VEKLURY- remdesivir injection VEKLURY- remdesivir injection, powder, lyophilized, for solution
Veklury by
Drug Labeling and Warnings
Veklury by is a Prescription medication manufactured, distributed, or labeled by Gilead Sciences, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VEKLURY safely and effectively. See full prescribing information for VEKLURY.
VEKLURY® (remdesivir) for injection, for intravenous use
Initial U.S. Approval: 2020RECENT MAJOR CHANGES
INDICATIONS AND USAGE
VEKLURY is a severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) nucleotide analog RNA polymerase inhibitor indicated for the treatment of coronavirus disease 2019 (COVID-19) in adults and pediatric patients (birth to less than 18 years of age weighing at least 1.5 kg) who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death. (1)
DOSAGE AND ADMINISTRATION
- Testing: In all patients, before starting VEKLURY and during treatment as clinically appropriate, perform hepatic laboratory testing. Assess prothrombin time before starting VEKLURY and monitor as clinically appropriate. (2.2)
- Recommended dosage:
- Adults and pediatric patients weighing at least 40 kg: a single loading dose of VEKLURY 200 mg on Day 1 followed by once-daily maintenance doses of VEKLURY 100 mg from Day 2 via intravenous infusion. (2.3)
- Pediatric patients (birth to less than 18 years of age) weighing 1.5 kg to less than 40 kg: Recommended dosage is based on weight. Refer to Table 1 of the full prescribing information for specific dosing guidelines based on body weight. (2.3)
- Hospitalized patients: The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made. (2.3)
- For hospitalized patients requiring invasive mechanical ventilation and/or ECMO, the recommended total treatment duration is 10 days. (2.3)
- For hospitalized patients not requiring invasive mechanical ventilation and/or ECMO, the recommended treatment duration is 5 days. If a patient does not demonstrate clinical improvement, treatment may be extended for up to 5 additional days for a total treatment duration of up to 10 days. (2.3)
- Non-hospitalized patients: The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made and within 7 days of symptom onset. (2.3)
- For non-hospitalized patients diagnosed with mild-to-moderate COVID-19 who are at high risk for progression to severe COVID-19, including hospitalization or death, the recommended total treatment duration is 3 days (2.3).
- Renal impairment: No dosage adjustment of VEKLURY is recommended in patients with any degree of renal impairment, including those on dialysis. (2.4)
- Administer VEKLURY via intravenous (IV) infusion over 30 to 120 minutes. (2.5)
- Dose preparation and administration: Refer to the full prescribing information for further details. (2.5)
- Storage of prepared dosages: VEKLURY contains no preservative. (2.6)
DOSAGE FORMS AND STRENGTHS
For injection: 100 mg of remdesivir as a lyophilized powder, in a single-dose vial. (3)
CONTRAINDICATIONS
VEKLURY is contraindicated in patients with a history of clinically significant hypersensitivity reactions to VEKLURY or any components of the product. (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity including infusion-related and anaphylactic reactions: Hypersensitivity reactions have been observed during and following administration of VEKLURY. Slower infusion rates, with a maximum infusion time of up to 120 minutes, can be considered to potentially prevent signs and symptoms of hypersensitivity. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate. If signs and symptoms of a clinically significant hypersensitivity reaction occur, immediately discontinue administration of VEKLURY and initiate appropriate treatment. (5.1)
- Increased risk of transaminase elevations: Transaminase elevations have been observed in healthy volunteers and have also been reported in patients with COVID-19 who received VEKLURY. Perform hepatic laboratory testing in all patients before starting VEKLURY and while receiving VEKLURY as clinically appropriate. Consider discontinuing VEKLURY if ALT levels increase to greater than 10 times the upper limit of normal. Discontinue VEKLURY if ALT elevation is accompanied by signs or symptoms of liver inflammation. (5.2)
- Risk of reduced antiviral activity when coadministered with chloroquine phosphate or hydroxychloroquine sulfate: Coadministration of VEKLURY and chloroquine phosphate or hydroxychloroquine sulfate is not recommended based on data from cell culture experiments demonstrating a potential antagonistic effect of chloroquine on the intracellular metabolic activation and antiviral activity of VEKLURY. (5.3)
ADVERSE REACTIONS
The most common adverse reactions (incidence greater than or equal to 5%, all grades) observed with treatment with VEKLURY are nausea, ALT increased, and AST increased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage and Administration Overview
2.2 Testing Before Starting and During Treatment with VEKLURY
2.3 Recommended Dosage in Adults and Pediatric Patients (Birth to Less than 18 Years of Age Weighing at Least 1.5 kg)
2.4 Renal Impairment
2.5 Dosage Preparation and Administration
2.6 Storage of Prepared Dosages
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Including Infusion-related and Anaphylactic Reactions
5.2 Increased Risk of Transaminase Elevations
5.3 Risk of Reduced Antiviral Activity When Coadministered with Chloroquine Phosphate or Hydroxychloroquine Sulfate
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on VEKLURY
7.2 Effects of VEKLURY on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Description of Clinical Trials
14.2 NIAID ACTT-1 Study in Hospitalized Subjects with Mild/Moderate and Severe COVID-19
14.3 Study GS-US-540-5773 in Hospitalized Subjects with Severe COVID-19
14.4 Study GS-US-540-5774 in Hospitalized Subjects with Moderate COVID-19
14.5 Study GS-US-540-9012 in Non-Hospitalized Subjects with Mild-to-Moderate COVID-19 and at High Risk for Progression to Severe Disease
14.6 Study GS-US-540-5823 in Hospitalized Pediatric Subjects with COVID-19
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
VEKLURY is indicated for the treatment of coronavirus disease 2019 (COVID-19) in adults and pediatric patients (birth to less than 18 years of age weighing at least 1.5 kg) who are [see Clinical Studies (14)]:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage and Administration Overview
- VEKLURY may only be administered in settings in which healthcare providers have immediate access to medications to treat a severe infusion or hypersensitivity reaction, such as anaphylaxis, and the ability to activate the emergency medical system (EMS), as necessary [see Dosage and Administration (2.5), Warnings and Precautions (5.1)].
- Administer VEKLURY for the treatment of COVID-19 in adults and pediatric patients (birth to less than 18 years of age weighing at least 1.5 kg) by intravenous infusion only. Do not administer by any other route.
- VEKLURY for injection must be reconstituted with Sterile Water for Injection prior to diluting with 0.9% sodium chloride injection.
2.2 Testing Before Starting and During Treatment with VEKLURY
Perform hepatic laboratory testing in all patients before starting VEKLURY and while receiving VEKLURY as clinically appropriate [see Warnings and Precautions (5.2) and Use in Specific Populations (8.7)].
Determine prothrombin time in all patients before starting VEKLURY and monitor while receiving VEKLURY as clinically appropriate [see Adverse Reactions (6.1)].
2.3 Recommended Dosage in Adults and Pediatric Patients (Birth to Less than 18 Years of Age Weighing at Least 1.5 kg)
- The recommended dosage for adults and pediatric patients weighing at least 40 kg is a single loading dose of VEKLURY 200 mg on Day 1 via intravenous infusion followed by once-daily maintenance doses of VEKLURY 100 mg from Day 2 via intravenous infusion.
- The recommended dosage for pediatric patients weighing 1.5 kg to less than 40 kg is presented in Table 1.
Table 1 Recommended Dosage in Pediatric Patients Including Term* Neonates and Infants Weighing 1.5 kg to Less than 40 kg Pediatric Patient Population Loading Dose Via Intravenous Infusion Maintenance Dose Via Intravenous Infusion - * Gestational age greater than 37 weeks.
Less than 28 days old and at least 1.5 kg VEKLURY 2.5 mg/kg on Day 1 VEKLURY 1.25 mg/kg once daily from Day 2 At least 28 days old and 1.5 kg to less than 3 kg At least 28 days old and 3 kg to less than 40 kg VEKLURY 5 mg/kg on Day 1 VEKLURY 2.5 mg/kg once daily from Day 2 The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made.
- The recommended total treatment duration for hospitalized patients requiring invasive mechanical ventilation and/or extracorporeal membrane oxygenation (ECMO) is 10 days.
- The recommended treatment duration for hospitalized patients not requiring invasive mechanical ventilation and/or ECMO is 5 days. If a patient does not demonstrate clinical improvement, treatment may be extended for up to 5 additional days for a total treatment duration of up to 10 days.
Non-hospitalized patients:
The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made and within 7 days of symptom onset.
- The recommended total treatment duration for non-hospitalized patients diagnosed with mild-to-moderate COVID-19 who are at high risk for progression to severe COVID-19, including hospitalization or death, is 3 days.
VEKLURY must be diluted prior to intravenous infusion. Refer to Dosage and Administration (2.5) for detailed preparation and administration instructions.
2.4 Renal Impairment
No dosage adjustment of VEKLURY is recommended in patients with any degree of renal impairment, including patients on dialysis. VEKLURY may be administered without regard to the timing of dialysis [see Dosage and Administration (2.3) and Use in Specific Populations (8.4, 8.6)].
2.5 Dosage Preparation and Administration
Reconstitution Instructions
Remove the required number of single-dose vial(s) from storage. For each vial:
- Inspect the vial to ensure the container closure is free from defects.
- Aseptically reconstitute VEKLURY lyophilized powder by adding 19 mL of Sterile Water for Injection using a suitably sized syringe and needle per vial, and insert the needle in the center of the vial stopper.
- Only use Sterile Water for Injection to reconstitute VEKLURY lyophilized powder.
- Discard the vial if a vacuum does not pull the Sterile Water for Injection into the vial.
- Immediately shake the vial for 30 seconds.
- Allow the contents of the vial to settle for 2 to 3 minutes. A clear, colorless to yellow solution, free of visible particles, should result.
- If the contents of the vial are not completely dissolved, shake the vial again for 30 seconds and allow the contents to settle for 2 to 3 minutes. Repeat this procedure as necessary until the contents of the vial are completely dissolved. Discard the vial if the contents are not completely dissolved.
- Following reconstitution, each vial contains 100 mg/20 mL (5 mg/mL) of remdesivir solution.
- Use reconstituted product immediately to prepare the diluted drug product. Detailed dilution and administration instruction based on patient’s weight is provided below.
Dilution Instructions for Adults and Pediatric Patients Weighing at Least 40 kg
Care should be taken during admixture to prevent inadvertent microbial contamination. As there is no preservative or bacteriostatic agent present in this product, aseptic technique must be used in preparation of the final parenteral solution. It is always recommended to administer intravenous medication immediately after preparation when possible.
- Reconstituted VEKLURY for injection, containing 100 mg/20 mL remdesivir solution, must be further diluted in either a 100 mL or 250 mL 0.9% sodium chloride injection infusion bag. Refer to Table 2 for instructions.
Table 2 Recommended Dilution Instructions—Reconstituted VEKLURY for Injection in Adults and Pediatric Patients Weighing at Least 40 kg VEKLURY dose 0.9% sodium chloride injection infusion bag volume to be used Volume to be withdrawn and discarded from 0.9% sodium chloride injection infusion bag Required volume of reconstituted VEKLURY for injection Loading dose
200 mg
(2 vials)250 mL 40 mL 40 mL (2 × 20 mL) 100 mL 40 mL 40 mL (2 × 20 mL) Maintenance dose
100 mg
(1 vial)250 mL 20 mL 20 mL 100 mL 20 mL 20 mL - Withdraw and discard the required volume of 0.9% sodium chloride injection from the bag following instructions in Table 2, using an appropriately sized syringe and needle.
- Inspect the reconstituted VEKLURY for injection vial to ensure the solution is free of particulate matter.
- Withdraw the required volume of reconstituted VEKLURY for injection from the VEKLURY vial following instructions in Table 2, using an appropriately sized syringe. Discard any unused portion remaining in the reconstituted vial.
- Transfer the required volume of reconstituted VEKLURY for injection to the selected infusion bag.
- Gently invert the bag 20 times to mix the solution in the bag. Do not shake.
- The prepared infusion solution can be stored for 24 hours at room temperature (20°C to 25°C [68°F to 77°F]) or 48 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]) prior to administration.
Administration Instructions for Adults and Pediatric Patients Weighing at Least 40 kg
Do not administer the prepared diluted solution simultaneously with any other medication. The compatibility of VEKLURY injection with intravenous solutions and medications other than 0.9% sodium chloride injection, USP is not known. Administer VEKLURY via intravenous infusion over 30 to 120 minutes.
Administration should be under conditions where management of severe hypersensitivity reactions, such as anaphylaxis, is possible. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate [see Warnings and Precautions (5.1)].
