ROPINIROLE HYDROCHLORIDE tablet, film coated
ropinirole hydrochloride by
Drug Labeling and Warnings
ropinirole hydrochloride by is a Prescription medication manufactured, distributed, or labeled by Zydus Pharmaceuticals (USA) Inc., Cadila Healthcare Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ROPINIROLE HYDROCHLORIDE TABLETS safely and effectively. See full prescribing information for ROPINIROLE HYDROCHLORIDE TABLETS.
ROPINIROLE HYDROCHLORIDE tablets, for oral use
Initial U.S. Approval: 1997
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
Ropinirole hydrochloride Tablets can be taken with or without food (2.1)
- Retitration of ropinirole may be warranted if therapy is interrupted (2.1)
Parkinson's Disease:
- The recommended starting dose is 0.25 mg taken three times daily; titrate to a maximum daily dose of 24 mg (2.2)
- Renal Impairment: The maximum recommended dose is 18 mg/day in patients with end-stage renal disease on hemodialysis (2.2)
Restless Legs Syndrome:
DOSAGE FORMS AND STRENGTHS
Tablets: 0.25 mg, 0.5 mg, 1 mg, 2 mg, 3 mg, 4 mg, and 5 mg (3)
CONTRAINDICATIONS
History of hypersensitivity/allergic reaction (including urticaria, angioedema, rash, pruritus) to ropinirole or to any of the excipients (4)
WARNINGS AND PRECAUTIONS
- Sudden onset of sleep and somnolence may occur (5.1)
- Syncope may occur (5.2)
- Hypotension, including orthostatic hypotension may occur (5.3)
- May cause hallucinations and psychotic-like behaviors (5.4)
- May cause or exacerbate dyskinesia (5.5)
- May cause problems with impulse control or compulsive behaviors (5.6)
ADVERSE REACTIONS
Most common adverse reactions (incidence with ropinirole at least 5% greater than placebo) in the respective indications were:
- Early PD: Nausea, somnolence, dizziness, syncope, asthenic condition, viral infection, leg edema, vomiting, and dyspepsia (6.1)
- Advanced PD: Dyskinesia, somnolence, nausea, dizziness, confusion, hallucinations, sweating, and headache (6.1)
- RLS: Nausea, vomiting, somnolence, dizziness, and asthenic condition (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Zydus Pharmaceuticals (USA) Inc. at 1-877-993-8779 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Inhibitors or inducers of CYP1A2: May alter the clearance of ropinirole; dose adjustment of ropinirole may be required (7.1,12.3)
- Hormone replacement therapy (HRT): Starting or stopping HRT may require dose adjustment of ropinirole (7.2,12.3)
- Dopamine antagonists (e.g., neuroleptics, metoclopramide): May reduce efficacy of ropinirole (7.3)
USE IN SPECIFIC POPULATIONS
Pregnancy: Based on animal data, may cause fetal harm. (8.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 12/2017
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
RECENT MAJOR CHANGES
Highlights
1 INDICATIONS AND USAGE
1.1 Parkinson’s Disease
1.2 Restless Legs Syndrome
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Recommendations
2.2 Dosing for Parkinson’s Disease
2.3 Dosing for Restless Legs Syndrome
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Falling Asleep during Activities of Daily Living and Somnolence
5.2 Syncope
5.3 Hypotension/Orthostatic Hypotension
5.4 Hallucinations/Psychotic-like Behavior
5.5 Dyskinesia
5.6 Impulse Control/Compulsive Behaviors
5.7 Withdrawal-Emergent Hyperpyrexia and Confusion
5.8 Melanoma
5.9 Augmentation and Early-Morning Rebound in Restless Legs Syndrome
5.10 Fibrotic Complications
5.11 Retinal Pathology
5.12 Binding to Melanin
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 CYP1A2 Inhibitors and Inducers
7.2 Estrogens
7.3 Dopamine Antagonists
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Parkinson’s Disease
14.2 Restless Legs Syndrome
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Recommendations
Ropinirole can be taken with or without food [see Clinical Pharmacology (12.3)].
If a significant interruption in therapy with ropinirole has occurred, retitration of therapy may be warranted.
2.2 Dosing for Parkinson’s Disease
The recommended starting dose of ropinirole hydrochloride tablets for Parkinson's disease is 0.25 mg three times daily. Based on individual patient therapeutic response and tolerability, if necessary, the dose should then be titrated with weekly increments as described in Table 1. After Week 4, if necessary, the daily dose may be increased by 1.5 mg/day on a weekly basis up to a dose of 9 mg/day, and then by up to 3 mg/day weekly up to a maximum recommended total daily dose of 24 mg/day (8 mg three times daily). Doses greater than 24 mg/day have not been tested in clinical trials.
Table 1 Ascending-Dose Schedule of Ropinirole for Parkinson's Disease Week
Dosage
Total Daily Dose
1
0.25 mg 3 times daily
0.75 mg
2
0.5 mg 3 times daily
1.5 mg
3
0.75 mg 3 times daily
2.25 mg
4
1 mg 3 times daily
3 mg
Ropinirole hydrochloride tablets should be discontinued gradually over a 7-day period in patients with Parkinson's disease. The frequency of administration should be reduced from three times daily to twice daily for 4 days. For the remaining 3 days, the frequency should be reduced to once daily prior to complete withdrawal of ropinirole hydrochloride tablets.
Renal Impairment
No dose adjustment is necessary in patients with moderate renal impairment (creatinine clearance of 30 to 50 mL/min). The recommended initial dose of ropinirole for patients with end-stage renal disease on hemodialysis is 0.25 mg three times a day. Further dose escalations should be based on tolerability and need for efficacy. The recommended maximum total daily dose is 18 mg/day in patients receiving regular dialysis. Supplemental doses after dialysis are not required. The use of ropinirole in patients with severe renal impairment without regular dialysis has not been studied.
2.3 Dosing for Restless Legs Syndrome
The recommended adult starting dose for RLS is 0.25 mg once daily 1 to 3 hours before bedtime. After 2 days, if necessary, the dose can be increased to 0.5 mg once daily, and to 1 mg once daily at the end of the first week of dosing, then as shown in Table 2 as needed to achieve efficacy. Titration should be based on individual patient therapeutic response and tolerability, up to a maximum recommended dose of 4 mg daily. For RLS, the safety and effectiveness of doses greater than 4 mg once daily have not been established.
Table 2 Dose Titration Schedule of Ropinirole for Restless Legs Syndrome Day/Week
Dose to be taken once daily,
1 to 3 hours before bedtime
Days 1 and 2
0.25 mg
Days 3 to 7
0.5 mg
Week 2
1 mg
Week 3
1.5 mg
Week 4
2 mg
Week 5
2.5 mg
Week 6
3 mg
Week 7
4 mg
When discontinuing ropinirole in patients with RLS, gradual reduction of the daily dose is recommended [see Warnings and Precautions (5.9)].
