OMEPRAZOLE AND SODIUM BICARBONATE- omeprazole, sodium bicarbonate powder, for suspension OMEPRAZOLE AND SODIUM BICARBONATE- omeprazole, sodium bicarbonate capsule
Omeprazole and Sodium Bicarbonate by
Drug Labeling and Warnings
Omeprazole and Sodium Bicarbonate by is a Prescription medication manufactured, distributed, or labeled by Oceanside Pharmaceuticals, Patheon Inc., Carton Service Incorporated. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use OMEPRAZOLE AND SODIUM BICARBONATE safely and effectively. See full prescribing information for OMEPRAZOLE AND SODIUM BICARBONATE.
OMEPRAZOLE AND SODIUM BICARBONATE powder for oral suspension
OMEPRAZOLE AND SODIUM BICARBONATE capsules for oral use
Initial U.S. Approval: 2004RECENT MAJOR CHANGES
- Warnings and Precautions, Fundic Gland Polyps (5.13) 06/2018
INDICATIONS AND USAGE
Omeprazole and Sodium Bicarbonate is a proton pump inhibitor (PPI) indicated for:
- Short-term treatment of active duodenal ulcer (1.1)
- Short-term treatment of active benign gastric ulcer (1.2)
- Treatment of gastroesophageal reflux disease (GERD) (1.3)
- Maintenance of healing of erosive esophagitis (1.4)
- Reduction of risk of upper GI bleeding in critically ill patients (1.5)
The safety and effectiveness of Omeprazole and Sodium Bicarbonate in pediatric patients (<18 years of age) have not been established. (8.4)
DOSAGE AND ADMINISTRATION
- Short-Term Treatment of Active Duodenal Ulcer: 20 mg once daily for 4 weeks (some patients may require an additional 4 weeks of therapy (14.1)) (2)
- Gastric Ulcer: 40 mg once daily for 4-8 weeks (2)
-
Gastroesophageal Reflux Disease (GERD) (2)
- Symptomatic GERD (with no esophageal erosions): 20 mg once daily for up to 4 weeks
- Erosive Esophagitis: 20 mg once daily for 4-8 weeks
- Maintenance of Healing of Erosive Esophagitis: 20 mg once daily* (2)
- Reduction of Risk of Upper Gastrointestinal Bleeding in Critically Ill Patients: (40 mg oral suspension only) 40 mg initially followed by 40 mg 6-8 hours later and 40 mg daily thereafter for 14 days (2)
*studied for 12 months
DOSAGE FORMS AND STRENGTHS
- Omeprazole and Sodium Bicarbonate is available as a capsule and as a powder for oral suspension in 20 mg and 40 mg strengths. (3)
CONTRAINDICATIONS
- Known hypersensitivity to any components of the formulation (4)
WARNINGS AND PRECAUTIONS
- Gastric Malignancy: In adults, symptomatic response does not preclude the presence of gastric malignancy. Consider additional follow-up and diagnostic testing. (5.1)
- Acute interstitial nephritis has been observed in patients taking PPIs. (5.2)
- Buffer Content: contains sodium bicarbonate (5.3)
- PPI therapy may be associated with increased risk of Clostridium difficile-associated diarrhea. (5.4)
- Bone Fracture: Long-term and multiple daily dose PPI therapy may be associated with an increased risk for osteoporosis-related fractures of the hip, wrist, or spine. (5.5)
- Cutaneous and Systemic Lupus Erythematosus: Mostly cutaneous; new onset or exacerbation of existing disease; discontinue Omeprazole and Sodium Bicarbonate and refer to specialist for evaluation. (5.6)
- Avoid concomitant use of Omeprazole and Sodium Bicarbonate with clopidogrel. (5.7)
- Cyanocobalamin (Vitamin B-12) Deficiency: Daily long-term use (e.g., longer than 3 years) may lead to malabsorption or a deficiency of cyanocobalamin. (5.8)
- Hypomagnesemia has been reported rarely with prolonged treatment with PPIs. (5.9)
- Avoid concomitant use of Omeprazole and Sodium Bicarbonate with St. John’s wort or rifampin due to the potential reduction in omeprazole concentrations. (5.10, 7.2)
- Interactions with Diagnostic Investigations for Neuroendocrine Tumors: Increases in intragastric pH may result in hypergastrinemia and enterochromaffin-like cell hyperplasia and increased Chromogranin A levels which may interfere with diagnostic investigations for neuroendocrine tumors. (5.11, 12.2)
- Interaction with Methotrexate: Concomitant use with PPIs may elevate and/or prolong serum concentrations of methotrexate and/or its metabolite, possibly leading to toxicity. With high dose methotrexate administration, consider a temporary withdrawal of Omeprazole and Sodium Bicarbonate. (5.12, 7.8)
- Fundic Gland Polyps: Risk increases with long-term use, especially beyond one year. Use the shortest duration of therapy. (5.13)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥2%) are: headache, abdominal pain, nausea, diarrhea, vomiting, and flatulence. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Oceanside Pharmaceuticals, a division of Valeant Pharmaceuticals North America LLC at 1-800-321-4576 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- May interfere with drugs for which gastric pH can affect bioavailability (e.g., ketoconazole, ampicillin esters, iron salts, erlotinib, digoxin, and mycophenolate mofetil. (7.1)
- Drugs Metabolized by Cytochrome P450 (e.g., diazepam, warfarin, phenytoin, cyclosporine, disulfiram, benzodiazepines): Omeprazole and Sodium Bicarbonate can prolong their elimination. Monitor to determine the need for possible dose adjustments when taken with Omeprazole and Sodium Bicarbonate. (7.2)
- Patients treated with proton pump inhibitors and warfarin concomitantly may need to be monitored for increases in INR and prothrombin time. (7.2)
- Voriconazole: May increase plasma levels of omeprazole. (7.2)
- Saquinavir: Omeprazole and Sodium Bicarbonate increases plasma levels of saquinavir. (7.3)
- Omeprazole and Sodium Bicarbonate may reduce plasma levels of atazanavir and nelfinavir. (7.3)
- Clopidogrel: Omeprazole and Sodium Bicarbonate decreases exposure to the active metabolite of clopidogrel. (7.5)
- Tacrolimus: Omeprazole and Sodium Bicarbonate may increase serum levels of tacrolimus. (7.6)
USE IN SPECIFIC POPULATIONS
- Pregnancy: Based upon animal data, may cause fetal harm. (8.1)
- The safety and effectiveness of Omeprazole and Sodium Bicarbonate in pediatric patients less than 18 years of age have not been established. (8.4)
- Hepatic Impairment: Consider dose reduction, particularly for maintenance of healing of erosive esophagitis. (12.3)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Duodenal Ulcer
1.2 Gastric Ulcer
1.3 Treatment of Gastroesophageal Reflux Disease (GERD)
1.4 Maintenance of Healing of Erosive Esophagitis
1.5 Reduction of Risk of Upper Gastrointestinal Bleeding in Critically Ill Patients (40 mg Oral Suspension Only)
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Presence of Gastric Malignancy
5.2 Acute Interstitial Nephritis
5.3 Buffer Content
5.4 Clostridium difficile-Associated Diarrhea
5.5 Bone Fracture
5.6 Cutaneous and Systemic Lupus Erythematosus
5.7 Interaction with Clopidogrel
5.8 Cyanocobalamin (Vitamin B-12) Deficiency
5.9 Hypomagnesemia
5.10 Concomitant Use of Omeprazole and Sodium Bicarbonate with St. John’s Wort or Rifampin
5.11 Interactions with Investigations for Neuroendocrine Tumors
5.12 Concomitant Use of Omeprazole and Sodium Bicarbonate with Methotrexate
5.13 Fundic Gland Polyps
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drugs for Which Gastric pH Can Affect Bioavailability
7.2 Drugs Metabolized by Cytochrome P450 (CYP)
7.3 Antiretroviral Agents
7.4 Combination Therapy with Clarithromycin
7.5 Clopidogrel
7.6 Tacrolimus
7.7 Interactions with Investigations of Neuroendocrine Tumors
7.8 Methotrexate
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
8.8 Asian Population
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Duodenal Ulcer Disease
14.2 Gastric Ulcer
14.3 Gastroesophageal Reflux Disease (GERD)
14.4 Long-Term Maintenance Treatment of Erosive Esophagitis
14.5 Reduction of Risk of Upper Gastrointestinal Bleeding in Critically Ill Patients
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Duodenal Ulcer
Omeprazole and Sodium Bicarbonate is indicated for short-term treatment of active duodenal ulcer. Most patients heal within four weeks. Some patients may require an additional four weeks of therapy. [See Clinical Studies (14.1).]
1.2 Gastric Ulcer
Omeprazole and Sodium Bicarbonate is indicated for short-term treatment (4-8 weeks) of active benign gastric ulcer. [See Clinical Studies (14.2).]
1.3 Treatment of Gastroesophageal Reflux Disease (GERD)
Symptomatic GERD
Omeprazole and Sodium Bicarbonate is indicated for the treatment of heartburn and other symptoms associated with GERD for up to 4 weeks. [See Clinical Studies (14.3).]
Erosive Esophagitis
Omeprazole and Sodium Bicarbonate is indicated for the short-term treatment (4-8 weeks) of erosive esophagitis which has been diagnosed by endoscopy.
The efficacy of Omeprazole and Sodium Bicarbonate used for longer than 8 weeks in these patients has not been established. If a patient does not respond to 8 weeks of treatment, it may be helpful to give up to an additional 4 weeks of treatment. If there is recurrence of erosive esophagitis or GERD symptoms (e.g., heartburn), an additional 4-8 week courses of Omeprazole and Sodium Bicarbonate may be considered. [See Clinical Studies (14.3).]
1.4 Maintenance of Healing of Erosive Esophagitis
Omeprazole and Sodium Bicarbonate is indicated to maintain healing of erosive esophagitis. Controlled studies do not extend beyond 12 months. [See Clinical Studies (14.4).]
1.5 Reduction of Risk of Upper Gastrointestinal Bleeding in Critically Ill Patients (40 mg Oral Suspension Only)
Omeprazole and Sodium Bicarbonate Powder for Oral Suspension 40 mg/1,680 mg is indicated for the reduction of risk of upper GI bleeding in critically ill patients. [See Clinical Studies (14.5).]
-
2 DOSAGE AND ADMINISTRATION
Omeprazole and Sodium Bicarbonate is available as a capsule and as a powder for oral suspension in 20 mg and 40 mg strengths of omeprazole for adult use. Directions for use for each indication are summarized in Table 1. All recommended doses throughout the labeling are based upon omeprazole.
Since both the 20 mg and 40 mg oral suspension packets contain the same amount of sodium bicarbonate (1,680 mg), two packets of 20 mg are not equivalent to one packet of Omeprazole and Sodium Bicarbonate 40 mg; therefore, two 20 mg packets of Omeprazole and Sodium Bicarbonate should not be substituted for one packet of Omeprazole and Sodium Bicarbonate 40 mg.
Since both the 20 mg and 40 mg capsules contain the same amount of sodium bicarbonate (1,100 mg), two capsules of 20 mg are not equivalent to one capsule of Omeprazole and Sodium Bicarbonate 40 mg; therefore, two 20 mg capsules of Omeprazole and Sodium Bicarbonate should not be substituted for one capsule of Omeprazole and Sodium Bicarbonate 40 mg.
Omeprazole and Sodium Bicarbonate should be taken on an empty stomach at least one hour before a meal.
For patients receiving continuous Nasogastric (NG)/Orogastric (OG) tube feeding, enteral feeding should be suspended approximately 3 hours before and 1 hour after administration of Omeprazole and Sodium Bicarbonate Powder for Oral Suspension.
Table 1: Recommended Doses of Omeprazole and Sodium Bicarbonate by Indication for Adults 18 Years and Older - * Most patients heal within 4 weeks. Some patients may require an additional 4 weeks of therapy. [See Clinical Studies (14.1).]
- † For additional information, [see Indications and Usage (1)].
- ‡ Controlled studies do not extend beyond 12 months. [See Clinical Studies (14).]