Administer the diluted solution with the infusion rate described in Table 3.
Table 3 Recommended Rate of Infusion—Diluted VEKLURY for Injection in Adults and Pediatric Patients Weighing at Least 40 kg Infusion bag volume Infusion time Rate of infusion 250 mL 30 min 8.33 mL/min 60 min 4.17 mL/min 120 min 2.08 mL/min 100 mL 30 min 3.33 mL/min 60 min 1.67 mL/min 120 min 0.83 mL/min Dilution Instructions for Pediatric Patients (Birth to Less than 18 Years of Age) Weighing 1.5 kg to Less than 40 kg
Care should be taken during admixture to prevent inadvertent microbial contamination. As there is no preservative or bacteriostatic agent present in this product, aseptic technique must be used in preparation of the final parenteral solution. It is always recommended to administer intravenous medication immediately after preparation when possible.
- Inspect the reconstituted VEKLURY for injection vial to ensure the reconstituted solution is free of particulate matter.
- For pediatric patients (birth to less than 18 years of age) weighing 1.5 kg to less than 40 kg, the 100 mg/20 mL (5 mg/mL) remdesivir reconstituted solution should be further diluted to a fixed concentration of 1.25 mg/mL using 0.9% sodium chloride injection.
- The final required infusion volume concentration of 1.25 mg/mL remdesivir diluted solution for infusion is based on the pediatric weight-based dosing regimens.
- Small 0.9% sodium chloride injection infusion bags (e.g., 25, 50, or 100 mL) or an appropriately sized syringe should be used for pediatric dosing. The recommended dose is administered via intravenous infusion in a total volume dependent on the dose to yield the target remdesivir concentration of 1.25 mg/mL.
- A syringe and syringe pump may be used for infusion volumes less than 50 mL.
Infusion with IV Bag
- Determine the total infusion volume needed to achieve a final infusion volume concentration of 1.25 mg/mL of remdesivir diluted solution based on the patient's calculated dose.
- Select an appropriately sized infusion bag (either prefilled with 0.9% sodium chloride injection or empty) to prepare VEKLURY diluted solution.
- If using a prefilled 0.9% sodium chloride injection infusion bag, withdraw and discard the amount of diluent equal to the volume of reconstituted VEKLURY solution needed per patient's dose plus a quantity sufficient to achieve a 1.25 mg/mL final volume concentration of remdesivir diluted solution.
- Withdraw the required volume of reconstituted VEKLURY solution into an appropriately sized syringe.
- Transfer the required volume of reconstituted VEKLURY solution to the 0.9% sodium chloride injection infusion bag.
- Gently invert the bag 20 times to mix the solution in the bag. Do not shake.
- If using an empty infusion bag, transfer the required volume of reconstituted VEKLURY solution to the bag, followed by a volume of 0.9% sodium chloride injection sufficient to achieve a 1.25 mg/mL final volume concentration of remdesivir diluted solution.
- The prepared infusion solution is stable for 24 hours at room temperature (20°C to 25°C [68°F to 77°F]) or 48 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]).
Infusion with Syringe
- Determine the total infusion volume needed to achieve a final infusion volume concentration of 1.25 mg/mL of remdesivir diluted solution based on patient's calculated dose.
- Select an appropriately sized syringe equal to or larger than the calculated total infusion volume of 1.25 mg/mL remdesivir solution needed.
- Withdraw the required volume of reconstituted VEKLURY solution from the vial into the syringe based on patient's calculated dose, followed by the required volume of 0.9% sodium chloride injection needed to achieve a 1.25 mg/mL final volume concentration of remdesivir diluted solution.
- Gently invert the syringe 20 times to mix the solution in the syringe. Do not shake.
- The prepared diluted solution should be used immediately.
Administration Instructions for Pediatric Patients (Birth to Less than 18 Years of Age) Weighing 1.5 kg to Less than 40 kg
The prepared diluted solution should not be administered simultaneously with any other medication. The compatibility of VEKLURY with IV solutions and medications other than 0.9% sodium chloride injection, USP is not known. Administer VEKLURY via intravenous infusion over 30 to 120 minutes. The rate of infusion (mL/min) should be calculated based on the total infusion volume and total infusion time.
Administration should be under conditions where management of severe hypersensitivity reactions, such as anaphylaxis, is possible. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate [see Warnings and Precautions (5.1)].
2.6 Storage of Prepared Dosages
After reconstitution, use vials immediately to prepare diluted solution. The diluted VEKLURY solution in the infusion bags can be stored up to 24 hours at room temperature (20°C to 25°C [68°F to 77°F]) prior to administration or 48 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]).
IMPORTANT:
This product contains no preservative. Any unused portion of a single-dose VEKLURY vial should be discarded after a diluted solution is prepared.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
VEKLURY is contraindicated in patients with a history of clinically significant hypersensitivity reactions to VEKLURY or any components of the product [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Including Infusion-related and Anaphylactic Reactions
Hypersensitivity reactions, including infusion-related and anaphylactic reactions, have been observed during and following administration of VEKLURY; most occurred within one hour. Signs and symptoms may include hypotension, hypertension, tachycardia, bradycardia, hypoxia, fever, dyspnea, wheezing, angioedema, rash, nausea, diaphoresis, and shivering. Slower infusion rates, with a maximum infusion time of up to 120 minutes, can be considered to potentially prevent these signs and symptoms. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate. If signs and symptoms of a clinically significant hypersensitivity reaction occur, immediately discontinue administration of VEKLURY and initiate appropriate treatment. The use of VEKLURY is contraindicated in patients with known hypersensitivity to VEKLURY or any components of the product [see Contraindications (4)].
5.2 Increased Risk of Transaminase Elevations
Transaminase elevations have been observed in healthy volunteers who received 200 mg of VEKLURY followed by 100 mg doses for up to 10 days; the transaminase elevations were mild (Grade 1) to moderate (Grade 2) in severity and resolved upon discontinuation of VEKLURY. Transaminase elevations have also been reported in patients with COVID-19 who received VEKLURY [see Adverse Reactions (6.1)]. Because transaminase elevations have been reported as a clinical feature of COVID-19, and the incidence was similar in patients receiving placebo versus VEKLURY in clinical trials of VEKLURY, discerning the contribution of VEKLURY to transaminase elevations in patients with COVID-19 can be challenging.
Perform hepatic laboratory testing in all patients before starting VEKLURY and while receiving VEKLURY as clinically appropriate [see Dosage and Administration (2.1) and Use in Specific Populations (8.7)].
- Consider discontinuing VEKLURY if ALT levels increase to greater than 10 times the upper limit of normal.
- Discontinue VEKLURY if ALT elevation is accompanied by signs or symptoms of liver inflammation.
5.3 Risk of Reduced Antiviral Activity When Coadministered with Chloroquine Phosphate or Hydroxychloroquine Sulfate
Coadministration of VEKLURY and chloroquine phosphate or hydroxychloroquine sulfate is not recommended based on data from cell culture experiments demonstrating a potential antagonistic effect of chloroquine on the intracellular metabolic activation and antiviral activity of VEKLURY [see Drug Interactions (7) and Microbiology (12.4)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Hypersensitivity Including Infusion-related and Anaphylactic Reactions [see Warnings and Precautions (5.1)]
- Increased Risk of Transaminase Elevations [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials in Adult Subjects
The safety of VEKLURY is based on data from four Phase 3 studies in 1,476 hospitalized adult subjects with COVID-19, one Phase 3 study in 279 non-hospitalized adult and pediatric subjects (12 years of age and older weighing at least 40 kg) with mild-to-moderate COVID-19, four Phase 1 studies in 131 healthy adults, and from patients with COVID-19 who received VEKLURY under the Emergency Use Authorization or in a compassionate use program.
Clinical Trials Experience in Adults with COVID-19
NIAID ACTT-1 was a randomized, double-blind, placebo-controlled clinical trial in hospitalized subjects with mild, moderate, and severe COVID-19 treated with VEKLURY (n=532) or placebo (n=516) for up to 10 days. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days [see Clinical Studies (14.1)]. The collection of adverse event data in this trial was limited to severe (Grade 3) or potentially life-threatening (Grade 4) adverse events, serious adverse events, adverse events leading to study drug discontinuation, and moderate (Grade 2) severity or higher hypersensitivity reactions. Rates of adverse reactions (≥ Grade 3), serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 4.
Table 4 Summary of Adverse Reaction Rates in Hospitalized Subjects with Mild, Moderate, or Severe COVID-19 in NIAID ACTT-1 Types of Adverse Reactions VEKLURY
N=532
n (%)Placebo
N=516
n (%)- * Seizure (n=1), infusion-related reaction (n=1).
- † Seizure (n=1), infusion-related reaction (n=1), transaminases increased (n=3), ALT increased and AST increased (n=1), GFR decreased (n=2), acute kidney injury (n=3).
Adverse reactions, Grades ≥3 41 (8%) 46 (9%) Serious adverse reactions 2 (0.4%)* 3 (0.6%) Adverse reactions leading to treatment discontinuation 11 (2%)† 15 (3%) Study GS-US-540-5773 was a randomized, open-label clinical trial in hospitalized subjects with severe COVID-19 treated with VEKLURY 200 mg on Day 1 and 100 mg once daily for 5 (n=200) or 10 days (n=197). Adverse reactions were reported in 33 (17%) subjects in the 5-day group and 40 (20%) subjects in the 10-day group [see Clinical Studies (14.2)]. The most common adverse reactions occurring in at least 5% of subjects in either the VEKLURY 5-day or 10-day group, respectively, were nausea (5% vs 3%), AST increased (3% vs 6%), and ALT increased (2% vs 7%). Rates of any adverse reactions, serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 5.
Table 5 Summary of Adverse Reaction Rates in Hospitalized Subjects with Severe COVID-19 in Study 5773 Types of Adverse Reactions VEKLURY
5 Days
N=200
n (%)VEKLURY
10 Days
N=197
n (%)- * Transaminases increased (n=5), hepatic enzyme increased (n=1), hypertransaminasaemia (n=1).
- † Transaminases increased (n=4), hepatic enzyme increased (n=2), LFT increased (n=2), hypertransaminasaemia (n=1), ALT increased (n=1), ALT increased and AST increased (n=2), injection site erythema (n=1), rash (n=1).
Any adverse reaction, all Grades 33 (17%) 40 (20%) Serious adverse reactions 3 (2%)* 4 (2%)* Adverse reactions leading to treatment discontinuation 5 (3%)† 9 (5%)† Study GS-US-540-5774 was a randomized, open-label clinical trial in hospitalized subjects with moderate COVID-19 treated with VEKLURY 200 mg on Day 1 and 100 mg daily for 5 (n=191) or 10 days (n=193), or standard of care (SOC) only (n=200) [see Clinical Studies (14.3)]. Adverse reactions were reported in 36 (19%) subjects in the 5-day group and 25 (13%) subjects in the 10-day group. The most common adverse reaction occurring in at least 5% of subjects in the VEKLURY groups was nausea (7% in the 5-day group, 4% in the 10-day group). Rates of any adverse reactions, serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 6.
Table 6 Summary of Adverse Reaction* Rates in Hospitalized Subjects with Moderate COVID-19 in Study 5774 Types of Adverse Reactions VEKLURY
5 Days
N=191
n (%)VEKLURY
10 Days
N=193
n (%)- * Attribution of events to study drug was not performed for the SOC group.
- † Heart rate decreased.
- ‡ ALT increased (n=2), ALT increased and AST increased (n=1), hypertransaminasaemia (n=1), blood alkaline phosphatase increased (n=1), rash (n=2), heart rate decreased (n=1).
Any adverse reaction, all Grades 36 (19%) 25 (13%) Serious adverse reactions 1 (<1%)† 0 Adverse reactions leading to treatment discontinuation 4 (2%)‡ 4 (2%)‡ Study GS-US-540-9012 was a randomized, double-blind, placebo-controlled clinical trial in subjects who were non-hospitalized, were symptomatic for COVID-19 for ≤7 days, had confirmed SARS-CoV-2 infection, and had at least one risk factor for progression to hospitalization treated with VEKLURY (n=279; 276 adults and 3 pediatric subjects 12 years of age and older weighing at least 40 kg) or placebo (n=283; 278 adults and 5 pediatric subjects 12 years of age and older weighing at least 40 kg) for 3 days. Of the 279 subjects treated with VEKLURY, 227 subjects received at least one dose of VEKLURY at an outpatient facility, 44 subjects received at least one dose of VEKLURY in a home healthcare setting, and 8 subjects received at least one dose of VEKLURY at a skilled nursing facility. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days [see Clinical Studies (14.4)]. Adverse reactions (all grades) were reported in 34 (12%) subjects in the VEKLURY group and 25 (9%) subjects in the placebo group. The most common adverse reaction occurring in at least 5% of subjects in the VEKLURY group was nausea (6%). There were no serious adverse reactions or adverse reactions leading to treatment discontinuation in either treatment group. Safety in subjects who received VEKLURY in a home healthcare setting was comparable to that observed in the overall GS-US-540-9012 study population, but these findings are based on limited data.