Renal Impairment
No dose adjustment is necessary in patients with moderate renal impairment (creatinine clearance of 30 to 50 mL/min). The recommended initial dose of ropinirole for patients with end-stage renal disease on hemodialysis is 0.25 mg once daily. Further dose escalations should be based on tolerability and need for efficacy. The recommended maximum total daily dose is 3 mg/day in patients receiving regular dialysis. Supplemental doses after dialysis are not required. The use of ropinirole in patients with severe renal impairment without regular dialysis has not been studied.
-
3 DOSAGE FORMS AND STRENGTHS
0.25 mg : White-colored, round-shaped, film-coated tablets, debossed with "ZF22" on one side and plain on the other side.
0.5 mg : Yellow-colored, round-shaped, film-coated tablets, debossed with "ZF23" on one side and plain on the other side.
1 mg : Green-colored, round-shaped, film-coated tablets, debossed with "ZF24" on one side and plain on the other side.
2 mg : Pink-colored, round-shaped, film-coated tablets, debossed with "ZF25" on one side and plain on the other side.
3 mg : Purple-colored, round-shaped, film-coated tablets, debossed with "ZF42" on one side and plain on the other side.
4 mg : Brown-colored, round-shaped, film-coated tablets, debossed with "ZF43" on one side and plain on the other side.
5 mg : Blue-colored, round-shaped, film-coated tablets, debossed with "ZF26" on one side and plain on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Falling Asleep during Activities of Daily Living and Somnolence
Patients treated with ropinirole have reported falling asleep while engaged in activities of daily living, including driving or operating machinery, which sometimes resulted in accidents. Although many of these patients reported somnolence while on ropinirole, some perceived that they had no warning signs, such as excessive drowsiness, and believed that they were alert immediately prior to the event. Some have reported these events more than 1 year after initiation of treatment.
In controlled clinical trials, somnolence was commonly reported in patients receiving ropinirole and was more frequent in Parkinson's disease (up to 40% ropinirole, 6% placebo) than in Restless Legs Syndrome (12% ropinirole, 6% placebo) [see Adverse Reactions (6.1)].
It has been reported that falling asleep while engaged in activities of daily living usually occurs in a setting of preexisting somnolence, although patients may not give such a history. For this reason, prescribers should reassess patients for drowsiness or sleepiness, especially since some of the events occur well after the start of treatment. Prescribers should also be aware that patients may not acknowledge drowsiness or sleepiness until directly questioned about drowsiness or sleepiness during specific activities.
Before initiating treatment with ropinirole, patients should be advised of the potential to develop drowsiness and specifically asked about factors that may increase the risk with ropinirole such as concomitant sedating medications or alcohol, the presence of sleep disorders (other than RLS), and concomitant medications that increase ropinirole plasma levels (e.g., ciprofloxacin) [see Drug Interactions (7.1)]. If a patient develops significant daytime sleepiness or episodes of falling asleep during activities that require active participation (e.g., driving a motor vehicle, conversations, eating), ropinirole should ordinarily be discontinued [see Dosage and Administration (2.2, 2.3)]. If a decision is made to continue ropinirole, patients should be advised to not drive and to avoid other potentially dangerous activities. There is insufficient information to establish that dose reduction will eliminate episodes of falling asleep while engaged in activities of daily living.
5.2 Syncope
Syncope, sometimes associated with bradycardia, was observed in association with treatment with ropinirole in both patients with Parkinson's disease and patients with RLS. In controlled clinical trials in patients with Parkinson's disease, syncope was observed more frequently in patients receiving ropinirole than in patients receiving placebo (early Parkinson's disease without L-dopa: ropinirole 12%, placebo 1%; advanced Parkinson's disease: ropinirole 3%, placebo 2%). Syncope was reported in 1% of patients treated with ropinirole for RLS in 12-week, placebo-controlled clinical trials compared with 0.2% of patients treated with placebo [see Adverse Reactions (6.1)]. Most cases occurred more than 4 weeks after initiation of therapy with ropinirole, and were usually associated with a recent increase in dose.
Because the trials conducted with ropinirole excluded patients with significant cardiovascular disease, patients with significant cardiovascular disease should be treated with caution.
Approximately 4% of patients with Parkinson's disease enrolled in Phase 1 trials had syncope following a 1-mg dose of ropinirole. In two trials in patients with RLS that used a forced-titration regimen and orthostatic challenge with intensive blood pressure monitoring, 2% of RLS patients treated with ropinirole compared with 0% of patients receiving placebo reported syncope.
In Phase 1 trials including healthy volunteers, the incidence of syncope was 2%. Of note, 1 subject with syncope developed hypotension, bradycardia, and sinus arrest; the subject recovered spontaneously without intervention.
5.3 Hypotension/Orthostatic Hypotension
Patients with Parkinson's disease may have impaired ability to respond normally to a fall in blood pressure after standing from lying down or seated position. Patients on ropinirole should be monitored for signs and symptoms of orthostatic hypotension, especially during dose escalation, and should be informed of the risk for syncope and hypotension [see Patient Counseling Information (17)].
Although the clinical trials were not designed to systematically monitor blood pressure, there were individual reported cases of orthostatic hypotension in early Parkinson's disease (without L-dopa) in patients treated with ropinirole. Most of these cases occurred more than 4 weeks after initiation of therapy with ropinirole and were usually associated with a recent increase in dose.
In 12-week, placebo-controlled trials of patients with RLS, the adverse event orthostatic hypotension was reported by 4 of 496 patients (0.8%) treated with ropinirole compared with 2 of 500 patients (0.4%) receiving placebo.
In two Phase 2 studies in patients with RLS, 14 of 55 patients (25%) receiving ropinirole experienced an adverse event of hypotension or orthostatic hypotension compared with none of the 27 patients receiving placebo. In these studies, 11 of the 55 patients (20%) receiving ropinirole and 3 of the 26 patients (12%) who had post-dose blood pressure assessments following placebo, experienced an orthostatic blood pressure decrease of at least 40 mm Hg systolic and/or at least 20 mm Hg diastolic.
In Phase 1 trials of ropinirole with healthy volunteers who received single doses on more than one occasion without titration, 7% had documented symptomatic orthostatic hypotension. These episodes appeared mainly at doses above 0.8 mg and these doses are higher than the starting doses recommended for patients with either Parkinson's disease or with RLS. In most of these individuals, the hypotension was accompanied by bradycardia but did not develop into syncope [see Warnings and Precautions (5.2)].
Although dizziness is not a specific manifestation of hypotension or orthostatic hypotension, patients with hypotension or orthostatic hypotension frequently reported dizziness. In controlled clinical trials, dizziness was a common adverse reaction in patients receiving ropinirole and was more frequent in patients with Parkinson's disease or with RLS receiving ropinirole than in patients receiving placebo (early Parkinson's disease without L-dopa: ropinirole 40%, placebo 22%; advanced Parkinson's disease: ropinirole 26%, placebo 16%; RLS: ropinirole 11%, placebo 5%). Dizziness of sufficient severity to cause trial discontinuation of ropinirole was 4% in patients with early Parkinson's disease without L-dopa, 3% in patients with advanced Parkinson's disease, and 1% in patients with RLS. [See Adverse Reactions (6.1).]