Indication
Recommended
Dose
Frequency
Short-Term Treatment of Active Duodenal Ulcer
20 mg
Benign Gastric Ulcer
40 mg
Gastroesophageal Reflux Disease (GERD)
- Symptomatic GERD (with no esophageal erosions)
20 mg
Once daily for up to 4 weeks†
- Erosive Esophagitis
20 mg
Once daily for 4-8 weeks†
Maintenance of Healing of Erosive Esophagitis
20 mg
Once daily‡
Reduction of Risk of Upper Gastrointestinal Bleeding in Critically Ill Patients
(40 mg oral suspension only)
40 mg
40 mg initially followed by
40 mg 6-8 hours later and
40 mg daily thereafter for
14 days‡
Special Populations
Hepatic Insufficiency
Consider dose reduction, particularly for maintenance of healing of erosive esophagitis. [See Clinical Pharmacology (12.3).]
Administration of Capsules
Omeprazole and Sodium Bicarbonate Capsules should be swallowed intact with water. DO NOT USE OTHER LIQUIDS. DO NOT OPEN CAPSULE AND SPRINKLE CONTENTS INTO FOOD.
Preparation and Administration of Suspension
Directions for use: Empty packet contents into a small cup containing 1-2 tablespoons of water. DO NOT USE OTHER LIQUIDS OR FOODS. Stir well and drink immediately. Refill cup with water and drink.
If Omeprazole and Sodium Bicarbonate is to be administered through a nasogastric (NG) or orogastric (OG) tube, the suspension should be constituted with approximately 20 mL of water. DO NOT USE OTHER LIQUIDS OR FOODS. Stir well and administer immediately. An appropriately-sized syringe should be used to instill the suspension in the tube. The suspension should be washed through the tube with 20 mL of water.
-
3 DOSAGE FORMS AND STRENGTHS
Omeprazole and Sodium Bicarbonate 20 mg Capsules: Each opaque, hard gelatin, white capsule, imprinted with the Santarus logo and “20”, contains 20 mg omeprazole and 1,100 mg sodium bicarbonate.
Omeprazole and Sodium Bicarbonate 40 mg Capsules: Each opaque, hard gelatin, colored dark blue and white capsule, imprinted with the Santarus logo and “40”, contains 40 mg omeprazole and 1,100 mg sodium bicarbonate.
Omeprazole and Sodium Bicarbonate Powder for Oral Suspension is a white to beige, flavored powder packaged in unit-dose packets. Each packet contains either 20 mg or 40 mg omeprazole and 1,680 mg sodium bicarbonate.
-
4 CONTRAINDICATIONS
Omeprazole and Sodium Bicarbonate is contraindicated in patients with known hypersensitivity to any components of the formulation. Hypersensitivity reactions may include anaphylaxis, anaphylactic shock, angioedema, bronchospasm, acute interstitial nephritis, and urticaria. [See Adverse Reactions (6).]
-
5 WARNINGS AND PRECAUTIONS
5.1 Presence of Gastric Malignancy
In adults, symptomatic response to therapy with Omeprazole and Sodium Bicarbonate does not preclude the presence of gastric malignancy. Consider additional follow-up and diagnostic testing in adult patients who have a suboptimal response or an early symptomatic relapse after completing treatment with a proton pump inhibitor (PPI). In older patients, also consider an endoscopy.
5.2 Acute Interstitial Nephritis
Acute interstitial nephritis has been observed in patients taking PPIs including Omeprazole and Sodium Bicarbonate. Acute interstitial nephritis may occur at any point during PPI therapy and is generally attributed to an idiopathic hypersensitivity reaction. Discontinue Omeprazole and Sodium Bicarbonate if acute interstitial nephritis develops. [See Contraindications (4).]
5.3 Buffer Content
Each Omeprazole and Sodium Bicarbonate Capsule contains 1,100 mg (13 mEq) of sodium bicarbonate. The total content of sodium in each capsule is 304 mg.
Each packet of Omeprazole and Sodium Bicarbonate Powder for Oral Suspension contains 1,680 mg (20 mEq) of sodium bicarbonate (equivalent to 460 mg of Na+).
The sodium content of Omeprazole and Sodium Bicarbonate products should be taken into consideration when administering to patients on a sodium restricted diet.
Because Omeprazole and Sodium Bicarbonate products contain sodium bicarbonate, they should be used with caution in patients with Bartter’s syndrome, hypokalemia, hypocalcemia, and problems with acid-base balance. Long-term administration of bicarbonate with calcium or milk can cause milk-alkali syndrome.
Chronic use of sodium bicarbonate may lead to systemic alkalosis, and increased sodium intake can produce edema and weight increase.
5.4 Clostridium difficile-Associated Diarrhea
Published observational studies suggest that PPI therapy like Omeprazole and Sodium Bicarbonate may be associated with an increased risk of Clostridium difficile-associated diarrhea, especially in hospitalized patients. This diagnosis should be considered for diarrhea that does not improve. [See Adverse Reactions (6.2).]
Patients should use the lowest dose and shortest duration of PPI therapy appropriate to the condition being treated.
5.5 Bone Fracture
Several published observational studies suggest that proton pump inhibitor (PPI) therapy may be associated with an increased risk for osteoporosis-related fractures of the hip, wrist, or spine. The risk of fracture was increased in patients who received high-dose, defined as multiple daily doses, and long-term PPI therapy (a year or longer). Patients should use the lowest dose and shortest duration of PPI therapy appropriate to the condition being treated. Patients at risk for osteoporosis-related fractures should be managed according to the established treatment guidelines. [See Dosage and Administration (2) and Adverse Reactions (6.2).]
5.6 Cutaneous and Systemic Lupus Erythematosus
Cutaneous lupus erythematosus (CLE) and systemic lupus erythematosus (SLE) have been reported in patients taking PPIs, including omeprazole. These events have occurred as both new onset and an exacerbation of existing autoimmune disease. The majority of PPI-induced lupus erythematous cases were CLE.
The most common form of CLE reported in patients treated with PPIs was subacute CLE (SCLE) and occurred within weeks to years after continuous drug therapy in patients ranging from infants to the elderly. Generally, histological findings were observed without organ involvement.
Systemic lupus erythematosus (SLE) is less commonly reported than CLE in patients receiving PPIs. PPI associated SLE is usually milder than non-drug induced SLE. Onset of SLE typically occurred within days to years after initiating treatment in patients ranging from young adults to the elderly. The majority of patients presented with rash; however, arthralgia and cytopenia were also reported.
Avoid administration of PPIs for longer than medically indicated. If signs or symptoms consistent with CLE or SLE are noted in patients receiving Omeprazole and Sodium Bicarbonate, discontinue the drug and refer the patient to the appropriate specialist for evaluation. Most patients improve with discontinuation of the PPI alone in 4 to 12 weeks. Serological testing (e.g., ANA) may be positive and elevated serological test results may take longer to resolve than clinical manifestations.
5.7 Interaction with Clopidogrel
Avoid concomitant use of Omeprazole and Sodium Bicarbonate with clopidogrel. Clopidogrel is a prodrug. Inhibition of platelet aggregation by clopidogrel is entirely due to an active metabolite. The metabolism of clopidogrel to its active metabolite can be impaired by use with concomitant medications, such as omeprazole, that interfere with CYP2C19 activity. Concomitant use of clopidogrel with 80 mg omeprazole reduces the pharmacological activity of clopidogrel, even when administered 12 hours apart. When using Omeprazole and Sodium Bicarbonate, consider alternative antiplatelet therapy. [See Drug Interactions (7.5) and Clinical Pharmacology (12.3).]
5.8 Cyanocobalamin (Vitamin B-12) Deficiency
Daily treatment with any acid-suppressing medications over a long period of time (e.g., longer than 3 years) may lead to malabsorption of cyanocobalamin (vitamin B-12) caused by hypo- or achlorhydria. Rare reports of cyanocobalamin deficiency occurring with acid-suppressing therapy have been reported in the literature. This diagnosis should be considered if clinical symptoms consistent with cyanocobalamin deficiency are observed.
5.9 Hypomagnesemia
Hypomagnesemia, symptomatic and asymptomatic, has been reported rarely in patients treated with PPIs for at least three months, in most cases after a year of therapy. Serious adverse events include tetany, arrhythmias, and seizures. In most patients, treatment of hypomagnesemia required magnesium replacement and discontinuation of the PPI.
For patients expected to be on prolonged treatment or who take PPIs with medications such as digoxin or drugs that may cause hypomagnesemia (e.g., diuretics), healthcare professionals may consider monitoring magnesium levels prior to initiation of PPI treatment and periodically. [See Adverse Reactions (6.2).]
5.10 Concomitant Use of Omeprazole and Sodium Bicarbonate with St. John’s Wort or Rifampin
Drugs which induce CYP2C19 or CYP3A4 (such as St. John’s wort or rifampin) can substantially decrease omeprazole concentrations [see Drug Interactions (7.2)]. Avoid concomitant use of Omeprazole and Sodium Bicarbonate with St. John’s wort or rifampin.
5.11 Interactions with Investigations for Neuroendocrine Tumors
Serum chromogranin A (CgA) levels increase secondary to drug-induced decreases in gastric acidity. The increased CgA level may cause false positive results in diagnostic investigations for neuroendocrine tumors. Providers should temporarily stop omeprazole treatment before assessing CgA levels and consider repeating the test if initial CgA levels are high. If serial tests are performed (e.g., for monitoring), the same commercial laboratory should be used for testing, as reference ranges between tests may vary. [See Clinical Pharmacology (12.2).]
5.12 Concomitant Use of Omeprazole and Sodium Bicarbonate with Methotrexate
Literature suggests that concomitant use of PPIs with methotrexate (primarily at high dose; see methotrexate prescribing information) may elevate and prolong serum levels of methotrexate and/or its metabolite, possibly leading to methotrexate toxicities. In high-dose methotrexate administration, a temporary withdrawal of the PPI may be considered in some patients. [See Drug Interactions (7.8).]
5.13 Fundic Gland Polyps
PPI use is associated with an increased risk of fundic gland polyps that increases with long-term use, especially beyond one year. Most PPIs users who developed fundic gland polyps were asymptomatic and fundic gland polyps were identified incidentally on endoscopy. Use the shortest duration of PPI therapy appropriate to the condition being treated.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described below and elsewhere in labeling:
- Acute Interstitial Nephritis [see Warnings and Precautions (5.2)]
- Clostridium difficile-Associated Diarrhea [see Warnings and Precautions (5.4)]
- Bone Fracture [see Warnings and Precautions (5.5)]
- Cutaneous and Systemic Lupus Erythematosus [see Warnings and Precautions (5.6)]
- Cyanocobalamin (Vitamin B-12) Deficiency [see Warnings and Precautions (5.8)]
- Hypomagnesemia [see Warnings and Precautions (5.9)]
- Fundic Gland Polyps [see Warnings and Precautions (5.13)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In the U.S. clinical trial population of 465 patients, the adverse reactions summarized in Table 2 were reported to occur in 1% or more of patients on therapy with omeprazole. Numbers in parentheses indicate percentages of the adverse reactions considered by investigators as possibly, probably or definitely related to the drug.
Table 2: Adverse Reactions Occurring in 1% or More of Patients on Omeprazole Therapy Omeprazole
(n = 465)
Placebo
(n = 64)
Ranitidine
(n = 195)
Headache
6.9 (2.4)
6.3
7.7 (2.6)
Diarrhea
3.0 (1.9)
3.1 (1.6)
2.1 (0.5)
Abdominal Pain
2.4 (0.4)
3.1
2.1
Nausea
2.2 (0.9)
3.1
4.1 (0.5)
URI
1.9
1.6
2.6
Dizziness
1.5 (0.6)
0.0
2.6 (1.0)
Vomiting
1.5 (0.4)
4.7
1.5 (0.5)
Rash
1.5 (1.1)
0.0
0.0
Constipation
1.1 (0.9)
0.0
0.0
Cough
1.1
0.0
1.5
Asthenia
1.1 (0.2)
1.6 (1.6)
1.5 (1.0)
Back Pain
1.1
0.0
0.5
Table 3 summarizes the adverse reactions that occurred in 1% or more of omeprazole-treated patients from international double-blind and open-label clinical trials in which 2,631 patients and subjects received omeprazole.