Less Common Adverse Reactions in Adults from Clinical Trials
Clinically significant adverse reactions that were reported in <2% of subjects exposed to VEKLURY in clinical trials are listed below:
- Hypersensitivity reactions [see Warnings and Precautions (5.1)].
- Generalized seizure
- Rash
Laboratory Abnormalities
Study GS-US-399-5505 was a Phase 1, randomized, blinded, placebo-controlled clinical trial in healthy volunteers administered VEKLURY 200 mg on Day 1 and 100 mg for either 4 days or 9 days. Mild (Grade 1, n=8) to moderate (Grade 2, n=1) elevations in ALT were observed in 9 of 20 subjects receiving 10 days of VEKLURY; the elevations in ALT resolved upon discontinuation of VEKLURY. No subjects (0 of 9) who received 5 days of VEKLURY had graded increases in ALT.
The frequencies of laboratory abnormalities (Grades 3–4) occurring in at least 3% of subjects with COVID-19 receiving VEKLURY in Trials NIAID ACTT-1, 5773, and 5774 are presented in Table 7, Table 8, and Table 9, respectively.
Table 7 Laboratory Abnormalities (Grades 3–4) Reported in ≥3% of Hospitalized Subjects with Mild, Moderate, or Severe COVID-19 in NIAID ACTT-1 Laboratory Parameter Abnormality* VEKLURY 10 Days
N=532Placebo
N=516- * Frequencies are based on treatment-emergent laboratory abnormalities. Graded per Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.1 dated July 2017.
- † Based on the Cockcroft-Gault formula.
ALT increased 3% 6% AST increased 6% 8% Bilirubin increased 2% 5% Creatinine clearance decreased† 18% 20% Creatinine increased 15% 16% eGFR decreased 18% 24% Glucose increased 12% 13% Hemoglobin decreased 15% 22% Lymphocytes decreased 11% 18% Prothrombin time increased 9% 4% Table 8 Laboratory Abnormalities (Grades 3–4) Reported in ≥3% of Hospitalized Subjects with Severe COVID-19 in Trial 5773 Laboratory Parameter Abnormality* VEKLURY
5 Days
N=200VEKLURY
10 Days
N=197- * Frequencies are based on treatment-emergent laboratory abnormalities. Graded per Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.1 dated July 2017.
- † Based on the Cockcroft-Gault formula.
ALT increased 6% 8% AST increased 7% 6% Creatinine clearance decreased† 10% 19% Creatinine increased 5% 15% Glucose increased 11% 8% Hemoglobin decreased 6% 8% Table 9 Laboratory Abnormalities (Grades 3–4) Reported in ≥3% of Hospitalized Subjects with Moderate COVID-19 in Trial 5774 Laboratory Parameter Abnormality* VEKLURY
5 Days
N=191VEKLURY 10 Days
N=193SOC
N=200SOC=Standard of care. - * Frequencies are based on treatment-emergent laboratory abnormalities. Graded per Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.1 dated July 2017.
- † Based on the Cockcroft-Gault formula.
ALT increased 2% 3% 8% Creatinine clearance decreased† 2% 5% 8% Glucose increased 4% 3% 2% Hemoglobin decreased 3% 1% 6% The frequencies of laboratory abnormalities (Grades 3–4) occurring in at least 2% of subjects with COVID-19 receiving VEKLURY in Trial GS-US-540-9012 are presented in Table 10.
Table 10 Laboratory Abnormalities (Grades 3–4) Reported in ≥2% of Non-Hospitalized Subjects in Trial 9012 Laboratory Parameter Abnormality* VEKLURY 3 Days
N=279Placebo
N=283- * Frequencies are based on treatment-emergent laboratory abnormalities. Graded per Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.1 dated July 2017.
- † Based on the Cockcroft-Gault formula.
Creatinine clearance decreased† 6% 2% Creatinine increased 3% 1% Glucose increased 6% 6% Lymphocytes decreased 2% 1% Prothrombin time increased 1% 2% Clinical Trials Experience in Adults with COVID-19 and Renal Impairment
Study GS-US-540-5912 was a randomized, double-blind, placebo-controlled clinical trial in which 163 hospitalized subjects with confirmed COVID-19 and acute kidney injury (AKI; N=60), chronic kidney disease (CKD; eGFR <30 mL/minute/1.73m2; N=44), or end-stage renal disease (ESRD; eGFR <15 mL/minute/1.73m2; N=59) on hemodialysis received VEKLURY for up to 5 days [see Use in Specific Populations (8.6)]. The adverse reactions observed were consistent with those observed in clinical trials of VEKLURY in adults. Adverse reactions (all grades) were reported in 13 (8%) subjects in the VEKLURY group and 3 (4%) subjects in the placebo group. The most common adverse reactions were nausea (1%), abdominal pain (1%), and diarrhea (1%). No subjects experienced serious adverse reactions. One subject permanently discontinued treatment due to an adverse reaction: lipase increased.
The frequencies of laboratory abnormalities (Grades 3–4) occurring in at least 3% of subjects with COVID-19 receiving VEKLURY in Trial GS-US-540-5912 are presented in Table 11.
Table 11 Laboratory Abnormalities (Grades 3–4) Reported in ≥3% of Hospitalized Subjects in Trial 5912 Laboratory Parameter Abnormality* VEKLURY 5 Days
N=163Placebo
N=80- * Frequencies are based on treatment-emergent laboratory abnormalities. Graded per Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.1 dated July 2017.
Lymphocytes decreased 27% 27% Hemoglobin decreased 25% 25% Glucose increased 15% 19% Uric acid increased 11% 4% Creatinine increased 12% 14% Albumin decreased 12% 10% Lipase increased 12% 7% Prothrombin time increased 11% 4% Prothrombin INR increased 7% 4% AST increased 6% 4% Thromboplastin time increased 5% 4% ALT increased 5% 6% Sodium increased 3% 3% Calcium increased 3% 0 Clinical Trials in Pediatric Subjects
Study GS-US-540-5823 was a Phase 2/3, single-arm, open-label clinical trial in hospitalized subjects from birth to <18 years of age and weighing at least 1.5 kg with mild, moderate, and severe COVID-19 treated with weight-based VEKLURY (n=58) for up to 10 days [see Clinical Studies (14.6)]:
- Cohorts 1, 8: Subjects ≥12 years and weighing ≥40 kg (n=12) and subjects <12 years and weighing ≥40 kg (n=5): Received 200 mg on Day 1 and 100 mg once daily on subsequent days.
- Cohorts 2–4: Subjects ≥28 days and weighing ≥20 to <40 kg (n=12); subjects ≥28 days and weighing ≥12 to <20 kg (n=12); and subjects ≥28 days and weighing ≥3 to <12 kg (n=12): Received 5 mg/kg on Day 1 and 2.5 mg/kg once daily on subsequent days.
- Cohort 5: Subjects 14 to <28 days old, gestational age (GA) >37 weeks, and weighing ≥2.5 kg (n=3): Received 5 mg/kg on Day 1 and 2.5 mg/kg once daily on subsequent days.
- Cohorts 6–7: Subjects <14 days old, GA >37 weeks, and weighing ≥2.5 kg at birth (n=1); and subjects <56 days old, GA ≤37 weeks, and weighing ≥1.5 kg at birth (n=1): Received 2.5 mg/kg on Day 1 and 1.25 mg/kg once daily on subsequent days.
The adverse reactions observed were consistent with those observed in clinical trials of VEKLURY in adults.
Infants, children, and adolescents; Cohorts 1–4, 8: Adverse reactions (all grades) were reported in 8 (15%) subjects. The most common adverse reaction occurring in at least 5% of subjects was ALT increased (6%). No subjects experienced serious adverse reactions. Two (4%) subjects permanently discontinued treatment due to adverse reactions (ALT increased [n=1], ALT increased and AST increased and hyperbilirubinemia [n=1]). Laboratory abnormalities (Grades 3–4) occurring in at least 3% of subjects with COVID-19 receiving VEKLURY in Trial 5823 and who had at least one post-baseline value for the specified test were hemoglobin decreased (18%, 9/51), eGFR decreased (18%, 7/40), creatinine increased (10%, 5/52), direct bilirubin increased (9%, 2/23), prothrombin time increased (7%, 3/46), APTT increased (7%, 3/45), lymphocytes decreased (6% 2/33), proteinuria (6%, 2/36), WBC decreased (4%, 2/51), ALT increased (4%, 2/51), glucose increased (4%, 2/52), glycosuria (4%, 2/46), potassium decreased (4%, 2/52).
Emergency Use Authorization Experience in Subjects with COVID-19
The following adverse reactions have been identified during use of VEKLURY under Emergency Use Authorization:
- General disorders and administration site conditions: Administration site extravasation
- Skin and subcutaneous tissue disorders: Rash
- Immune system disorders: Anaphylaxis, angioedema, infusion-related reactions, hypersensitivity
- Investigations: Transaminase elevations
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on VEKLURY
Due to potential antagonism based on data from cell culture experiments, concomitant use of VEKLURY with chloroquine phosphate or hydroxychloroquine sulfate is not recommended [see Warnings and Precautions (5.3) and Microbiology (12.4)].
Based on drug interaction studies conducted with VEKLURY, no clinically significant drug interactions are expected with inducers of cytochrome P450 (CYP) 3A4 or inhibitors of Organic Anion Transporting Polypeptides (OATP) 1B1/1B3 and, P-glycoprotein (P-gp) [see Clinical Pharmacology (12.3)].
7.2 Effects of VEKLURY on Other Drugs
Based on drug interaction studies conducted with VEKLURY, it is a weak inhibitor of CYP3A and does not inhibit OATP1B1/1B3 [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from a clinical trial (IMPAACT 2032), published reports, the COVID-PR pregnancy exposure registry, and compassionate use of remdesivir in pregnant individuals have not identified a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes following exposure in the second and third trimester. However, there are insufficient pregnancy data available to evaluate the risk of remdesivir exposure during the first trimester. A study evaluating the pharmacokinetics of remdesivir during pregnancy demonstrated no clinically relevant differences between pregnant and non-pregnant individuals. No dose adjustments are recommended in patients who receive VEKLURY during pregnancy (see Data) and [see Clinical Pharmacology (12.3)]. In nonclinical reproductive toxicity studies, remdesivir demonstrated no adverse effect on embryo-fetal development when administered to pregnant animals at systemic exposures (AUC) of the predominant circulating metabolite of remdesivir (GS-441524) that were 4 times (rats and rabbits) the exposure in humans at the recommended human dose (RHD) (see Data). There are maternal and fetal risks associated with untreated COVID-19 in pregnancy (see Clinical Considerations).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
A non-randomized, open-label clinical study (IMPAACT 2032) evaluated pharmacokinetics and safety of up to 10 days of treatment with VEKLURY in 25 hospitalized pregnant and 28 hospitalized non-pregnant individuals of childbearing potential. Subjects received VEKLURY 200 mg once daily for 1 day followed by VEKLURY 100 mg once daily on subsequent days via intravenous infusion. Subjects were enrolled prior to their fourth VEKLURY infusion. Assessments occurred at the following intervals: Screening; Pre-infusion (defined as 48 hours prior to start of first infusion); each infusion day; 48 hours after the last infusion; 7 days after the last infusion; 4 weeks after the last infusion. Assessments also occurred 24 hours post-delivery in subjects who delivered. Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to the completion of 10 days of treatment.
Of the 25 pregnant subjects, median age was 33 years (Q1, Q3: 27 years, 37 years); 40% were White, 24% were Black, and 48% were Hispanic or Latino. A total of 9 subjects (36%) were on high-flow oxygen; 12 subjects (48%) were on low-flow oxygen; and 1 subject (4%) was on room air, at baseline. Three subjects (12%) did not have data available on baseline oxygen status. The overall median (Q1, Q3) duration of symptoms prior to hospitalization was 7 (6, 9) days. The overall median (Q1, Q3) duration of symptoms prior to first dose of VEKLURY was 8 (6, 9) days.