5.4 Hallucinations/Psychotic-like Behavior
In double-blind, placebo-controlled, early-therapy trials in patients with Parkinson's disease who were not treated with L-dopa, 5.2% (8 of 157) of patients treated with ropinirole reported hallucinations, compared with 1.4% of patients on placebo (2 of 147). Among those patients receiving both ropinirole and L-dopa in advanced Parkinson's disease studies, 10.1% (21 of 208) were reported to experience hallucinations, compared with 4.2% (5 of 120) of patients treated with placebo and L-dopa.
The incidence of hallucination was increased in elderly patients (i.e., older than 65 years) treated with extended-release ropinirole [see Use in Specific Populations (8.5)].
Postmarketing reports indicate that patients may experience new or worsening mental status and behavioral changes, which may be severe, including psychotic-like behavior during treatment with ropinirole or after starting or increasing the dose of ropinirole. Other drugs prescribed to improve the symptoms of Parkinson's disease can have similar effects on thinking and behavior. This abnormal thinking and behavior can consist of one or more of a variety of manifestations including paranoid ideation, delusions, hallucinations, confusion, psychotic-like behavior, disorientation, aggressive behavior, agitation, and delirium.
Patients with a major psychotic disorder should ordinarily not be treated with ropinirole because of the risk of exacerbating the psychosis. In addition, certain medications used to treat psychosis may exacerbate the symptoms of Parkinson's disease and may decrease the effectiveness of ropinirole [see Drug Interactions (7.3)].
5.5 Dyskinesia
Ropinirole may cause exacerbate pre-existing dyskinesia in patients treated with L-dopa for Parkinson's disease. In double-blind, placebo-controlled trials in advanced Parkinson's disease, dyskinesia was much more common in patients treated with ropinirole than in those treated with placebo. Among those patients receiving both ropinirole and L-dopa in advanced Parkinson's disease trials, 34% were reported to experience dyskinesia, compared with 13% of patients treated with placebo [see Adverse Reactions (6.1)].
Decreasing the dose of dopaminergic medications may ameliorate this adverse reaction.
5.6 Impulse Control/Compulsive Behaviors
Reports suggest that patients can experience intense urges to gamble, increased sexual urges, intense urges to spend money, binge or compulsive eating, and/or other intense urges, and the inability to control these urges while taking one or more of the medications, including ropinirole, that increase central dopaminergic tone and that are generally used for the treatment of Parkinson's disease and RLS. In some cases, although not all, these urges were reported to have stopped when the dose was reduced or the medication was discontinued. Because patients may not recognize these behaviors as abnormal, it is important for prescribers to specifically ask patients or their caregivers about the development of new or increased gambling urges, sexual urges, uncontrolled spending, binge or compulsive eating, or other urges while being treated with ropinirole. Physicians should consider dose reduction or stopping the medication if a patient develops such urges while taking ropinirole.
5.7 Withdrawal-Emergent Hyperpyrexia and Confusion
A symptom complex resembling the neuroleptic malignant syndrome (characterized by elevated temperature, muscular rigidity, altered consciousness, and autonomic instability), with no other obvious etiology, has been reported in association with rapid dose reduction of, withdrawal of, or changes in dopaminergic therapy. It is recommended that the dose be tapered at the end of treatment with ropinirole as a prophylactic measure [see Dosage and Administration (2.2, 2.3)].
5.8 Melanoma
Epidemiological studies have shown that patients with Parkinson's disease have a higher risk (2- to approximately 6-fold higher) of developing melanoma than the general population. Whether the increased risk observed was due to Parkinson's disease or other factors, such as drugs used to treat Parkinson's disease, is unclear.
For the reasons stated above, patients and providers are advised to monitor for melanomas frequently and on a regular basis when using ropinirole for any indication. Ideally, periodic skin examinations should be performed by appropriately qualified individuals (e.g., dermatologists).
5.9 Augmentation and Early-Morning Rebound in Restless Legs Syndrome
Augmentation is a phenomenon in which dopaminergic medication causes a worsening of symptom severity above and beyond the level at the time the medication was started. The symptoms of augmentation may include the earlier onset of symptoms in the evening (or even the afternoon), increase in symptoms, and spread of symptoms to involve other extremities. Augmentation has been described during therapy for RLS. Rebound refers to new onset of symptoms in the early morning hours. Augmentation and/or early-morning rebound have been observed in a postmarketing trial of ropinirole. If augmentation or early-morning rebound occurs, the use of ropinirole should be reviewed and dosage adjustment or discontinuation of treatment should be considered. When discontinuing ropinirole in patients with RLS, gradual reduction of the daily dose is recommended whenever possible [see Dosage and Administration 2.3].
5.10 Fibrotic Complications
Cases of retroperitoneal fibrosis, pulmonary infiltrates, pleural effusion, pleural thickening, pericarditis, and cardiac valvulopathy have been reported in some patients treated with ergot-derived dopaminergic agents. While these complications may resolve when the drug is discontinued, complete resolution does not always occur.
Although these adverse reactions are believed to be related to the ergoline structure of these compounds, whether other, non-ergot-derived dopamine agonists such as ropinirole can cause them is unknown.
Cases of possible fibrotic complications, including pleural effusion, pleural fibrosis, interstitial lung disease, and cardiac valvulopathy have been reported in the development program and postmarketing experience for ropinirole. While the evidence is not sufficient to establish a causal relationship between ropinirole and these fibrotic complications, a contribution of ropinirole cannot be excluded.
5.11 Retinal Pathology
Retinal degeneration was observed in albino rats in the 2-year carcinogenicity study at all doses tested The lowest dose tested (1.5 mg/kg/day) is less than the maximum recommended human dose (MRHD) for Parkinson's disease (24 mg/day) on a mg/m2basis. Retinal degeneration was not observed in a 3-month study in pigmented rats, in a 2-year carcinogenicity study in albino mice, or in 1-year studies in monkeys or albino rats. The significance of this effect for humans has not been established, but involves disruption of a mechanism that is universally present in vertebrates (e.g., disk shedding).
Ocular electroretinogram (ERG) assessments were conducted during a 2-year, double-blind, multicenter, flexible dose, L-dopa–controlled clinical trial of ropinirole in patients with Parkinson's disease; 156 patients (78 on ropinirole, mean dose: 11.9 mg/day, and 78 on L-dopa, mean dose: 555.2 mg/day) were evaluated for evidence of retinal dysfunction through electroretinograms. There was no clinically meaningful difference between the treatment groups in retinal function over the duration of the trial.