Table 3: Incidence of Adverse Reactions ≥1% Causal Relationship Not Assessed Omeprazole
(n = 2,631)
Placebo
(n = 120)
Body as a Whole, Site Unspecified
- Abdominal Pain
5.2
3.3
- Asthenia
1.3
0.8
Digestive System
- Constipation
1.5
0.8
- Diarrhea
3.7
2.5
- Flatulence
2.7
5.8
- Nausea
4.0
6.7
- Vomiting
3.2
10.0
- Acid Regurgitation
1.9
3.3
Nervous System/Psychiatric
- Headache
2.9
2.5
A controlled clinical trial was conducted in 359 critically ill patients, comparing Omeprazole and Sodium Bicarbonate 40 mg/1,680 mg suspension once daily to I.V. cimetidine 1,200 mg/day for up to 14 days. The incidence and total number of AEs experienced by ≥3% of patients in either group are presented in Table 4 by body system and preferred term.
Table 4: Number (%) of Critically Ill Patients with Frequently Occurring (≥3%) Adverse Events by Body System and Preferred Term - * Clinically significant upper gastrointestinal bleeding was considered a serious adverse event but it is not included in this table.
Omeprazole and
Sodium Bicarbonate
(N=178)
Cimetidine
(N=181)
MedDRA
Body System
All AEs
All AEs
- Preferred Term
n (%)
n (%)
BLOOD AND LYMPHATIC SYSTEM DISORDERS
- Anemia NOS
14 (7.9)
14 (7.7)
- Anemia NOS Aggravated
4 (2.2)
7 (3.9)
- Thrombocytopenia
18 (10.1)
11 (6.1)
CARDIAC DISORDERS
- Atrial Fibrillation
11 (6.2)
7 (3.9)
- Bradycardia NOS
7 (3.9)
5 (2.8)
- Supraventricular Tachycardia
6 (3.4)
2 (1.1)
- Tachycardia NOS
6 (3.4)
6 (3.3)
- Ventricular Tachycardia
8 (4.5)
6 (3.3)
GASTROINTESTINAL DISORDERS*
- Constipation
8 (4.5)
8 (4.4)
- Diarrhea NOS
7 (3.9)
15 (8.3)
- Gastric Hypomotility
3 (1.7)
6 (3.3)
GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONS
- Hyperpyrexia
8 (4.5)
3 (1.7)
- Edema NOS
5 (2.8)
11 (6.1)
- Pyrexia
36 (20.2)
29 (16.0)
INFECTIONS AND INFESTATIONS
- Candidal Infection NOS
3 (1.7)
7 (3.9)
- Oral Candidiasis
7 (3.9)
1 (0.6)
- Sepsis NOS
9 (5.1)
9 (5.0)
- Urinary Tract Infection NOS
4 (2.2)
6 (3.3)
INVESTIGATIONS
- Liver Function Tests NOS Abnormal
3 (1.7)
6 (3.3)
METABOLISM AND NUTRITION DISORDERS
- Fluid Overload
9 (5.1)
14 (7.7)
- Hyperglycemia NOS
19 (10.7)
21 (11.6)
- Hyperkalemia
4 (2.2)
6 (3.3)
- Hypernatremia
3 (1.7)
9 (5.0)
- Hypocalcemia
11 (6.2)
10 (5.5)
- Hypoglycemia NOS
6 (3.4)
8 (4.4)
- Hypokalemia
22 (12.4)
24 (13.3)
- Hypomagnesemia
18 (10.1)
18 (9.9)
- Hyponatremia
7 (3.9)
5 (2.8)
- Hypophosphatemia
11 (6.2)
7 (3.9)
PSYCHIATRIC DISORDERS
- Agitation
6 (3.4)
16 (8.8)
RESPIRATORY, THORACIC AND MEDIASTINAL DISORDERS
- Acute Respiratory Distress Syndrome
6 (3.4)
7 (3.9)
- Nosocomial Pneumonia
20 (11.2)
17 (9.4)
- Pneumothorax NOS
1 (0.6)
8 (4.4)
- Respiratory Failure
3 (1.7)
6 (3.3)
SKIN AND SUBCUTANEOUS TISSUE DISORDERS
- Decubitus Ulcer
6 (3.4)
5 (2.8)
- Rash NOS
10 (5.6)
11 (6.1)
VASCULAR DISORDERS
- Hypertension NOS
14 (7.9)
6 (3.3)
- Hypotension NOS
17 (9.6)
12 (6.6)
NOS = Not otherwise specified
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of omeprazole. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Body as a Whole: Hypersensitivity reactions, including anaphylaxis, anaphylactic shock, angioedema, bronchospasm, interstitial nephritis, urticaria (see also Skin below), fever, pain, fatigue, malaise, and systemic lupus erythematosus.
Cardiovascular: Chest pain or angina, tachycardia, bradycardia, palpitation, elevated blood pressure, and peripheral edema.
Gastrointestinal: Pancreatitis (some fatal), anorexia, irritable colon, flatulence, fecal discoloration, esophageal candidiasis, mucosal atrophy of the tongue, dry mouth, stomatitis, abdominal swelling and fundic gland polyps. Gastroduodenal carcinoids have been reported in patients with Zollinger-Ellison syndrome on long-term treatment with omeprazole. This finding is believed to be a manifestation of the underlying condition, which is known to be associated with such tumors.
Hepatic: Mild and, rarely, marked elevations of liver function tests [ALT (SGPT), AST (SGOT), ᵞ-glutamyl transpeptidase, alkaline phosphatase, and bilirubin (jaundice)]. In rare instances, overt liver disease has occurred, including hepatocellular, cholestatic, or mixed hepatitis, liver necrosis (some fatal), hepatic failure (some fatal), and hepatic encephalopathy.
Infections and Infestations: Clostridium difficile-associated diarrhea.
Metabolism and Nutritional Disorders: Hyponatremia, hypoglycemia, hypomagnesemia, and weight gain.
Musculoskeletal: Muscle cramps, myalgia, muscle weakness, joint pain, bone fracture, and leg pain.
Nervous System/Psychiatric: Psychic disturbances including depression, agitation, aggression, hallucinations, confusion, insomnia, nervousness, tremors, apathy, somnolence, anxiety, dream abnormalities; vertigo; paresthesia; and hemifacial dysesthesia.
Respiratory: Epistaxis, pharyngeal pain.
Skin: Severe generalized skin reactions including toxic epidermal necrolysis (TEN; some fatal), Stevens-Johnson syndrome, cutaneous lupus erythematosus and erythema multiforme (some severe); purpura and/or petechiae (some with rechallenge); skin inflammation, urticaria, angioedema, pruritus, photosensitivity, alopecia, dry skin, and hyperhidrosis.
Special Senses: Tinnitus, taste perversion.
Ocular: Blurred vision, ocular irritation, dry eye syndrome, optic atrophy, anterior ischemic optic neuropathy, optic neuritis, and double vision.
Urogenital: Interstitial nephritis (some with positive rechallenge), urinary tract infection, microscopic pyuria, urinary frequency, elevated serum creatinine, proteinuria, hematuria, glycosuria, testicular pain, and gynecomastia.
Hematologic: Rare instances of pancytopenia, agranulocytosis (some fatal), thrombocytopenia, neutropenia, leukopenia, anemia, leukocytosis, and hemolytic anemia have been reported.
The incidence of clinical adverse experiences in patients greater than 65 years of age was similar to that in patients 65 years of age or less.
Additional adverse reactions that could be caused by sodium bicarbonate include metabolic alkalosis, seizures, and tetany.
-
7 DRUG INTERACTIONS
7.1 Drugs for Which Gastric pH Can Affect Bioavailability
Due to its effects on gastric acid secretion, omeprazole can reduce the absorption of drugs where gastric pH is an important determinant of their bioavailability. Like with other drugs that decrease the intragastric acidity, the absorption of drugs such as ketoconazole, atazanavir, iron salts, erlotinib, and mycophenolate mofetil (MMF) can decrease, while the absorption of drugs such as digoxin can increase during treatment with omeprazole.
Concomitant treatment with omeprazole (20 mg daily) and digoxin in healthy subjects increased the bioavailability of digoxin by 10% (30% in two subjects). Coadministration of digoxin with Omeprazole and Sodium Bicarbonate is expected to increase the systemic exposure of digoxin. Therefore, patients may need to be monitored when digoxin is taken concomitantly with Omeprazole and Sodium Bicarbonate.
Coadministration of omeprazole in healthy subjects and in transplant patients receiving MMF has been reported to reduce the exposure to the active metabolite, mycophenolic acid (MPA), possibly due to a decrease in MMF solubility at an increased gastric pH. The clinical relevance of reduced MPA exposure on organ rejection has not been established in transplant patients receiving Omeprazole and Sodium Bicarbonate and MMF. Use Omeprazole and Sodium Bicarbonate with caution in transplant patients receiving MMF. [See Clinical Pharmacology (12.3).]
7.2 Drugs Metabolized by Cytochrome P450 (CYP)
Omeprazole can prolong the elimination of diazepam, warfarin and phenytoin, drugs that are metabolized by oxidation in the liver. There have been reports of increased international normalized ratio (INR) and prothrombin time in patients receiving proton pump inhibitors, including omeprazole, and warfarin concomitantly. Increases in INR and prothrombin time may lead to abnormal bleeding and even death. Patients treated with proton pump inhibitors and warfarin may need to be monitored for increases in INR and prothrombin time.
Although in normal subjects no interaction with theophylline or propranolol was found, there have been clinical reports of interaction with other drugs metabolized via the cytochrome P450 system (e.g., cyclosporine, disulfiram, benzodiazepines). Patients should be monitored to determine if it is necessary to adjust the dosage of these drugs when taken concomitantly with Omeprazole and Sodium Bicarbonate.
Concomitant administration of omeprazole and voriconazole (a combined inhibitor of CYP2C19 and CYP3A4) resulted in more than doubling of the omeprazole exposure. Dose adjustment of omeprazole is not normally required. When voriconazole (400 mg every 12 hours for one day, then 200 mg for 6 days) was given with omeprazole (40 mg once daily for 7 days) to healthy subjects, it significantly increased the steady-state Cmax and AUC0-24 of omeprazole, an average of 2 times (90% CI: 1.8, 2.6) and 4 times (90% CI: 3.3, 4.4) respectively as compared to when omeprazole was given without voriconazole.
Drugs known to induce CYP2C19 or CYP3A4 (such as rifampin) may lead to decreased omeprazole serum levels. In a crossover study in 12 healthy male subjects, St. John’s wort (300 mg three times daily for 14 days), an inducer of CYP3A4, decreased the systemic exposure of omeprazole in CYP2C19 poor metabolizers (Cmax and AUC decreased by 37.5% and 37.9%, respectively) and extensive metabolizers (Cmax and AUC decreased by 49.6% and 43.9%, respectively). Avoid concomitant use of St. John’s wort or rifampin with omeprazole.
7.3 Antiretroviral Agents
Concomitant administration of atazanavir and proton pump inhibitors is not recommended. Coadministration of atazanavir with proton pump inhibitors is expected to substantially decrease atazanavir plasma concentrations and thereby reduce its therapeutic effect.
Omeprazole has been reported to interact with some antiretroviral drugs. The clinical importance and the mechanisms behind these interactions are not always known. Increased gastric pH during omeprazole treatment may change the absorption of the antiretroviral drug. Other possible interaction mechanisms are via CYP2C19. For some antiretroviral drugs, such as atazanavir and nelfinavir, decreased serum levels have been reported when given together with omeprazole. Following multiple doses of nelfinavir (1,250 mg, twice daily) and omeprazole (40 mg, daily), AUC was decreased by 36% and 92%, Cmax by 37% and 89% and Cmin by 39% and 75% respectively for nelfinavir and M8. Following multiple doses of atazanavir (400 mg, daily) and omeprazole (40 mg, daily, 2 hours before atazanavir), AUC was decreased by 94%, Cmax by 96%, and Cmin by 95%. Concomitant administration with omeprazole and drugs such as atazanavir and nelfinavir is therefore not recommended.
Increased Concentration of Saquinavir
For other antiretroviral drugs, such as saquinavir, elevated serum levels have been reported with an increase in AUC by 82%, in Cmax by 75% and in Cmin by 106% following multiple dosing of saquinavir/ritonavir (1,000/100 mg) twice daily for 15 days with omeprazole 40 mg daily coadministered days 11 to 15. Dose reduction of saquinavir should be considered from the safety perspective for individual patients. There are also some antiretroviral drugs of which unchanged serum levels have been reported when given with omeprazole.
