NULOJIX- belatacept injection, powder, lyophilized, for solution
NULOJIX by
Drug Labeling and Warnings
NULOJIX by is a Prescription medication manufactured, distributed, or labeled by E.R. Squibb & Sons, L.L.C.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NULOJIX safely and effectively. See full prescribing information for NULOJIX.
NULOJIX (belatacept) for injection, for intravenous use
Initial U.S. Approval: 2011WARNING: POST-TRANSPLANT LYMPHOPROLIFERATIVE DISORDER, OTHER MALIGNANCIES, AND SERIOUS INFECTIONS
See full prescribing information for complete boxed warning.
- Increased risk for developing post-transplant lymphoproliferative disorder (PTLD), predominantly involving the central nervous system (CNS). Recipients without immunity to Epstein-Barr virus (EBV) are at a particularly increased risk; therefore, use in EBV seropositive patients only. Do not use NULOJIX in transplant recipients who are EBV seronegative or with unknown serostatus. (4, 5.1)
- Only physicians experienced in immunosuppressive therapy and management of kidney transplant patients should prescribe NULOJIX. (5.2)
- Increased susceptibility to infection and the possible development of malignancies may result from immunosuppression. (5.1, 5.3, 5.4, 5.5)
- Use in liver transplant patients is not recommended due to an increased risk of graft loss and death. (5.6)
RECENT MAJOR CHANGES
Warnings and Precautions (5.9) 5/2017
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- Use of higher than recommended or more frequent dosing is not recommended due to increased risk of serious infections and malignancy. (5.1, 5.4, 6.1)
- For complete dosing instructions, see full prescribing information. (2.1)
Dosing of NULOJIX for Kidney Transplant Recipients (2.1) Dosing for Initial Phase
Dose
Day 1 (day of transplantation, prior to implantation) and Day 5 (approximately 96 hours after Day 1 dose)
10 mg per kg
End of Week 2 and Week 4 after transplantation
10 mg per kg
End of Week 8 and Week 12 after transplantation
10 mg per kg
Dosing for Maintenance Phase
Dose
End of Week 16 after transplantation and every 4 weeks (plus or minus 3 days) thereafter
5 mg per kg
DOSAGE FORMS AND STRENGTHS
Lyophilized powder for injection: 250 mg per vial (3)
CONTRAINDICATIONS
Patients who are EBV seronegative or with unknown EBV serostatus. (4)
WARNINGS AND PRECAUTIONS
- Post-Transplant Lymphoproliferative Disorder (PTLD): increased risk, predominantly involving the CNS; monitor for new or worsening neurological, cognitive, or behavioral signs and symptoms. (Boxed Warning, 4, 5.1, 5.6)
- Other malignancies: increased risk with all immunosuppressants; appears related to intensity and duration of use. Avoid prolonged exposure to UV light and sunlight. (5.3)
- Progressive Multifocal Leukoencephalopathy (PML): increased risk; consider in the diagnosis of patients reporting new or worsening neurological, cognitive, or behavioral signs and symptoms. Recommended doses of immunosuppressants should not be exceeded. (5.4)
- Other serious infections: increased risk of bacterial, viral, fungal, and protozoal infections, including opportunistic infections and tuberculosis. Some infections were fatal. Polyoma virus-associated nephropathy can lead to kidney graft loss; consider reduction in immunosuppression. Evaluate for tuberculosis and initiate treatment for latent infection prior to NULOJIX use. Cytomegalovirus and pneumocystis prophylaxis are recommended after transplantation. (5.1, 5.4, 5.5)
- Liver transplant: use is not recommended. (5.6)
- Acute Rejection and Graft Loss with Corticosteroid Minimization: corticosteroid utilization should be consistent with the NULOJIX clinical trial experience. (2.1, 5.7, 14.1)
- Immunizations: avoid use of live vaccines during treatment. (5.8)
- Coadministration with Anti-Thymocyte Globulin: in de novo kidney transplant recipients, especially those with other predisposing risk factors for venous thrombosis of the renal allograft, coadministration (at the same or nearly the same time) with anti-thymocyte globulin may pose a risk for venous thrombosis of the renal allograft. (5.9, 6.2, 7.3).
ADVERSE REACTIONS
Most common adverse reactions (≥20% on NULOJIX treatment) are anemia, diarrhea, urinary tract infection, peripheral edema, constipation, hypertension, pyrexia, graft dysfunction, cough, nausea, vomiting, headache, hypokalemia, hyperkalemia, and leukopenia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: POST-TRANSPLANT LYMPHOPROLIFERATIVE DISORDER, OTHER MALIGNANCIES, AND SERIOUS INFECTIONS
1 INDICATIONS AND USAGE
1.1 Adult Kidney Transplant Recipients
1.2 Limitations of Use
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Adult Kidney Transplant Recipients
2.2 Preparation and Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Post-Transplant Lymphoproliferative Disorder
5.2 Management of Immunosuppression
5.3 Other Malignancies
5.4 Progressive Multifocal Leukoencephalopathy
5.5 Other Serious Infections
5.6 Liver Transplant
5.7 Acute Rejection and Graft Loss with Corticosteroid Minimization
5.8 Immunizations
5.9 Coadministration with Anti-Thymocyte Globulin
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
6.2 Postmarketing Experience
6.3 Long-Term Extension Studies
7 DRUG INTERACTIONS
7.1 Mycophenolate Mofetil (MMF)
7.2 Cytochrome P450 Substrates
7.3 Anti-Thymocyte Globulin
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Prevention of Organ Rejection in Kidney Transplant Recipients
14.2 Long-Term Extension (LTE) of Study 1 and Study 2
14.3 Follow-Up Data of Patients with Complete 7-Year Patient and Graft Survival
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 Storage
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: POST-TRANSPLANT LYMPHOPROLIFERATIVE DISORDER, OTHER MALIGNANCIES, AND SERIOUS INFECTIONS
Increased risk for developing post-transplant lymphoproliferative disorder (PTLD), predominantly involving the central nervous system (CNS). Recipients without immunity to Epstein-Barr virus (EBV) are at a particularly increased risk; therefore, use in EBV seropositive patients only. Do not use NULOJIX in transplant recipients who are EBV seronegative or with unknown EBV serostatus [see Contraindications (4) and Warnings and Precautions (5.1)].
Only physicians experienced in immunosuppressive therapy and management of kidney transplant patients should prescribe NULOJIX. Patients receiving the drug should be managed in facilities equipped and staffed with adequate laboratory and supportive medical resources. The physician responsible for maintenance therapy should have complete information requisite for the follow-up of the patient [see Warnings and Precautions (5.2)].
Increased susceptibility to infection and the possible development of malignancies may result from immunosuppression [see Warnings and Precautions (5.1, 5.3, 5.4, 5.5)].
Use in liver transplant patients is not recommended due to an increased risk of graft loss and death [see Warnings and Precautions (5.6)].
-
1 INDICATIONS AND USAGE
1.1 Adult Kidney Transplant Recipients
NULOJIX® (belatacept) is indicated for prophylaxis of organ rejection in adult patients receiving a kidney transplant. NULOJIX is to be used in combination with basiliximab induction, mycophenolate mofetil, and corticosteroids.
1.2 Limitations of Use
Use NULOJIX only in patients who are EBV seropositive [see Contraindications (4) and Warnings and Precautions (5.1)].
Use of NULOJIX for the prophylaxis of organ rejection in transplanted organs other than kidney has not been established [see Warnings and Precautions (5.6)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Adult Kidney Transplant Recipients
NULOJIX should be administered in combination with basiliximab induction, mycophenolate mofetil (MMF), and corticosteroids. In clinical trials the median (25th to 75th percentile) corticosteroid doses were tapered to approximately 15 mg (10 to 20 mg) per day by the first 6 weeks and remained at approximately 10 mg (5 to 10 mg) per day for the first 6 months post-transplant. Corticosteroid utilization should be consistent with the NULOJIX clinical trial experience [see Warnings and Precautions (5.7) and Clinical Studies (14.1)].
Due to an increased risk of post-transplant lymphoproliferative disorder (PTLD) predominantly involving the central nervous system (CNS), progressive multifocal leukoencephalopathy (PML), and serious CNS infections, administration of higher than the recommended doses or more frequent dosing of NULOJIX is not recommended [see Warnings and Precautions (5.1, 5.4, 5.5) and Adverse Reactions (6.1)].
NULOJIX is for intravenous infusion only. Patients do not require premedication prior to administration of NULOJIX.
Dosing instructions are provided in Table 1.
- The total infusion dose of NULOJIX should be based on the actual body weight of the patient at the time of transplantation, and should not be modified during the course of therapy, unless there is a change in body weight of greater than 10%.
-
The prescribed dose of NULOJIX must be evenly divisible by 12.5 mg in order for the dose to be prepared accurately using the reconstituted solution and the silicone-free disposable syringe provided. Evenly divisible increments are 0, 12.5, 25, 37.5, 50, 62.5, 75, 87.5, and 100. For example:
- - A patient weighs 64 kg. The dose is 10 mg per kg.
- - Calculated Dose: 64 kg × 10 mg per kg = 640 mg
- - The closest doses evenly divisible by 12.5 mg below and above 640 mg are 637.5 mg and 650 mg.
- - The nearest dose to 640 mg is 637.5 mg.
- - Therefore, the actual prescribed dose for the patient should be 637.5 mg.
Table 1: Dosinga,b of NULOJIX for Kidney Transplant Recipients a [See Clinical Studies (14.1)]
b The dose prescribed for the patient must be evenly divisible by 12.5 mg (see instructions above; eg, evenly divisible increments are 0, 12.5, 25, 37.5, 50, 62.5, 75, 87.5, and 100).Dosing for Initial Phase
Dose
Day 1 (day of transplantation, prior to implantation) and Day 5 (approximately 96 hours after Day 1 dose)
10 mg per kg
End of Week 2 and Week 4 after transplantation
10 mg per kg
End of Week 8 and Week 12 after transplantation
10 mg per kg
Dosing for Maintenance Phase
Dose
End of Week 16 after transplantation and every 4 weeks (plus or minus 3 days) thereafter
5 mg per kg
2.2 Preparation and Administration Instructions
NULOJIX is for intravenous infusion only.
Caution: NULOJIX must be reconstituted/prepared using only the silicone-free disposable syringe provided with each vial.
If the silicone-free disposable syringe is dropped or becomes contaminated, use a new silicone-free disposable syringe from inventory.
Preparation for Administration
- 1. Calculate the number of NULOJIX vials required to provide the total infusion dose. Each vial contains 250 mg of belatacept lyophilized powder.
- 2.
Reconstitute the contents of each vial of NULOJIX with 10.5 mL of a suitable diluent using the silicone-free disposable syringe provided with each vial and an 18- to 21-gauge needle. Suitable diluents include: sterile water for injection (SWFI), 0.9% sodium chloride (NS), or 5% dextrose in water (D5W).
Note: If the NULOJIX powder is accidentally reconstituted using a different syringe than the one provided, the solution may develop a few translucent particles. Discard any solutions prepared using siliconized syringes. - 3. To reconstitute the NULOJIX powder, remove the flip-top from the vial and wipe the top with an alcohol swab. Insert the syringe needle into the vial through the center of the rubber stopper and direct the stream of diluent (10.5 mL of SWFI, NS, or D5W) to the glass wall of the vial.
- 4. To minimize foam formation, rotate the vial and invert with gentle swirling until the contents are completely dissolved. Avoid prolonged or vigorous agitation. Do not shake.
- 5. The reconstituted solution contains a belatacept concentration of 25 mg/mL and should be clear to slightly opalescent and colorless to pale yellow. Do not use if opaque particles, discoloration, or other foreign particles are present.
- 6.
Calculate the total volume of the reconstituted 25 mg/mL NULOJIX solution required to provide the total infusion dose.
Volume of 25 mg/mL NULOJIX solution (in mL) = Prescribed Dose (in mg) ÷ 25 mg/mL - 7.
Prior to intravenous infusion, the required volume of the reconstituted NULOJIX solution must be further diluted with a suitable infusion fluid (NS or D5W). NULOJIX reconstituted with:
- SWFI should be further diluted with either NS or D5W
- NS should be further diluted with NS
- D5W should be further diluted with D5W
- 8.
From the appropriate size infusion bag or bottle, withdraw a volume of infusion fluid that is equal to the volume of the reconstituted NULOJIX solution required to provide the prescribed dose. With the same silicone-free disposable syringe used for reconstitution, withdraw the required amount of belatacept solution from the vial, inject it into the infusion bag or bottle, and gently rotate the infusion bag or bottle to ensure mixing.