-
6 ADVERSE REACTIONS
The following adverse reactions are described in more detail in other sections of the label:
- Hypersensitivity [see Contraindications (4)]
- Falling Asleep during Activities of Daily Living and Somnolence [see Warnings and Precautions (5.1)]
- Syncope [see Warnings and Precautions (5.2)]
- Hypotension/Orthostatic Hypotension [see Warnings and Precautions (5.3)]
- Hallucinations/Psychotic-like Behavior [see Warnings and Precautions (5.4)]
- Dyskinesia [see Warnings and Precautions (5.5)]
- Impulse Control/Compulsive Behaviors [see Warnings and Precautions (5.6)]
- Withdrawal-Emergent Hyperpyrexia and Confusion [see Warnings and Precautions (5.7)]
- Melanoma [see Warnings and Precautions (5.8)]
Augmentation and Early-Morning Rebound in RLS [see Warnings and Precautions (5.9)]
Fibrotic Complications [see Warnings and Precautions (5.10)]
Retinal Pathology[see Warnings and Precautions (5.11)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug (or of another development program of a different formulation of the same drug) and may not reflect the rates observed in practice.
Parkinson's Disease
During the premarketing development of ropinirole, patients received ropinirole either without L-dopa (early Parkinson's disease trials) or as concomitant therapy with L-dopa (advanced Parkinson's disease trials). Because these two populations may have differential risks for various adverse reactions, this section will in general present adverse reaction data for these two populations separately.
Early Parkinson's Disease (without L-dopa)
In the double-blind, placebo-controlled trials in patients with early-stage Parkinson's disease, the most commonly observed adverse reactions in patients treated with ropinirole (incidence at least 5% greater than placebo) were nausea, somnolence, dizziness, syncope, asthenic condition (i.e., asthenia, fatigue, and/or malaise), viral infection, leg edema, vomiting, and dyspepsia.
Approximately 24% of patients treated with ropinirole who participated in the double-blind, placebo-controlled early Parkinson's disease (without L-dopa) trials discontinued treatment due to adverse reactions compared with 13% of patients who received placebo. The most common adverse reactions in patients treated with ropinirole (incidence at least 2% greater than placebo) of sufficient severity to cause discontinuation were nausea and dizziness.
Table 3 lists treatment-emergent adverse reactions that occurred in at least 2% of patients with early Parkinson's disease (without L-dopa) treated with ropinirole participating in the double-blind, placebo-controlled trials and were numerically more common than the incidence for placebo-treated patients. In these trials, either ropinirole or placebo was used as early therapy (i.e., without L-dopa).
Table 3 Treatment-Emergent Adverse Reaction Incidence in Double-blind, Placebo-Controlled Early Parkinson's Disease (without L-dopa) Trials (Events ≥ 2% of Patients Treated with Ropinirole and Numerically More Frequent than the Placebo Group)a a Patients may have reported multiple adverse reactions during the trial or at discontinuation; thus, patients may be included in more than one category.
b Asthenic condition (i.e., asthenia, fatigue, and/or malaise).
Body System/Adverse Reaction
Ropinirole
(n = 157)
(%)
Placebo
(n = 147)
(%)
Autonomic nervous system
Flushing
3
1
Dry mouth
5
3
Increased sweating
6
4
Body as a whole
Asthenic conditionb
16
5
Chest pain
4
2
Dependent edema
6
3
Leg edema
7
1
Pain
8
4
Cardiovascular general
Hypertension
5
3
Hypotension
2
0
Orthostatic symptoms
6
5
Syncope
12
1
Central/peripheral nervous system
Dizziness
40
22
Hyperkinesia
2
1
Hypesthesia
4
2
Vertigo
2
0
Gastrointestinal
Abdominal pain
6
3
Anorexia
4
1
Dyspepsia
10
5
Flatulence
3
1
Nausea
60
22
Vomiting
12
7
Heart rate/rhythm
Extrasystoles
2
1
Atrial fibrillation
2
0
Palpitation
3
2
Tachycardia
2
0
Metabolic/nutritional
3
1
Increased alkaline phosphatase
Psychiatric
Amnesia
3
1
Impaired concentration
2
0
Confusion
5
1
Hallucination
5
1
Somnolence
40
6
Yawning
3
0
Reproductive male
Impotence
3
1
Resistance mechanism
Viral infection
11
3
Respiratory
Bronchitis
3
1
Dyspnea
3
0
Pharyngitis
6
4
Rhinitis
4
3
Sinusitis
4
3
Urinary
Urinary tract infection
5
4
Vascular extracardiac
Peripheral ischemia
3
0
Vision
Eye abnormality
3
1
Abnormal vision
6
3
Xerophthalmia
2
0
Advanced Parkinson's Disease (with L-dopa)
In the double-blind, placebo-controlled trials in patients with advanced-stage Parkinson's disease, the most commonly observed adverse reactions in patients treated with ropinirole (incidence at least 5 % greater than placebo) were dyskinesia, somnolence, nausea, dizziness, confusion, hallucinations, increased sweating, and headache.
Approximately 24% of patients who received ropinirole in the double-blind, placebo-controlled advanced Parkinson's disease (with L-dopa) trials discontinued treatment due to adverse reactions compared with 18% of patients who received placebo. The most common adverse reaction in patients treated with ropinirole (incidence at least 2% greater than placebo) of sufficient severity to cause discontinuation was dizziness.
Table 4 lists treatment-emergent adverse reactions that occurred in at least 2% of patients with advanced Parkinson's disease (with L-dopa) treated with ropinirole who participated in the double-blind, placebo-controlled trials and were numerically more common than the incidence for placebo-treated patients. In these trials, either ropinirole or placebo was used as an adjunct to L-dopa.
Table 4 Treatment-Emergent Adverse Reaction Incidence in Double-blind, Placebo-Controlled Advanced Parkinson's Disease (with L-dopa) Trials (Events ≥ 2% of Patients Treated with Ropinirole and Numerically More Frequent than the Placebo Group)a aPatients may have reported multiple adverse reactions during the trial or at discontinuation; thus, patients may be included in more than one category.
Body System/Adverse Reaction
Ropinirole
(n = 208)
(%)
Placebo
(n = 120)
(%)
Autonomic nervous system
Dry mouth
5
1
Increased sweating
7
2
Body as a whole
Increased drug level
7
3
Pain
5
3
Cardiovascular general
Hypotension
2
1
Syncope
3
2
Central/peripheral nervous system
Dizziness
26
16
Dyskinesia
34
13
Falls
10
7
Headache
17
12
Hypokinesia
5
4
Paresis
3
0
Paresthesia
5
3
Tremor
6
3
Gastrointestinal
Abdominal pain
9
8
Constipation
6
3
Diarrhea
5
3
Dysphagia
2
1
Flatulence
2
1
Nausea
30
18
Increased saliva
2
1
Vomiting
7
4
Metabolic/nutritional
Weight decrease
2
1
Musculoskeletal
Arthralgia
7
5
Arthritis
3
1
Psychiatric
Amnesia
5
1
Anxiety
6
3
Confusion
9
2
Abnormal dreaming
3
2
Hallucination
10
4
Nervousness
5
3
Somnolence
20
8
Red blood cell
Anemia
2
0
Resistance mechanism
Upper respiratory tract infection
9
8
Respiratory
Dyspnea
3
2
Urinary
Pyuria
2
1
Urinary incontinence
2
1
Urinary tract infection
6
3
Vision
Diplopia
2
1
In the double-blind, placebo-controlled trials in patients with RLS, the most commonly observed adverse reactions in patients treated with ropinirole (incidence at least 5% greater than placebo) were nausea, vomiting, somnolence, dizziness, and asthenic condition (i.e., asthenia, fatigue, and/or malaise).