7.4 Combination Therapy with Clarithromycin
Concomitant administration of clarithromycin with other drugs can lead to serious adverse reactions due to drug interaction [see Warnings and Precautions in prescribing information for clarithromycin]. Because of these drug interactions, clarithromycin is contraindicated for coadministration with certain drugs [see Contraindications in prescribing information for clarithromycin].
7.5 Clopidogrel
Omeprazole is an inhibitor of CYP2C19 enzyme. Clopidogrel is metabolized to its active metabolite in part by CYP2C19. Concomitant use of omeprazole 80 mg results in reduced plasma concentrations of the active metabolite of clopidogrel and a reduction in platelet inhibition. Avoid concomitant administration of Omeprazole and Sodium Bicarbonate with clopidogrel. When using Omeprazole and Sodium Bicarbonate, consider use of alternative antiplatelet therapy. [See Clinical Pharmacology (12.3).]
7.6 Tacrolimus
Concomitant administration of omeprazole and tacrolimus may increase the serum levels of tacrolimus.
7.7 Interactions with Investigations of Neuroendocrine Tumors
Drug-induced decrease in gastric acidity results in enterochromaffin-like cell hyperplasia and increased Chromogranin A levels which may interfere with investigations for neuroendocrine tumors. [See Clinical Pharmacology (12).]
7.8 Methotrexate
Case reports, published population pharmacokinetic studies, and retrospective analyses suggest that concomitant administration of PPIs and methotrexate (primarily at high dose; see methotrexate prescribing information) may elevate and prolong serum levels of methotrexate and/or its metabolite hydroxymethotrexate. However, no formal drug interaction studies of methotrexate with PPIs have been conducted. [See Warnings and Precautions (5.12).]
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C
Risk Summary
There are no adequate and well-controlled studies on the use of Omeprazole and Sodium Bicarbonate in pregnant women. Available epidemiologic data fail to demonstrate an increased risk of major congenital malformations or other adverse pregnancy outcomes with first trimester omeprazole use. Teratogenicity was not observed in animal reproduction studies with administration of oral esomeprazole magnesium in rats and rabbits with doses about 68 times and 42 times, respectively, an oral human dose of 40 mg (based on body surface area basis for a 60 kg person). However, changes in bone morphology were observed in offspring of rats dosed through most of pregnancy and lactation at doses equal to or greater than approximately 33.6 times an oral human dose of 40 mg (see Animal Data). Because of the observed effect at high doses of esomeprazole magnesium on developing bone in rat studies, Omeprazole and Sodium Bicarbonate should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Human Data
Four published epidemiological studies compared the frequency of congenital abnormalities among infants born to women who used omeprazole during pregnancy with the frequency of abnormalities among infants of women exposed to H2-receptor antagonists or other controls.
A population-based retrospective cohort epidemiological study from the Swedish Medical Birth Register, covering approximately 99% of pregnancies, from 1995-99, reported on 955 infants (824 exposed during the first trimester with 39 of these exposed beyond first trimester, and 131 exposed after the first trimester) whose mothers used omeprazole during pregnancy. The number of infants exposed in utero to omeprazole that had any malformation, low birth weight, low Apgar score, or hospitalization was similar to the number observed in this population. The number of infants born with ventricular septal defects and the number of stillborn infants was slightly higher in the omeprazole-exposed infants than the expected number in this population.
A population-based retrospective cohort study covering all live births in Denmark from 1996-2009, reported on 1,800 live births whose mothers used omeprazole during the first trimester of pregnancy and 837,317 live births whose mothers did not use any proton pump inhibitor. The overall rate of birth defects in infants born to mothers with first trimester exposure to omeprazole was 2.9% and 2.6% in infants born to mothers not exposed to any proton pump inhibitor during the first trimester.
A retrospective cohort study reported on 689 pregnant women exposed to either H2-blockers or omeprazole in the first trimester (134 exposed to omeprazole) and 1,572 pregnant women unexposed to either during the first trimester. The overall malformation rate in offspring born to mothers with first trimester exposure to omeprazole, an H2-blocker, or were unexposed was 3.6%, 5.5%, and 4.1% respectively.
A small prospective observational cohort study followed 113 women exposed to omeprazole during pregnancy (89% first trimester exposures). The reported rate of major congenital malformations was 4% in the omeprazole group, 2% in controls exposed to non-teratogens, and 2.8% in disease-paired controls. Rates of spontaneous and elective abortions, preterm deliveries, gestational age at delivery, and mean birth weight were similar among the groups.
Several studies have reported no apparent adverse short-term effects on the infant when single-dose oral or intravenous omeprazole was administered to over 200 pregnant women as premedication for cesarean section under general anesthesia.
Animal Data
Reproductive studies conducted with omeprazole in rats at oral doses up to 138 mg/kg/day (about 33.6 times an oral human dose of 40 mg on a body surface area basis) and in rabbits at doses up to 69 mg/kg/day (about 33.6 times an oral human dose of 40 mg on a body surface area basis) did not disclose any evidence for a teratogenic potential of omeprazole. In rabbits, omeprazole in a dose range of 6.9 to 69.1 mg/kg/day (about 3.36 to 33.6 times an oral human dose of 40 mg on a body surface area basis) produced dose-related increases in embryo-lethality, fetal resorptions, and pregnancy disruptions. In rats, dose-related embryo/fetal toxicity and postnatal developmental toxicity were observed in offspring resulting from parents treated with omeprazole at 13.8 to 138.0 mg/kg/day (about 3.36 to 33.6 times an oral human dose of 40 mg on a body surface area basis).
Reproduction studies have been performed with esomeprazole magnesium in rats at oral doses up to 280 mg/kg/day (about 68 times an oral human dose of 40 mg on a body surface area basis) and in rabbits at oral doses up to 86 mg/kg/day (about 42 times an oral human dose of 40 mg on a body surface area basis) and have revealed no evidence of impaired fertility or harm to the fetus due to esomeprazole magnesium.
A pre- and postnatal developmental toxicity study in rats with additional endpoints to evaluate bone development were performed with the S-enantiomer, esomeprazole magnesium at oral doses of 14 to 280 mg/kg/day (about 3.4 to 68 times an oral human dose of 40 mg of esomeprazole on a body surface area basis). Neonatal/early postnatal (birth to weaning) survival was decreased at doses equal to or greater than 138 mg/kg/day (about 33.6 times an oral human dose of 40 mg on a body surface area basis). Body weight and body weight gain were reduced and neurobehavioral or general developmental delays in the immediate post-weaning timeframe were evident at doses equal to or greater than 69 mg/kg/day (about 16.8 times an oral human dose of 40 mg on a body surface area basis). In addition, decreased femur length, width and thickness of cortical bone, decreased thickness of the tibial growth plate and minimal to mild bone marrow hypocellularity were noted at doses of esomeprazole magnesium equal to or greater than 14 mg/kg/day (about 3.4 times an oral human dose of 40 mg on a body surface area basis). Physeal dysplasia in the femur was observed in offspring of rats treated with oral doses of esomeprazole magnesium at doses equal to or greater than 138 mg/kg/day (about 33.6 times an oral human dose of 40 mg on a body surface area basis).
Effects on maternal bone were observed in pregnant and lactating rats in a pre- and postnatal toxicity study when esomeprazole magnesium was administered at oral doses of 14 to 280 mg/kg/day (about 3.4 to 68 times an oral human dose of 40 mg on a body surface area basis). When rats were dosed from gestational Day 7 through weaning on postnatal Day 21, a statistically significant decrease in maternal femur weight of up to 14% (as compared to placebo treatment) was observed at doses of esomeprazole magnesium equal to or greater than 138 mg/kg/day (about 33.6 times an oral human dose of 40 mg on a body surface area basis).
A pre- and postnatal development study in rats with esomeprazole strontium (using equimolar doses compared to esomeprazole magnesium study) produced similar results in dams and pups as described above.
8.3 Nursing Mothers
Omeprazole concentrations have been measured in breast milk of a woman following oral administration of 20 mg. The peak concentration of omeprazole in breast milk was less than 7% of the peak serum concentration. The concentration will correspond to 0.004 mg of omeprazole in 200 mL of milk. Because omeprazole is excreted in human milk, because of the potential for serious adverse reactions in nursing infants from omeprazole, and because of the potential for tumorigenicity shown for omeprazole in rat carcinogenicity studies, a decision should be made to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. In addition, sodium bicarbonate should be used with caution in nursing mothers.
8.4 Pediatric Use
Safety and effectiveness of Omeprazole and Sodium Bicarbonate have not been established in pediatric patients less than 18 years of age.
Juvenile Animal Data
In a juvenile rat toxicity study, esomeprazole was administered with both magnesium and strontium salts at oral doses about 34 to 68 times a daily human dose of 40 mg on a body surface area basis. Increases in death were seen at the high dose, and at all doses of esomeprazole, there were decreases in body weight, body weight gain, femur weight and femur length, and decreases in overall growth. [See Nonclinical Toxicology (13.2).]
8.5 Geriatric Use
Omeprazole was administered to over 2,000 elderly individuals (≥65 years of age) in clinical trials in the U.S. and Europe. There were no differences in safety and effectiveness between the elderly and younger subjects. Other reported clinical experience has not identified differences in response between the elderly and younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
Pharmacokinetic studies with buffered omeprazole have shown the elimination rate was somewhat decreased in the elderly and bioavailability was increased. The plasma clearance of omeprazole was 250 mL/min (about half that of young subjects). The plasma half-life averaged one hour, about twice that in nonelderly, healthy subjects taking Omeprazole and Sodium Bicarbonate. However, no dosage adjustment is necessary in the elderly. [See Clinical Pharmacology (12.3).]
8.6 Hepatic Impairment
Consider dose reduction, particularly for maintenance of healing of erosive esophagitis. [See Clinical Pharmacology (12.3).]
8.8 Asian Population
Recommend dose reduction, particularly for maintenance of healing of erosive esophagitis. [See Clinical Pharmacology (12.3).]
-
10 OVERDOSAGE
Reports have been received of overdosage with omeprazole in humans. Doses ranged up to 2,400 mg (120 times the usual recommended clinical dose). Manifestations were variable, but included confusion, drowsiness, blurred vision, tachycardia, nausea, vomiting, diaphoresis, flushing, headache, dry mouth, and other adverse reactions similar to those seen in normal clinical experience [see Adverse Reactions (6)]. Symptoms were transient, and no serious clinical outcome has been reported when omeprazole was taken alone. No specific antidote for omeprazole overdosage is known. Omeprazole is extensively protein bound and is, therefore, not readily dialyzable. In the event of overdosage, treatment should be symptomatic and supportive.
As with the management of any overdose, the possibility of multiple drug ingestion should be considered. For current information on treatment of any drug overdose, a certified Regional Poison Control Center should be contacted. Telephone numbers are listed in the Physicians’ Desk Reference (PDR) or local telephone book.
Single oral doses of omeprazole at 1,350, 1,339, and 1,200 mg/kg were lethal to mice, rats, and dogs, respectively. Animals given these doses showed sedation, ptosis, tremors, convulsions, and decreased activity, body temperature, and respiratory rate and increased depth of respiration.
In addition, a sodium bicarbonate overdose may cause hypocalcemia, hypokalemia, hypernatremia, and seizures.
-
11 DESCRIPTION
Omeprazole and Sodium Bicarbonate is a combination of omeprazole, a proton-pump inhibitor, and sodium bicarbonate, an antacid. Omeprazole is a substituted benzimidazole, 5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulfinyl]-1H-benzimidazole, a racemic mixture of two enantiomers that inhibits gastric acid secretion. Its empirical formula is C17H19N3O3S, with a molecular weight of 345.42. The structural formula is:
Omeprazole is a white to off-white crystalline powder which melts with decomposition at about 155°C. It is a weak base, freely soluble in ethanol and methanol, slightly soluble in acetone and isopropanol and very slightly soluble in water. The stability of omeprazole is a function of pH; it is rapidly degraded in acid media but has acceptable stability under alkaline conditions.