The final belatacept concentration in the infusion bag or bottle should range from 2 mg/mL to 10 mg/mL. Typically, an infusion volume of 100 mL will be appropriate for most patients and doses, but total infusion volumes ranging from 50 mL to 250 mL may be used. Any unused solution remaining in the vials must be discarded. - 9. Prior to administration, the NULOJIX infusion should be inspected visually for particulate matter and discoloration. Discard the infusion if any particulate matter or discoloration is observed.
- 10.
The entire NULOJIX infusion should be administered over a period of 30 minutes and must be administered with an infusion set and a sterile, non-pyrogenic, low-protein-binding filter (with a pore size of 0.2 to 1.2 µm).
- The reconstituted solution should be transferred from the vial to the infusion bag or bottle immediately. The NULOJIX infusion must be completed within 24 hours of reconstitution of the NULOJIX lyophilized powder. If not used immediately, the infusion solution may be stored under refrigeration conditions: 2°C to 8°C (36°F to 46°F) and protected from light for up to 24 hours (a maximum of 4 hours of the total 24 hours can be at room temperature: 20°C to 25°C [68°F to 77°F] and room light).
- Infuse NULOJIX in a separate line from other concomitantly infused agents. NULOJIX should not be infused concomitantly in the same intravenous line with other agents. No physical or biochemical compatibility studies have been conducted to evaluate the coadministration of NULOJIX with other agents [see Warnings and Precautions (5.9)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
NULOJIX is contraindicated in transplant recipients who are Epstein-Barr virus (EBV) seronegative or with unknown EBV serostatus due to the risk of post-transplant lymphoproliferative disorder (PTLD), predominantly involving the central nervous system (CNS) [see Boxed Warning and Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Post-Transplant Lymphoproliferative Disorder
NULOJIX-treated patients have an increased risk for developing post-transplant lymphoproliferative disorder (PTLD), predominantly involving the CNS, compared to patients on a cyclosporine-based regimen [see Adverse Reactions (6.1), Clinical Studies (14.2)]. As the total burden of immunosuppression is a risk factor for PTLD, higher than the recommended doses or more frequent dosing of NULOJIX and higher than recommended doses of concomitant immunosuppressive agents are not recommended [see Dosage and Administration (2.1) and Warnings and Precautions (5.6)]. Physicians should consider PTLD in patients reporting new or worsening neurological, cognitive, or behavioral signs or symptoms.
EBV Serostatus
The risk of PTLD was higher in EBV seronegative patients compared to EBV seropositive patients. EBV seropositive patients are defined as having evidence of acquired immunity shown by the presence of IgG antibodies to viral capsid antigen (VCA) and EBV nuclear antigen (EBNA).
Epstein-Barr virus serology should be ascertained before starting administration of NULOJIX, and only patients who are EBV seropositive should receive NULOJIX. Transplant recipients who are EBV seronegative, or with unknown serostatus, should not receive NULOJIX [see Boxed Warning and Contraindications (4)].
Other Risk Factors
Other known risk factors for PTLD include cytomegalovirus (CMV) infection and T cell-depleting therapy. T cell-depleting therapies to treat acute rejection should be used cautiously. CMV prophylaxis is recommended for at least 3 months after transplantation [see Warnings and Precautions (5.5)].
Patients who are EBV seropositive and CMV seronegative may be at increased risk for PTLD compared to patients who are EBV seropositive and CMV seropositive [see Adverse Reactions (6.1)]. Since CMV seronegative patients are at increased risk for CMV disease (a known risk factor for PTLD), the clinical significance of CMV serology for PTLD remains to be determined; however, these findings should be considered when prescribing NULOJIX.
5.2 Management of Immunosuppression
Only physicians experienced in management of systemic immunosuppressant therapy in transplantation should prescribe NULOJIX. Patients receiving the drug should be managed in facilities equipped and staffed with adequate laboratory and supportive medical resources. The physician responsible for the maintenance therapy should have complete information requisite for the follow-up of the patient [see Boxed Warning].
5.3 Other Malignancies
Patients receiving immunosuppressants, including NULOJIX, are at increased risk of developing malignancies, in addition to PTLD, including the skin [see Boxed Warning and Warnings and Precautions (5.1)]. Exposure to sunlight and ultraviolet (UV) light should be limited by wearing protective clothing and using a sunscreen with a high protection factor.
5.4 Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) is an often rapidly progressive and fatal opportunistic infection of the CNS that is caused by the JC virus, a human polyoma virus. In clinical trials with NULOJIX, two cases of PML were reported in patients receiving NULOJIX at higher cumulative doses and more frequently than the recommended regimen, along with mycophenolate mofetil (MMF) and corticosteroids; one case occurred in a kidney transplant recipient and the second case occurred in a liver transplant recipient [see Warnings and Precautions (5.6), Adverse Reactions (6.1), Clinical Studies (14.2)]. As PML has been associated with high levels of overall immunosuppression, the recommended doses and frequency of NULOJIX and concomitant immunosuppressives, including MMF, should not be exceeded.
Physicians should consider PML in the differential diagnosis in patients with new or worsening neurological, cognitive, or behavioral signs or symptoms. PML is usually diagnosed by brain imaging, cerebrospinal fluid (CSF) testing for JC viral DNA by polymerase chain reaction (PCR), and/or brain biopsy. Consultation with a specialist (eg, neurologist and/or infectious disease) should be considered for any suspected or confirmed cases of PML.
If PML is diagnosed, consideration should be given to reduction or withdrawal of immunosuppression taking into account the risk to the allograft.
5.5 Other Serious Infections
Patients receiving immunosuppressants, including NULOJIX, are at increased risk of developing bacterial, viral (cytomegalovirus [CMV] and herpes), fungal, and protozoal infections, including opportunistic infections. These infections may lead to serious, including fatal, outcomes [see Boxed Warning and Adverse Reactions (6.1)].
Prophylaxis for cytomegalovirus is recommended for at least 3 months after transplantation. Prophylaxis for Pneumocystis jiroveci is recommended after transplantation.
Tuberculosis
Tuberculosis was more frequently observed in patients receiving NULOJIX than cyclosporine in clinical trials [see Adverse Reactions (6.1)]. Patients should be evaluated for tuberculosis and tested for latent infection prior to initiating NULOJIX. Treatment of latent tuberculosis infection should be initiated prior to NULOJIX use.
Polyoma Virus Nephropathy
In addition to cases of JC virus-associated PML [see Warnings and Precautions (5.4)], cases of polyoma virus-associated nephropathy (PVAN), mostly due to BK virus infection, have been reported. PVAN is associated with serious outcomes; including deteriorating renal function and kidney graft loss [see Adverse Reactions (6.1)]. Patient monitoring may help detect patients at risk for PVAN. Reductions in immunosuppression should be considered for patients who develop evidence of PVAN. Physicians should also consider the risk that reduced immunosuppression represents to the functioning allograft.
5.6 Liver Transplant
Use of NULOJIX in liver transplant patients is not recommended [see Boxed Warning]. In a clinical trial of liver transplant patients, use of NULOJIX regimens with more frequent administration of belatacept than any of those studied in kidney transplant, along with mycophenolate mofetil (MMF) and corticosteroids, was associated with a higher rate of graft loss and death compared to the tacrolimus control arms. In addition, two cases of PTLD involving the liver allograft (one fatal) and one fatal case of PML were observed among the 147 patients randomized to NULOJIX. The two cases of PTLD were reported among the 140 EBV seropositive patients (1.4%). The fatal case of PML was reported in a patient receiving higher than recommended doses of NULOJIX and MMF [see Warnings and Precautions (5.4)].
5.7 Acute Rejection and Graft Loss with Corticosteroid Minimization
In postmarketing experience, use of NULOJIX in conjunction with basiliximab induction, MMF, and corticosteroid minimization to 5 mg per day between Day 3 and Week 6 post-transplant was associated with an increased rate and grade of acute rejection, particularly Grade III rejection. These Grade III rejections occurred in patients with 4 to 6 HLA mismatches. Graft loss was a consequence of Grade III rejection in some patients.
Corticosteroid utilization should be consistent with the NULOJIX clinical trial experience [see Dosage and Administration (2.1) and Clinical Studies (14.1)].
5.8 Immunizations
The use of live vaccines should be avoided during treatment with NULOJIX, including but not limited to the following: intranasal influenza, measles, mumps, rubella, oral polio, BCG, yellow fever, varicella, and TY21a typhoid vaccines.
5.9 Coadministration with Anti-Thymocyte Globulin
In postmarketing experience in de novo kidney transplant recipients, some with other predisposing risk factors for venous thrombosis of the renal allograft, venous thrombosis of the renal allograft has occurred when the initial dose of anti-thymocyte globulin, as immunosuppressive induction, was coadministered (at the same or nearly the same time) with the first dose of belatacept. In such patients, the coadministration (at the same or nearly the same time) of anti-thymocyte globulin and belatacept may pose a risk for venous thrombosis of the renal allograft. If anti-thymocyte globulin (or any other cell-depleting induction treatment) and belatacept will be administered concomitantly, a twelve-hour interval between the two administrations is suggested.
-
6 ADVERSE REACTIONS
The most serious adverse reactions reported with NULOJIX are:
- PTLD, predominantly CNS PTLD, and other malignancies [see Boxed Warning and Warnings and Precautions (5.1, 5.3)]
- Serious infections, including JC virus-associated PML and polyoma virus nephropathy [see Warnings and Precautions (5.4, 5.5, 5.6)]
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other trials and may not reflect the rates observed in clinical practice.
The data described below primarily derive from two randomized, active-controlled three-year trials of NULOJIX in de novo kidney transplant patients. In Study 1 and Study 2, NULOJIX was studied at the recommended dose and frequency [see Dosage and Administration (2.1)] in a total of 401 patients compared to a cyclosporine control regimen in a total of 405 patients. These two trials also included a total of 403 patients treated with a NULOJIX regimen of higher cumulative dose and more frequent dosing than recommended [see Clinical Studies (14.1)]. All patients also received basiliximab induction, mycophenolate mofetil, and corticosteroids. Patients were treated and followed for three years.
CNS PTLD, PML, and other CNS infections were more frequently observed in association with a NULOJIX regimen of higher cumulative dose and more frequent dosing compared to the recommended regimen; therefore, administration of higher than the recommended doses and/or more frequent dosing of NULOJIX is not recommended [see Dosage and Administration (2.1), Clinical Studies (14.2)].
The average age of patients in Studies 1 and 2 in the NULOJIX recommended dose and cyclosporine control regimens was 49 years, ranging from 18 to 79 years. Approximately 70% of patients were male; 67% were white, 11% were black, and 22% other races. About 25% of patients were from the United States and 75% from other countries.
The most commonly reported adverse reactions occurring in ≥20% of patients treated with the recommended dose and frequency of NULOJIX were anemia, diarrhea, urinary tract infection, peripheral edema, constipation, hypertension, pyrexia, graft dysfunction, cough, nausea, vomiting, headache, hypokalemia, hyperkalemia, and leukopenia.
The proportion of patients who discontinued treatment due to adverse reactions was 13% for the recommended NULOJIX regimen and 19% for the cyclosporine control arm through three years of treatment. The most common adverse reactions leading to discontinuation in NULOJIX-treated patients were cytomegalovirus infection (1.5%) and complications of transplanted kidney (1.5%).
Information on selected significant adverse reactions observed during clinical trials is summarized below.
Post-Transplant Lymphoproliferative Disorder
Reported cases of post-transplant lymphoproliferative disorder (PTLD) up to 36 months post-transplant were obtained for NULOJIX by pooling both dosage regimens of NULOJIX in Studies 1 and 2 (804 patients) with data from a third study in kidney transplantation (Study 3, 145 patients) which evaluated two NULOJIX dosage regimens similar, but slightly different, from those of Studies 1 and 2 (see Table 2). The total number of NULOJIX patients from these three studies (949) was compared to the pooled cyclosporine control groups from all three studies (476 patients).
Among 401 patients in Studies 1 and 2 treated with the recommended regimen of NULOJIX and the 71 patients in Study 3 treated with a very similar (but non-identical) NULOJIX regimen, there were 5 cases of PTLD: 3 in EBV seropositive patients and 2 in EBV seronegative patients. Two of the 5 cases presented with CNS involvement.