Approximately 5% of patients treated with ropinirole who participated in the double-blind, placebo-controlled trials in the treatment of RLS discontinued treatment due to adverse reactions compared with 4% of patients who received placebo. The most common adverse reaction in patients treated with ropinirole (incidence at least 2% greater than placebo) of sufficient severity to cause discontinuation was nausea.
Table 5 lists treatment-emergent adverse reactions that occurred in at least 2% of patients with RLS treated with ropinirole participating in the 12-week, double-blind, placebo-controlled trials and were numerically more common than the incidence for placebo-treated patients.
Table 5 Treatment-Emergent Adverse Reaction Incidence in Double-blind, Placebo-Controlled RLS Trials (Events ≥ 2% of Patients Treated with Ropinirole and Numerically More Frequent than the Placebo Group)a a Patients may have reported multiple adverse reactions during the trial or at discontinuation; thus, patients may be included in more than one category.
b Asthenic condition (i.e., asthenia, fatigue, and/or malaise).
Body System/Adverse Reaction
Ropinirole
(n = 496)
(%)
Placebo
(n = 500)
(%)
Ear and labyrinth
Vertigo
2
1
Gastrointestinal
Nausea
40
8
Vomiting
11
2
Diarrhea
5
3
Dyspepsia
4
3
Dry mouth
3
2
Abdominal pain upper
3
1
General disorders and administration site conditions
Asthenic conditionb
9
4
Edema peripheral
2
1
Infections and infestations
Nasopharyngitis
9
8
Influenza
3
2
Musculoskeletal and connective tissue
Arthralgia
4
3
Muscle cramps
3
2
Pain in extremity
3
2
Nervous system
Somnolence
12
6
Dizziness
11
5
Paresthesia
3
1
Respiratory, thoracic, and mediastinal
Cough
3
2
Nasal congestion
2
1
Skin and subcutaneous tissue
Hyperhidrosis
3
1
-
7 DRUG INTERACTIONS
7.1 CYP1A2 Inhibitors and Inducers
In vitro metabolism studies showed that CYP1A2 is the major enzyme responsible for the metabolism of ropinirole. There is thus the potential for inducers or inhibitors of this enzyme to alter the clearance of ropinirole. Therefore, if therapy with a drug known to be a potent inducer or inhibitor of CYP1A2 is stopped or started during treatment with ropinirole, adjustment of the dose of ropinirole may be required. Coadministration of ciprofloxacin, an inhibitor of CYP1A2, increases the AUC and Cmax of ropinirole [see Clinical Pharmacology (12.3)]. Cigarette smoking is expected to increase the clearance of ropinirole since CYP1A2 is known to be induced by smoking [see Clinical Pharmacology (12.3)].
7.2 Estrogens
Population pharmacokinetic analysis revealed that higher doses of estrogens (usually associated with hormone replacement therapy [HRT]) reduced the clearance of ropinirole. Starting or stopping HRT may require adjustment of dosage of ropinirole [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
There are no adequate data on the developmental risk associated with the use of ropinirole in pregnant women. In animal studies, ropinirole had adverse effects on development when administered to pregnant rats at doses similar to (neurobehavioral impairment) or greater than (teratogenicity and embryolethality at >36 times) the maximum recommended human dose (MRHD) for Parkinson's disease. Ropinirole doses associated with teratogenicity and embryolethality in pregnant rats were associated with maternal toxicity. In pregnant rabbits, ropinirole potentiated the teratogenic effects of L-dopa when these drugs were administered in combination [see Data].
In the U.S. general population, the estimated background risk of major birth defects and of miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The background risk of major birth defects and miscarriage in the indicated populations is unknown.
Data
Animal Data:
Oral administration of ropinirole (0, 20, 60, 90, 120, or 150 mg/kg/day) to pregnant rats during organogenesis resulted in embryolethality, increased incidence of fetal malformations (digit, cardiovascular, and neural tube defects) and variations, and decreased fetal weight at the two highest doses. These doses were also associated with maternal toxicity. The highest no-effect dose for adverse effects on embryofetal development (90 mg/kg/day) is approximately 36 times the MRHD for Parkinson's disease (24 mg/day) on a body surface area (mg/m2) basis.
No effect on embryofetal development was observed in rabbits when ropinirole was administered alone during organogenesis at oral doses of 0, 1, 5, or 20 mg/kg/day (up to 16 times the MRHD on a mg/m2basis). In pregnant rabbits, there was a greater incidence and severity of fetal malformations (primarily digit defects) when ropinirole (10 mg/kg/day) was administered orally during gestation in combination with L-dopa (250 mg/kg/day) than when L-dopa was administered alone. This drug combination was also associated with maternal toxicity.
Oral administration of ropinirole (0, 0.1, 1, or 10 mg/kg/day) to rats during late gestation and continuing throughout lactation resulted in neurobehavioral impairment (decreased startle response) and decreased body weight in offspring at the highest dose. The no-effect dose of 1 mg/kg/day is less than the MRHD on a mg/m2basis.
8.2 Lactation
There are no data on the presence of ropinirole in human milk, the effects of ropinirole on the breastfed infant, or the effects of ropinirole on milk production. However, inhibition of lactation is expected because ropinirole inhibits secretion of prolactin in humans. Ropinirole or metabolites, or both, are present in rat milk.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ropinirole and any potential adverse effects on the breastfed infant from ropinirole or from the underlying maternal condition.
8.5 Geriatric Use
Dose adjustment is not necessary in elderly (65 years and older) patients, as the dose of ropinirole is individually titrated to clinical therapeutic response and tolerability. Pharmacokinetic trials conducted in patients demonstrated that oral clearance of ropinirole is reduced by 15% in patients older than 65 years compared with younger patients [see Clinical Pharmacology (12.3)].
In flexible-dose clinical trials of extended-release ropinirole for Parkinson's disease, 387 patients were 65 years and older and 107 patients were 75 years and older. Among patients receiving extended-release ropinirole, hallucination was more common in elderly patients (10%) compared with non-elderly patients (2%). In these trials, the incidence of overall adverse reactions increased with increasing age for both patients receiving extended-release ropinirole and placebo.
In the fixed-dose clinical trials of extended-release ropinirole, 176 patients were 65 years and older and 73 were 75 and older. Among patients with advanced Parkinson's disease receiving extended-release ropinirole, vomiting and nausea were more common in patients greater than 65 years (5% and 9%, respectively) compared with patients less than 65 (1% and 7%, respectively).