Omeprazole and Sodium Bicarbonate is supplied as immediate-release capsules and unit-dose packets as powder for oral suspension. Each capsule contains either 40 mg or 20 mg of omeprazole and 1,100 mg of sodium bicarbonate with the following excipients: croscarmellose sodium and sodium stearyl fumarate. Packets of powder for oral suspension contain either 40 mg or 20 mg of omeprazole and 1,680 mg of sodium bicarbonate with the following excipients: xylitol, sucrose, sucralose, xanthan gum, and flavorings.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Omeprazole belongs to a class of antisecretory compounds, the substituted benzimidazoles, that do not exhibit anticholinergic or H2 histamine antagonistic properties, but that suppress gastric acid secretion by specific inhibition of the H+/K+ ATPase enzyme system at the secretory surface of the gastric parietal cell. Because this enzyme system is regarded as the acid (proton) pump within the gastric mucosa, omeprazole has been characterized as a gastric acid-pump inhibitor, in that it blocks the final step of acid production. This effect is dose related and leads to inhibition of both basal and stimulated acid secretion irrespective of the stimulus. Animal studies indicate that after rapid disappearance from plasma, omeprazole can be found within the gastric mucosa for a day or more.
Omeprazole is acid labile and thus rapidly degraded by gastric acid. Omeprazole and Sodium Bicarbonate Capsules and Powder for Oral Suspension are immediate-release formulations that contain sodium bicarbonate which raises the gastric pH and thus protects omeprazole from acid degradation.
12.2 Pharmacodynamics
Antisecretory Activity
Results from a PK/PD study of the antisecretory effect of repeated once-daily dosing of 40 mg and 20 mg of Omeprazole and Sodium Bicarbonate Oral Suspension in healthy subjects are shown in Table 5 below.
Table 5: Effect of Omeprazole and Sodium Bicarbonate Oral Suspension on Intragastric pH, Day 7 - * P < 0.05 20 mg vs. 40 mg
Omeprazole/Sodium Bicarbonate
Parameter
40 mg/1,680 mg
(n = 24)
20 mg/1,680 mg
(n = 28)
% Decrease from Baseline for Integrated Gastric Acidity (mmol●hr/L)
84%
82%
- Coefficient of Variation
20%
24%
% Time Gastric pH > 4*
(Hours)*
77%
(18.6 h)
51%
(12.2 h)
- Coefficient of Variation
27%
43%
Median pH
5.2
4.2
- Coefficient of Variation
17%
37%
Note: Values represent medians. All parameters were measured over a 24-hour period.
Results from a separate PK/PD study of antisecretory effect on repeated once-daily dosing of 40 mg/1,100 mg and 20 mg/1,100 mg of Omeprazole and Sodium Bicarbonate Capsules in healthy subjects show similar effects in general on the above three PD parameters as those for Omeprazole and Sodium Bicarbonate 40 mg/1,680 mg and 20 mg/1,680 mg Oral Suspension, respectively.
The antisecretory effect lasts longer than would be expected from the very short (1 hour) plasma half-life, apparently due to irreversible binding to the parietal H+/K+ ATPase enzyme.
Enterochromaffin-like (ECL) Cell Effects
In 24-month carcinogenicity studies in rats, a dose-related significant increase in gastric carcinoid tumors and ECL cell hyperplasia was observed in both male and female animals [see Nonclinical Toxicology (13.1)]. Carcinoid tumors have also been observed in rats subjected to fundectomy or long-term treatment with other proton pump inhibitors or high doses of H2-receptor antagonists. Human gastric biopsy specimens have been obtained from more than 3,000 patients treated with omeprazole in long-term clinical trials. The incidence of ECL cell hyperplasia in these studies increased with time; however, no case of ECL cell carcinoids, dysplasia, or neoplasia has been found in these patients. These studies are of insufficient duration and size to rule out the possible influence of long-term administration of omeprazole on the development of any premalignant or malignant conditions.
Serum Gastrin Effects
In studies involving more than 200 patients, serum gastrin levels increased during the first 1 to 2 weeks of once-daily administration of therapeutic doses of omeprazole in parallel with inhibition of acid secretion. No further increase in serum gastrin occurred with continued treatment. In comparison with histamine H2-receptor antagonists, the median increases produced by 20 mg doses of omeprazole were higher (1.3 to 3.6-fold vs. 1.1- to 1.8-fold increase). Gastrin values returned to pretreatment levels, usually within 1 to 2 weeks after discontinuation of therapy.
Increased gastrin causes enterochromaffin-like cell hyperplasia and increased serum Chromogranin A (CgA) levels. The increased CgA levels may cause false positive results in diagnostic investigations for neuroendocrine tumors.
Other Effects
Systemic effects of omeprazole in the CNS, cardiovascular and respiratory systems have not been found to date. Omeprazole, given in oral doses of 30 or 40 mg for 2 to 4 weeks, had no effect on thyroid function, carbohydrate metabolism, or circulating levels of parathyroid hormone, cortisol, estradiol, testosterone, prolactin, cholecystokinin or secretin.
No effect on gastric emptying of the solid and liquid components of a test meal was demonstrated after a single dose of omeprazole 90 mg. In healthy subjects, a single I.V. dose of omeprazole (0.35 mg/kg) had no effect on intrinsic factor secretion. No systematic dose-dependent effect has been observed on basal or stimulated pepsin output in humans. However, when intragastric pH is maintained at 4.0 or above, basal pepsin output is low, and pepsin activity is decreased.
As do other agents that elevate intragastric pH, omeprazole administered for 14 days in healthy subjects produced a significant increase in the intragastric concentrations of viable bacteria. The pattern of the bacterial species was unchanged from that commonly found in saliva. All changes resolved within three days of stopping treatment.
The course of Barrett’s esophagus in 106 patients was evaluated in a U.S. double-blind controlled study of omeprazole 40 mg twice daily for 12 months followed by 20 mg twice daily for 12 months or ranitidine 300 mg twice daily for 24 months. No clinically significant impact on Barrett’s mucosa by antisecretory therapy was observed. Although neosquamous epithelium developed during antisecretory therapy, complete elimination of Barrett’s mucosa was not achieved. No significant difference was observed between treatment groups in development of dysplasia in Barrett’s mucosa, and no patient developed esophageal carcinoma during treatment. No significant differences between treatment groups were observed in development of ECL cell hyperplasia, corpus atrophic gastritis, corpus intestinal metaplasia, or colon polyps exceeding 3 mm in diameter.
12.3 Pharmacokinetics
Absorption
In separate in vivo bioavailability studies, when Omeprazole and Sodium Bicarbonate Oral Suspension and Capsules are administered on an empty stomach 1 hour prior to a meal, the absorption of omeprazole is rapid, with mean peak plasma levels (% CV) of omeprazole being 1,954 ng/mL (33%) and 1,526 ng/mL (49%), respectively, and time to peak of approximately 30 minutes (range 10-90 min) after a single-dose or repeated-dose administration. Absolute bioavailability of Omeprazole and Sodium Bicarbonate Powder for Oral Suspension (compared to I.V. administration) is about 30-40% at doses of 20 to 40 mg, due in large part to presystemic metabolism.
When Omeprazole and Sodium Bicarbonate Oral Suspension 40 mg/1,680 mg was administered in a two-dose loading regimen, the omeprazole AUC (0-inf) (ng●hr/mL) was 1,665 after Dose 1 and 3,356 after Dose 2, while Tmax was approximately 30 minutes for both Dose 1 and Dose 2.
Following single or repeated once-daily dosing, peak plasma concentrations of omeprazole from Omeprazole and Sodium Bicarbonate are approximately proportional from 20 to 40 mg doses, but a greater than linear mean AUC (three-fold increase) is observed when doubling the dose to 40 mg. The bioavailability of omeprazole from Omeprazole and Sodium Bicarbonate increases upon repeated administration.
When Omeprazole and Sodium Bicarbonate is administered 1 hour after a meal, the omeprazole AUC is reduced by approximately 24% relative to administration 1 hour prior to a meal.
Distribution
Omeprazole is bound to plasma proteins. Protein binding is approximately 95%.
Metabolism
Following single-dose oral administration of omeprazole, the majority of the dose (about 77%) is eliminated in urine as at least six metabolites. Two metabolites have been identified as hydroxyomeprazole and the corresponding carboxylic acid. The remainder of the dose was recoverable in feces. This implies a significant biliary excretion of the metabolites of omeprazole. Three metabolites have been identified in plasma – the sulfide and sulfone derivatives of omeprazole, and hydroxyomeprazole. These metabolites have very little or no antisecretory activity.
Excretion
Following single-dose oral administration of omeprazole, little, if any, unchanged drug is excreted in urine. The mean plasma omeprazole half-life in healthy subjects is approximately 1 hour (range 0.4 to 3.2 hours), and the total body clearance is 500-600 mL/min.
Concomitant Use with Clopidogrel
In a crossover clinical study, 72 healthy subjects were administered clopidogrel (300 mg loading dose followed by 75 mg per day) alone and with omeprazole (80 mg at the same time as clopidogrel) for 5 days. The exposure to the active metabolite of clopidogrel was decreased by 46% (Day 1) and 42% (Day 5) when clopidogrel and omeprazole were administered together.
Results from another crossover study in healthy subjects showed a similar pharmacokinetic interaction between clopidogrel (300 mg loading dose/75 mg daily maintenance dose) and omeprazole 80 mg daily when coadministered for 30 days. Exposure to the active metabolite of clopidogrel was reduced by 41% to 46% over this time period.
In another study, 72 healthy subjects were given the same doses of clopidogrel and 80 mg omeprazole, but the drugs were administered 12 hours apart; the results were similar, indicating that administering clopidogrel and omeprazole at different times does not prevent their interaction.
Concomitant Use with Mycophenolate Mofetil
Administration of omeprazole 20 mg twice daily for 4 days and a single 1,000 mg dose of MMF approximately one hour after the last dose of omeprazole to 12 healthy subjects in a crossover study resulted in a 52% reduction in the Cmax and 23% reduction in the AUC of MPA.
Special Populations
Geriatric
The elimination rate of omeprazole was somewhat decreased in the elderly, and bioavailability was increased. Omeprazole was 76% bioavailable when a single 40 mg oral dose of omeprazole (buffered solution) was administered to healthy elderly subjects versus 58% in young subjects given the same dose. Nearly 70% of the dose was recovered in urine as metabolites of omeprazole, and no unchanged drug was detected. The plasma clearance of omeprazole was 250 mL/min (about half that of young subjects), and its plasma half-life averaged one hour, similar to that of young healthy subjects.
Pediatric
The pharmacokinetics of Omeprazole and Sodium Bicarbonate has not been studied in patients <18 years of age.
Gender
There are no known differences in the absorption or excretion of omeprazole between males and females.
Hepatic Insufficiency
In patients with chronic hepatic disease, the bioavailability of omeprazole from a buffered solution increased to approximately 100% compared to an I.V. dose, reflecting decreased first-pass effect, and the mean plasma half-life of the drug increased to nearly 3 hours compared to the mean half-life of 1 hour in normal subjects. Plasma clearance averaged 70 mL/min, compared to a value of 500-600 mL/min in normal subjects. Dose reduction, particularly where maintenance of healing of erosive esophagitis is indicated, for the hepatically impaired should be considered.
Renal Insufficiency
In patients with chronic renal impairment, whose creatinine clearance ranged between 10 and 62 mL/min/1.73 m2, the disposition of omeprazole from a buffered solution was very similar to that in healthy subjects, although there was a slight increase in bioavailability. Because urinary excretion is a primary route of excretion of omeprazole metabolites, their elimination slowed in proportion to the decreased creatinine clearance. No dose reduction is necessary in patients with renal impairment.