Among the 477 patients in Studies 1, 2, and 3 treated with the NULOJIX regimen of higher cumulative dose and more frequent dosing than recommended, there were 8 cases of PTLD: 2 in EBV seropositive patients and 6 in EBV seronegative or serostatus unknown patients. Six of the 8 cases presented with CNS involvement. Therefore, administration of higher than the recommended doses or more frequent dosing of NULOJIX is not recommended [see Dosage and Administration (2.1) and Warnings and Precautions (5.1)].
One of the 476 patients treated with cyclosporine developed PTLD, without CNS involvement.
All cases of PTLD reported up to 36 months post-transplant in NULOJIX- or cyclosporine-treated patients presented within 18 months of transplantation.
Overall, the rate of PTLD in 949 patients treated with any of the NULOJIX regimens was 9-fold higher in those who were EBV seronegative or EBV serostatus unknown (8/139) compared to those who were EBV seropositive (5/810 patients). Therefore NULOJIX is recommended for use only in patients who are EBV seropositive [see Boxed Warning and Contraindications (4)].
Table 2: Summary of PTLD Reported in Studies 1, 2, and 3 Through Three Years of Treatment NULOJIX
Nonrecommended
Regimena
(N=477)NULOJIX
Recommended
Regimenb
(N=472)Cyclosporine
(N=476)Trial EBV
Positive
(n=406)EBV
Negative
(n=43)EBV
Unknown
(n=28)EBV
Positive
(n=404)EBV
Negative
(n=48)EBV
Unknown
(n=20)EBV
Positive
(n=399)EBV
Negative
(n=57)EBV
Unknown
(n=20)a Regimen with higher cumulative dose and more frequent dosing than the recommended NULOJIX regimen.
b In Studies 1 and 2 the NULOJIX regimen is identical to the recommended regimen, but is slightly different in Study 3.Study 1
CNS PTLD
1
1
Non-CNS
PTLD1
2
1
Study 2
CNS PTLD
1
1
1
1
Non-CNS
PTLD1
Study 3
CNS PTLD
2
Non-CNS
PTLD1
Total (%)
2 (0.5)
5 (11.6)
1 (3.6)
3 (0.7)
2 (4.1)
0
0
1 (1.8)
0
PTLD in EBV Seropositive Subpopulation
Among the 806 EBV seropositive patients with known CMV serostatus treated with either NULOJIX regimen in Studies 1, 2, and 3, 2% (4/210) of CMV seronegative patients developed PTLD compared to 0.2% (1/596) of CMV seropositive patients. Among the 404 EBV seropositive recipients treated with the recommended dosage regimen of NULOJIX, 3 PTLD cases were detected among 99 CMV seronegative patients (3%) and there was no case detected among 303 CMV seropositive patients. The clinical significance of CMV serology as a risk factor for PTLD remains to be determined; however, these findings should be considered when prescribing NULOJIX [see Warnings and Precautions (5.1)].
Other Malignancies
Malignancies, excluding non-melanoma skin cancer and PTLD, were reported in Study 1 and Study 2 in 3.5% (14/401) of patients treated with the recommended NULOJIX regimen and 3.7% (15/405) of patients treated with the cyclosporine control regimen. Non-melanoma skin cancer was reported in 1.5% (6/401) of patients treated with the recommended NULOJIX regimen and in 3.7% (15/405) of patients treated with cyclosporine [see Warnings and Precautions (5.3)].
Progressive Multifocal Leukoencephalopathy
Two fatal cases of progressive multifocal leukoencephalopathy (PML) have been reported among 1096 patients treated with a NULOJIX-containing regimen: 1 patient in clinical trials of kidney transplant (Studies 1, 2, and 3 described above) and 1 patient in a trial of liver transplant (trial of 250 patients). No cases of PML were reported in patients treated with the recommended NULOJIX regimen or the control regimen in these trials.
The kidney transplant recipient was treated with the NULOJIX regimen of higher cumulative dose and more frequent dosing than recommended, mycophenolate mofetil (MMF), and corticosteroids for two years. The liver transplant recipient was treated with 6 months of a NULOJIX dosage regimen that was more intensive than that studied in kidney transplant recipients, MMF at doses higher than the recommended dose, and corticosteroids [see Warnings and Precautions (5.4)].
Bacterial, Mycobacterial, Viral, and Fungal Infections
Adverse reactions of infectious etiology were reported based on clinical assessment by physicians. The causative organisms for these reactions are identified when provided by the physician. The overall number of infections, serious infections, and select infections with identified etiology reported in patients treated with the NULOJIX recommended regimen or the cyclosporine control in Studies 1 and 2 are shown in Table 3. Fungal infections were reported in 18% of patients receiving NULOJIX compared to 22% receiving cyclosporine, primarily due to skin and mucocutaneous fungal infections. Tuberculosis and herpes infections were reported more frequently in patients receiving NULOJIX than cyclosporine. Of the patients who developed tuberculosis through three years, all but one NULOJIX patient lived in countries with a high prevalence of tuberculosis [see Warnings and Precautions (5.5)].
Table 3: Overall Infections and Select Infections with Identified Etiology by Treatment Group Following One and Three Years of Treatment in Studies 1 and 2a Up to Year 1 Up to Year 3b NULOJIX
Recommended
Regimen
N=401
n (%)Cyclosporine
N=405
n (%)NULOJIX
Recommended
Regimen
N=401
n (%)Cyclosporine
N=405
n (%)a Studies 1 and 2 were not designed to support comparative claims for NULOJIX for the adverse reactions reported in this table.
b Median exposure in days for pooled studies: 1203 for NULOJIX recommended regimen and 1163 for cyclosporine in Studies 1 and 2.
c All infections include bacterial, viral, fungal, and other organisms. For infectious adverse reactions, the causative organism is reported if specified by the physician in the clinical trials.
d A medically important event that may be life-threatening or result in death or hospitalization or prolongation of existing hospitalization. Infections not meeting these criteria are considered non-serious.
e BK virus-associated nephropathy was reported in 6 NULOJIX patients (4 of which resulted in graft loss) and 6 cyclosporine patients (none of which resulted in graft loss) by Year 3.
f Most herpes infections were non-serious and 1 led to treatment discontinuation.All infectionsc
287 (72)
299 (74)
329 (82)
327 (81)
Serious infectionsd
98 (24)
113 (28)
144 (36)
157 (39)
CMV
44 (11)
52 (13)
53 (13)
56 (14)
Polyoma viruse
10 (3)
23 (6)
17 (4)
27 (7)
Herpesf
27 (7)
26 (6)
55 (14)
46 (11)
Tuberculosis
2 (1)
1 (<1)
6 (2)
1 (<1)
Infections Reported in the CNS
Following three years of treatment in Studies 1 and 2, cryptococcal meningitis was reported in 1 patient out of 401 patients treated with the NULOJIX recommended regimen (0.2%) and 1 patient out of the 405 treated with the cyclosporine control (0.2%).
Six patients out of the 403 who were treated with the NULOJIX regimen of higher cumulative dose and more frequent dosing than recommended in Studies 1 and 2 (1.5%) were reported to have developed CNS infections, including 2 cases of cryptococcal meningitis, 1 case of Chagas encephalitis with cryptococcal meningitis, 1 case of cerebral aspergillosis, 1 case of West Nile encephalitis, and 1 case of PML (discussed above).
Infusion Reactions
There have been no reports of anaphylaxis or drug hypersensitivity in patients treated with NULOJIX in Studies 1 and 2 with up to three years of follow-up. However, milder infusion-related reactions within one hour of infusion were reported in 5% of patients treated with the recommended dose of NULOJIX, similar to the placebo rate. The most frequent reactions were hypotension and hypertension. A case of anaphylaxis was reported in the postmarketing experience [see Adverse Reactions (6.2)].
Proteinuria
At Month 1 after transplantation in Studies 1 and 2, the frequency of 2+ proteinuria on urine dipstick in patients treated with the NULOJIX recommended regimen was 33% (130/390) and 28% (107/384) in patients treated with the cyclosporine control regimen. The frequency of 2+ proteinuria was similar between the two treatment groups between one and three years after transplantation (<10% in both studies). There were no differences in the occurrence of 3+ proteinuria (<4% in both studies) at any time point, and no patients experienced 4+ proteinuria. The clinical significance of this increase in early proteinuria is unknown.
Immunogenicity
Antibodies directed against the belatacept molecule were assessed in 398 patients treated with the NULOJIX recommended regimen in Studies 1 and 2 (212 of these patients were treated for at least two years). Of the 372 patients with immunogenicity assessment at baseline (prior to receiving belatacept treatment), 29 patients tested positive for anti-belatacept antibodies; 13 of these patients had antibodies to the modified cytotoxic T lymphocyte-associated antigen 4 (CTLA-4). Anti-belatacept antibody titers did not increase during treatment in these 29 patients.
Eight (2%) patients developed antibodies during treatment with the NULOJIX recommended regimen. In the patients who developed antibodies during treatment, the median titer (by dilution method) was 8, with a range of 5 to 80. Of 56 patients who tested negative for antibodies during treatment and reassessed approximately 7 half-lives after discontinuation of NULOJIX, 1 tested antibody positive. Anti-belatacept antibody development was not associated with altered clearance of belatacept.
Samples from 6 patients with confirmed binding activity to the modified cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) region of the belatacept molecule were assessed by an in vitro bioassay for the presence of neutralizing antibodies. Three of these 6 patients tested positive for neutralizing antibodies. However, the development of neutralizing antibodies may be underreported due to lack of assay sensitivity.
The clinical impact of anti-belatacept antibodies (including neutralizing anti-belatacept antibodies) could not be determined in the studies.
The data reflect the percentage of patients whose test results were positive for antibodies to belatacept in specific assays. The observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to belatacept with the incidence of antibodies to other products may be misleading.
New-Onset Diabetes After Transplantation
The incidence of new-onset diabetes after transplantation (NODAT) was defined in Studies 1 and 2 as use of an antidiabetic agent for ≥30 days or ≥2 fasting plasma glucose values ≥126 mg/dL (7.0 mmol/L) post-transplantation. Of the patients treated with the NULOJIX recommended regimen, 5% (14/304) developed NODAT by the end of one year compared to 10% (27/280) of patients on the cyclosporine control regimen. However, by the end of the third year, the cumulative incidence of NODAT was 8% (24/304) in patients treated with the NULOJIX recommended regimen and 10% (29/280) in patients treated with the cyclosporine regimen.
Hypertension
Blood pressure and use of antihypertensive medications were reported in Studies 1 and 2. By Year 3, one or more antihypertensive medications were used in 85% of NULOJIX-treated patients and 92% of cyclosporine-treated patients. At one year after transplantation, systolic blood pressures were 8 mmHg lower and diastolic blood pressures were 3 mmHg lower in patients treated with the NULOJIX recommended regimen compared to the cyclosporine control regimen. At three years after transplantation, systolic blood pressures were 6 mmHg lower and diastolic blood pressures were 3 mmHg lower in NULOJIX-treated patients compared to cyclosporine-treated patients. Hypertension was reported as an adverse reaction in 32% of NULOJIX-treated patients and 37% of cyclosporine-treated patients (see Table 4).
Dyslipidemia
Mean values of total cholesterol, HDL, LDL, and triglycerides were reported in Studies 1 and 2. At one year after transplantation these values were 183 mg/dL, 50 mg/dL, 102 mg/dL, and 151 mg/dL, respectively, in 401 patients treated with the NULOJIX recommended regimen and 196 mg/dL, 48 mg/dL, 108 mg/dL, and 195 mg/dL, respectively, in 405 patients treated with the cyclosporine control regimen. At three years after transplantation, the total cholesterol, HDL, LDL, and triglycerides were 176 mg/dL, 49 mg/dL, 100 mg/dL, and 141 mg/dL, respectively, in NULOJIX-treated patients compared to 193 mg/dL, 48 mg/dL, 106 mg/dL, and 180 mg/dL in cyclosporine-treated patients.
The clinical significance of the lower mean triglyceride values in NULOJIX-treated patients at one and three years is unknown.
Other Adverse Reactions
Adverse reactions that occurred at a frequency of ≥10% in patients treated with the NULOJIX recommended regimen or cyclosporine control regimen in Studies 1 and 2 through three years are summarized by preferred term in decreasing order of frequency within Table 4.