8.6 Renal Impairment
No dose adjustment is necessary in patients with moderate renal impairment (creatinine clearance of 30 to 50 mL/min). For patients with end-stage renal disease on hemodialysis, a reduced maximum dose is recommended [see Dosage and Administration (2.2, 2.3), Clinical Pharmacology (12.3)].
The use of ropinirole in patients with severe renal impairment (creatinine clearance less than 30 mL/min) without regular dialysis has not been studied.
-
10 OVERDOSAGE
The symptoms of overdose with ropinirole are related to its dopaminergic activity. General supportive measures are recommended. Vital signs should be maintained, if necessary.
In clinical trials, there have been patients who accidentally or intentionally took more than their prescribed dose of ropinirole. The largest overdose reported with ropinirole in clinical trials was 435 mg taken over a 7-day period (62.1 mg/day). Of patients who received a dose greater than 24 mg/day, reported symptoms included adverse events commonly reported during dopaminergic therapy (nausea, dizziness), as well as visual hallucinations, hyperhidrosis, claustrophobia, chorea, palpitations, asthenia, and nightmares. Additional symptoms reported in cases of overdose included vomiting, increased coughing, fatigue, syncope, vasovagal syncope, dyskinesia, agitation, chest pain, orthostatic hypotension, somnolence, and confusional state.
-
11 DESCRIPTION
Ropinirole hydrochloride tablets contain ropinirole, a non-ergoline dopamine agonist, as the hydrochloride salt. The chemical name of ropinirole hydrochloride is 4-[2-(dipropylamino)ethyl]-1,3-dihydro-2H-indol-2-one and the molecular formula is C16H24N2OHCl. The molecular weight is 296.84 (260.38 as the free base).
The structural formula is:
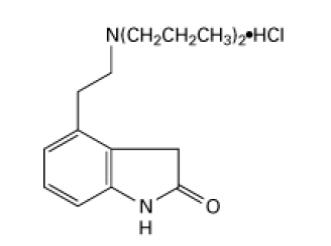
Ropinirole hydrochloride is a white to cream-colored crystalline powder with a melting range of 241° to 245°C and a solubility of 133 mg/mL in water.
Each ropinirole hydrochloride tablet intended for oral administration contains ropinirole hydrochloride equivalent to 0.25 mg or 0.5 mg or 1 mg or 2 mg or 3 mg or 4 mg or 5 mg of ropinirole. In addition, each tablet contains the following inactive ingredients: croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol and titanium dioxide. Additionally, each 0.5 mg tablet contains: FD&C blue# 2, iron oxide red and iron oxide yellow; each 1 mg tablet contains: FD&C blue#2 and iron oxide yellow; each 2 mg tablet contains: iron oxide red and iron oxide yellow; each 3 mg tablet contains carmine, FD&C blue # 2 and FD&C yellow # 6; each 4 mg tablet contains: iron oxide black, iron oxide red and iron oxide yellow and each 5 mg tablet contains: FD&C blue# 2.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ropinirole is a non-ergoline dopamine agonist. The precise mechanism of action of ropinirole as a treatment for Parkinson's disease is unknown, although it is thought to be related to its ability to stimulate dopamine D2 receptors within the caudate-putamen in the brain. The precise mechanism of action of ropinirole as a treatment for Restless Legs Syndrome is unknown, although it is thought to be related to its ability to stimulate dopamine receptors.
12.2 Pharmacodynamics
Clinical experience with dopamine agonists, including ropinirole, suggests an association with impaired ability to regulate blood pressure resulting in orthostatic hypotension, especially during dose escalation. In some patients in clinical trials, blood pressure changes were associated with the emergence of orthostatic symptoms, bradycardia, and, in one case in a healthy volunteer, transient sinus arrest with syncope [see Warnings and Precautions (5.2, 5.3)].
The mechanism of orthostatic hypotension induced by ropinirole is presumed to be due to a D2-mediated blunting of the noradrenergic response to standing and subsequent decrease in peripheral vascular resistance. Nausea is a common concomitant symptom of orthostatic signs and symptoms.
At oral doses as low as 0.2 mg, ropinirole suppressed serum prolactin concentrations in healthy male volunteers.
Ropinirole had no dose-related effect on ECG wave form and rhythm in young, healthy, male volunteers in the range of 0.01 to 2.5 mg.
Ropinirole had no dose- or exposure-related effect on mean QT intervals in healthy male and female volunteers titrated to doses up to 4 mg/day. The effect of ropinirole on QTc intervals at higher exposures achieved either due to drug interactions, hepatic impairment, or at higher doses has not been systematically evaluated.
12.3 Pharmacokinetics
Ropinirole displayed linear kinetics over the dosing range of 1 to 8 mg three times daily. Steady-state concentrations are expected to be achieved within 2 days of dosing. Accumulation upon multiple dosing is predictive from single dosing.
Absorption
Ropinirole is rapidly absorbed after oral administration, reaching peak concentration in approximately 1 to 2 hours. In clinical trials, more than 88% of a radiolabeled dose was recovered in urine and the absolute bioavailability was 45% to 55%, indicating approximately 50% first-pass effect.
Relative bioavailability from a tablet compared with an oral solution is 85%. Food does not affect the extent of absorption of ropinirole, although its Tmax is increased by 2.5 hours and its Cmax is decreased by approximately 25% when the drug is taken with a high-fat meal.
Distribution
Ropinirole is widely distributed throughout the body, with an apparent volume of distribution of 7.5 L/kg. It is up to 40% bound to plasma proteins and has a blood-to-plasma ratio of 1:1.
Metabolism
Ropinirole is extensively metabolized by the liver. The major metabolic pathways are N-despropylation and hydroxylation to form the inactive N-despropyl metabolite and hydroxy metabolites. The N-despropyl metabolite is converted to carbamyl glucuronide, carboxylic acid, and N-despropyl hydroxy metabolites. The hydroxy metabolite of ropinirole is rapidly glucuronidated.
In vitro studies indicate that the major cytochrome P450 enzyme involved in the metabolism of ropinirole is CYP1A2, an enzyme known to be induced by smoking and omeprazole and inhibited by, for example, fluvoxamine, mexiletine, and the older fluoroquinolones such as ciprofloxacin and norfloxacin.
Elimination
The clearance of ropinirole after oral administration is 47 L/h and its elimination half-life is approximately 6 hours. Less than 10% of the administered dose is excreted as unchanged drug in urine. N-despropyl ropinirole is the predominant metabolite found in urine (40%), followed by the carboxylic acid metabolite (10%), and the glucuronide of the hydroxy metabolite (10%).
Drug Interactions
Digoxin: Coadministration of ropinirole (2 mg three times daily) with digoxin (0.125 to 0.25 mg once daily) did not alter the steady-state pharmacokinetics of digoxin in 10 patients.
Theophylline: Administration of theophylline (300 mg twice daily, a substrate of CYP1A2) did not alter the steady-state pharmacokinetics of ropinirole (2 mg three times daily) in 12 patients with Parkinson's disease. Ropinirole (2 mg three times daily) did not alter the pharmacokinetics of theophylline (5 mg/kg intravenously) in 12 patients with Parkinson's disease.