Asian Population
In pharmacokinetic studies of single 20 mg omeprazole doses, an increase in AUC of approximately four-fold was noted in Asian subjects compared to Caucasians. Dose adjustment, particularly where maintenance of healing of erosive esophagitis is indicated, for Asian subjects should be considered.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In two 24-month carcinogenicity studies in rats, omeprazole at daily doses of 1.7, 3.4, 13.8, 44.0 and 140.8 mg/kg/day (approximately 0.4 to 34.2 times the human dose of 40 mg/day on a body surface area basis) produced gastric ECL cell carcinoids in a dose-related manner in both male and female rats; the incidence of this effect was markedly higher in female rats, which had higher blood levels of omeprazole. Gastric carcinoids seldom occur in the untreated rat. In addition, ECL cell hyperplasia was present in all treated groups of both sexes. In one of these studies, female rats were treated with 13.8 mg omeprazole/kg/day (approximately 3.36 times the human dose of 40 mg/day on a body surface area basis) for one year, then followed for an additional year without the drug. No carcinoids were seen in these rats. An increased incidence of treatment-related ECL cell hyperplasia was observed at the end of one year (94% treated versus 10% controls). By the second year the difference between treated and control rats was much smaller (46% versus 26%) but still showed more hyperplasia in the treated group. Gastric adenocarcinoma was seen in one rat (2%). No similar tumor was seen in male or female rats treated for two years. For this strain of rat no similar tumor has been noted historically, but a finding involving only one tumor is difficult to interpret. In a 52-week toxicity study in Sprague Dawley rats, brain astrocytomas were found in a small number of males that received omeprazole at dose levels of 0.4, 2, and 16 mg/kg/day (about 0.1 to 3.9 times the human dose of 40 mg/day on a body surface area basis). No astrocytomas were observed in female rats in this study. In a 2-year carcinogenicity study in Sprague Dawley rats, no astrocytomas were found in males and females at the high dose of 140.8 mg/kg/day (about 34 times the human dose of 40 mg/day on a body surface area basis). A 78-week mouse carcinogenicity study of omeprazole did not show increased tumor occurrence, but the study was not conclusive. A 26-week p53 (+/-) transgenic mouse carcinogenicity study was not positive.
Omeprazole was positive for clastogenic effects in an in vitro human lymphocyte chromosomal aberration assay, in one of two in vivo mouse micronucleus tests, and in an in vivo bone marrow cell chromosomal aberration assay. Omeprazole was negative in the in vitro Ames test, an in vitro mouse lymphoma cell forward mutation assay and an in vivo rat liver DNA damage assay.
In a 24-month carcinogenicity studies in rats, a dose-related significant increase in gastric carcinoid tumors and ECL cell hyperplasia was observed in both male and female animals [see Warnings and Precautions (5)]. Carcinoid tumors have also been observed in rats subjected to fundectomy or long-term treatment with other proton pump inhibitors or high doses of H2-receptor antagonists.
Omeprazole at oral doses up to 138 mg/kg/day (about 33.6 times the human dose of 40 mg/day on a body surface area basis) was found to have no effect on the fertility and general reproductive performance in rats.
13.2 Animal Toxicology and/or Pharmacology
Reproductive Toxicology Studies
Reproduction studies conducted in pregnant rats with omeprazole at doses up to 138 mg/kg/day (about 33.6 times an oral human dose of 40 mg on a body surface area basis) and in pregnant rabbits at doses up to 69 mg/kg/day (about 33.6 times an oral human dose of 40 mg on a body surface area basis) did not disclose any evidence for a teratogenic potential of omeprazole.
In rabbits, omeprazole in a dose range of 6.9 to 69 mg/kg/day (about 3.3 to 33.6 times the human dose of 40 mg/day on a body surface area basis) produced dose-related increases in embryo-lethality, fetal resorptions and pregnancy disruptions. In rats, dose-related embryo/fetal toxicity and postnatal developmental toxicity were observed in offspring resulting from parents treated with omeprazole at 13.8 to 138.0 mg/kg/day (about 3.3 to 33.6 times the human dose of 40 mg/day on a body surface area basis).
Juvenile Animal Study
A 28-day toxicity study with a 14-day recovery phase was conducted in juvenile rats with esomeprazole magnesium at doses of 70 to 280 mg/kg/day (about 17 to 68 times a daily oral human dose of 40 mg on a body surface area basis). An increase in the number of deaths at the high dose of 280 mg/kg/day was observed when juvenile rats were administered esomeprazole magnesium from postnatal Day 7 through postnatal Day 35. In addition, doses equal to or greater than 140 mg/kg/day (about 34 times a daily oral human dose of 40 mg on a body surface area basis), produced treatment-related decreases in body weight (approximately 14%) and body weight gain, decreases in femur weight and femur length, and affected overall growth. Comparable findings described above have also been observed in this study with another esomeprazole salt, esomeprazole strontium, at equimolar doses of esomeprazole.
-
14 CLINICAL STUDIES
14.1 Duodenal Ulcer Disease
Active Duodenal Ulcer – In a multicenter, double-blind, placebo-controlled study of 147 patients with endoscopically documented duodenal ulcer, the percentage of patients healed (per protocol) at 2 and 4 weeks was significantly higher with omeprazole 20 mg once a day than with placebo (p ≤ 0.01). (See Table 6.)
Table 6: Treatment of Active Duodenal Ulcer % of Patients Healed - * (p ≤ 0.01)
Omeprazole
20 mg a.m.
(n = 99)
Placebo
a.m.
(n = 48)
Week 2
41*
13
Week 4
75*
27
Complete daytime and nighttime pain relief occurred significantly faster (p ≤ 0.01) in patients treated with omeprazole 20 mg than in patients treated with placebo. At the end of the study, significantly more patients who had received omeprazole had complete relief of daytime pain (p ≤ 0.05) and nighttime pain (p ≤ 0.01).
In a multicenter, double-blind study of 293 patients with endoscopically documented duodenal ulcer, the percentage of patients healed (per protocol) at 4 weeks was significantly higher with omeprazole 20 mg once a day than with ranitidine 150 mg twice daily (p < 0.01). (See Table 7.)
Table 7: Treatment of Active Duodenal Ulcer % of Patients Healed - * (p < 0.01)
Omeprazole
20 mg a.m.
(n = 145)
Ranitidine
150 mg twice daily
(n = 148)
Week 2
42
34
Week 4
82*
63
Healing occurred significantly faster in patients treated with omeprazole than in those treated with ranitidine 150 mg twice daily (p < 0.01).
In a foreign multinational randomized, double-blind study of 105 patients with endoscopically documented duodenal ulcer, 40 mg and 20 mg of omeprazole were compared to 150 mg twice daily of ranitidine at 2, 4 and 8 weeks. At 2 and 4 weeks both doses of omeprazole were statistically superior (per protocol) to ranitidine, but 40 mg was not superior to 20 mg of omeprazole, and at 8 weeks there was no significant difference between any of the active drugs. (See Table 8.)
14.2 Gastric Ulcer
In a U.S. multicenter, double-blind study of omeprazole 40 mg once a day, 20 mg once a day, and placebo in 520 patients with endoscopically diagnosed gastric ulcer, the following results were obtained. (See Table 9.)
Table 9: Treatment of Gastric Ulcer % of Patients Healed (All Patients Treated) - * (p < 0.01) omeprazole 40 mg or 20 mg versus placebo
- † (p < 0.05) omeprazole 40 mg versus 20 mg
Omeprazole
40 mg once daily
(n = 214)
Omeprazole
20 mg once daily
(n = 202)
Placebo
(n = 104)
Week 4
55.6*
47.5*
30.8
Week 8
74.8*
48.1
For the stratified groups of patients with ulcer size less than or equal to 1 cm, no difference in healing rates between 40 mg and 20 mg was detected at either 4 or 8 weeks. For patients with ulcer size greater than 1 cm, 40 mg was significantly more effective than 20 mg at 8 weeks.
In a foreign, multinational, double-blind study of 602 patients with endoscopically diagnosed gastric ulcer, omeprazole 40 mg once a day, 20 mg once a day, and ranitidine 150 mg twice a day were evaluated. (See Table 10.)
Table 10: Treatment of Gastric Ulcer % of Patients Healed (All Patients Treated) - * (p < 0.01) omeprazole 40 mg versus ranitidine
- † (p < 0.01) omeprazole 40 mg versus 20 mg
Omeprazole
40 mg once daily
(n = 187)
Omeprazole
20 mg once daily
(n = 200)
Ranitidine
150 mg twice daily
(n = 199)
Week 4
63.5
56.3
Week 8
81.5
78.4
14.3 Gastroesophageal Reflux Disease (GERD)
Symptomatic GERD – A placebo-controlled study was conducted in Scandinavia to compare the efficacy of omeprazole 20 mg or 10 mg once daily for up to 4 weeks in the treatment of heartburn and other symptoms in GERD patients without erosive esophagitis. Results are shown in Table 11.
Table 11: % Successful Symptomatic Outcome* - * Defined as complete resolution of heartburn
- † (p < 0.005) versus 10 mg
- ‡ (p < 0.005) versus placebo
Omeprazole
20 mg a.m.
Omeprazole
10 mg a.m.
Placebo
a.m.
All Patients
(n = 205)
31‡
(n = 199)
13
(n = 105)
Patients with Confirmed GERD
(n = 115)
36‡
(n = 109)
14
(n = 59)
Erosive Esophagitis - In a U.S. multicenter, double-blind, placebo-controlled study of 40 mg or 20 mg of omeprazole delayed-release capsules in patients with symptoms of GERD and endoscopically diagnosed erosive esophagitis of grade 2 or above, the percentage healing rates (per protocol) were as shown in Table 12.
Table 12: % Patients Healed - * (p < 0.01) omeprazole versus placebo.
Omeprazole
40 mg
(n = 87)
Omeprazole
20 mg
(n = 83)
Placebo
(n = 43)
Week 4
45*
39*
7
Week 8
75*
74*
14
In this study, the 40 mg dose was not superior to the 20 mg dose of omeprazole in the percentage healing rate. Other controlled clinical trials have also shown that omeprazole is effective in severe GERD. In comparisons with histamine H2-receptor antagonists in patients with erosive esophagitis, grade 2 or above, omeprazole in a dose of 20 mg was significantly more effective than the active controls. Complete daytime and nighttime heartburn relief occurred significantly faster (p < 0.01) in patients treated with omeprazole than in those taking placebo or histamine H2-receptor antagonists.
In this and five other controlled GERD studies, significantly more patients taking 20 mg omeprazole (84%) reported complete relief of GERD symptoms than patients receiving placebo (12%).
14.4 Long-Term Maintenance Treatment of Erosive Esophagitis
In a U.S. double-blind, randomized, multicenter, placebo-controlled study; two-dose regimens of omeprazole were studied in patients with endoscopically confirmed healed esophagitis. Results to determine maintenance of healing of erosive esophagitis are shown in Table 13.
Table 13: Life Table Analysis - * (p < 0.01) omeprazole 20 mg once daily versus omeprazole 20 mg 3 consecutive days per week or placebo.
Omeprazole
20 mg once daily
(n = 138)
Omeprazole
20 mg 3 days per week
(n = 137)
Placebo
(n = 131)
Percent in Endoscopic Remission at 6 Months
70*
34
11
In an international, multicenter, double-blind study, omeprazole 20 mg daily and 10 mg daily were compared to ranitidine 150 mg twice daily in patients with endoscopically confirmed healed esophagitis. Table 14 provides the results of this study for maintenance of healing of erosive esophagitis.
Table 14: Life Table Analysis - * (p = 0.01) omeprazole 20 mg once daily versus omeprazole 10 mg once daily or Ranitidine.
- † (p = 0.03) omeprazole 10 mg once daily versus Ranitidine.
Omeprazole
20 mg once daily
(n = 131)
Omeprazole
10 mg once daily
(n = 133)
Ranitidine
150 mg twice daily
(n = 128)
Percent in Endoscopic Remission at 12 Months
77*
58†
46
In patients who initially had grades 3 or 4 erosive esophagitis, for maintenance after healing 20 mg daily of omeprazole was effective, while 10 mg did not demonstrate effectiveness.