Table 4: Adverse Reactions Reported by ≥10% of Patients Treated with Either the NULOJIX Recommended Regimen or Control in Studies 1 and 2 Through Three Yearsa,b Adverse Reaction NULOJIX
Recommended Regimen
N=401
%Cyclosporine
N=405
%a All randomized and transplanted patients in Studies 1 and 2.
b Studies 1 and 2 were not designed to support comparative claims for NULOJIX for the adverse reactions reported in this table.Infections and Infestations
Urinary tract infection
37
36
Upper respiratory infection
15
16
Nasopharyngitis
13
16
Cytomegalovirus infection
12
12
Influenza
11
8
Bronchitis
10
7
Gastrointestinal Disorders
Diarrhea
39
36
Constipation
33
35
Nausea
24
27
Vomiting
22
20
Abdominal pain
19
16
Abdominal pain upper
9
10
Metabolism and Nutrition Disorders
Hyperkalemia
20
20
Hypokalemia
21
14
Hypophosphatemia
19
13
Dyslipidemia
19
24
Hyperglycemia
16
17
Hypocalcemia
13
11
Hypercholesterolemia
11
11
Hypomagnesemia
7
10
Hyperuricemia
5
12
Procedural Complications
Graft dysfunction
25
34
General Disorders
Peripheral edema
34
42
Pyrexia
28
26
Blood and Lymphatic System Disorders
Anemia
45
44
Leukopenia
20
23
Renal and Urinary Disorders
Hematuria
16
18
Proteinuria
16
12
Dysuria
11
11
Renal tubular necrosis
9
13
Vascular Disorders
Hypertension
32
37
Hypotension
18
12
Respiratory, Thoracic, and Mediastinal Disorders
Cough
24
18
Dyspnea
12
15
Investigations
Blood creatinine increased
15
20
Musculoskeletal and Connective Tissue Disorders
Arthralgia
17
13
Back pain
13
13
Nervous System Disorders
Headache
21
18
Dizziness
9
10
Tremor
8
17
Skin and Subcutaneous Tissue Disorders
Acne
8
11
Psychiatric Disorders
Insomnia
15
18
Anxiety
10
11
Selected adverse reactions occurring in <10% from NULOJIX-treated patients in either regimen through three years in Studies 1 and 2 are listed below:
Immune System Disorders: Guillain-Barré syndrome
Infections and Infestations: see Table 3
Gastrointestinal Disorders: stomatitis, including aphthous stomatitis
Injury, Poisoning, and Procedural Complications: chronic allograft nephropathy, complications of transplanted kidney, including wound dehiscence, arteriovenous fistula thrombosis
Blood and Lymphatic System Disorders: neutropenia
Renal and Urinary Disorders: renal impairment, including acute renal failure, renal artery stenosis, urinary incontinence, hydronephrosis
Vascular Disorders: hematoma, lymphocele
Musculoskeletal and Connective Tissue Disorders: musculoskeletal pain
Skin and Subcutaneous Tissue Disorders: alopecia, hyperhidrosis
Cardiac Disorders: atrial fibrillation
6.2 Postmarketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorder: Anaphylaxis
Spontaneous reports during the postmarketing experience included a case of anaphylaxis, which was observed in a kidney transplant patient whose belatacept therapy had been interrupted for two months during treatment of a systemic varicella infection. When belatacept therapy was resumed, within five minutes after the start of the belatacept infusion the patient developed a generalized rash, pruritus, hypotension, atrial fibrillation, respiratory distress, and syncope, requiring medical treatment. Another belatacept infusion was attempted one month later, but was terminated when the patient experienced more pronounced symptoms of anaphylaxis and required medical treatment.
Vascular Disorder: Venous Thrombosis of the Renal Allograft
In postmarketing experience in de novo kidney transplant recipients, some with other predisposing risk factors for venous thrombosis of the renal allograft, venous thrombosis of the renal allograft has occurred when the initial dose of anti-thymocyte globulin, as immunosuppressive induction, was coadministered (at the same or nearly the same time) with the first dose of belatacept [see Warnings and Precautions (5.9)].
6.3 Long-Term Extension Studies
After completion of the 36-month studies, patients remaining on randomized therapy in Study 1 and Study 2 were eligible for enrollment in the long-term extension studies [see Clinical Studies (14.2, 14.3)]. No new adverse reactions were observed in the extension studies.
-
7 DRUG INTERACTIONS
7.1 Mycophenolate Mofetil (MMF)
Monitor for a need to adjust concomitant mycophenolate mofetil (MMF) dosage when a patient’s therapy is switched between cyclosporine and NULOJIX, as cyclosporine decreases mycophenolic acid (MPA) exposure by preventing enterohepatic recirculation of MPA while NULOJIX does not [see Clinical Pharmacology (12.3)]:
- A higher MMF dosage may be needed after switching from NULOJIX to cyclosporine, since this may result in lower MPA concentrations and increase the risk of graft rejection.
- A lower MMF dosage may be needed after switching from cyclosporine to NULOJIX, since this may result in higher MPA concentrations and increase the risk for adverse reactions related to MPA (review the Full Prescribing Information for MMF).
7.2 Cytochrome P450 Substrates
No dosage adjustments are needed for drugs metabolized via CYP1A2, CYP2C9, CYP2D6, CYP3A, and CYP2C19 when coadministered with NULOJIX [see Clinical Pharmacology (12.3)].
7.3 Anti-Thymocyte Globulin
Coadministration (at the same or nearly the same time) of anti-thymocyte globulin (or any other cell-depleting induction treatment) and belatacept in de novo kidney transplant recipients, especially those with other predisposing risk factors for venous thrombosis of the renal allograft, may pose a risk for venous thrombosis of the renal allograft [see Warnings and Precautions (5.9)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
To monitor maternal-fetal outcomes of pregnant women who have received immunosuppressants including NULOJIX or whose partners have received NULOJIX, healthcare providers are strongly encouraged to register pregnant patients in the Transplant Pregnancy Registry International (TPR) by calling 1-877-955-6877.
Risk Summary
The data with NULOJIX use in pregnant women are insufficient to inform on drug-associated risk. Belatacept is known to cross the placenta of animals. Administration of belatacept to pregnant rats and rabbits during the period of organogenesis was not teratogenic at exposures approximately 16 and 19 times greater than that observed at the maximum recommended human dose (MRHD) of 10 mg per kg body weight administered over the first month of treatment, based on area under the concentration-time curve (AUC). In a pre- and postnatal development study in rats, treatment-related infections in dams were associated with increased pup mortality, presumably secondary to deteriorating maternal health, at exposures 3 times higher than that observed at MRHD [see Animal Data].
The background risk of major birth defects and miscarriage for the indicated population is unknown; however, in the U.S. general population, the estimated background risk of major birth defects is 2 to 4% and of miscarriage is 15 to 20% of clinically recognized pregnancies.
Data
Animal Data
In embryo-fetal development studies, daily intravenous administration of belatacept to pregnant rats and rabbits throughout the period of organogenesis did not produce adverse fetal effects at doses up to 200 mg per kg and 100 mg per kg, respectively (16 and 19 times the MRHD exposure, based on AUC). In a pre- and postnatal development study, daily intravenous administration of belatacept to rats from Day 6 of gestation through Day 20 of the lactation period was associated with maternal toxicity (infections) in a small percentage of dams at doses ≥20 mg per kg (3 times the MRHD exposure, based on AUC) resulting in increased pup mortality (up to 100% pup mortality in some dams). In pups that survived, there were no abnormalities or malformations at doses up to 200 mg per kg (19 times the MRHD exposure, based on AUC).
In vitro data indicate that belatacept has lower binding affinity to CD80/CD86 and lower potency in rodents than in humans. Although the rat toxicity studies with belatacept were done at pharmacologically saturating doses, the in vivo difference in potency between rats and humans is unknown. Therefore, the relevance of the rat toxicities to humans and the significance of the magnitude of the relative exposures (rats:humans) are unknown.
Abatacept, a fusion protein that differs from belatacept by two amino acids, binds to the same ligands (CD80/CD86) and blocks T cell costimulation like belatacept, but is more active than belatacept in rodents. Therefore, toxicities identified with abatacept in rodents, including infections and autoimmunity, may be predictive of adverse effects in humans treated with belatacept [see Nonclinical Toxicology (13.2)].
Autoimmunity was observed in 1 rat offspring exposed to abatacept in utero and/or during lactation and in juvenile rats after treatment with abatacept. However, the clinical relevance of autoimmunity in rats to patients or a fetus exposed in utero is unknown [see Nonclinical Toxicology (13.2)].
8.2 Lactation
Risk Summary
There are no data on the presence of NULOJIX in human milk or the effects of NULOJIX on breastfed infants or human milk production to inform risk of NULOJIX to an infant during lactation. Belatacept is excreted in rat milk after intravenous administration, and it is possible that the drug will be present in human milk. However, absorption of intact belatacept from the nursing infant’s gastrointestinal tract has not been studied. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for NULOJIX and any potential adverse effects on the breastfed child from NULOJIX or from the underlying maternal conditions.
8.4 Pediatric Use
The safety and efficacy of NULOJIX in patients under 18 years of age have not been established. Because T cell development continues into the teenage years, the potential concern for autoimmunity in neonates applies to pediatric use as well [see Use in Specific Populations (8.1)].
8.5 Geriatric Use
Of 401 patients treated with the recommended dosage regimen of NULOJIX, 15% were 65 years of age and older, while 3% were 75 and older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, but greater sensitivity or less efficacy in older individuals cannot be ruled out.
-
10 OVERDOSAGE
Single doses up to 20 mg per kg of NULOJIX have been administered to healthy subjects without apparent toxic effect. The administration of NULOJIX of higher cumulative dose and more frequent dosing than recommended in kidney transplant patients resulted in a higher frequency of CNS-related adverse reactions [see Adverse Reactions (6.1)]. In case of overdosage, it is recommended that the patient be monitored for any signs or symptoms of adverse reactions and appropriate symptomatic treatment instituted.
-
11 DESCRIPTION
NULOJIX® (belatacept), a selective T cell costimulation blocker, is a soluble fusion protein consisting of the modified extracellular domain of CTLA-4 fused to a portion (hinge-CH2-CH3 domains) of the Fc domain of a human immunoglobulin G1 antibody. Belatacept is produced by recombinant DNA technology in a mammalian cell expression system. Two amino acid substitutions (L104 to E; A29 to Y) were made in the ligand binding region of CTLA-4. As a result of these modifications, belatacept binds CD80 and CD86 more avidly than abatacept, the parent CTLA4-Immunoglobulin (CTLA4-Ig) molecule from which it is derived. The molecular weight of belatacept is approximately 90 kilodaltons.
NULOJIX is supplied as a sterile, white or off-white lyophilized powder for intravenous administration. Prior to use, the lyophile is reconstituted with a suitable fluid to obtain a clear to slightly opalescent, colorless to pale yellow solution, with a pH in the range of 7.2 to 7.8. Suitable fluids for constitution of the lyophile include SWFI, 0.9% NS, or D5W [see Dosage and Administration (2.2)]. Each 250 mg single-use vial of NULOJIX also contains: monobasic sodium phosphate (34.5 mg), sodium chloride (5.8 mg), and sucrose (500 mg).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Belatacept, a selective T cell (lymphocyte) costimulation blocker, binds to CD80 and CD86 on antigen-presenting cells thereby blocking CD28 mediated costimulation of T lymphocytes. In vitro, belatacept inhibits T lymphocyte proliferation and the production of the cytokines interleukin-2, interferon-γ, interleukin-4, and TNF-α. Activated T lymphocytes are the predominant mediators of immunologic rejection.
In non-human primate models of renal transplantation, belatacept monotherapy prolonged graft survival and decreased the production of anti-donor antibodies, compared to vehicle.
12.2 Pharmacodynamics
Belatacept-mediated costimulation blockade results in the inhibition of cytokine production by T cells required for antigen-specific antibody production by B cells. In clinical trials, greater reductions in mean immunoglobulin (IgG, IgM, and IgA) concentrations were observed from baseline to Month 6 and Month 12 post-transplant in belatacept-treated patients compared to cyclosporine-treated patients. In an exploratory subset analysis, a trend of decreasing IgG concentrations with increasing belatacept trough concentrations was observed at Month 6. Also in this exploratory subset analysis, belatacept-treated patients with CNS PTLD, CNS infections including PML, other serious infections, and malignancies were observed to have a higher incidence of IgG concentrations below the lower limit of the normal range (<694 mg/dL) at Month 6 than those patients who did not experience these adverse events. This observation was more pronounced with the higher than recommended dose of belatacept. A similar trend was also observed for cyclosporine-treated patients with serious infections and malignancies.
However, it is unclear whether any causal relationship between an IgG concentration below the lower level of normal and these adverse events exists, as the analysis may have been confounded by other factors (eg, age greater than 60 years, receipt of an extended criteria donor kidney, exposure to lymphocyte-depleting agents) which were also associated with IgG below the lower level of normal at Month 6 in these trials.