Of the 25 pregnant subjects, median gestational age was 28 weeks at baseline (range 22 to 33 weeks) and about half of subjects were in each of the second and third trimester of pregnancy. No clinically relevant differences in the pharmacokinetics of remdesivir or its metabolites (GS-704277 and GS-441524) were observed between pregnant (n=21) and non-pregnant (n=22) individuals [see Clinical Pharmacology (12.3)]. No difference in pharmacokinetics of remdesivir or its metabolites is expected between the first and second/third trimesters. The adverse reactions observed were consistent with those observed in clinical trials of VEKLURY in adults [see Adverse Reactions (6.1)]. There were no adverse reactions in infants born during the study (n=16).
Animal Data
Remdesivir was administered via intravenous injection to pregnant rats and rabbits (up to 20 mg/kg/day) on Gestation Days 6 through 17, and 7 through 20, respectively, and also to rats from Gestation Day 6 to Lactation/Post-partum Day 20. No adverse effects on embryo-fetal (rats and rabbits) or pre/postnatal (rats) development were observed in rats and rabbits at nontoxic doses in pregnant animals. During organogenesis, exposures to the predominant circulating metabolite (GS-441524) were 4 times higher (rats and rabbits) than the exposure in humans at the RHD. In a pre/postnatal development study, exposures to the predominant circulating metabolite of remdesivir (GS-441524) were similar to the human exposures at the RHD.
8.2 Lactation
Risk Summary
A published case report describes the presence of remdesivir and active metabolite GS-441524 in human milk. Available data (n=11) from pharmacovigilance reports do not indicate adverse effects on breastfed infants from exposure to remdesivir and its metabolite through breastmilk. There are no available data on the effects of remdesivir on milk production. In animal studies, remdesivir and metabolites have been detected in the nursing pups of mothers given remdesivir, likely due to the presence of remdesivir in milk (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VEKLURY and any potential adverse effects on the breastfed child from VEKLURY or from the underlying maternal condition. Breastfeeding individuals with COVID-19 should follow practices according to clinical guidelines to avoid exposing the infant to COVID-19.
Data
Remdesivir and its metabolites were detected in the plasma of nursing rat pups, likely due to the presence of remdesivir and/or its metabolites in milk, following daily intravenous administration of remdesivir to pregnant rats from Gestation Day 6 to Lactation Day 20. Exposures in nursing pups were approximately 1% that of maternal exposure on Lactation Day 10. The concentration of remdesivir in animal milk does not necessarily predict the concentration of drug in human milk.
8.4 Pediatric Use
The safety and effectiveness of VEKLURY for the treatment of COVID-19 have been established in pediatric patients from birth to less than 18 years of age and weighing at least 1.5 kg, who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
Use in this age group is supported by the following:
- Trials in adults [see Clinical Studies (14.1, 14.2, 14.3, 14.4, 14.5)]
- An open-label trial (Study 5823) in 58 hospitalized pediatric subjects [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.6)].
Use of VEKLURY in pediatric patients from birth to less than 18 years of age and weighing at least 1.5 kg is supported by Study 5823 where 58 hospitalized pediatric subjects were treated with weight-based VEKLURY for up to 10 days in the following cohorts:
- Cohorts 1–4, 8; infants, children, and adolescents: Subjects ≥12 years and weighing ≥40 kg (n=12); subjects <12 years and weighing ≥40 kg (n=5); subjects ≥28 days and weighing ≥20 to <40 kg (n=12); subjects ≥28 days and weighing ≥12 to <20 kg (n=12); and subjects ≥28 days and weighing ≥3 to <12 kg (n=12);
- Cohorts 5–7; neonates and infants: Subjects 14 to <28 days old, GA >37 weeks, and weighing ≥2.5 kg (n=3); subjects <14 days old, GA >37 weeks, and weighing ≥2.5 kg at birth (n=1); and subjects <56 days old, GA ≤37 weeks, and weighing ≥1.5 kg at birth (n=1).
The safety and pharmacokinetic results in pediatric subjects were similar to those in adults [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.6)].
Use of VEKLURY in pediatric patients weighing at least 40 kg is further supported by a clinical trial of VEKLURY in non-hospitalized subjects that included 3 pediatric subjects 12 years and older, and by clinical trials in hospitalized subjects that included 30 adult subjects weighing 40 to 50 kg. The safety in this weight group was comparable to adult subjects weighing greater than 50 kg. Thirty-nine pediatric patients 12 years and older and weighing at least 40 kg received VEKLURY in a compassionate use program in hospitalized subjects; the available clinical data from these patients are limited [see Adverse Reactions (6.1) and Clinical Studies (14)].
Use of VEKLURY in pediatric patients with renal impairment is supported by safety data in adults [see Adverse Reactions (6.1), Use in Specific Populations (8.6)]. Limited data are available regarding the safety of VEKLURY in pediatric patients with mild or moderate renal impairment. No data are available regarding the safety of VEKLURY in pediatric patients with severe renal impairment. In adults with severe renal impairment, including those requiring dialysis, exposures of GS-441524 and GS-704277, the metabolites of remdesivir, and betadex sulfobutyl ether sodium (SBECD) are increased [see Clinical Pharmacology (12.3)]. VEKLURY contains SBECD which, when administered intravenously, is eliminated through glomerular filtration and therefore when administered to pediatric patients with renal immaturity or renal impairment, may result in higher exposure to SBECD.
The safety and effectiveness of VEKLURY have not been established in pediatric patients weighing less than 1.5 kg.
8.5 Geriatric Use
Of the 1,062 hospitalized subjects with SARS-CoV-2 infection randomized in ACTT-1, 36% were 65 years or older. Of the 397 hospitalized subjects with SARS-CoV-2 infection randomized in Study GS-US-540-5773, 42% were 65 years or older. Of the 584 hospitalized subjects with SARS-CoV-2 infection randomized in Study GS-US-540-5774, 27% were 65 years or older. Of the 562 non-hospitalized subjects with SARS-CoV-2 infection randomized in Study GS-US-540-9012, 17% were 65 years or older. Reported clinical experience has not identified differences in responses between the elderly and younger patients [see Clinical Studies (14)]. No dosage adjustment is required in patients over the age of 65 years. In general, appropriate caution should be exercised in the administration of VEKLURY and monitoring of elderly patients, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
Use of VEKLURY in patients with COVID-19 and renal impairment, including those on dialysis, is supported by safety and pharmacokinetic data from the following:
- a randomized, double-blind, placebo-controlled trial (Study 5912) in adults [see Adverse Reactions (6.1) and Clinical Pharmacology (12.3)].
- an open-label, parallel-group, single-dose trial in subjects with normal renal function and renal impairment (Study 9015) [see Clinical Pharmacology (12.3)].
The pharmacokinetics and safety of VEKLURY in patients with COVID-19 and renal impairment, including those on dialysis, were evaluated in 163 subjects in a randomized, double-blind, placebo-controlled trial, Study GS-US-540-5912 [see Adverse Reactions (6.1) and Clinical Pharmacology (12.3)].
Study GS-US-540-5912 evaluated VEKLURY 200 mg once daily for 1 day followed by VEKLURY 100 mg once daily for 4 days (for a total of up to 5 days of intravenously administered therapy) in 243 hospitalized adult subjects with confirmed COVID-19 and renal impairment. The trial included 90 subjects (37%) with AKI (defined as a 50% increase in serum creatinine within a 48-hour period that was sustained for ≥6 hours despite supportive care), 64 subjects (26%) with CKD (eGFR <30 mL/minute/1.73m2), and 89 subjects (37%) with ESRD (eGFR <15 mL/minute/1.73m2) requiring hemodialysis. Subjects were randomized in a 2:1 manner, stratified by ESRD, high-flow oxygen requirement, and region (US vs ex-US) to receive VEKLURY (n=163) or placebo (n=80), plus standard of care.
At baseline, mean age was 69 years (with 62% of subjects aged 65 or older); 57% of subjects were male, 67% were White, 26% were Black, and 3% were Asian. The most common baseline risk factors were hypertension (89%), diabetes mellitus (79%), and cardiovascular or cerebrovascular disease (51%); the distribution of risk factors was similar between the two treatment groups. A total of 45 subjects (19%) were on high-flow oxygen, 144 (59%) were on low-flow oxygen, and 54 (22%) were on room air at baseline; no subjects were on invasive mechanical ventilation (IMV). A total of 182 subjects (75%) were not on renal replacement therapy, and 31 subjects (13%) had received a COVID-19 vaccine.
The safety results in subjects with COVID-19 and renal impairment, including those on dialysis, were consistent with those observed in clinical trials of VEKLURY in adults [see Adverse Reactions (6.1)]. Study GS-US-540-5912 closed prematurely due to feasibility issues and was underpowered to assess for efficacy because of lower than expected enrollment.
The pharmacokinetics and safety of VEKLURY in subjects with normal renal function and renal impairment, including those on dialysis, were evaluated in 75 subjects (43 subjects with renal impairment plus 32 matched control subjects with normal renal function) in an open-label, parallel-group, single-dose trial, Study GS-US-540-9015 [see Clinical Pharmacology (12.3)].
In studies GS-US-540-5912 and GS-US-540-9015, exposures of GS-441524 and GS-704277, the metabolites of remdesivir, and SBECD are increased in subjects with mild to severe renal impairment, including those requiring dialysis, relative to subjects with normal renal function [see Clinical Pharmacology (12.3)].
No dosage adjustment of VEKLURY is recommended for patients with any degree of renal impairment, including those on dialysis [see Dosage and Administration (2.2, 2.4), Use in Specific Populations (8.4)].
8.7 Hepatic Impairment
No dosage adjustment of VEKLURY is recommended for patients with mild, moderate, or severe hepatic impairment (Child-Pugh Class A, B, or C) [see Clinical Pharmacology (12.3)].
Perform hepatic laboratory testing in all patients before starting VEKLURY and while receiving VEKLURY as clinically appropriate [see Dosage and Administration (2.2) and Warnings and Precautions (5.2)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
VEKLURY contains remdesivir, a SARS-CoV-2 nucleotide analog RNA polymerase inhibitor. The chemical name for remdesivir is 2-ethylbutyl N-{(S)-[2-C-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-2,5-anhydro-d-altrononitril-6-O-yl]phenoxyphosphoryl}-L-alaninate. It has a molecular formula of C27H35N6O8P and a molecular weight of 602.6 g/mol. Remdesivir has the following structural formula:
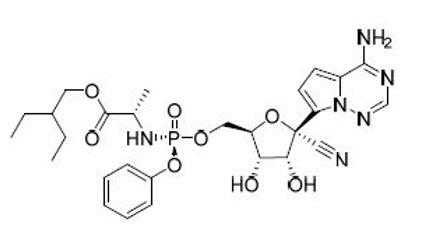
VEKLURY for injection contains 100 mg of remdesivir as a sterile, preservative-free lyophilized white to off-white to yellow powder in a single-dose clear glass vial. It requires reconstitution and then further dilution prior to administration by intravenous infusion [see Dosage and Administration (2.5)]. The inactive ingredients are 3 g betadex sulfobutyl ether sodium and may include hydrochloric acid and/or sodium hydroxide for pH adjustment.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Remdesivir is an antiviral drug with activity against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [see Microbiology (12.4)].
12.2 Pharmacodynamics
12.3 Pharmacokinetics
The pharmacokinetic (PK) properties of remdesivir and metabolites are provided in Table 12. The multiple dose PK parameters of remdesivir and metabolites in adults with COVID-19 are provided in Table 13.
Table 12 Pharmacokinetic Properties of Remdesivir and Metabolites (GS-441524 and GS-704277) Remdesivir GS-441524 GS-704277 ND=not detected - * Remdesivir administered as a 30-minute IV infusion (Study GS-US-399-5505); range of median observed on Day 1 and Day 5 or 10.
- † Range of protein binding for remdesivir from 2 independent experiments show no evidence of concentration-dependent protein binding for remdesivir.
- ‡ Median (Study GS-US-399-4231).
- § Mean (Study GS-US-399-4231).
Absorption Tmax (h)* 0.67–0.68 1.51–2.00 0.75–0.75 Distribution % bound to human plasma proteins 88–93.6† 2 1 Blood-to-plasma ratio 0.68–1.0 1.19 0.56 Elimination t1/2 (h)‡ 1 27 1.3 Metabolism Metabolic pathway(s) CES1 (80%)
Cathepsin A (10%)
CYP3A (10%)Not significantly metabolized HINT1 Excretion Major route of elimination Metabolism Glomerular filtration and active tubular secretion Metabolism % of dose excreted in urine§ 10 49 2.9 % of dose excreted in feces§ ND 0.5 ND Table 13 Multiple Dose PK Parameters* of Remdesivir and Metabolites (GS-441524 and GS-704277) Following IV Administration of VEKLURY 100 mg to Adults with COVID-19 Parameter
Mean† (95% CI)Remdesivir GS-441524 GS-704277 CI=Confidence Interval; ND=Not detectable (at 24 hours post-dose) - * Population PK estimates for 30-minute IV infusion of remdesivir for 3 days (Study GS-US-540-9012, n=147).