14.5 Reduction of Risk of Upper Gastrointestinal Bleeding in Critically Ill Patients
A double-blind, multicenter, randomized, non-inferiority clinical trial was conducted to compare Omeprazole and Sodium Bicarbonate Oral Suspension 40 mg/1,680 mg and I.V. cimetidine for the reduction of risk of upper gastrointestinal (GI) bleeding in critically ill patients (mean APACHE II score = 23.7). The primary endpoint was significant upper GI bleeding defined as bright red blood which did not clear after adjustment of the nasogastric tube and a 5 to 10 minute lavage, or persistent Gastroccult positive coffee grounds for 8 consecutive hours which did not clear with 100 cc lavage. Omeprazole and Sodium Bicarbonate Oral Suspension 40 mg/1,680 mg (two doses administered 6 to 8 hours apart on the first day via orogastric or nasogastric tube, followed by 40 mg once daily thereafter) was compared to continuous I.V. cimetidine (300 mg bolus, and 50 to 100 mg/hr continuously thereafter) for up to 14 days (mean = 6.8 days). A total of 359 patients were studied, age range 16 to 91 (mean = 56 yrs), 58.5% were males, and 64% were Caucasians. The results of the study showed that Omeprazole and Sodium Bicarbonate was non-inferior to I.V. cimetidine, 10/181 (5.5%) patients in the cimetidine group vs. 7/178 (3.9%) patients in the Omeprazole and Sodium Bicarbonate group experienced clinically significant upper GI bleeding.
-
15 REFERENCES
- 1. Friedman JM and Polifka JE. Omeprazole. In: Teratogenic Effects of Drugs: A Resource for Clinicians (TERIS). 2nd ed. Baltimore, MD: The Johns Hopkins University Press 2000; p. 516.
- 2. Källén BA. Use of omeprazole during pregnancy – no hazard demonstrated in 955 infants exposed during pregnancy. Eur J Obstet Gynecol Reprod Biol 2001 May;96(1):63-8.
- 3. Ruigómez A, García Rodríquez LA, Cattaruzzi C, et al. Use of cimetidine, omeprazole, and ranitidine in pregnant women and pregnancy outcomes. Am J Epidemiol. 1999 Sep 1;150:476-81.
- 4. Lalkin A, Loebstein R, Addis A, et al. The safety of omeprazole during pregnancy: a multicenter prospective controlled study. Am J Obstet Gynecol. 1998 Sep 1:179 (3 Pt 1):727-30.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Omeprazole and Sodium Bicarbonate 20 mg Capsules: Each opaque, hard gelatin, white capsule, imprinted with the Santarus logo and “20”, contains 20 mg omeprazole and 1,100 mg sodium bicarbonate.
- NDC: 68682-102-30 Bottles of 30 capsules
Omeprazole and Sodium Bicarbonate 40 mg Capsules: Each opaque, hard gelatin, colored dark blue and white capsule, imprinted with the Santarus logo and “40”, contains 40 mg omeprazole and 1,100 mg sodium bicarbonate.
- NDC: 68682-104-30 Bottles of 30 capsules
Omeprazole and Sodium Bicarbonate Powder for Oral Suspension is a white, flavored powder packaged in unit-dose packets. Each packet contains either 20 mg or 40 mg omeprazole and 1,680 mg sodium bicarbonate.
- NDC: 68682-990-30 Cartons of 30: 20 mg unit-dose packets
- NDC: 68682-991-30 Cartons of 30: 40 mg unit-dose packets
Storage
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
Keep container tightly closed. Protect from light and moisture.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Adverse Reactions
Advise patients to report to their healthcare provider if they experience any signs or symptoms consistent with:- Hypersensitivity Reactions [see Contraindications (4)]
- Acute Interstitial Nephritis [see Warnings and Precautions (5.2)]
- Clostridium difficile-Associated Diarrhea [see Warnings and Precautions (5.4)]
- Bone Fracture [see Warnings and Precautions (5.5)]
- Cutaneous and Systemic Lupus Erythematosus [see Warnings and Precautions (5.6)]
- Cyanocobalamin (Vitamin B-12) Deficiency [see Warnings and Precautions (5.8)]
- Hypomagnesemia [see Warnings and Precautions (5.9)]
Sodium Content
Patients on a sodium-restricted diet or patients at risk of developing congestive heart failure (CHF) should be informed of the sodium content of Omeprazole and Sodium Bicarbonate Capsules (304 mg per capsule) and Omeprazole and Sodium Bicarbonate Powder (460 mg per packet). Patients should be informed that chronic use of sodium bicarbonate may cause problems and increased sodium intake can cause swelling and weight gain. If this occurs, they should contact their healthcare provider. [See Warnings and Precautions (5.3).]Drug Interactions
Advise patients to report to their healthcare provider if they start treatment with clopidogrel, St. John’s wort or rifampin, or if they take high-dose methotrexate. [See Warnings and Precautions (5.7, 5.10, 5.12).]Administration
Instruct patients that Omeprazole and Sodium Bicarbonate should be taken on an empty stomach at least one hour prior to a meal. [See Dosage and Administration (2).]Instruct patients in Directions for Use as follows:
Capsules: Swallow intact capsule with water. DO NOT USE OTHER LIQUIDS. DO NOT OPEN CAPSULE AND SPRINKLE CONTENTS INTO FOOD.
Powder for Oral Suspension: Empty packet contents into a small cup containing 1-2 tablespoons of water. DO NOT USE OTHER LIQUIDS OR FOODS. Stir well and drink immediately. Refill cup with water and drink.
Omeprazole and Sodium Bicarbonate is available either as 40 mg or 20 mg capsules with 1,100 mg sodium bicarbonate. Omeprazole and Sodium Bicarbonate is also available either as 40 mg or 20 mg single-dose packets of powder for oral suspension with 1,680 mg sodium bicarbonate.
Patients should be instructed not to substitute Omeprazole and Sodium Bicarbonate Capsules or Suspension for other Omeprazole and Sodium Bicarbonate dosage forms because different dosage forms contain different amounts of sodium bicarbonate and magnesium hydroxide. [See Dosage and Administration (2)]
Patients should be advised that since both the 20 mg and 40 mg oral suspension packets contain the same amount of sodium bicarbonate (1,680 mg), two packets of 20 mg are not equivalent to one packet of Omeprazole and Sodium Bicarbonate 40 mg; therefore, two 20 mg packets of Omeprazole and Sodium Bicarbonate should not be substituted for one packet of Omeprazole and Sodium Bicarbonate 40 mg. Conversely ½ of a 40 mg packet should not be substituted for one 20 mg packet. [See Dosage and Administration (2).]
Patients should be advised that since both the 20 mg and 40 mg capsules contain the same amount of sodium bicarbonate (1,100 mg), two capsules of 20 mg are not equivalent to one capsule of Omeprazole and Sodium Bicarbonate 40 mg; therefore, two 20 mg capsules of Omeprazole and Sodium Bicarbonate should not be substituted for one capsule of Omeprazole and Sodium Bicarbonate 40 mg. [See Dosage and Administration (2).]
Distributed by:
Oceanside Pharmaceuticals, a division of
Valeant Pharmaceuticals North America LLC
Bridgewater, NJ 08807 USAAll other product/brand names and/or logos are trademarks of the respective owners.
© Valeant Pharmaceuticals North America LLC
9513002 2000010802
9654400 70013886
-
Medication Guide
Omeprazole and Sodium Bicarbonate
Powder for Oral Suspension and Capsules
Read this Medication Guide before you start taking Omeprazole and Sodium Bicarbonate and each time you get a refill. There may be new information. This information does not take the place of talking to your doctor about your medical condition or treatment.
What is the most important information I should know about Omeprazole and Sodium Bicarbonate?
Omeprazole and Sodium Bicarbonate may help with your acid-related symptoms, but you could still have serious stomach problems. Talk with your doctor.
Omeprazole and Sodium Bicarbonate can cause serious side effects, including:
- A type of kidney problem (acute interstitial nephritis). Some people who take proton pump inhibitor (PPI) medicines, including Omeprazole and Sodium Bicarbonate, may develop a kidney problem called acute interstitial nephritis that can happen at any time during treatment with Omeprazole and Sodium Bicarbonate. Call your doctor if you have a decrease in the amount that you urinate or if you have blood in your urine.
- Omeprazole and Sodium Bicarbonate contains sodium bicarbonate. Tell your doctor if you are on a sodium restricted diet or if you have Bartter’s Syndrome (a rare kidney disorder).
- Tell your doctor right away if you have confusion, shaking hands, dizziness, muscle twitching, nausea, vomiting, and numbness or tingling in the face, arms, or legs.
- Diarrhea. Omeprazole and Sodium Bicarbonate may increase your risk of getting severe diarrhea. This diarrhea may be caused by an infection (Clostridium difficile) in your intestines.
- Call your doctor right away if you have watery stool, stomach pain, and fever that do not go away.
- Bone fractures. People who take multiple daily doses of PPI medicines for a long period of time (a year or longer) may have
- an increased risk of fractures of the hip, wrist or spine. You should take Omeprazole and Sodium Bicarbonate exactly as prescribed, at the lowest dose possible for your treatment and for the shortest time needed. Talk to your doctor about your risk of bone fracture if you take Omeprazole and Sodium Bicarbonate.
- Certain types of lupus erythematosus. Lupus erythematosus is an autoimmune disorder (the body’s immune cells attack other cells or organs in the body). Some people who take PPI medicines, including Omeprazole and Sodium Bicarbonate, may develop certain types of lupus erythematosus or have worsening of the lupus they already have. Call your doctor right away if you have new or worsening joint pain or a rash on your cheeks or arms that gets worse in the sun.
Omeprazole and Sodium Bicarbonate can have other serious side effects. See “What are the possible side effects of Omeprazole and Sodium Bicarbonate?”
What is Omeprazole and Sodium Bicarbonate?
Omeprazole and Sodium Bicarbonate is a prescription medicine called a proton pump inhibitor (PPI). Omeprazole and Sodium Bicarbonate reduces the amount of acid in your stomach.
Omeprazole and Sodium Bicarbonate is used in adults:
- for 4 weeks to heal ulcers in the first part of the small bowel (duodenal ulcers). Your doctor may prescribe another 4 weeks of Omeprazole and Sodium Bicarbonate.
- for up to 8 weeks for healing stomach ulcers.
- for up to 4 weeks to treat heartburn and other symptoms that happen with gastroesophageal reflux disease (GERD).
- GERD happens when acid from the stomach backs up into the tube (esophagus) that connects your mouth to your stomach. This may cause a burning feeling in your chest or throat, sour taste, or burping.
- for up to 8 weeks to heal acid-related damage to the lining of the esophagus (called erosive esophagitis or EE).
- to maintain healing of the esophagus. It is not known if Omeprazole and Sodium Bicarbonate is safe and effective if used longer than 12 months (1 year).
- to lower the risk of stomach bleeding in critically ill people (40 mg Oral Suspension only).
It is not known if Omeprazole and Sodium Bicarbonate is safe and effective in children less than 18 years of age.
Who should not take Omeprazole and Sodium Bicarbonate?
Do not take Omeprazole and Sodium Bicarbonate if you:
- are allergic to omeprazole or any of the ingredients in Omeprazole and Sodium Bicarbonate. See the end of this Medication Guide for a complete list of ingredients in Omeprazole and Sodium Bicarbonate.
- are allergic to any other proton pump inhibitor (PPI) medicine.
What should I tell my doctor before I take Omeprazole and Sodium Bicarbonate?
Before you take Omeprazole and Sodium Bicarbonate, tell your doctor if you:
- have been told that you have low magnesium, calcium, or potassium levels in your blood
- have liver problems
- have heart failure
- have Bartter’s syndrome (a rare kidney disorder)
- have any allergies
- have any other medical conditions
- are pregnant or plan to become pregnant. It is not known if Omeprazole and Sodium Bicarbonate can harm your unborn baby.
- are breastfeeding or plan to breastfeed. Omeprazole and Sodium Bicarbonate can pass into your breast milk and may harm your baby. You and your doctor should decide if you will take Omeprazole and Sodium Bicarbonate or breastfeed. You should not do both. Talk with your doctor about the best way to feed your baby if you take Omeprazole and Sodium Bicarbonate.
Tell your doctor about all the medicines you take, including prescription and non-prescription drugs, anti-cancer drugs, vitamins and herbal supplements. Omeprazole and Sodium Bicarbonate may affect how other medicines work, and other medicines may affect how Omeprazole and Sodium Bicarbonate works.