12.3 Pharmacokinetics
Table 5 summarizes the pharmacokinetic parameters of belatacept in healthy adult subjects after a single 10 mg per kg intravenous infusion; and in kidney transplant patients after the 10 mg per kg intravenous infusion at Week 12, and after 5 mg per kg intravenous infusion every four weeks at Month 12 post-transplant or later.
Table 5: Pharmacokinetic Parameters (Mean±SD [Range]) of Belatacept in Healthy Subjects and Kidney Transplant Patients After 5 and 10 mg per kg Intravenous Infusions Administered Over 30 Minutes Pharmacokinetic Parameter Healthy Subjects
(After 10 mg per kg
Single Dose)
N=15Kidney Transplant
Patients
(After 10 mg per kg
Multiple Doses)
N=10Kidney Transplant
Patients
(After 5 mg per kg
Multiple Doses)
N=14a AUC=AUC (INF) after single dose and AUC (TAU) after multiple dose, where TAU=4 weeks Peak concentration (Cmax) [µg/mL]
300±77
(190-492)247±68
(161-340)139±28
(80-176)AUCa
[μgh/mL]26398±5175
(18964-40684)22252±7868
(13575-42144)14090±3860
(7906-20510)Terminal half-life (t1/2) [days]
9.8±2.8
(6.4-15.6)9.8±3.2
(6.1-15.1)8.2±2.4
(3.1-11.9)Systemic clearance (CL) [mL/h/kg]
0.39±0.07
(0.25-0.53)0.49±0.13
(0.23-0.70)0.51±0.14
(0.33-0.75)Volume of distribution (Vss) [L/kg]
0.09±0.02
(0.07-0.15)0.11±0.03
(0.067-0.17)0.12±0.03
(0.09-0.17)In healthy subjects, the pharmacokinetics of belatacept was linear and the exposure to belatacept increased proportionally after a single intravenous infusion dose of 1 to 20 mg per kg. The pharmacokinetics of belatacept in de novo kidney transplant patients and healthy subjects are comparable. Following the recommended regimen, the mean belatacept serum concentration reached steady-state by Week 8 in the initial phase following transplantation and by Month 6 during the maintenance phase. Following once monthly intravenous infusion of 10 mg per kg and 5 mg per kg, there was about 20% and 10% systemic accumulation of belatacept in kidney transplant patients, respectively.
Based on population pharmacokinetic analysis of 924 kidney transplant patients up to one year post-transplant, the pharmacokinetics of belatacept were similar at different time periods post-transplant. In clinical trials, trough concentrations of belatacept were consistently maintained from Month 6 up to three years post-transplant. Population pharmacokinetic analyses in kidney transplant patients revealed that there was a trend toward higher clearance of belatacept with increasing body weight. Age, gender, race, renal function (measured by calculated glomerular filtration rate [GFR]), hepatic function (measured by albumin), diabetes, and concomitant dialysis did not affect the clearance of belatacept.
Drug Interactions
Mycophenolate Mofetil
In a pharmacokinetic substudy of Studies 1 and 2, the plasma concentrations of MPA were measured in 41 patients who received fixed MMF doses of 500 to 1500 mg twice daily with either 5 mg per kg of NULOJIX or cyclosporine. The mean dose-normalized MPA Cmax and AUC0-12 were approximately 20% and 40% higher, respectively, with NULOJIX coadministration than with cyclosporine coadministration [see Drug Interactions (7.1)].
Cytochrome P450 Substrates
The potential of NULOJIX to alter the systemic concentrations of drugs that are CYP450 substrates was investigated in healthy subjects following administration of a cocktail of probe drugs given concomitantly with, and at three days and at seven days following a single intravenous 10 mg per kg dose of NULOJIX. NULOJIX did not alter the pharmacokinetics of drugs that are substrates of CYP1A2 (caffeine), CYP2C9 (losartan), CYP2D6 (dextromethorphan), CYP3A (midazolam), and CYP2C19 (omeprazole) [see Drug Interactions (7.2)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A carcinogenicity study was not conducted with belatacept. However, a murine carcinogenicity study was conducted with abatacept (a more active analog in rodents) to determine the carcinogenic potential of CD28 blockade. Weekly subcutaneous injections of 20, 65, or 200 mg per kg of abatacept were associated with increases in the incidence of malignant lymphomas (all doses) and mammary gland tumors (intermediate- and high-dose in females) at clinically relevant exposures. The mice in this study were infected with endogenous murine leukemia and mouse mammary tumor viruses which are associated with an increased incidence of lymphomas and mammary gland tumors, respectively, in immunosuppressed mice. Although the precise relevance of these findings to the clinical use of NULOJIX is unknown, cases of PTLD (a premalignant or malignant proliferation of B lymphocytes) were reported in clinical trials.
Genotoxicity testing is not required for protein therapeutics; therefore, no genotoxicity studies were conducted with belatacept.
Belatacept had no adverse effects on male or female fertility in rats at doses up to 200 mg per kg daily (25 times the MRHD exposure).
13.2 Animal Toxicology and/or Pharmacology
Abatacept, a fusion protein that differs from belatacept by two amino acids, binds to the same ligands (CD80/CD86) and blocks T cell costimulation like belatacept, but is more active than belatacept in rodents. Therefore, toxicities identified with abatacept in rodents may be predictive of adverse effects in humans treated with belatacept.
Studies in rats exposed to abatacept have shown immune system abnormalities including a low incidence of infections leading to death (observed in juvenile rats and pregnant rats), as well as autoimmunity of the thyroid and pancreas (observed in rats exposed in utero, as juveniles or as adults). Studies of abatacept in adult mice and monkeys, as well as belatacept in adult monkeys, have not demonstrated similar findings.
The increased susceptibility to opportunistic infections observed in juvenile rats is likely associated with the exposure to abatacept before the complete development of memory immune responses. In pregnant rats, the increased susceptibility to opportunistic infections may be due to the inherent lapses in immunity that occur in rats during late pregnancy/lactation. Infections related to NULOJIX have been observed in human clinical trials [see Warnings and Precautions (5.5)].
Administration of abatacept to rats was associated with a significant decrease in T regulatory cells (up to 90%). Deficiency of T regulatory cells in humans has been associated with autoimmunity. The occurrence of autoimmune events across the core clinical trials was infrequent. However, the possibility that patients administered NULOJIX could develop autoimmunity (or that fetuses exposed to NULOJIX in utero could develop autoimmunity) cannot be excluded.
In a 6-month toxicity study with belatacept in cynomolgus monkeys administered weekly doses up to 50 mg per kg (6 times the MRHD exposure) and in a 1-year toxicity study with abatacept in adult cynomolgus monkeys administered weekly doses up to 50 mg per kg, no significant drug-related toxicities were observed. Reversible pharmacological effects consisted of minimal transient decreases in serum IgG and minimal to severe lymphoid depletion of germinal centers in the spleen and/or lymph nodes.
Following 5 doses (10 mg per kg or 50 mg per kg, once a week for five weeks) of systemic administration, belatacept was not detected in brain tissue of normal healthy cynomolgus monkeys. The number of cells expressing major histocompatibility complex (MHC) class-II antigens (potential marker of immune cell activation) in the brain were increased in monkeys administered belatacept compared to vehicle control. However, distribution of some other cells expressing CD68, CD20, CD80, and CD86, typically expressed on MHC class II-positive cells, was not altered and there were no other histological changes in the brain. The clinical relevance of the findings is unknown.
-
14 CLINICAL STUDIES
14.1 Prevention of Organ Rejection in Kidney Transplant Recipients
The efficacy and safety of NULOJIX in de novo kidney transplantation were assessed in two open-label, randomized, multicenter, active-controlled trials (Study 1 and Study 2). These trials evaluated two dose regimens of NULOJIX, the recommended dosage regimen [see Dosage and Administration (2.1)] and a regimen with higher cumulative doses and more frequent dosing than the recommended dosage regimen, compared to a cyclosporine control regimen. All treatment groups also received basiliximab induction, mycophenolate mofetil (MMF), and corticosteroids.
Treatment Regimen
The NULOJIX recommended regimen consisted of a 10 mg per kg dose administered on Day 1 (the day of transplantation, prior to implantation), Day 5 (approximately 96 hours after the Day 1 dose), end of Weeks 2 and 4; then every four weeks through Week 12 after transplantation. Starting at Week 16 after transplantation, NULOJIX was administered at the maintenance dose of 5 mg per kg every four weeks (plus or minus three days). NULOJIX was administered as an intravenous infusion over 30 minutes [see Dosage and Administration (2.1)].
Basiliximab 20 mg was administered intravenously on the day of transplantation and four days later.
The initial dose of MMF was 1 gram twice daily and was adjusted as needed, based on clinical signs of adverse events or efficacy failure.
The protocol-specified dosing of corticosteroids in Studies 1 and 2 at Day 1 was methylprednisolone (as sodium succinate) 500 mg IV on arrival in the operating room, Day 2, methylprednisolone 250 mg IV, and Day 3, prednisone 100 mg orally. Actual median corticosteroid doses used with the NULOJIX recommended regimen from Week 1 through Month 6 are summarized in the table below (Table 6).
Table 6: Actual Corticosteroida Dosing in Studies 1 and 2 Day of Dosing Median (Q1–Q3) Daily Doseb,c Study 1 Study 2 a Corticosteroid = prednisone or prednisolone.
b The protocols allowed for flexibility in determining corticosteroid dose and rapidity of taper after Day 15. It is not possible to distinguish corticosteroid doses used to treat acute rejection versus doses used in a maintenance regimen.
c Q1 and Q3 are the 25th and 75th percentiles of daily corticosteroid doses, respectively.Week 1
31.7 mg (26.7-50 mg)
30 mg (26.7-50 mg)
Week 2
25 mg (20-30 mg)
25 mg (20-30 mg)
Week 4
20 mg (15-20 mg)
20 mg (15-22.5 mg)
Week 6
15 mg (10-20 mg)
16.7 mg (12.5-20 mg)
Month 6
10 mg (5-10 mg)
10 mg (5-12.5 mg)
Study 1 enrolled recipients of living donor and standard criteria deceased donor organs and Study 2 enrolled recipients of extended criteria donor organs. Standard criteria donor organs were defined as organs from a deceased donor with anticipated cold ischemia time of <24 hours and not meeting the definition of extended criteria donor organs. Extended criteria donors were defined as deceased donors with at least one of the following: (1) donor age ≥60 years; (2) donor age ≥50 years and other donor comorbidities (≥2 of the following: stroke, hypertension, serum creatinine >1.5 mg/dL); (3) donation of organ after cardiac death; or (4) anticipated cold ischemia time of the organ of ≥24 hours. Study 1 excluded recipients undergoing a first transplant whose current Panel Reactive Antibodies (PRA) were ≥50% and recipients undergoing a retransplantation whose current PRA were ≥30%; Study 2 excluded recipients with a current PRA ≥30%. Both studies excluded recipients with HIV, hepatitis C, or evidence of current hepatitis B infection; recipients with active tuberculosis; and recipients in whom intravenous access was difficult to obtain.
Efficacy data are presented for the NULOJIX recommended regimen and cyclosporine regimen in Studies 1 and 2.
The NULOJIX regimen with higher cumulative doses and more frequent dosing of belatacept was associated with more efficacy failures. Higher doses and/or more frequent dosing of NULOJIX are not recommended [see Dosage and Administration (2.1), Warnings and Precautions (5.1), and Adverse Reactions (6.1)].
Study 1: Recipients of Living Donor and Standard Criteria Deceased Donor Kidneys
In Study 1 (NCT00256750), 666 patients were enrolled, randomized, and transplanted: 226 to the NULOJIX recommended regimen, 219 to the NULOJIX regimen with higher cumulative doses and more frequent dosing than recommended, and 221 to cyclosporine control regimen. The median age was 45 years; 58% of organs were from living donors; 3% were re-transplanted; 69% of the study population was male; 61% of patients were white, 8% were black/African-American, 31% were categorized as of other races; 16% had PRA ≥10%; 41% had 4 to 6 HLA mismatches; and 27% had diabetes prior to transplant. The incidence of delayed graft function was similar in all treatment arms (14% to 18%).
Premature discontinuation from treatment at the end of the first year occurred in 19% of patients receiving the NULOJIX recommended regimen and 19% of patients on the cyclosporine regimen. Among the patients who received the NULOJIX recommended regimen, 10% discontinued due to lack of efficacy, 5% due to adverse events, and 4% for other reasons. Among the patients who received the cyclosporine regimen, 9% discontinued due to adverse events, 5% due to lack of efficacy, and 5% for other reasons.