- † Geometric mean estimates.
Cmax
(nanogram per mL)2700 (2440, 2990) 143 (135, 152) 198 (180, 218) AUCtau
(nanogram∙h per mL)1710 (1480, 1980) 2410 (2250, 2580) 392 (348, 442) Ctrough
(nanogram per mL)ND 61.5 (56.5, 66.8) ND Specific Populations
Pharmacokinetic differences based on sex, race, age, and renal function on the exposures of remdesivir were evaluated using population pharmacokinetic analysis. Sex and race did not affect the pharmacokinetics of remdesivir and its metabolites (GS-441524 and GS-704277).
Pregnant Individuals
The pharmacokinetics of remdesivir and its circulating metabolites (GS-441524 and GS-704277) were evaluated in pregnant individuals with COVID-19. Exposures (AUCtau, Cmax, and Ctau) of remdesivir and its circulating metabolites during pregnancy were similar to those in non-pregnant individuals (see Table 14).
Table 14 Multiple Dose PK Parameters* of Remdesivir and Metabolites (GS-441524 and GS-704277) Following Intravenous Administration of VEKLURY to Pregnant and Non-Pregnant Individuals with COVID-19 Parameter
Mean† (90% CI)Pregnant Individuals
(N=21)Non-Pregnant Individuals
(N=22)CI=Confidence Interval - * Study CO-US-590-5961 (IMPAACT).
- † Geometric mean estimates.
- ‡ N=18
- § N=17
- ¶ N=20
- # N=21
Remdesivir Cmax (nanogram per mL) 1360 (978, 1890) 1240 (891, 1720) AUCtau (nanogram∙h per mL) 1250 (916, 1700)‡ 1300 (1070, 1590)§ GS-441524 Cmax (nanogram per mL) 113 (102, 126) 121 (108, 136) AUCtau (nanogram∙h per mL) 1840 (1630, 2070)¶ 2050 (1780, 2350)# Ctau (nanogram per mL) 51.6 (44.7, 59.6)¶ 57.1 (48.7, 66.9)# GS-704277 Cmax (nanogram per mL) 217 (187, 252) 213 (188, 240) AUCtau (nanogram∙h per mL) 454 (406, 508)¶ 437 (384, 497) Patients with Renal Impairment
The pharmacokinetics of remdesivir and its metabolites (GS-441524 and GS-704277) and excipient SBECD were evaluated in healthy subjects, those with mild (eGFR 60–89 mL/minute/1.73m2), moderate (eGFR 30–59 mL/minute/1.73m2), severe (eGFR 15–29 mL/minute/1.73m2) renal impairment, or kidney failure (eGFR <15 mL/minute/1.73m2) on dialysis or not on dialysis following a single dose of up to 100 mg of VEKLURY (see Table 15); and in COVID-19 patients with severely reduced kidney function (AKI [defined as a 50% increase in serum creatinine within a 48-hour period that was sustained for ≥6 hours despite supportive care]; CKD [eGFR <30 mL/minute/1.73m2]; or ESRD [eGFR <15 mL/minute/1.73m2] requiring hemodialysis) receiving VEKLURY 200 mg loading dose on Day 1 followed by 100 mg from Day 2 to Day 5 (see Table 16). Pharmacokinetic exposures of remdesivir were not affected by renal function or timing of VEKLURY administration around dialysis.
Exposures of GS-441524, GS-704277, and SBECD were up to 7.9-fold, 2.8-fold, and 21-fold higher, respectively, in those with renal impairment compared to those with normal renal function (see Table 15 and Table 16). These changes are not considered to be clinically significant [see Adverse Reactions (6.1) and Use in Specific Populations (8.6)].
Remdesivir was not efficiently removed through hemodialysis. Average hemodialysis clearance of GS-441524 and GS-704277 was 149 mL/minute and 92.6 mL/minute, respectively.
Table 15 Comparison of PK Parameters* of Remdesivir and Metabolites (GS-441524 and GS-704277) Following IV Administration of Single Dose VEKLURY to Adults with Renal Impairment† as Compared to Adults with Normal Renal Function Mean Ratio (90% CI)‡ 60–89 mL per minute†
N=1030–59 mL per minute†
N=1015–29 mL per minute†
N=10<15 mL per minute† Pre-hemodialysis
N=6Post-hemodialysis
N=6No dialysis
N=3CI=Confidence Interval - * Exposures were estimated using noncompartmental analysis from a dedicated Phase 1 renal impairment Study GS-US-540-9015; single doses up to 100 mg were administered; each subject with renal impairment had a matched control participant enrolled with normal renal function (eGFR ≥90 mL/minute/1.73m2), same sex, and similar BMI (± 20%) and age (± 10 years).
- † eGFR was calculated using Modification of Diet in Renal Disease equation and values represent mL/minute/1.73m2.
- ‡ No effect=1.0 (0.5–2.0)
Remdesivir Cmax 0.96
(0.71, 1.31)1.20
(1.01, 1.42)0.97
(0.83, 1.13)0.89
(0.67, 1.18)1.13
(0.79, 1.60)0.94
(0.65, 1.35)AUCinf 1.00
(0.75, 1.32)1.22
(0.98, 1.52)0.94
(0.83, 1.07)0.80
(0.59, 1.08)1.08
(0.72, 1.63)0.89
(0.55, 1.43)GS-441524 Cmax 1.07
(0.90, 1.26)1.44
(1.13, 1.85)1.68
(1.28, 2.20)2.27
(1.72, 2.99)3.07
(2.21, 4.26)3.00
(2.63, 3.42)AUCinf 1.19
(0.97, 1.47)2.02
(1.57, 2.62)3.26
(2.39, 4.46)4.97
(3.65, 6.77)6.22
(4.44, 8.71)7.87
(6.49, 9.53)GS-704277 Cmax 2.25
(1.20, 4.20)1.83
(1.34, 2.49)1.27
(0.96, 1.68)1.43
(1.00, 2.05)1.23
(0.84, 1.80)1.76
(1.19, 2.61)AUCinf 1.39
(1.13, 1.71)2.01
(1.48, 2.73)1.78
(1.27, 2.49)2.18
(1.61, 2.95)2.06
(1.42, 2.97)2.81
(1.79, 4.43)Table 16 Comparison of PK Parameters of Remdesivir and Metabolites (GS-441524 and GS-704277) Following IV Administration of VEKLURY (200 mg on Day 1 Followed by 100 mg Daily on Days 2–5) in Adults with COVID-19 with* or without† Severely Reduced Kidney Function‡ Mean Ratio (90% CI)§ Remdesivir GS-441524 GS-704277 CI=Confidence Interval; ND=Not detectable (at 24 hours post-dose) - * Population PK estimates for 30-minute IV infusion of remdesivir for 5 days (Study GS-US-540-5912, n=90).
- † Population PK estimates for 30-minute IV infusion of remdesivir for 3 days (Study GS-US-540-9012, n=148).
- ‡ AKI (defined as a 50% increase in serum creatinine within a 48-hour period that was sustained for ≥6 hours despite supportive care); CKD (eGFR <30 mL/minute/1.73m2); or ESRD (eGFR <15 mL/minute/1.73m2) requiring hemodialysis.
- § No effect=1.0 (0.5–2.0)
Cmax 1.39 (1.25, 1.54) 4.98 (4.61, 5.38) 1.84 (1.63, 2.08) AUCtau 1.79 (1.59, 2.01) 6.59 (6.05, 7.18) 3.94 (3.50, 4.43) Ctau ND 5.82 (5.25, 6.45) ND Patients with Hepatic Impairment
The pharmacokinetics of remdesivir and GS-441524 were evaluated in healthy subjects and those with moderate or severe hepatic impairment (Child-Pugh Class B or C) following a single dose of 100 mg of VEKLURY (see Table 17). Relative to subjects with normal hepatic function, mean exposures (AUCinf, Cmax) of remdesivir and GS-441524 were similar in subjects with moderate hepatic impairment and higher in subjects with severe hepatic impairment. The exposure differences in subjects with severe hepatic impairment are not considered to be clinically significant [see Use in Specific Populations (8.7)].
Table 17 Comparison of PK Parameters of Remdesivir and GS-441524 Following IV Administration of Single Dose VEKLURY to Adults with Hepatic Impairment as Compared to Adults with Normal Hepatic Function Mean Ratio (90% CI)* Moderate Hepatic Impairment
N=10Severe Hepatic Impairment
N=6CI=Confidence Interval - * No effect=1.0 (0.5–2.0)
Remdesivir AUCinf 1.21 (0.87, 1.67) 1.56 (1.20, 2.03) Cmax 1.10 (0.75, 1.60) 1.03 (0.70, 1.51) Unbound AUCinf 1.15 (0.86, 1.54) 2.44 (1.93, 3.08) Unbound Cmax 1.04 (0.73, 1.48) 1.57 (1.08, 2.29) GS-441524 AUCinf 0.90 (0.69, 1.17) 1.31 (0.93, 1.84) Cmax 1.09 (0.86, 1.38) 1.48 (1.17, 1.86) C24 0.93 (0.69, 1.24) 1.16 (0.76, 1.77) Pediatric Patients
Population pharmacokinetic models for remdesivir and its circulating metabolites (GS-441524 and GS-704277), developed using pooled data from studies in healthy subjects and in adult and pediatric patients with COVID-19, were used to estimate pharmacokinetic exposures in pediatric patients aged from birth to <18 years and weighing ≥1.5 kg (Study 5823). Geometric mean estimated exposures (AUCtau, Cmax, and Ctau) for patients ≥28 days to <18 years old and weighing ≥3 kg (Cohorts 1–4 and 8, n=50) at the doses administered were 33% to 130% higher for remdesivir, 3% lower to 60% higher for GS-441524, and 32% to 124% higher for GS-704277 as compared to those in adult patients with COVID-19; however, the increases were not considered clinically significant. Individual estimated exposures (AUCtau, Cmax, and Ctau) for patients 14 to <28 days old, GA >37 weeks, and weighing ≥2.5 kg (Cohort 5, n=3); patients <14 days old, GA >37 weeks, and weighing ≥2.5 kg at birth (Cohort 6, n=1); and patients <56 days old, GA ≤37 weeks, and weighing ≥1.5 kg at birth (Cohort 7, n=1) at the doses administered were higher for remdesivir, GS-441524, and GS-704277 as compared to median exposures in adult patients with COVID-19; however, the increases were not considered clinically significant. As limited PK data were available in Cohorts 5–7, additional analyses were conducted using a simulated population.
Using age and weight distributions from pediatric growth charts, simulated population datasets were created for Cohorts 5–6. Modeling and simulation incorporating maturation functions that account for renal function and drug metabolizing enzyme ontogeny with age were used to predict exposures for subjects <28 days old, GA >37 weeks, and weighing ≥1.5 kg and subjects ≥28 days old and weighing ≥1.5 to <3 kg. Predicted geometric mean exposures (AUCtau, Cmax, and Ctau) at the recommended doses were 10% to 96% higher for remdesivir, 15% lower to 3% higher for GS-441524, and 14% lower to 132% higher for GS-704277 as compared to those in adult patients with COVID-19; however, changes in exposure were not considered clinically significant. Results of simulated population led to the recommended dosing regimen as they more closely align with adult exposures compared to the doses studied.
Plasma exposures of excipient SBECD were generally similar for all pediatric patients at the doses administered in Study GS-US-540-5823 and were similar compared to adults with normal renal function, although data are very limited [see Use in Specific Populations (8.4)].
The multiple dose PK parameters of remdesivir and metabolites in pediatric patients with COVID-19 in Cohorts 1–4 and 8 are provided in Table 18.
Table 18 Multiple Dose PK Parameters* of Remdesivir and Metabolites (GS-441524 and GS-704277) Following Intravenous Administration of VEKLURY 100 mg (Cohorts 1 and 8) or 2.5 mg/kg (Cohorts 2–4) to Pediatric Patients with COVID-19 Cohort 1 Cohort 8 Cohort 2 Cohort 3 Cohort 4 Parameter
Mean† (95% CI)12 to <18 Years and Weighing ≥40 kg
(N=12)<12 Years and Weighing ≥40 kg
(N=5)28 Days to <18 Years and Weighing 20 to <40 kg
(N=12)28 Days to <18 Years and Weighing 12 to <20 kg
(N=11)28 Days to <18 Years and Weighing 3 to <12 kg
(N=10)CI=Confidence Interval - * Population PK estimates for 30-minutes IV infusion of remdesivir for up to 10 days (Study GS-US-540-5823).