Ciprofloxacin: Coadministration of ciprofloxacin (500 mg twice daily), an inhibitor of CYP1A2, with ropinirole (2 mg three times daily) increased ropinirole AUC by 84% on average and Cmax by 60% (n = 12 patients).
Estrogens: Population pharmacokinetic analysis revealed that estrogens (mainly ethinylestradiol: intake 0.6 to 3 mg over 4-month to 23-year period) reduced the oral clearance of ropinirole by 36% in 16 patients.
L-dopa: Coadministration of carbidopa + L-dopa (10/100 mg twice daily) with ropinirole (2 mg three times daily) had no effect on the steady-state pharmacokinetics of ropinirole (n = 28 patients). Oral administration of ropinirole 2 mg three times daily increased mean steady-state Cmax of L-dopa by 20%, but its AUC was unaffected (n = 23 patients).
Commonly Administered Drugs: Population analysis showed that commonly administered drugs, e.g., selegiline, amantadine, tricyclic antidepressants, benzodiazepines, ibuprofen, thiazides, antihistamines, and anticholinergics, did not affect the clearance of ropinirole. An in vitro study indicates that ropinirole is not a substrate for P-glycoprotein. Ropinirole and its circulating metabolites do not inhibit or induce P450 enzymes; therefore, ropinirole is unlikely to affect the pharmacokinetics of other drugs by a P450 mechanism.
Specific Populations
Because therapy with ropinirole is initiated at a low dose and gradually titrated upward according to clinical tolerability to obtain the optimum therapeutic effect, adjustment of the initial dose based on gender, weight, or age is not necessary.
Age: Oral clearance of ropinirole is reduced by 15% in patients older than 65 years compared with younger patients. Dosage adjustment is not necessary in the elderly (older than 65 years), as the dose of ropinirole is to be individually titrated to clinical response.
Gender: Female and male patients showed similar clearance.
Race: The influence of race on the pharmacokinetics of ropinirole has not been evaluated.
Cigarette Smoking: Smoking is expected to increase the clearance of ropinirole since CYP1A2 is known to be induced by smoking. In a trial in patients with RLS, smokers (n = 7) had an approximately 30% lower Cmax and a 38% lower AUC than did nonsmokers (n = 11) when those parameters were normalized for dose.
Renal Impairment: Based on population pharmacokinetic analysis, no difference was observed in the pharmacokinetics of ropinirole in subjects with moderate renal impairment (creatinine clearance between 30 to 50 mL/min) compared with an age-matched population with creatinine clearance above 50 mL/min. Therefore, no dosage adjustment is necessary in patients with moderate renal impairment.
A trial of ropinirole in subjects with end-stage renal disease on hemodialysis has shown that clearance of ropinirole was reduced by approximately 30%. The recommended maximum dose is lower in these patients [see Dosage and Administration (2.2, 2.3)].
The use of ropinirole in subjects with severe renal impairment (creatinine clearance less than 30 mL/min) without regular dialysis has not been studied.
Hepatic Impairment: The pharmacokinetics of ropinirole have not been studied in patients with hepatic impairment. Because ropinirole is extensively metabolized by the liver, these patients may have higher plasma levels and lower clearance of ropinirole than patients with normal hepatic function.
Other Diseases: Population pharmacokinetic analysis revealed no change in the clearance of ropinirole in patients with concomitant diseases such as hypertension, depression, osteoporosis/arthritis, and insomnia compared with patients with Parkinson's disease only.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year carcinogenicity studies of ropinirole were conducted in mice at oral doses of 0, 5, 15, and 50 mg/kg/day and in rats at oral doses of 0, 1.5, 15, and 50 mg/kg/day.
In rats, there was an increase in testicular Leydig cell adenomas at all doses tested. The lowest dose tested (1.5 mg/kg/day) is less than the MRHD for Parkinson's disease (24 mg/day) on a mg/m2 basis. The endocrine mechanisms believed to be involved in the production of these tumors in rats are not considered relevant to humans.
In mice, there was an increase in benign uterine endometrial polyps at a dose of 50 mg/kg/day. The highest dose not associated with this finding (15 mg/kg/day) is 3 times the MRHD on a mg/m2 basis.
Mutagenesis
Ropinirole was not mutagenic or clastogenic in in vitro (Ames, chromosomal aberration in human lymphocytes, mouse lymphoma tk) assays, or in the in vivo mouse micronucleus test.
Impairment of Fertility
When administered to female rats prior to and during mating and throughout pregnancy, ropinirole caused disruption of implantation at oral doses of 20 mg/kg/day (8 times the MRHD on a mg/m2 basis) or greater. This effect in rats is thought to be due to the prolactin-lowering effect of ropinirole. In rat studies using a low oral dose (5 mg/kg) during the prolactin-dependent phase of early pregnancy (gestation days 0 to 8), ropinirole did not affect female fertility at oral doses up to 100 mg/kg/day (40 times the MRHD on a mg/m2 basis). No effect on male fertility was observed in rats at oral doses up to 125 mg/kg/day (50 times the MRHD on a mg/m2 basis).
-
14 CLINICAL STUDIES
14.1 Parkinson’s Disease
The effectiveness of ropinirole in the treatment of Parkinson's disease was evaluated in a multinational drug development program consisting of 11 randomized, controlled trials. Four trials were conducted in patients with early Parkinson's disease and no concomitant L-dopa and seven trials were conducted in patients with advanced Parkinson's disease with concomitant L-dopa.
Three placebo-controlled trials provide evidence of effectiveness of ropinirole in the management of patients with Parkinson's disease who were and were not receiving concomitant L-dopa. Two of these three trials enrolled patients with early Parkinson's disease (without L-dopa) and one enrolled patients receiving L-dopa.
In these trials a variety of measures were used to assess the effects of treatment (e.g., the Unified Parkinson's Disease Rating Scale [UPDRS], Clinical Global Impression [CGI] scores, patient diaries recording time "on" and "off," tolerability of L-dopa dose reductions).
In both trials of patients with early Parkinson's disease (without L-dopa), the motor component (Part III) of the UPDRS was the primary outcome assessment. The UPDRS is a multi-item rating scale intended to evaluate mentation (Part I), activities of daily living (Part II), motor performance (Part III), and complications of therapy (Part IV). Part III of the UPDRS contains 14 items designed to assess the severity of the cardinal motor findings in patients with Parkinson's disease (e.g., tremor, rigidity, bradykinesia, postural instability) scored for different body regions and has a maximum (worst) score of 108. In the trial of patients with advanced Parkinson's disease (with L-dopa), both reduction in percent awake time spent "off" and the ability to reduce the daily use of L-dopa were assessed as a combined endpoint and individually.