Especially tell your doctor if you take:
- mycophenolate mofetil (Cellcept)
- diazepam (Valium)
- warfarin (Coumadin, Jantoven)
- phenytoin (Dilantin)
- cyclosporine (Gengraf, Neoral, Sandimmune)
- disulfiram (Antabuse)
- a benzodiazepine medicine
- ketoconazole (Nizoral)
- an antibiotic that contains ampicillin
- products that contain iron
- digoxin (Lanoxin)
- voriconazole (Vfend)
- atazanavir (Reyataz)
- nelfinavir (Viracept)
- tacrolimus (Prograf)
- saquinavir (Fortovase)
- clarithromycin (Biaxin, Biaxin XL)
- clopidogrel (Plavix)
- St. John’s wort (Hypericum perforatum)
- rifampin (Rifater, Rifamate, Rimactane, Rifadin)
- methotrexate
Ask your doctor or pharmacist for a list of these medicines, if you are not sure.
Know the medicines that you take. Keep a list of them to show your doctor and pharmacist when you get a new medicine.
How should I take Omeprazole and Sodium Bicarbonate?
- Take Omeprazole and Sodium Bicarbonate exactly as prescribed by your doctor.
- Do not change your dose or stop taking Omeprazole and Sodium Bicarbonate without talking to your doctor. Take Omeprazole and Sodium Bicarbonate on an empty stomach at least one hour before a meal.
- Empty the contents of a packet of Omeprazole and Sodium Bicarbonate Powder for Oral Suspension into a small cup containing 1 to 2 tablespoons of water. Do not use other liquids or foods. Stir well and drink immediately. Refill cup with water and drink.
- Swallow Omeprazole and Sodium Bicarbonate Capsules whole with water. Do not use other liquids. Do not crush or chew the capsule. Do not open the capsule and sprinkle contents into food.
- If you miss a dose of Omeprazole and Sodium Bicarbonate, take it as soon as you remember. If it is almost time for your next dose, do not take the missed dose. Take the next dose at your regular time. Do not take two doses to make up for a missed dose.
- Do not substitute two 20 mg packets for one 40 mg packet of Omeprazole and Sodium Bicarbonate Powder for Oral Suspension because you will receive twice the amount of sodium bicarbonate. Talk to your doctor if you have questions.
- Do not substitute two 20 mg capsules for one 40 mg capsule of Omeprazole and Sodium Bicarbonate because you will receive twice the amount of sodium bicarbonate. Talk to your doctor if you have questions.
- If you take too much Omeprazole and Sodium Bicarbonate, call your doctor or Poison Control Center right away, or go to the nearest hospital emergency room.
- Your doctor may prescribe antibiotic medicines with Omeprazole and Sodium Bicarbonate to help treat a stomach infection and heal stomach-area (duodenal) ulcers that are caused by bacteria called H. pylori. Make sure you read the patient information that comes with an antibiotic before you start taking it.
- See the ”Instructions for Use” at the end of this Medication Guide for instructions on how to mix and give Omeprazole and Sodium Bicarbonate Powder for Oral Suspension through a nasogastric tube or orogastric tube.
What are the possible side effects of Omeprazole and Sodium Bicarbonate?
Omeprazole and Sodium Bicarbonate may cause serious side effects, including:
- See “What is the most important information I should know about Omeprazole and Sodium Bicarbonate?”
- Vitamin B-12 deficiency. Omeprazole and Sodium Bicarbonate reduces the amount of acid in your stomach. Stomach acid is needed to absorb vitamin B-12 properly. Talk with your doctor about the possibility of vitamin B-12 deficiency if you have been on Omeprazole and Sodium Bicarbonate for a long time (more than 3 years).
-
Low magnesium levels in your body. This problem can be serious. Low magnesium can happen in some people who take a PPI medicine for at least 3 months. If low magnesium levels happen, it is usually after a year of treatment. You may or may not have symptoms of low magnesium.
Tell your doctor right away if you develop any of these symptoms:- seizures
- dizziness
- abnormal or fast heartbeat
- jitteriness
- jerking movements or shaking (tremors)
- muscle weakness
- spasms of the hands and feet
- cramps or muscle aches
- spasm of the voice box
Your doctor may check the level of magnesium in your body before you start taking Omeprazole and Sodium Bicarbonate, or during treatment, if you will be taking Omeprazole and Sodium Bicarbonate for a long period of time.
- Stomach growths (fundic gland polyps). People who take PPI medicines for a long time have an increased risk of developing a certain type of stomach growths called fundic gland polyps, especially after taking PPI medicines for more than 1 year.
The most common side effects with Omeprazole and Sodium Bicarbonate include:
- headache
- abdominal pain
- nausea
- diarrhea
- vomiting
- gas
- Other side effects:
-
Serious allergic reactions. Tell your doctor if you get any of the following symptoms with Omeprazole and Sodium Bicarbonate.
- rash
- face swelling
- throat tightness
- difficulty breathing
Your doctor may stop Omeprazole and Sodium Bicarbonate if these symptoms happen.
Using Omeprazole and Sodium Bicarbonate for a long time may cause problems such as swelling and weight gain. Tell your doctor if this happens.
If you are on a low-sodium diet or at risk of developing congestive heart failure (CHF), you and your doctor should decide if you will take Omeprazole and Sodium Bicarbonate.
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of Omeprazole and Sodium Bicarbonate. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Omeprazole and Sodium Bicarbonate?
- Store Omeprazole and Sodium Bicarbonate at room temperature between 59° to 86°F (15° to 30°C).
- Keep Omeprazole and Sodium Bicarbonate Capsules in a tightly closed container.
- Keep Omeprazole and Sodium Bicarbonate in a dry place and out of light.
Keep Omeprazole and Sodium Bicarbonate and all medicines out of reach of children.
General information about Omeprazole and Sodium Bicarbonate
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use Omeprazole and Sodium Bicarbonate for any condition for which it was not prescribed by your doctor. Do not give Omeprazole and Sodium Bicarbonate to other people, even if they have the same symptoms as you. It may harm them.
This Medication Guide summarizes the most important information about Omeprazole and Sodium Bicarbonate. If you would like more information, talk to your doctor. You can also ask your doctor or pharmacist for information about Omeprazole and Sodium Bicarbonate that is written for healthcare professionals.
What are the ingredients in Omeprazole and Sodium Bicarbonate?
Active ingredients: Omeprazole and Sodium Bicarbonate
Inactive ingredients of Omeprazole and Sodium Bicarbonate Powder for Oral Suspension: xylitol, sucrose, sucralose, xanthan gum, and flavorings.
Inactive ingredients of Omeprazole and Sodium Bicarbonate Capsules: croscarmellose sodium and sodium stearyl fumarate.
Instructions for Use
For instructions on taking Omeprazole and Sodium Bicarbonate Capsules and Omeprazole and Sodium Bicarbonate Powder for Oral Suspension by mouth, see “How should I take Omeprazole and Sodium Bicarbonate?”
Giving Omeprazole and Sodium Bicarbonate Powder for Oral Suspension through a nasogastric tube (NG tube) or orogastric tube:
- Add 20 mL of water to a catheter tipped syringe and then add the contents of a packet as prescribed by your doctor. Use only a catheter tipped syringe to give Omeprazole and Sodium Bicarbonate through an NG tube or orogastric tube.
- Shake the syringe to dissolve the powder.
- Give the medicine through the NG or orogastric tube into the stomach right away.
- Refill the syringe with an equal amount of water.
- Shake and flush any remaining contents from the NG tube or orogastric tube into the stomach.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Manufactured for:
Oceanside Pharmaceuticals, a division of
Valeant Pharmaceuticals North America LLC
Bridgewater, NJ 08807 USA
© Valeant Pharmaceuticals North America LLC
Rev. 06/2018
9513002 2000010802
9654400 70013886
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL- 20 mg carton
NDC: 68682-990-30
Rx only
Contains 30 single
20-mg dose patchesOmeprazole and
Sodium Bicarbonate
Powder for
Oral Suspension
20 mg/1680 mg
PHARMACIST: Dispense the enclosed
Medication Guide to each patient.For more information,
call 1-800-321-4576.OCEANSIDE
PHARMACEUTICALS
-
Package/Label Display Panel- 40 mg carton
NDC: 68682-991-30
Rx only
Contains 30 single
40-mg dose packetsOmeprazole and
Sodium Bicarbonate
Powder for
Oral Suspension
40 mg/1680 mg
PHARMACIST: Dispense the enclosed
Medication Guide to each patient.For more information,
call 1-800-321-4576.OCEANSIDE
PHARMACEUTICALS
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL- 40mg Bottle Label
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL-20mg Bottle Label
-
INGREDIENTS AND APPEARANCE
OMEPRAZOLE AND SODIUM BICARBONATE
omeprazole, sodium bicarbonate powder, for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68682-990 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OMEPRAZOLE (UNII: KG60484QX9) (OMEPRAZOLE - UNII:KG60484QX9) OMEPRAZOLE 20 mg SODIUM BICARBONATE (UNII: 8MDF5V39QO) (BICARBONATE ION - UNII:HN1ZRA3Q20) SODIUM BICARBONATE 1680 mg Inactive Ingredients Ingredient Name Strength XYLITOL (UNII: VCQ006KQ1E) SUCROSE (UNII: C151H8M554) SUCRALOSE (UNII: 96K6UQ3ZD4) XANTHAN GUM (UNII: TTV12P4NEE) Product Characteristics Color Score Shape Size Flavor PEACH, PEPPERMINT Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68682-990-30 30 in 1 CARTON 06/15/2004 1 1 in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA authorized generic NDA021636 06/15/2004 OMEPRAZOLE AND SODIUM BICARBONATE
omeprazole, sodium bicarbonate powder, for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68682-991 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OMEPRAZOLE (UNII: KG60484QX9) (OMEPRAZOLE - UNII:KG60484QX9) OMEPRAZOLE 40 mg SODIUM BICARBONATE (UNII: 8MDF5V39QO) (BICARBONATE ION - UNII:HN1ZRA3Q20) SODIUM BICARBONATE 1680 mg Inactive Ingredients Ingredient Name Strength XYLITOL (UNII: VCQ006KQ1E) SUCROSE (UNII: C151H8M554) SUCRALOSE (UNII: 96K6UQ3ZD4) XANTHAN GUM (UNII: TTV12P4NEE) Product Characteristics Color Score Shape Size Flavor PEACH, PEPPERMINT Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68682-991-30 30 in 1 CARTON 06/15/2004 1 1 in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA authorized generic NDA021636 06/15/2004 OMEPRAZOLE AND SODIUM BICARBONATE
omeprazole, sodium bicarbonate capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68682-102 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OMEPRAZOLE (UNII: KG60484QX9) (OMEPRAZOLE - UNII:KG60484QX9) OMEPRAZOLE 20 mg SODIUM BICARBONATE (UNII: 8MDF5V39QO) (BICARBONATE ION - UNII:HN1ZRA3Q20) SODIUM BICARBONATE 1100 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) Product Characteristics Color WHITE Score no score Shape CAPSULE Size 23mm Flavor Imprint Code 20 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68682-102-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 02/27/2006 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA authorized generic NDA021849 02/27/2006 OMEPRAZOLE AND SODIUM BICARBONATE
omeprazole, sodium bicarbonate capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68682-104 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OMEPRAZOLE (UNII: KG60484QX9) (OMEPRAZOLE - UNII:KG60484QX9) OMEPRAZOLE 40 mg SODIUM BICARBONATE (UNII: 8MDF5V39QO) (BICARBONATE ION - UNII:HN1ZRA3Q20) SODIUM BICARBONATE 1100 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) Product Characteristics Color BLUE Score no score Shape CAPSULE Size 23mm Flavor Imprint Code 40 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68682-104-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 02/27/2007 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA authorized generic NDA021849 02/27/2006 Labeler - Oceanside Pharmaceuticals (832011691) Establishment Name Address ID/FEI Business Operations Patheon Inc. 205475333 MANUFACTURE(68682-104, 68682-102, 68682-990, 68682-991) Establishment Name Address ID/FEI Business Operations Carton Service Incorporated 928861723 PACK(68682-990, 68682-991, 68682-102, 68682-104)
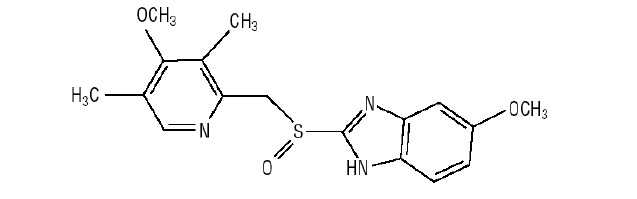


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.