- † Geometric mean estimates.
Remdesivir Cmax (nanogram per mL) 3890 (3110, 4870) 3920 (2260, 6820) 5730 (4660, 7050) 5570 (4250, 7300) 4870 (3750, 6340) AUCtau (nanogram∙h per mL) 2470 (1920, 3160) 2270 (1200, 4310) 3510 (2560, 4820) 3930 (2140, 7210) 2910 (1880, 4510) GS-441524 Cmax (nanogram per mL) 196 (122, 315) 163 (57.6, 461) 183 (129, 260) 171 (130, 223) 205 (174, 241) AUCtau (nanogram∙h per mL) 3430 (1980, 5920) 2640 (767, 9100) 2370 (1500, 3740) 2410 (1740, 3340) 2850 (2290, 3540) Ctau (nanogram per mL) 98.5 (59.1, 164) 76.2 (23.9, 243) 59.9 (34.2, 105) 68.9 (47.4, 100) 79.7 (59.5, 107) GS-704277 Cmax (nanogram per mL) 308 (211, 450) 266 (137, 514) 419 (306, 575) 444 (335, 587) 385 (294, 504) AUCtau (nanogram∙h per mL) 819 (474, 1420) 518 (192, 1400) 753 (542, 1050) 733 (504, 1060) 687 (484, 973) Drug Interaction Studies
In vitro, remdesivir is a substrate for enzymes CYP3A, carboxylesterase 1 (CES1), and cathepsin A (CatA) and OATP1B1 and P-gp transporters; GS-704277 is a substrate for OATP1B1 and OATP1B3. In vitro, remdesivir is an inhibitor of CYP3A, UGT1A1, OATP1B1, OATP1B3, and MATE1; however, no clinically significant effects on substrates of UGT1A1 or MATE1 are expected. No inhibitory interactions were identified for GS-704277 or GS-441524 in vitro.
Remdesivir is not a substrate for CYP1A1, 1A2, 2B6, 2C9, 2C19, or OATP1B3. GS-704277 and GS-441524 are not substrates for CYP1A1, 1A2, 2B6, 2C8, 2C9, 2D6, or 3A5. GS-441524 is also not a substrate for CYP2C19 or 3A4. GS-704277 and GS-441524 are not substrates for OAT1, OAT3, OCT1, OCT2, MATE1, or MATE2k. GS-441524 is also not a substrate for OATP1B1 or OATP1B3.
Drug-drug interaction studies were conducted with VEKLURY. Table 19 summarizes the pharmacokinetic effects of other drugs on remdesivir and metabolites GS-704277 and GS-441524. Table 20 summarizes the effects of remdesivir on the pharmacokinetics of other drugs.
Table 19 Effect of Other Drugs on Remdesivir and Metabolites GS-704277 and GS-441524 Coadministered Drug Dose of Coadministered Drug (mg) Remdesivir Dose
(mg)N
Mean Ratio (90% CI) of Remdesivir, GS-704277, and GS-441524 PK With/Without Coadministered Drug
No Effect = 1.00 (0.70–1.43)Cmax AUCinf C24 CI=confidence interval - * Interaction study conducted in healthy volunteers.
Cyclosporin A* 400 single dose 100 single dose 9 remdesivir 1.49
(1.38–1.60)1.89
(1.77–2.02)- GS-704277 2.51
(2.26–2.78)2.97
(2.75–3.20)- GS-441524 1.17
(1.12–1.22)1.03
(0.99–1.08)1.02
(0.95–1.10)Carbamazepine* 300 twice daily 100 single dose 8 remdesivir 0.87
(0.78–0.97)0.92
(0.83–1.02)- GS-704277 0.96
(0.84–1.10)0.98
(0.92–1.05)- GS-441524 0.97
(0.88–1.07)0.83
(0.78–0.89)0.71
(0.64–0.78)Table 20 Effect of Remdesivir on the Pharmacokinetics of Other Drugs Coadministered Drug Dose of Coadministered Drug (mg) Remdesivir Dose (mg) N Mean Ratio (90% CI) of Coadministered Drug PK With/Without Remdesivir No Effect = 1.00 (0.80–1.25) Cmax AUCinf CI=confidence Interval - * Interaction study conducted in healthy volunteers.
- † Midazolam administered with last dose of remdesivir.
Midazolam* 2.5 single dose 200 single dose 19 1.29 (1.19–1.41) 1.20 (1.14–1.26) Midazolam* 2.5 single dose 200 single dose followed by 100 once daily (10 doses)† 14 1.45 (1.23– 1.70) 1.30 (1.16– 1.45) Pitavastatin* 2 single dose 200 single dose 20 1.05 (0.92–1.20) 1.17 (1.09–1.24) 12.4 Microbiology
Mechanism of Action
Remdesivir is an inhibitor of the SARS-CoV-2 RNA-dependent RNA polymerase (RdRp), which is essential for viral replication. Remdesivir is an adenosine nucleotide prodrug that distributes into cells where it is metabolized to a nucleoside monophosphate intermediate by carboxyesterase 1 and/or cathepsin A, depending upon the cell type. The nucleoside monophosphate is subsequently phosphorylated by cellular kinases to form the pharmacologically active nucleoside triphosphate metabolite (GS-443902). Remdesivir triphosphate (RDV-TP) acts as an analog of adenosine triphosphate (ATP) and competes with high selectivity (3.65-fold) over the natural ATP substrate for incorporation into nascent RNA chains by the SARS-CoV-2 RdRp, which results in delayed chain termination (position i+3) during replication of the viral RNA. In a biochemical assay assessing RDV-TP incorporation by the MERS-CoV RdRp complex, RDV-TP inhibited RNA synthesis with an IC50 value of 0.032 µM. RDV-TP can also inhibit viral RNA synthesis following its incorporation into the template viral RNA as a result of read-through by the viral polymerase that may occur at higher nucleotide concentrations. When remdesivir nucleotide is present in the viral RNA template, the efficiency of incorporation of the complementary natural nucleotide is compromised, thereby inhibiting viral RNA synthesis. Remdesivir triphosphate is a weak inhibitor of mammalian DNA and RNA polymerases, including human mitochondrial RNA polymerase.
Antiviral Activity
In Cell Culture
Remdesivir exhibited cell culture antiviral activity against a clinical isolate of SARS‑CoV‑2 in primary human airway epithelial (HAE) cells with a 50% effective concentration (EC50) of 9.9 nM after 48 hours of treatment. Remdesivir inhibited the replication of SARS-CoV-2 in the continuous human lung epithelial cell lines Calu-3 and A549-hACE2 with EC50 values of 280 nM after 72 hours of treatment and 115 nM after 48 hours of treatment, respectively.
Remdesivir EC50 values for SARS-CoV-2 in A549-hACE2 cells were not different when combined with chloroquine phosphate or hydroxychloroquine sulfate at concentrations up to 2.5 µM. In a separate study, the antiviral activity of remdesivir was antagonized by chloroquine phosphate in a dose-dependent manner when the two drugs were co-incubated at clinically relevant concentrations in HEp-2 cells infected with respiratory syncytial virus (RSV). Higher remdesivir EC50 values were observed with increasing concentrations of chloroquine phosphate. Increasing concentrations of chloroquine phosphate or hydroxychloroquine sulfate reduced formation of remdesivir triphosphate in A549-hACE2, HEp-2, and normal human bronchial epithelial cells.
Based on cell culture susceptibility testing by virus yield reduction assay and/or N protein ELISA assay, remdesivir retained similar antiviral activity against clinical isolates of SARS-CoV-2 variants compared to an earlier lineage SARS-CoV-2 (lineage A) isolate, including Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), Epsilon (B.1.429), Zeta (P.2), Iota (B.1.526), Kappa (B.1.617.1), Lambda (C.37), and Omicron variants (including B.1.1.529/BA.1, BA.2, BA.2.12.1, BA.2.75, BA.2.86, BA.4, BA.4.6, BA.5, BF.5, BF.7, BQ.1, BQ.1.1, CH.1.1, EG.1.2, EG.5.1, EG.5.1.4, FL.22, HK.3, HV.1, JN.1, XBB, XBB.1.5, XBB.1.5.72, XBB.1.16, XBB.2.3.2, XBC.1.6, and XBF). For these variants, the EC50 fold change values ranged between 0.2 and 2.3 compared to an earlier lineage SARS-CoV-2 (lineage A) isolate. Using the SARS-CoV-2 replicon system, remdesivir retained similar antiviral activity against Omicron subvariants JN.1.7, JN.1.18, KP.2, KP.3, LB.1, and XBB.1.9.2 compared to the wildtype reference replicon (lineage B).
In Clinical Trials
SARS-CoV-2 RNA shedding results from GS-US-540-5776 (ACTT-1) indicate that remdesivir does not significantly reduce the amount of detectable SARS-CoV-2 RNA in oropharyngeal or nasopharyngeal swabs or plasma samples in hospitalized patients compared to placebo, and SARS-CoV-2 RNA shedding results from GS-US-540-9012 indicate that remdesivir does not significantly reduce the amount of detectable SARS-CoV-2 RNA in nasopharyngeal swabs in non-hospitalized patients compared to placebo.
Resistance
In Cell Culture
SARS-CoV-2 isolates with reduced susceptibility to remdesivir have been selected in cell culture. In a selection with GS-441524, the parent nucleoside of remdesivir, virus pools emerged expressing amino acid substitutions at V166A, N198S, S759A, V792I, C799F, and C799R in the viral RdRp (nsp12). When these substitutions were individually introduced into a wild-type recombinant virus by site-directed mutagenesis, 1.7- to 3.5-fold reductions in susceptibility to remdesivir were observed. In a cell culture resistance selection experiment with remdesivir, nsp12 substitution E802D emerged, resulting in a 1.4- to 2.5-fold reduction in susceptibility to remdesivir. In another selection with remdesivir using a SARS-CoV-2 isolate containing the P323L substitution in the viral polymerase, a single amino acid substitution at V166L emerged. Recombinant SARS-CoV-2 with substitutions at P323L alone or P323L+V166L in combination exhibited 1.3- and 1.5-fold reductions in remdesivir susceptibility, respectively.
Cell culture resistance profiling of remdesivir using the rodent CoV murine hepatitis virus identified two substitutions (F476L and V553L) in the viral RdRp (nsp12) at residues conserved across CoVs. Introduction of the corresponding substitutions (F480L and V557L) into SARS-CoV resulted in 6-fold reduction in susceptibility to remdesivir in cell culture and attenuated SARS-CoV pathogenesis in a mouse model. When individually introduced into a SARS-CoV-2 recombinant virus, the corresponding substitutions at F480L and V557L each conferred 2-fold reduced susceptibility to remdesivir.
In Clinical Studies
In a literature publication, the SARS-CoV-2 nsp12 E802D substitution previously identified in a resistance selection experiment emerged in one individual treated with remdesivir. The E802D substitution resulted in a 1.4- to 2.5-fold increase in the remdesivir EC50 value.
In Study CO-US-540-5776 (ACTT-1), among 61 subjects with baseline and post-baseline sequencing data available, the rate of emerging substitutions in the viral RdRp (nsp12) was similar in subjects treated with VEKLURY compared to placebo. Two subjects treated with VEKLURY had an emergent substitution previously identified in resistance selection experiments (nsp12 V792I in one and C799F in the other). These substitutions are associated with 2.2- and 2.5-fold decreases in remdesivir susceptibility, respectively, based on assessments of clinical isolates. In one subject treated with VEKLURY, nsp12 V792F emerged at low frequency and was associated with a 1.8-fold decrease in remdesivir susceptibility.
In Study GS-US-540-5773, among 19 subjects treated with VEKLURY with baseline and post-baseline sequencing data available, the V792F substitution in viral RdRp (nsp12) emerged at low frequency in one subject.
In Study GS-US-540-9012, among 244 subjects with baseline and post-baseline sequencing data available, the rate of emerging substitutions in the viral RdRp (nsp12) was similar in subjects treated with VEKLURY compared to placebo. In one subject treated with VEKLURY, one substitution in the RdRp (nsp12 A376V) emerged and was associated with a 12.6-fold decrease in remdesivir susceptibility in a subgenomic replicon assay. This subject was not hospitalized and showed alleviation of all baseline symptoms, except loss of taste and smell, prior to or on Day 14.