At the end of three years, 25% of patients receiving the NULOJIX recommended regimen and 34% of patients receiving the cyclosporine regimen had discontinued from treatment. Among the patients who received the NULOJIX recommended regimen, 12% discontinued due to lack of efficacy, 7% due to adverse events, and 6% for other reasons. Among the patients who received the cyclosporine regimen, 15% discontinued due to adverse events, 8% due to lack of efficacy, and 11% for other reasons.
Assessment of Efficacy
Table 7 summarizes the results of Study 1 following one and three years of treatment with the NULOJIX recommended dosage regimen and the cyclosporine control regimen. Efficacy failure at one year was defined as the occurrence of biopsy proven acute rejection (BPAR), graft loss, death, or lost to follow-up. BPAR was defined as histologically confirmed acute rejection by a central pathologist on a biopsy done for any reason, whether or not accompanied by clinical signs of rejection. Patient and graft survival was also assessed separately.
Table 7: Efficacy Outcomes by Years 1 and 3 for Study 1: Recipients of Living and Standard Criteria Deceased Donor Kidneys Parameter NULOJIX
Recommended Regimen
N=226
n (%)Cyclosporine
(CSA)
N=221
n (%)NULOJIX-CSA
(97.3% CI)a Patients may have experienced more than one event.
b Patients known to be alive with a functioning graft.Efficacy Failure by Year 1
49 (21.7)
37 (16.7)
4.9 (−3.3, 13.2)
Components of Efficacy Failurea
Biopsy Proven Acute Rejection
45 (19.9)
23 (10.4)
Graft Loss
5 (2.2)
8 (3.6)
Death
4 (1.8)
7 (3.2)
Lost to follow-up
0
1 (0.5)
Efficacy Failure by Year 3
58 (25.7)
57 (25.8)
−0.1 (−9.3, 9)
Components of Efficacy Failurea
Biopsy Proven Acute Rejection
50 (22.1)
31 (14)
Graft Loss
9 (4)
10 (4.5)
Death
10 (4.4)
15 (6.8)
Lost to follow-up
2 (0.9)
5 (2.3)
Patient and graft survivalb
Year 1
218 (96.5)
206 (93.2)
3.2 (−1.5, 8.4)
Year 3
206 (91.2)
192 (86.9)
4.3 (−2.2, 10.8)
In Study 1, the rate of BPAR at one year and three years was higher in patients treated with the NULOJIX recommended regimen than the cyclosporine regimen. Of the patients who experienced BPAR with NULOJIX, 70% experienced BPAR by Month 3, and 84% experienced BPAR by Month 6. By three years, recurrent BPAR occurred with similar frequency across treatment groups (<3%). The component of BPAR determined by biopsy only (subclinical protocol-defined acute rejection) was 5% in both treatment groups.
Patients treated with the NULOJIX recommended regimen experienced episodes of BPAR classified as Banff grade IIb or higher (6% [14/226] at one year and 7% [15/226] at three years) more frequently compared to patients treated with the cyclosporine regimen (2% [4/221] at one year and 2% [5/221] at three years). Also, T cell-depleting therapy was used more frequently to treat episodes of BPAR in NULOJIX-treated patients (10%; 23/226) compared to cyclosporine-treated patients (2%; 5/221). At Month 12, the difference in mean calculated glomerular filtration rate (GFR) between patients with and without history of BPAR was 19 mL/min/1.73 m2 among NULOJIX-treated patients compared to 7 mL/min/1.73 m2 among cyclosporine-treated patients. By three years, 22% (11/50) of NULOJIX-treated patients with a history of BPAR experienced graft loss and/or death compared to 10% (3/31) of cyclosporine-treated patients with a history of BPAR; at that time point, 10% (5/50) of NULOJIX-treated patients experienced graft loss and 12% (6/50) of NULOJIX-treated patients had died following an episode of BPAR, whereas 7% (2/31) of cyclosporine-treated patients experienced graft loss and 7% (2/31) of cyclosporine-treated patients had died following an episode of BPAR. The overall prevalence of donor-specific antibodies was 5% and 11% for the NULOJIX recommended regimen and cyclosporine, respectively, up to 36 months post-transplant.
While the difference in GFR in patients with BPAR versus those without BPAR was greater in patients treated with NULOJIX than cyclosporine, the mean GFR following BPAR was similar in NULOJIX (49 mL/min/1.73 m2) and cyclosporine treated patients (43 mL/min/1.73 m2) at one year. The relationship between BPAR, GFR, and patient and graft survival is unclear due to the limited number of patients who experienced BPAR, differences in renal hemodynamics (and, consequently, GFR) across maintenance immunosuppression regimens, and the high rate of switching treatment regimens after BPAR.
Assessment of Efficacy in the EBV Seropositive Subpopulation
NULOJIX is recommended for use only in EBV seropositive patients [see Indications and Usage (1.2)].
In Study 1, approximately 87% of patients were EBV seropositive prior to transplant. Efficacy results in the EBV seropositive subpopulation were consistent with those in the total population studied.
By one year, the efficacy failure rate in the EBV seropositive population was 21% (42/202) in patients treated with the NULOJIX recommended regimen and 17% (31/184) in patients treated with cyclosporine (difference=4%, 97.3% CI [−4.8, 12.8]). Patient and graft survival was 98% (198/202) in NULOJIX-treated patients and 92% (170/184) in cyclosporine-treated patients (difference=5.6%, 97.3% CI [0.8, 10.4]).
By three years, efficacy failure was 25% in both treatment groups and patient and graft survival was 94% (187/202) in NULOJIX-treated patients compared with 88% (162/184) in cyclosporine-treated patients (difference=4.6%, 97.3% CI [−2.1, 11.3]).
Assessment of Glomerular Filtration Rate (GFR)
Glomerular Filtration Rate (GFR) was measured at one and two years and was calculated using the Modification of Diet in Renal Disease (MDRD) formula at one, two, and three years after transplantation. As shown in Table 8, both measured and calculated GFR was higher in patients treated with the NULOJIX recommended regimen compared to patients treated with the cyclosporine control regimen at all time points. As shown in Figure 1, the differences in GFR were apparent in the first month after transplant and were maintained up to three years (36 months). An analysis of change of calculated mean GFR between three and 36 months demonstrated an increase of 0.8 mL/min/year (95% CI [−0.2, 1.8]) for NULOJIX-treated patients and a decrease of 2.2 mL/min/year (95% CI [−3.2, −1.2]) for cyclosporine-treated patients.
Table 8: Measured and Calculated GFR for Study 1: Recipients of Living and Standard Criteria Deceased Donor Kidneys Parameter NULOJIX
Recommended Regimen
N=226Cyclosporine
(CSA)
N=221NULOJIX-CSA
(97.3% CI)a GFR was measured using the cold-iothalamate method.
b Measured GFR was not assessed at Year 3.
c GFR was calculated using the MDRD formula.Measured GFRa mL/min/1.73 m2 mean (SD)
Year 1
63.4 (27.7)
50.4 (18.7)
13.0 (7.3, 18.7)
(n=206)
(n=199)
Year 2b
67.9 (29.9)
50.5 (20.5)
17.4 (11.5, 23.4)
(n=199)
(n=185)
Calculated GFRc mL/min/1.73 m2 mean (SD)
Year 1
65.4 (22.9)
50.1 (21.1)
15.3 (10.3, 20.3)
(n=200)
(n=199)
Year 2
65.4 (25.2)
47.9 (23)
17.5 (12, 23.1)
(n=201)
(n=182)
Year 3
65.8 (27)
44.4 (23.6)
21.4 (15.4, 27.4)
(n=190)
(n=171)
Figure 1: Calculated (MDRD) GFR Through Month 36; Study 1: Recipients of Living and Standard Criteria Deceased Donor Kidneys
Assessment of Chronic Allograft Nephropathy (CAN)
The prevalence of chronic allograft nephropathy (CAN) at one year, as defined by the Banff ’97 classification system, was 24% (54/226) in patients treated with the NULOJIX recommended regimen and in 32% (71/219) of patients treated with the cyclosporine control regimen. CAN was not evaluated after the first year following transplantation. The clinical significance of this finding is unknown.
Study 2: Recipients of Extended Criteria Donor Kidneys
In Study 2 (NCT00114777), 543 patients were enrolled, randomized, and transplanted: 175 to the NULOJIX recommended regimen, 184 to the NULOJIX regimen with higher cumulative doses and more frequent dosing than recommended, and 184 to the cyclosporine control regimen. The median age was 58 years; 67% of the study population was male; 75% of patients were white, 13% were black/African-American, 12% were categorized as of other races; 3% had PRA ≥10%; 53% had 4 to 6 HLA mismatches; and 29% had diabetes prior to transplantation. The incidence of delayed graft function was similar in all treatment arms (47% to 49%).
Premature discontinuation from treatment at the end of the first year occurred in 25% of patients receiving the NULOJIX recommended regimen and 30% of patients receiving the cyclosporine control regimen. Among the patients who received the NULOJIX recommended regimen, 14% discontinued due to adverse events, 9% due to lack of efficacy, and 2% for other reasons. Among the patients who received the cyclosporine regimen, 17% discontinued due to adverse events, 7% due to lack of efficacy, and 6% for other reasons.
At the end of three years, 35% of patients receiving the NULOJIX recommended regimen and 44% of patients receiving the cyclosporine regimen had discontinued from treatment. Among the patients who received the NULOJIX recommended regimen, 20% discontinued due to adverse events, 9% due to lack of efficacy, and 6% for other reasons. Among the patients who received the cyclosporine regimen, 25% discontinued due to adverse events, 10% due to lack of efficacy, and 10% for other reasons.
Assessment of Efficacy
Table 9 summarizes the results of Study 2 following one and three years of treatment with the NULOJIX recommended dosage regimen and the cyclosporine control regimen. Efficacy failure at one year was defined as the occurrence of biopsy proven acute rejection (BPAR), graft loss, death, or lost to follow-up. BPAR was defined as histologically confirmed acute rejection by a central pathologist on a biopsy done for any reason, whether or not accompanied by clinical signs of rejection. Patient and graft survival was also assessed.
Table 9: Efficacy Outcomes by Years 1 and 3 for Study 2: Recipients of Extended Criteria Donor Kidneys Parameter NULOJIX
Recommended Regimen
N=175
n (%)Cyclosporine
(CSA)
N=184
n (%)NULOJIX-CSA
(97.3% CI)a Patients may have experienced more than one event.
b Patients known to be alive with a functioning graft.Efficacy Failure by Year 1
51 (29.1)
52 (28.3)
0.9 (−9.7, 11.5)
Components of Efficacy Failurea
Biopsy Proven Acute Rejection
37 (21.1)
34 (18.5)
Graft Loss
16 (9.1)
20 (10.9)
Death
5 (2.9)
8 (4.3)
Lost to follow-up
0
2 (1.1)
Efficacy Failure by Year 3
63 (36)
68 (37)
−1.0 (−12.1, 10.3)
Components of Efficacy Failurea
Biopsy Proven Acute Rejection
42 (24)
42 (22.8)
Graft Loss
21 (12)
23 (12.5)
Death
15 (8.6)
17 (9.2)
Lost to follow-up
1 (0.6)
5 (2.7)
Patient and graft survivalb
Year 1
155 (88.6)
157 (85.3)
3.2 (−4.8, 11.3)
Year 3
143 (81.7)
143 (77.7)
4.0 (−5.4, 13.4)
In Study 2, the rate of BPAR at one year and three years was similar in patients treated with NULOJIX and cyclosporine. Of the patients who experienced BPAR with NULOJIX, 62% experienced BPAR by Month 3, and 76% experienced BPAR by Month 6. By three years, recurrent BPAR occurred with similar frequency across treatment groups (<3%). The component of BPAR determined by biopsy only (subclinical protocol-defined acute rejection) was 5% in both treatment groups.
A similar proportion of patients in the NULOJIX recommended regimen group experienced BPAR classified as Banff grade IIb or higher (5% [9/175] at one year and 6% [10/175] at three years) compared to patients treated with the cyclosporine regimen (4% [7/184] at one year and 5% [9/184] at three years). Also, T cell-depleting therapy was used with similar frequency to treat any episode of BPAR in NULOJIX-treated patients (5% or 9/175) compared to cyclosporine-treated patients (4% or 7/184). At Month 12, the difference in mean calculated GFR between patients with and without a history of BPAR was 10 mL/min/1.73 m2 among NULOJIX-treated patients compared to 14 mL/min/1.73 m2 among cyclosporine-treated patients. By three years, 24% (10/42) of NULOJIX-treated patients with a history of BPAR experienced graft loss and/or death compared to 31% (13/42) of cyclosporine-treated patients with a history of BPAR; at that time point, 17% (7/42) of NULOJIX-treated patients experienced graft loss and 14% (6/42) of NULOJIX-treated patients had died following an episode of BPAR, whereas 19% (8/42) of cyclosporine-treated patients experienced graft loss and 19% (8/42) of cyclosporine-treated patients had died following an episode of BPAR. The overall prevalence of donor-specific antibodies was 6% and 15% for the NULOJIX recommended regimen and cyclosporine, respectively, up to 36 months post-transplant.
