms you have. It may harm them.
This Patient Information summarizes the most important information about DUAVEE. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about DUAVEE that is written for health professionals.
For more information, go to www.DUAVEE.com, or call 1-800-438-1985.
What are the ingredients in DUAVEE?
Active Ingredients: conjugated estrogens and bazedoxifene. Conjugated estrogens are a mixture of sodium estrone sulfate and sodium equilin sulfate and other components, including sodium sulfate conjugates, 17α-dihydroequilin, 17α-estradiol, and 17β-dihydroequilin.
Inactive Ingredients: calcium phosphate tribasic, hydroxypropyl cellulose, microcrystalline cellulose, powdered cellulose, hypromellose, lactose monohydrate, magnesium stearate, polyethylene glycol, sucrose, ascorbic acid, sucrose palmitic acid ester, hydroxyethylcellulose, titanium dioxide, red iron oxide, yellow iron oxide, black iron oxide, povidone, polydextrose, maltitol, poloxamer 188, propylene glycol, isopropyl alcohol.
This Patient Information has been approved by the U.S. Food and Drug Administration.
This product's label may have been updated. For current full prescribing information, please visit www.pfizer.com.

LAB-0583-1.0
October 2013
-
PRINCIPAL DISPLAY PANEL - 7 Tablet Blister Card
PROFESSIONAL SAMPLE - NOT FOR SALE
NDC: 63539-122-07
Pfizer
Duavee™
(conjugated estrogens/
bazedoxifene)
tabletsper tablet
0.45 mg/20 mg*
*Each tablet contains bazedoxifene
equivalent to 22.6 mg of
bazedoxifene acetate7 tablets
After opening foil pouch, product must be used within 60 days.
Date foil pouch opened:
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to
15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
DOSAGE AND USE: See accompanying prescribing information.
Store product in original package to keep from moisture.
Tablets should not be removed from blisters until immediately
before administration. Do not place medication in pill boxes.Distributed by
U.S. Pharmaceuticals
Pfizer Inc, NY, NY 10017
MADE IN IRELANDRx only
PAA042983
LOT:
EXP:
-
PRINCIPAL DISPLAY PANEL - 7 Tablet Blister Pouch
PROFESSIONAL SAMPLE - NOT FOR SALE
NDC: 63539-122-07
Pfizer
Duavee™
(conjugated estrogens/
bazedoxifene)
tablets0.45 mg/20 mg*
After opening foil pouch, product must be used
within 60 days.Dispense and store product in original package
to protect from moisture.7 tablets
Rx onlyPackage includes 1 blister card
containing 7 tablets.
-
PRINCIPAL DISPLAY PANEL - 7 Tablet Carton
PROFESSIONAL SAMPLE - NOT FOR SALE
NDC: 63539-122-07Pfizer Duavee™ 0.45 mg/20 mg*
(conjugated estrogens/
bazedoxifene)
tabletsAfter opening foil pouch, product must be used
within 60 days.Dispense and store product in original package
to protect from moisture.Rx only
7 tablets
Package includes 1 blister card
containing 7 tablets.
-
INGREDIENTS AND APPEARANCE
DUAVEE
conjugated estrogens/bazedoxifene tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 63539-112 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ESTROGENS, CONJUGATED (UNII: IU5QR144QX) (ESTROGENS, CONJUGATED - UNII:IU5QR144QX) ESTROGENS, CONJUGATED 0.45 mg BAZEDOXIFENE ACETATE (UNII: J70472UD3D) (BAZEDOXIFENE - UNII:Q16TT9C5BK) BAZEDOXIFENE 20 mg Inactive Ingredients Ingredient Name Strength TRIBASIC CALCIUM PHOSPHATE (UNII: 91D9GV0Z28) HYDROXYPROPYL CELLULOSE (TYPE H) (UNII: RFW2ET671P) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POWDERED CELLULOSE (UNII: SMD1X3XO9M) HYPROMELLOSE 2208 (100000 MPA.S) (UNII: VM7F0B23ZI) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) SUCROSE (UNII: C151H8M554) ASCORBIC ACID (UNII: PQ6CK8PD0R) SUCROSE PALMITATE (UNII: 3OSQ643ZK5) HYDROXYETHYL CELLULOSE (100 MPA.S AT 2%) (UNII: R33S7TK2EP) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERROSOFERRIC OXIDE (UNII: XM0M87F357) POVIDONES (UNII: FZ989GH94E) POLYDEXTROSE (UNII: VH2XOU12IE) MALTITOL (UNII: D65DG142WK) POLOXAMER 188 (UNII: LQA7B6G8JG) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) ISOPROPYL ALCOHOL (UNII: ND2M416302) Product Characteristics Color PINK (pink) Score no score Shape OVAL (oval) Size 12mm Flavor Imprint Code 045;20 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 63539-112-07 1 in 1 CARTON 1 7 in 1 BLISTER PACK Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022247 11/15/2013 Labeler - U.S. Pharmaceuticals (829076905) Registrant - Wyeth Pharmaceuticals Inc. (113008515) Establishment Name Address ID/FEI Business Operations Wyeth Pharmaceuticals Inc., a subsidiary of Pfizer Inc. 828831441 ANALYSIS(63539-112) , API MANUFACTURE(63539-112) Establishment Name Address ID/FEI Business Operations Pfizer Ireland Pharmaceuticals 985098505 ANALYSIS(63539-112) , LABEL(63539-112) , MANUFACTURE(63539-112) , PACK(63539-112) Establishment Name Address ID/FEI Business Operations Pfizer Pharmaceuticals LLC 829084552 LABEL(63539-112) , PACK(63539-112)
Trademark Results [Duavee]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 DUAVEE 86618211 4836834 Live/Registered |
Wyeth LLC 2015-05-04 |
 DUAVEE 85840812 4544493 Live/Registered |
WYETH LLC 2013-02-05 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.