In Study GS-US-540-5912, among 60 subjects with baseline and post-baseline sequencing data available, substitutions in the viral RdRp (nsp12) emerged in 8 subjects treated with VEKLURY. In 4 subjects treated with VEKLURY, three substitutions in the RdRp (nsp12 E136V, M794I, or C799F) emerged and were associated with 2.9-, 2.9-, and 3.4-fold reduced susceptibility to remdesivir in a subgenomic replicon assay.
In Study GS-US-540-5823, among pediatric subjects with baseline and post-baseline sequencing data available, treatment-emergent substitutions in the viral RdRp (nsp12) were observed in 3 of 27 subjects treated with VEKLURY and were evaluated for susceptibility to remdesivir. In one subject, two substitutions (nsp12 substitutions V166L and V792I) emerged and were associated with 1.85- and 3.6-fold decreases in remdesivir susceptibility relative to reference, respectively. This subject was hospitalized at baseline, recovered from COVID-19, and was released from the hospital on Day 13. None of the substitutions observed in any of the other genes (nsp9–10, nsp13–14) encoding for proteins of the viral replication-transcription complex have been associated with reduced susceptibility to remdesivir.
The relationship between the level of reduced susceptibility to remdesivir observed in subgenomic replicon assays and the inhibition of SARS-CoV-2 replication by remdesivir in humans has not been fully established.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
Given the short-term administration of VEKLURY for the treatment of COVID-19, long-term animal studies to evaluate the carcinogenic potential of remdesivir were not conducted.
Remdesivir was not genotoxic in a battery of assays, including bacterial mutagenicity, chromosome aberration using human peripheral blood lymphocytes, and in vivo rat micronucleus assays.
Impairment of Fertility
Nonclinical toxicity studies in rats demonstrated no adverse effect on male fertility at exposures of the predominant circulating metabolite (GS-441524) approximately 2 times the exposure in humans at the RHD.
Reproductive toxicity, including decreases in corpora lutea, numbers of implantation sites, and viable embryos, was seen when remdesivir was administered by daily intravenous administration at a systemically toxic dose (10 mg/kg) in female rats 14 days prior to mating and during conception; exposures of the predominant circulating metabolite (GS-441524) were 1.3 times the exposure in humans at the RHD.
13.2 Animal Toxicology and/or Pharmacology
Intravenous administration (slow bolus) of remdesivir to male rhesus monkeys at dosage levels of 5, 10, and 20 mg/kg/day for 7 days resulted, at all dose levels, in increased mean urea nitrogen and increased mean creatinine, renal tubular atrophy, and basophilia and casts.
Intravenous administration (slow bolus) of remdesivir to rats at dosage levels of ≥3 mg/kg/day for up to 4 weeks resulted in findings indicative of kidney injury and/or dysfunction.
Kidney-related effects in rats and monkeys were observed at exposures of the predominant circulating metabolite (GS-441524) that are lower than the exposure in humans at the RHD.
-
14 CLINICAL STUDIES
14.1 Description of Clinical Trials
The efficacy and safety of VEKLURY were evaluated in the trials summarized in Table 21.
Table 21 Trials Conducted with VEKLURY in Subjects with COVID-19 Trial Population Trial Arms (N) Timepoint COVID-19: coronavirus disease 2019 - * Randomized, double-blind, placebo-controlled trial.
- † Randomized, open-label trial.
- ‡ Open-label trial, descriptive outcome analyses.
NIAID ACTT-1*
(NCT04280705)Hospitalized with mild/moderate and severe COVID-19 VEKLURY 10 Days (532)
Placebo (516)29 Days after Randomization GS-US-540-5773†
(NCT04292899)Hospitalized with severe COVID-19 VEKLURY 5 Days (200)
VEKLURY 10 Days (197)Day 14 GS-US-540-5774†
(NCT04292730)Hospitalized with moderate COVID-19 VEKLURY 5 Days (191)
VEKLURY 10 Days (193)
Standard of care (200)Day 11 GS-US-540-9012*
(NCT04501952)Non-hospitalized with mild-to-moderate COVID-19 and at high risk for progression to severe disease VEKLURY 3 Days (279)
Placebo (283)Day 28 GS-US-540-5823 (Cohorts 1–8)‡
(NCT04431453)Hospitalized pediatric subjects from birth to <18 years of age and weighing at least 1.5 kg with COVID-19 VEKLURY up to 10 Days (58) Day 10 14.2 NIAID ACTT-1 Study in Hospitalized Subjects with Mild/Moderate and Severe COVID-19
A randomized, double-blind, placebo-controlled clinical trial (ACTT-1) of hospitalized adult subjects with confirmed SARS-CoV-2 infection and mild, moderate, or severe COVID-19 compared treatment with VEKLURY for 10 days (n=541) with placebo (n=521). Mild/moderate disease was defined as SpO2 >94% and respiratory rate <24 breaths/minute without supplemental oxygen; severe disease was defined as an SpO2 ≤94% on room air, a respiratory rate ≥24 breaths/minute, an oxygen requirement, or a requirement for mechanical ventilation. Subjects had to have at least one of the following to be enrolled in the trial: radiographic infiltrates by imaging, SpO2 ≤94% on room air, a requirement for supplemental oxygen, or a requirement for mechanical ventilation. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days, for 10 days of treatment via intravenous infusion. Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to the completion of 10 days of treatment.
At baseline, mean age was 59 years (with 36% of subjects aged 65 or older); 64% of subjects were male, 53% were White, 21% were Black, and 13% were Asian; 24% were Hispanic or Latino; 105 subjects had mild/moderate disease (10% in both treatment groups); 957 subjects had severe disease (90% in both treatment groups). Subjects in this trial were unvaccinated. A total of 285 subjects (27%) (n=131 received VEKLURY) were on invasive mechanical ventilation or ECMO. The most common comorbidities were hypertension (51%), obesity (45%), and type 2 diabetes mellitus (31%); the distribution of comorbidities was similar between the two treatment groups.
The primary clinical endpoint was time to recovery within 29 days after randomization. Recovery was defined as discharged from the hospital without limitations on activities, discharged from the hospital with limitations on activities and/or requiring home oxygen, or hospitalized but not requiring supplemental oxygen and no longer requiring ongoing medical care. The median time to recovery was 10 days in the VEKLURY group compared to 15 days in the placebo group (recovery rate ratio 1.29 [95% CI 1.12 to 1.49], p<0.001). Among subjects with mild/moderate disease at enrollment (n=105), the median time to recovery was 5 days in both the VEKLURY and placebo groups (recovery rate ratio 1.22 [95% CI 0.82 to 1.81]). Among subjects with severe disease at enrollment (n=957), the median time to recovery was 11 days in the VEKLURY group compared to 18 days in the placebo group (recovery rate ratio 1.31 [95% CI 1.12 to 1.52]).
A key secondary endpoint was clinical status on Day 15 assessed on an 8-point ordinal scale consisting of the following categories:
- not hospitalized, no limitations on activities;
- not hospitalized, limitation on activities and/or requiring home oxygen;
- hospitalized, not requiring supplemental oxygen - no longer requires ongoing medical care;
- hospitalized, not requiring supplemental oxygen - requiring ongoing medical care (COVID-19 related or otherwise);
- hospitalized, requiring supplemental oxygen;
- hospitalized, on noninvasive ventilation or high-flow oxygen devices;
- hospitalized, on invasive mechanical ventilation or ECMO; and
- death.
Overall, the odds of improvement in the ordinal scale were higher in the VEKLURY group at Day 15 when compared to the placebo group (odds ratio 1.54 [95% CI 1.25 to 1.91]).
Overall, 29-day mortality was 11% for the VEKLURY group vs 15% for the placebo group (hazard ratio 0.73 [95% CI 0.52 to 1.03]).
14.3 Study GS-US-540-5773 in Hospitalized Subjects with Severe COVID-19
A randomized, open-label multi-center clinical trial (Study 5773) in adult subjects with confirmed SARS-CoV-2 infection, an SpO2 of ≤94% on room air, and radiological evidence of pneumonia compared 200 subjects who received VEKLURY for 5 days with 197 subjects who received VEKLURY for 10 days. Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to completion of their protocol-defined duration of treatment. Subjects on mechanical ventilation at screening were excluded. All subjects received 200 mg of VEKLURY on Day 1 and 100 mg once daily on subsequent days via intravenous infusion, plus standard of care.
At baseline, the median age of subjects was 61 years (range, 20 to 98 years); 64% were male, 75% were White, 12% were Black, and 12% were Asian; 22% were Hispanic or Latino. More subjects in the 10-day group than the 5-day group required invasive mechanical ventilation or ECMO (5% vs 2%), or high-flow oxygen support (30% vs 25%), at baseline. Subjects in this trial were unvaccinated. Median duration of symptoms and hospitalization prior to first dose of VEKLURY were similar across treatment groups.
The primary endpoint was clinical status on Day 14 assessed on a 7-point ordinal scale consisting of the following categories:
- death;
- hospitalized, receiving invasive mechanical ventilation or ECMO;
- hospitalized, receiving noninvasive ventilation or high-flow oxygen devices;
- hospitalized, requiring low-flow supplemental oxygen;
- hospitalized, not requiring supplemental oxygen but receiving ongoing medical care (related or not related to COVID-19);
- hospitalized, requiring neither supplemental oxygen nor ongoing medical care (other than that specified in the protocol for remdesivir administration); and
- not hospitalized.
Overall, after adjusting for between-group differences at baseline, subjects receiving a 5-day course of VEKLURY had similar clinical status at Day 14 as those receiving a 10-day course (odds ratio for improvement 0.75 [95% CI 0.51 to 1.12]). There were no statistically significant differences in recovery rates or mortality rates in the 5-day and 10-day groups once adjusted for between-group differences at baseline. All-cause mortality at Day 28 was 12% vs 14% in the 5- and 10-day treatment groups, respectively.
14.4 Study GS-US-540-5774 in Hospitalized Subjects with Moderate COVID-19
A randomized, open-label multi-center clinical trial (Study 5774) of hospitalized adult subjects with confirmed SARS-CoV-2 infection, SpO2 >94% and radiological evidence of pneumonia compared treatment with VEKLURY for 5 days (n=191) and treatment with VEKLURY for 10 days (n=193) with standard of care (n=200). Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to completion of their protocol-defined duration of treatment. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days via intravenous infusion.
At baseline, the median age of subjects was 57 years (range, 12 to 95 years); 61% were male, 61% were White, 19% were Black, and 19% were Asian; 18% were Hispanic or Latino. Subjects in this trial were unvaccinated. Baseline clinical status, oxygen support status, and median duration of symptoms and hospitalization prior to first dose of VEKLURY were similar across treatment groups.
The primary endpoint was clinical status on Day 11 assessed on a 7-point ordinal scale consisting of the following categories:
- death;
- hospitalized, receiving invasive mechanical ventilation or ECMO;
- hospitalized, receiving noninvasive ventilation or high-flow oxygen devices;
- hospitalized, requiring low-flow supplemental oxygen;
- hospitalized, not requiring supplemental oxygen but receiving ongoing medical care (related or not related to COVID-19);
- hospitalized, requiring neither supplemental oxygen nor ongoing medical care (other than that specified in the protocol for remdesivir administration); and
- not hospitalized.
Overall, the odds of improvement in the ordinal scale were higher in the 5-day VEKLURY group at Day 11 when compared to those receiving only standard of care (odds ratio 1.65 [95% CI 1.09 to 2.48], p=0.017). The odds of improvement in clinical status with the 10-day treatment group when compared to those receiving only standard of care were not statistically significant (odds ratio 1.31 [95% CI 0.88 to 1.95]). All-cause mortality at Day 28 was ≤2% in all treatment groups.
14.5 Study GS-US-540-9012 in Non-Hospitalized Subjects with Mild-to-Moderate COVID-19 and at High Risk for Progression to Severe Disease
A randomized, double-blind, placebo-controlled, clinical trial (Study 9012) evaluated VEKLURY 200 mg once daily for 1 day followed by VEKLURY 100 mg once daily for 2 days (for a total of 3 days of intravenously administered therapy) in 554 adult and 8 pediatric subjects (12 years of age and older and weighing at least 40 kg) who were non-hospitalized, had mild-to-moderate COVID-19, were symptomatic for COVID-19 for ≤7 days, had confirmed SARS-CoV-2 infection, and had at least one risk factor for progression to hospitalization. Risk factors for progression to hospitalization included age ≥60 years, obesity (BMI ≥30), chronic lung disease, hypertension, cardiovascular or cerebrovascular disease, diabetes mellitus, immunocompromised state, chronic mild or moderate kidney disease, chronic liver disease, current cancer, and sickle cell disease. Subjects who received, required, or were expected to require supplemental oxygen were excluded from the trial. Subjects were randomized in a 1:1 manner, stratified by residence in a skilled nursing facility (yes/no), age (<60 vs ≥60 years), and region (US vs ex-US) to receive VEKLURY (n=279) or placebo (n=283), plus standard of care.