The mean GFR following BPAR was 36 mL/min/1.73 m2 in NULOJIX patients and 24 mL/min/1.73 m2 in cyclosporine-treated patients at one year. The relationship between BPAR, GFR, and patient and graft survival is unclear due to the limited number of patients who experienced BPAR, differences in renal hemodynamics (and, consequently, GFR) across maintenance immunosuppression regimens, and the high rate of switching treatment regimens after BPAR.
Assessment of Efficacy in the EBV Seropositive Subpopulation
NULOJIX is recommended for use only in EBV seropositive patients [see Indications and Usage (1.2)].
In Study 2, approximately 91% of the patients were EBV seropositive prior to transplant. Efficacy results in the EBV seropositive subpopulation were consistent with those in the total population studied.
By one year, the efficacy failure rate in the EBV seropositive population was 29% (45/156) in patients treated with the NULOJIX recommended regimen and 28% (47/168) in patients treated with cyclosporine (difference=0.8%, 97.3% CI [−10.3, 11.9]). Patient and graft survival rate in the EBV seropositive population was 89% (139/156) in the NULOJIX-treated patients and 86% (144/168) in cyclosporine-treated patients (difference=3.4%, 97.3% CI [−4.7, 11.5]).
By three years, efficacy failure was 35% (54/156) in NULOJIX-treated patients and 36% (61/168) in cyclosporine-treated patients. Patient and graft survival was 83% (130/156) in NULOJIX-treated patients compared with 77% (130/168) in cyclosporine-treated patients (difference=5.9%, 97.3% CI [−3.8, 15.6]).
Assessment of Glomerular Filtration Rate (GFR)
Glomerular Filtration Rate (GFR) was measured at one and two years and was calculated using the Modification of Diet in Renal Disease (MDRD) formula at one, two, and three years after transplantation. As shown in Table 10, both measured and calculated GFR was higher in patients treated with the NULOJIX recommended regimen compared to patients treated with the cyclosporine control regimen at all time points. As shown in Figure 2, the differences in GFR were apparent in the first month after transplant and were maintained up to three years (36 months). An analysis of change of calculated mean GFR between Month 3 and Month 36 demonstrated a decrease of 0.8 mL/min/year (95% CI [−1.9, 0.3]) for NULOJIX-treated patients and a decrease of 2.0 mL/min/year (95% CI [−3.1, −0.8]) for cyclosporine-treated patients.
Table 10: Measured and Calculated GFR for Study 2: Recipients of Extended Criteria Donor Kidneys Parameter NULOJIX
Recommended Regimen
N=175Cyclosporine
(CSA)
N=184NULOJIX-CSA
(97.3% CI)a GFR was measured using the cold-iothalamate method.
b Measured GFR was not assessed at Year 3.
c GFR was calculated using the MDRD formula.Measured GFRa mL/min/1.73 m2 mean (SD)
Year 1
49.6 (25.8)
45.2 (21.1)
4.3 (−1.5, 10.2)
(n=151)
(n=154)
Year 2b
49.7 (23.7)
45.0 (27.2)
4.7 (−1.8, 11.3)
(n=139)
(n=136)
Calculated GFRc mL/min/1.73 m2 mean (SD)
Year 1
44.5 (21.8)
36.5 (21.1)
8.0 (2.5, 13.4)
(n=158)
(n=159)
Year 2
42.8 (24.1)
34.9 (21.6)
8.0 (1.9, 14)
(n=158)
(n=154)
Year 3
42.2 (25.2)
31.5 (22.1)
10.7 (4.3, 17.2)
(n=154)
(n=143)
Figure 2: Calculated (MDRD) GFR Through Month 36; Study 2: Recipients of Extended Criteria Donor Kidneys
Assessment of Chronic Allograft Nephropathy (CAN)
The prevalence of chronic allograft nephropathy (CAN) at one year, as defined by the Banff ’97 classification system, was 46% (80/174) in patients treated with the NULOJIX recommended regimen and 52% (95/184) of patients treated with the cyclosporine control regimen. CAN was not evaluated after the first year following transplantation. The clinical significance of this finding is unknown.
14.2 Long-Term Extension (LTE) of Study 1 and Study 2
Although initially designed as three-year studies, Studies 1 and 2 were subsequently extended to seven years to provide descriptive long-term safety and efficacy data. Only patients who completed the assigned treatment for three years and consented to remain on the assigned treatment from three to seven years were eligible for the long-term extension (LTE) studies.
Long-Term Extension of Study 1
In the LTE of Study 1, of the 666 originally randomized and transplanted patients, 457 (69%) patients enrolled into the LTE study: 73% (166/226) in the NULOJIX recommended regimen group, 71% (155/219) in the NULOJIX nonrecommended regimen group, and 62% (136/221) in the cyclosporine group. Fourteen (2%) patients who completed the assigned treatment at the end of Year 3 did not enroll into the LTE study: 4 in the NULOJIX recommended regimen group, 3 in the NULOJIX nonrecommended regimen group, and 7 in the cyclosporine group.
Of the 457 patients enrolled in the LTE study, 356 (79%) patients completed the assigned treatment at the end of Year 7: 82% (136/166) in the NULOJIX recommended regimen group, 83% (128/155) in the NULOJIX nonrecommended regimen group, and 68% (92/136) in the cyclosporine group. The most common reasons for discontinuation from the LTE study included adverse events and death.
Seven (4.2%) deaths and 2 (1.2%) graft losses were reported in the NULOJIX recommended regimen group while 7 (4.5%) deaths and no graft loss were reported in the NULOJIX nonrecommended regimen group, and 10 (7.4%) deaths and 6 (4.4%) graft losses were reported in the cyclosporine group.
No PTLD was reported in the NULOJIX groups while 1 case of non-CNS PTLD was reported in the cyclosporine group, in a patient who was EBV seropositive at the time of transplant.
No PML was reported in the NULOJIX groups while 1 case of PML was reported in the cyclosporine group at 82 months post-transplant (56 days after discontinuing therapy).
The higher calculated GFR observed in NULOJIX-treated patients compared to cyclosporine-treated patients during the first three years was maintained during the LTE period.
Table 11: Events Reported in Long-Term Extension from 36 to 84 Months Post-Transplant of Study 1: Recipients of Living and Standard Criteria Deceased Donor Kidneys NULOJIX
Recommended
Regimen
N=166
n (%)NULOJIX
Nonrecommended
Regimen
N=155
n (%)
Cyclosporine
N=136
n (%)a This patient was EBV seropositive at the time of transplant. Death
7 (4.2)
7 (4.5)
10 (7.4)
Graft Loss
2 (1.2)
0 (0)
6 (4.4)
Death or Graft Loss
9 (5.4)
7 (4.5)
14 (10.3)
PTLD
0 (0)
0 (0)
1a (0.7)
PML
0 (0)
0 (0)
1 (0.7)
Long-Term Extension of Study 2
In the LTE of Study 2, of the 543 originally randomized and transplanted patients, 304 (56%) patients enrolled into the LTE study: 65% (113/175) in the NULOJIX recommended regimen group, 57% (104/184) in the NULOJIX nonrecommended regimen group, and 47% (87/184) in the cyclosporine group. Nineteen (3.5%) patients who completed the assigned treatment at the end of Year 3 did not enroll into the LTE study: 1 in the NULOJIX recommended regimen group, 5 in the NULOJIX nonrecommended regimen group, and 13 in the cyclosporine group.
Of the 304 patients enrolled in the LTE study, 215 (71%) patients completed the assigned treatment at the end of Year 7: 74% (84/113) in the NULOJIX recommended regimen group, 71% (74/104) in the NULOJIX nonrecommended regimen group, and 66% (57/87) in the cyclosporine group. The most common reasons for discontinuation from the LTE study included adverse events and death.
Twenty-one (18.6%) deaths and 1 (0.9%) graft loss were reported in the NULOJIX recommended regimen group while 14 (13.5%) deaths and 2 (1.9%) graft losses were reported in the NULOJIX nonrecommended regimen group, and 9 (10.3%) deaths and 6 (6.9%) graft losses were reported in the cyclosporine group.
Six cases of PTLD were reported among the three treatment groups: 4 in the NULOJIX recommended regimen group, 1 in the NULOJIX nonrecommended regimen group, and 1 in the cyclosporine group. Three of these cases (1 in each treatment group) occurred in patients who were EBV seropositive at the time of transplant and the other 3 cases (in NULOJIX recommended regimen) occurred in patients who were EBV seronegative. No case of PML was reported among the three treatment groups.
The higher mean calculated GFR observed in NULOJIX-treated patients compared to cyclosporine-treated patients during the first three years was maintained during the LTE period.
Table 12: Events Reported in Long-Term Extension from 36 to 84 Months Post-Transplant of Study 2: Recipients of Extended Criteria Donor Kidneys NULOJIX
Recommended
Regimen
N=113
n (%)NULOJIX
Nonrecommended
Regimen
N=104
n (%)
Cyclosporine
N=87
n (%)a Three of these patients were EBV seropositive at the time of transplant. Death
21 (18.6)
14 (13.5)
9 (10.3)
Graft Loss
1 (0.9)
2 (1.9)
6 (6.9)
Death or Graft Loss
22 (19.5)
16 (15.4)
14 (16.1)
PTLD
4a (3.6)
1 (1.0)
1 (1.2)
PML
0 (0)
0 (0)
0 (0)
14.3 Follow-Up Data of Patients with Complete 7-Year Patient and Graft Survival
In Study 1, of the original intention-to-treat (ITT) population (N=666), 72% (163/226) of patients in the NULOJIX recommended regimen group, 70% (153/219) of patients in the NULOJIX nonrecommended regimen group and 60% (132/221) of patients in the cyclosporine group had complete 7-year patient and graft survival follow-up data. Among these completers, the proportion of patients who died or had graft loss was 17% (27/163) in the NULOJIX recommended regimen group, 16% (25/153) in the NULOJIX nonrecommended regimen group and 30% (40/132) in the cyclosporine group.
In Study 2, of the original ITT population (N=543), 79% (138/175) of patients in the NULOJIX recommended regimen group, 128/184 (70%) of patients in the NULOJIX nonrecommended regimen group and 59% (108/184) of patients in the cyclosporine group had complete 7-year patient and graft survival follow-up data. Among these completers, the proportion of patients who died or had graft loss was 39% (54/138) in the NULOJIX recommended regimen group, 42% (54/128) in the NULOJIX nonrecommended regimen group and 48% (52/108) in the cyclosporine group.
Table 13: Events Reported in Patients with Complete 7-Year Patient and Graft Survival Follow-Up Data NULOJIX
Recommended
Regimen
n (%)NULOJIX
Nonrecommended
Regimen
n (%)
Cyclosporine
n (%)Study 1
N=163
N=153
N=132
Death
17 (10%)
17 (11%)
26 (20%)
Graft Loss
11 (7%)
10 (7%)
17 (13%)
Death or Graft Loss
27 (17%)
25 (16%)
40 (30%)
Study 2
N=138
N=128
N=108
Death
37 (27%)
37 (29%)
29 (27%)
Graft Loss
23 (17%)
21 (16%)
29 (27%)
Death or Graft Loss
54 (39%)
54 (42%)
52 (48%)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
NULOJIX® (belatacept) lyophilized powder for intravenous infusion is supplied as a single-use vial with a silicone-free disposable syringe in the following packaging configuration:
Description
NDC Number
One 250-mg vial
One 12 mL Syringe
0003-0371-13
16.1 Storage
NULOJIX lyophilized powder is stored refrigerated at 2°C to 8°C (36°F to 46°F). Protect NULOJIX from light by storing in the original package until time of use.