At baseline, mean age was 50 years (with 30% of subjects aged 60 or older); 52% were male, 80% were White, 8% were Black, and 2% were Asian; 44% were Hispanic or Latino; median body mass index was 30.7 kg/m2. Subjects in this trial were unvaccinated. VEKLURY or placebo was first administered to subjects in outpatient facilities (84%), home healthcare settings (13%), or skilled nursing facilities (3%). The most common comorbidities were diabetes mellitus (62%), obesity (56%), and hypertension (48%). Median (Q1, Q3) duration of symptoms prior to treatment was 5 (3, 6) days; median viral load was 6.3 log10 copies/mL at baseline. The baseline demographics and disease characteristics were well balanced across the VEKLURY and placebo treatment groups.
The primary endpoint was the proportion of subjects with COVID-19 related hospitalization (defined as at least 24 hours of acute care) or all-cause mortality through Day 28. Events occurred in 2 (0.7%) subjects treated with VEKLURY compared to 15 (5.3%) subjects concurrently randomized to placebo (hazard ratio 0.134 [95% CI 0.031 to 0.586]; p=0.0076). No deaths were observed through Day 28.
14.6 Study GS-US-540-5823 in Hospitalized Pediatric Subjects with COVID-19
The primary objectives of this Phase 2/3 single-arm, open-label clinical trial (Study GS-US-540-5823) were to evaluate pharmacokinetics and safety of up to 10 days of treatment with VEKLURY in pediatric subjects. A total of 58 pediatric subjects from birth (including preterm to term infants) to <18 years of age and weighing at least 1.5 kg with confirmed SARS-CoV-2 infection and mild, moderate, or severe COVID-19 was evaluated in eight cohorts:
- Cohorts 1–4, 8; infants, children, and adolescents: Subjects ≥12 years and weighing ≥40 kg (n=12); subjects <12 years and weighing ≥40 kg (n=5); subjects ≥28 days and weighing ≥20 to <40 kg (n=12); subjects ≥28 days and weighing ≥12 to <20 kg (n=12); and subjects ≥28 days and weighing ≥3 to <12 kg (n=12). Subjects weighing ≥40 kg received 200 mg of VEKLURY on Day 1 followed by VEKLURY 100 mg once daily on subsequent days; subjects weighing ≥3 kg to <40 kg received VEKLURY 5 mg/kg on Day 1 followed by VEKLURY 2.5 mg/kg once daily on subsequent days;
- Cohorts 5–7; neonates and infants: Subjects 14 to <28 days old, GA >37 weeks, and weighing ≥2.5 kg (n=3); subjects <14 days old, GA >37 weeks, and weighing ≥2.5 kg at birth (n=1); and subjects <56 days old, GA ≤37 weeks, and weighing ≥1.5 kg at birth (n=1). Subjects 14 to <28 days old, GA >37 weeks, and weighing ≥2.5 kg received VEKLURY 5 mg/kg on Day 1 followed by VEKLURY 2.5 mg/kg once daily on subsequent days. Subjects <14 days old, GA >37 weeks, and weighing at least 2.5 kg at birth, and subjects <56 days old, GA ≤37 weeks, and weighing ≥1.5 kg at birth, received VEKLURY 2.5 mg/kg on Day 1 followed by VEKLURY 1.25 mg/kg once daily on subsequent days.
Assessments occurred at the following intervals: Screening; Day 1 (Baseline); Days 2–10, or until discharge, whichever came earlier; Follow-Up on Day 30 (±5). Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to the completion of 10 days of treatment.
Infants, children, and adolescents: At baseline, median age was 7 years (Q1, Q3: 2 years, 12 years); 57% were female, 70% were White, 30% were Black, and 44% were Hispanic or Latino; median weight was 25 kg (range: 4 to 192 kg). Subjects in this trial were unvaccinated. A total of 12 subjects (23%) were on invasive mechanical ventilation, 18 (34%) were on non-invasive ventilation or high-flow oxygen; 10 (19%) were on low-flow oxygen; and 13 (25%) were on room air, at baseline. The overall median (Q1, Q3) duration of symptoms and hospitalization prior to first dose of VEKLURY was 5 (3, 7) days and 1 (1, 3) day, respectively.
The descriptive outcome analyses showed treatment with VEKLURY for up to 10 days resulted in an overall median (Q1, Q3) change from baseline in clinical status (assessed on a 7-point ordinal scale ranging from death [score of 1] to ventilatory support and decreasing levels of oxygen to hospital discharge [score of 7]) of +2.0 (1.0, 4.0) points on Day 10.
Recovery (defined as an improvement from a baseline clinical status score of 2 through 5 to a score of 6 or 7, or an improvement from a baseline score of 6 to a score of 7) was reported for 62% of subjects on Day 10; median (Q1, Q3) time to recovery was 7 (5, 16) days.
Overall, 60% of subjects were discharged by Day 10, and 83% of subjects were discharged by Day 30. Three subjects (6%) from Cohorts 1–4 and Cohort 8 died during the study.
Neonates and infants: At baseline, subjects ranged in age from 12 to 30 days; 3/5 were female, 4/5 were White, 1/5 was Black; weight ranged from 2.2 to 3.5 kg. Three subjects were on invasive mechanical ventilation and 2 were on high-flow oxygen. The duration of symptoms and hospitalization prior to first dose of VEKLURY ranged from 2 to 9 days and 1 to 9 days, respectively.
The descriptive outcome analyses showed treatment with VEKLURY for up to 10 days resulted in recovery (defined as an improvement from a baseline clinical status score of 2 through 5 to a score of 6 or 7, or an improvement from a baseline score of 6 to a score of 7) for 3 subjects, including for one subject by Day 10. Time to recovery ranged from 9 to 19 days.
Overall, a total of 3 subjects were discharged by Day 30, of which one subject was discharged by Day 10. No subjects from Cohorts 5–7 died during the study.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
VEKLURY for injection, 100 mg (NDC: 61958-2901-2) is supplied as a single-dose vial containing a sterile, preservative-free white to off-white to yellow lyophilized powder. It requires reconstitution and further dilution prior to administration by intravenous infusion [see Dosage and Administration (2.5)]. Discard unused portion. The container closure is not made with natural rubber latex.
Storage and Handling
These products contain no preservative; therefore, partially used vials should be discarded [see Dosage and Administration (2.6)].
Store VEKLURY for injection, 100 mg vials below 30°C (below 86°F) until required for use.
After reconstitution, use vials immediately to prepare diluted solution. Dilute the reconstituted solution in 0.9% sodium chloride injection, USP within the same day as administration. The diluted VEKLURY solution in the infusion bags can be stored up to 24 hours at room temperature (20°C to 25°C [68°F to 77°F]) prior to administration or 48 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Hypersensitivity Reactions
Inform patients that hypersensitivity reactions have been seen in patients receiving VEKLURY during and after infusion. Advise patients to inform their healthcare provider if they experience any of the following: changes in heart rate; fever; shortness of breath, wheezing; swelling of the lips, face, or throat; rash; nausea; sweating; or shivering [see Warnings and Precautions (5.1)].
Increased Risk of Transaminase Elevations
Inform patients that VEKLURY may increase the risk of hepatic laboratory abnormalities. Advise patients to alert their healthcare provider immediately if they experience any symptoms of liver inflammation [see Warnings and Precaution (5.2)].
Drug Interactions
Inform patients that VEKLURY may interact with other drugs. Advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products, including chloroquine phosphate or hydroxychloroquine sulfate [see Warnings and Precautions (5.3), Drug Interactions (7), and Microbiology (12.4)].
Pregnancy
Inform patients to notify their healthcare provider in the event of a pregnancy [see Use in Specific Populations (8.1)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 10/2025 PATIENT INFORMATION VEKLURY® (VEK-lur-ee)
(remdesivir)
for injectionWhat is VEKLURY?
VEKLURY is a prescription medicine used for the treatment of coronavirus disease 2019 (COVID-19) in adults and children weighing at least 3 pounds (1.5 kg) who are:- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
Do not take VEKLURY if you are allergic to remdesivir or any of the ingredients in VEKLURY. See the end of this leaflet for a complete list of ingredients in VEKLURY. Before receiving VEKLURY, tell your healthcare provider about all of your medical conditions, including if you: - have liver problems
- are pregnant or plan to become pregnant. It is not known if VEKLURY may harm your unborn baby if taken during the first trimester of pregnancy. Tell your healthcare provider right away if you are or if you become pregnant.
- are breastfeeding or plan to breastfeed. VEKLURY can pass into your breast milk. Talk to your healthcare provider about the best way to feed your baby.
Especially tell your healthcare provider if you are taking the medicines chloroquine phosphate or hydroxychloroquine sulfate.How will I receive VEKLURY? - Hospitalized: VEKLURY is given to you through a vein by intravenous (IV) infusion one time each day for up to 10 days. Your healthcare provider will decide how many doses you need.
- Not hospitalized: VEKLURY is given to you through a vein by intravenous (IV) infusion one time each day for 3 days.
- Your healthcare provider will do certain blood tests before starting and during treatment with VEKLURY.
What are the possible side effects of VEKLURY?
VEKLURY may cause serious side effects, including:- Allergic reactions. Allergic reactions can happen during or after infusion with VEKLURY. Your healthcare provider will monitor you for signs and symptoms of allergic reactions during your infusion and for at least 1 hour after your infusion. Tell your healthcare provider right away if you get any of the following signs and symptoms of an allergic reaction:
- changes in your heart rate
- fever
- shortness of breath, wheezing
- swelling of the lips, face, or throat
- rash
- nausea
- sweating
- shivering
- Increase in liver enzymes. Increases in liver enzymes are common in people who have received VEKLURY and may be a sign of liver injury. Your healthcare provider will do blood tests to check your liver enzymes before and during treatment with VEKLURY as needed. Your healthcare provider may stop treatment with VEKLURY if you develop liver problems.
These are not all of the possible side effects of VEKLURY.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of VEKLURY.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about VEKLURY that is written for healthcare professionals.What are the ingredients in VEKLURY?
Active ingredient: remdesivir
Inactive ingredients: betadex sulfobutyl ether sodium and may include hydrochloric acid and/or sodium hydroxide for pH adjustment.
Manufactured and distributed by: Gilead Sciences, Inc., Foster City, CA 94404
VEKLURY is a trademark of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners.
© 2025 Gilead Sciences, Inc. All rights reserved.
214787-GS-022
For more information, call 1-800-445-3235 or go to www.VEKLURY.com. - PRINCIPAL DISPLAY PANEL - 100 mg Vial Label
-
PRINCIPAL DISPLAY PANEL - 100 mg Vial Carton
NDC: 61958-2901-2
Rx onlyVeklury®
(remdesivir) for injection
100 mg/vialSingle-Dose Vial: Discard
Unused PortionMust be reconstituted and
further diluted prior to use.
For Intravenous Infusion.GILEAD
-
INGREDIENTS AND APPEARANCE
VEKLURY
remdesivir injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-2902 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength REMDESIVIR (UNII: 3QKI37EEHE) (REMDESIVIR - UNII:3QKI37EEHE) REMDESIVIR 5 mg in 1 mL Inactive Ingredients Ingredient Name Strength BETADEX SULFOBUTYL ETHER SODIUM (UNII: 2PP9364507) WATER (UNII: 059QF0KO0R) HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-2902-2 1 in 1 CARTON 03/25/2022 10/07/2025 1 20 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA214787 03/25/2022 10/07/2025 VEKLURY
remdesivir injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-2901 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength REMDESIVIR (UNII: 3QKI37EEHE) (REMDESIVIR - UNII:3QKI37EEHE) REMDESIVIR 100 mg Inactive Ingredients Ingredient Name Strength BETADEX SULFOBUTYL ETHER SODIUM (UNII: 2PP9364507) WATER (UNII: 059QF0KO0R) HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-2901-2 1 in 1 CARTON 11/01/2020 1 1 in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA214787 11/01/2020 Labeler - Gilead Sciences, Inc. (185049848)

Trademark Results [Veklury]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VEKLURY 86497165 5588555 Live/Registered |
Gilead Sciences Ireland UC 2015-01-07 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.