The reconstituted solution should be transferred from the vial to the infusion bag or bottle immediately. The NULOJIX infusion must be completed within 24 hours of constitution of the NULOJIX lyophilized powder. If not used immediately, the infusion solution may be stored under refrigeration conditions: 2°C to 8°C (36°F to 46°F) and protected from light for up to 24 hours (a maximum of 4 hours of the total 24 hours can be at room temperature: 20°C to 25°C [68°F to 77°F] and room light) [see Dosage and Administration (2.2)].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Post-Transplant Lymphoproliferative Disorder
The overall risk of PTLD, especially CNS PTLD, was elevated in NULOJIX-treated patients. Instruct patients to immediately report any of the following neurological, cognitive, or behavioral signs and symptoms during and after therapy with NULOJIX [see Boxed Warning and Warnings and Precautions (5.1)]:
- changes in mood or usual behavior
- confusion, problems thinking, loss of memory
- changes in walking or talking
- decreased strength or weakness on one side of the body
- changes in vision
Other Malignancies
Inform patients about the increased risk of malignancies, in addition to PTLD, while taking immunosuppressive therapy, especially skin cancer. Instruct patients to limit exposure to sunlight and UV light by wearing protective clothing and using a sunscreen with a high protection factor. Instruct patients to look for any signs and symptoms of skin cancer, such as suspicious moles or lesions [see Warnings and Precautions (5.3)].
Progressive Multifocal Leukoencephalopathy
Cases of PML have been reported in NULOJIX-treated patients. Instruct patients to immediately report any of the following neurological, cognitive, or behavioral signs and symptoms during and after therapy with NULOJIX [see Warnings and Precautions (5.4)]:
- changes in mood or usual behavior
- confusion, problems thinking, loss of memory
- changes in walking or talking
- decreased strength or weakness on one side of the body
- changes in vision
Other Serious Infections
Inform patients about the increased risk of infection while taking immunosuppressive therapy. Instruct patients to adhere to antimicrobial prophylaxis regimens as prescribed. Tell patients to immediately report any signs and symptoms of infection during therapy with NULOJIX [see Warnings and Precautions (5.5)].
Immunizations
Inform patients that vaccinations may be less effective while they are being treated with NULOJIX. Advise patients that live vaccines should be avoided [see Warnings and Precautions (5.8)].
Pregnant Women and Nursing Mothers
Inform patients that NULOJIX has not been studied in pregnant women or nursing mothers so the effects of NULOJIX on pregnant women or nursing infants are not known. Instruct patients to tell their healthcare provider if they are pregnant, become pregnant, or are thinking about becoming pregnant [see Use in Specific Populations (8.1)]. Instruct patients to tell their healthcare provider if they plan to breast-feed their infant [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
NULOJIX® (noo-LOJ-jiks)
(belatacept)
For Injection, For Intravenous Use
Read this Medication Guide before you start receiving NULOJIX and before each treatment. There may be new information. This Medication Guide does not take the place of talking with your doctor about your medical condition or your treatment.
What is the most important information I should know about NULOJIX?
NULOJIX increases your risk of serious side effects, including:
-
Post-transplant lymphoproliferative disorder (PTLD). PTLD is a condition that can happen if certain white blood cells grow out of control after an organ transplant because your immune system is weak. PTLD can get worse and become a type of cancer. PTLD can lead to death.
People treated with NULOJIX have a higher risk of getting PTLD. If you get PTLD with NULOJIX you are at especially high risk of getting it in your brain. Your risk for PTLD is also higher if you:- have never been exposed to the Epstein-Barr virus (EBV). Your doctor should test you for EBV. Do not receive NULOJIX unless you are EBV positive (you have been exposed to EBV).
- get an infection with a virus called cytomegalovirus (CMV).
- receive treatment for transplant rejection that lowers certain white blood cells called T lymphocytes.
- Increased risk of getting cancers other than PTLD. People who take medicines that weaken the immune system, including NULOJIX, have a higher risk of getting other cancers, including skin cancer. Talk to your doctor about your risk for cancer. See "What should I avoid while receiving NULOJIX?"
- Progressive multifocal leukoencephalopathy (PML). PML is a rare, serious brain infection caused by JC virus. People with weakened immune systems are at risk for getting PML. PML can result in death or severe disability. There is no known prevention, treatment, or cure for PML.
- Increased risk of getting other serious infections, including tuberculosis (TB) and other infections caused by bacteria, viruses, or fungi. These serious infections may lead to death. Also, a virus called BK virus can affect how your kidney works and cause your transplanted kidney to fail.
Tell your doctor right away if you get any of the following symptoms during treatment with NULOJIX:
- change in mood or your usual behavior
- confusion or problems thinking or with memory
- change in the way you walk or talk
- decreased strength or weakness on one side of your body
- change in vision
- fever, night sweats, or tiredness that does not go away
- weight loss
- swollen glands
- flu, cold symptoms, or cough
- stomach-area pain
- vomiting or diarrhea
- tenderness over your transplanted kidney
- change in the amount of urine that you make, blood in your urine, pain or burning on urination
- a new skin lesion or bump, or change in size or color of a mole
See "What are the possible side effects of NULOJIX?" for more information about side effects.
Liver transplant patients should not receive NULOJIX because of an increased risk of losing the transplanted liver (graft loss) and death. Talk to your doctor if you would like more information about this risk.
What is NULOJIX?
NULOJIX is a prescription medicine used in adults to prevent transplant rejection in people who have received a kidney transplant. Transplant rejection happens when the body’s immune system senses that the new transplanted kidney is different or foreign, and attacks it. NULOJIX is used with corticosteroids and certain other medicines to help prevent rejection of your new kidney.
It is not known if NULOJIX is safe and effective in children under 18 years of age.
NULOJIX is only used in people who have been exposed to the EBV virus.
It is not known if NULOJIX is safe and effective in people who receive an organ transplant other than a kidney transplant.
Who should not receive NULOJIX?
Do not receive treatment with NULOJIX if you are EBV negative. Your doctor will do a test to see if you were exposed to EBV in the past.
What should I tell my doctor before receiving NULOJIX?
Before receiving NULOJIX, tell your doctor if you:
- plan to receive any vaccines. Talk to your doctor about which vaccines are safe for you to receive during your treatment with NULOJIX. See “What should I avoid while receiving NULOJIX?”
- have any other medical conditions
-
are pregnant or plan to become pregnant. It is not known if NULOJIX will harm your unborn baby. If you become pregnant while taking NULOJIX:
- Tell your doctor right away. You and your doctor should decide if you will keep receiving NULOJIX while you are pregnant.
- Talk with your doctor about enrolling in the National Transplant Pregnancy Registry (NTPR). This Registry collects information about pregnancies in women who have received NULOJIX or if their partner has received NULOJIX, and had a transplant. You can also enroll by calling 1-877-955-6877.
- are breast-feeding or plan to breast-feed. It is not known if NULOJIX passes into your breast milk. You and your doctor should decide if you will receive NULOJIX or breast-feed. You should not do both.
Tell your doctor about all of the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
Know the medicines you take. Keep a list of them to show your doctor and pharmacist when you get a new medicine. Do not take any new medicine without talking with your transplant doctor first.
How will I receive NULOJIX?
- To help prevent rejection of your new kidney, you will receive NULOJIX regularly as prescribed by your doctor. It is important for you to keep all your appointments for NULOJIX treatment and follow up.
- You will receive NULOJIX as an intravenous (IV) infusion in your arm. Each IV infusion takes about 30 minutes.
- During treatment with NULOJIX, your doctor will test your blood and urine to check how your kidney is working.
- Take all the medicines prescribed by your doctor to prevent infection or transplant rejection. Take them exactly as your doctor tells you. Talk to your doctor or pharmacist if you have any questions about how to take your medicines.
What should I avoid while receiving NULOJIX?
- Limit the amount of time you spend in sunlight. Avoid using tanning beds or sunlamps. People who take medicines that weaken the immune system, including NULOJIX, have a higher risk of getting cancer, including skin cancer. Wear protective clothing and use sunscreen with a high protection factor (SPF) when you have to be in the sun.
- Avoid receiving live vaccines during treatment with NULOJIX. Talk to your doctor to find out which vaccines are safe for you during this time. Some vaccines may not work as well while you are receiving NULOJIX. See “What should I tell my doctor before receiving NULOJIX?”
What are the possible side effects of NULOJIX?
NULOJIX increases your risk of serious side effects that can cause death. See "What is the most important information I should know about NULOJIX?"
Common side effects of NULOJIX include:
- low red blood count (anemia)
- diarrhea
- kidney or bladder infection
- swollen legs, feet, or ankles
- constipation
- high blood pressure
- fever
- new kidney not working well
- cough
- nausea or vomiting
- headache
- low potassium or high potassium in your blood
- low white blood cell count
Tell your doctor about any side effect that bothers you or that does not go away. These are not all the possible side effects of NULOJIX. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to BMS at 1-800-321-1335.
General information about NULOJIX
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. This Medication Guide summarizes the most important information about NULOJIX. If you would like more information about NULOJIX, talk with your doctor. You can ask your pharmacist or doctor for information about NULOJIX that is written for healthcare professionals.
For more information, go to www.NULOJIX.com or call 1-800-321-1335.
What are the ingredients in NULOJIX?
Active ingredient: belatacept
Inactive ingredients: monobasic sodium phosphate, sodium chloride, and sucrose
This Medication Guide has been approved by the U.S. Food and Drug Administration.
-
Post-transplant lymphoproliferative disorder (PTLD). PTLD is a condition that can happen if certain white blood cells grow out of control after an organ transplant because your immune system is weak. PTLD can get worse and become a type of cancer. PTLD can lead to death.
- SPL UNCLASSIFIED SECTION
-
NULOJIX 250 mg per vial Representative Packaging
See How Supplied section for a complete list of available packages of NULOJIX.
Contains 1 Vial and 1 Syringe
NDC: 0003-0371-13
Nulojix®
(belatacept)
Rx only
For Injection
For Intravenous Infusion
250 mg per vial
ONLY USE THE SILICONE-FREE DISPOSABLE SYRINGE INCLUDED IN THIS PACKAGE FOR RECONSTITUTION
Provide enclosed Medication Guide to the patient.
1-888-NULOJIX
www.nulojix.com
Bristol-Myers Squibb -
INGREDIENTS AND APPEARANCE
NULOJIX
belatacept injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0003-0371 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength belatacept (UNII: E3B2GI648A) (belatacept - UNII:E3B2GI648A) belatacept 250 mg Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) 34.5 mg sodium chloride (UNII: 451W47IQ8X) 5.8 mg sucrose (UNII: C151H8M554) 500 mg Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0003-0371-13 1 in 1 CARTON 06/15/2011 1 1 in 1 VIAL, SINGLE-USE; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125288 06/15/2011 Labeler - E.R. Squibb & Sons, L.L.C. (011550092)
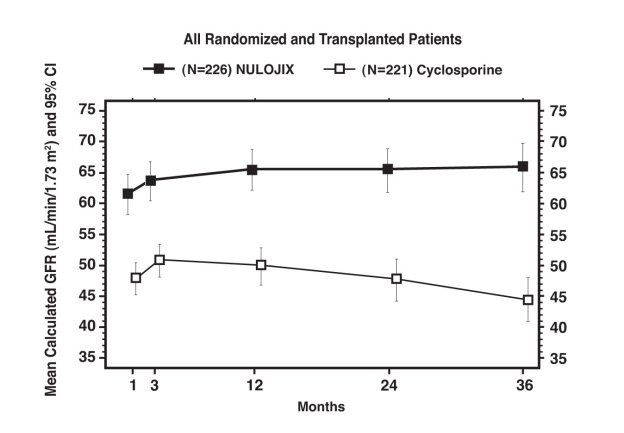
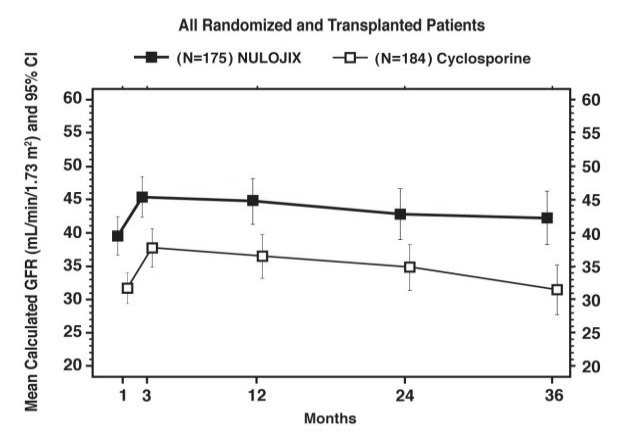

Trademark Results [NULOJIX]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 NULOJIX 77046754 3890407 Live/Registered |
Bristol-Myers Squibb Company 2006-11-17 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.