Trials in Patients with Early Parkinson's Disease (without L-dopa)
Trial 1 was a 12-week multicenter trial in which 63 patients with idiopathic Parkinson's disease receiving concomitant anti-Parkinson medication (but not L-dopa) were enrolled and 41 were randomized to ropinirole and 22 to placebo. Patients had a mean disease duration of approximately 2 years. Patients were eligible for enrollment if they presented with bradykinesia and at least tremor, rigidity, or postural instability. In addition, they must have been classified as Hoehn & Yahr Stage I-IV. This scale, ranging from I = unilateral involvement with minimal impairment to V = confined to wheelchair or bed, is a standard instrument used for staging patients with Parkinson's disease. The primary outcome measure in this trial was the proportion of patients experiencing a decrease (compared with baseline) of at least 30% in the UPDRS motor score.
Patients were titrated for up to 10 weeks, starting at 0.5 mg twice daily, with weekly increments of 0.5 mg twice daily to a maximum of 5 mg twice daily. Once patients reached their maximally tolerated dose (or 5 mg twice daily), they were maintained on that dose through 12 weeks. The mean dose achieved by patients at trial endpoint was 7.4 mg/day. Mean baseline UPDRS motor score was 18.6 for patients treated with ropinirole and 19.9 for patients treated with placebo. At the end of 12 weeks, the percentage of responders was greater on ropinirole than on placebo and the difference was statistically significant (Table 6).
Table 6 Percent Responders for UPDRS Motor Score in Trial 1 (Intent-to-Treat Population) % Responders
Difference from
Placebo
Placebo
41%
NA
Ropinirole
71%
30%
Trial 2 in patients with early Parkinson's disease (without L-dopa) was a double-blind, randomized, placebo-controlled, 6-month trial. In this trial, 241 patients were enrolled and 116 were randomized to ropinirole and 125 to placebo. Patients were essentially similar to those in the trial described above; concomitant use of selegiline was allowed, but patients were not permitted to use anticholinergics or amantadine during the trial. Patients had a mean disease duration of 2 years and limited (not more than a 6-week period) or no prior exposure to L-dopa. The starting dosage of ropinirole in this trial was 0.25 mg three times daily. The dosage was titrated at weekly intervals by increments of 0.25 mg three times daily to a dosage of 1 mg three times daily. Further titrations at weekly intervals were at increments of 0.5 mg three times daily up to a dosage of 3 mg three times daily, and then weekly at increments of 1 mg three times daily. Patients were to be titrated to a dosage of at least 1.5 mg three times daily and then to their maximally tolerated dosage, up to a maximum of 8 mg three times daily. The mean dose attained in patients at trial endpoint was 15.7 mg/day.
The primary measure of effectiveness was the mean percent reduction (improvement) from baseline in the UPDRS motor score. At the end of the 6-month trial, patients treated with ropinirole showed improvement in motor score compared with placebo and the difference was statistically significant (Table 7).
Table 7 Mean Percentage Change from Baseline in UPDRS Motor Score at End of Treatment in Trial 2 (Intent-to-Treat Population) Treatment
Baseline UPDRS Motor Score
Mean Change from Baseline
Difference from Placebo
Placebo
17.7
+4%
NA
Ropinirole
17.9
-22%
-26%
Trial in Patients with Advanced Parkinson's Disease (with L-dopa)
Trial 3 was a double-blind, randomized, placebo-controlled, 6-month trial that randomized 149 patients (Hoehn & Yahr II-IV) who were not adequately controlled on L-dopa. Ninety-five patients were randomized to ropinirole and 54 were randomized to placebo. Patients in this trial had a mean disease duration of approximately 9 years, had been exposed to L-dopa for approximately 7 years, and had experienced "on-off" periods with L-dopa therapy. Patients previously receiving stable doses of selegiline, amantadine, and/or anticholinergic agents could continue on these agents during the trial. Patients were started at a dosage of 0.25 mg three times daily of ropinirole and titrated upward by weekly intervals until an optimal therapeutic response was achieved. The maximum dosage of trial medication was 8 mg three times daily. All patients had to be titrated to at least a dosage of 2.5 mg three times daily. Patients could then be maintained on this dosage level or higher for the remainder of the trial. Once a dosage of 2.5 mg three times daily was achieved, patients underwent a mandatory reduction in their L-dopa dosage, to be followed by additional mandatory reductions with continued escalation of the dosage of ropinirole. Reductions in the dosage of L-dopa were also allowed if patients experienced adverse reactions that the investigator considered related to dopaminergic therapy. The mean dose attained at trial endpoint was 16.3 mg/day. The primary outcome was the proportion of responders, defined as patients who were able both to achieve a decrease (compared with baseline) of at least 20% in their L-dopa dosage and a decrease of at least 20% in the proportion of the time awake in the "off" condition (a period of time during the day when patients are particularly immobile), as determined by subject diary. In addition, the mean change in "off" time from baseline and the percent change from baseline in daily L-dopa dosage were examined.
At the end of 6 months, the percentage of responders was greater on ropinirole than on placebo and the difference was statistically significant (Table 8).
Based on the protocol-mandated reductions in L-dopa dosage with escalating doses of ropinirole, patients treated with ropinirole had a 19.4% mean reduction in L-dopa dosage while patients treated with placebo had a 3% reduction. Mean daily L-dopa dosage at baseline was 759 mg for patients treated with ropinirole and 843 mg for patients treated with placebo.
The mean number of daily "off" hours at baseline was 6.4 hours for patients treated with ropinirole and 7.3 hours for patients treated with placebo. At the end of the 6-month trial, there was a mean reduction of 1.5 hours of "off" time in patients treated with ropinirole and a mean reduction of 0.9 hours of "off" time in patients treated with placebo, resulting in a treatment difference of 0.6 hours of "off" time.
Table 8 Mean Responder Percentage of Patients Reducing Daily L-Dopa Dosage by at Least 20% and Daily Proportion of "Off" Time by at Least 20% at End of Treatment in Trial 3 (Intent-to-Treat Population) Treatment
% Responders
Difference from Placebo
Placebo
11%
NA
Ropinirole
28%
17%
14.2 Restless Legs Syndrome
The effectiveness of ropinirole in the treatment of RLS was demonstrated in randomized, double-blind, placebo-controlled trials in adults diagnosed with RLS using the International Restless Legs Syndrome Study Group diagnostic criteria. Patients were required to have a history of a minimum of 15 RLS episodes/month during the previous month and a total score of ≥ 15 on the International RLS Rating Scale (IRLS scale) at baseline. Patients with RLS secondary to other conditions (e.g., pregnancy, renal failure, anemia) were excluded. All trials employed flexible dosing, with patients initiating therapy at 0.25 mg ropinirole once daily. Patients were titrated based on clinical response and tolerability over 7 weeks to a maximum of 4 mg once daily. All doses were taken between 1 and 3 hours before bedtime.
A variety of measures were used to assess the effects of treatment, including the IRLS scale and Clinical Global Impression-Global Improvement (CGI-I) scores. The IRLS scale contains 10 items designed to assess the severity of sensory and motor symptoms, sleep disturbance, daytime somnolence, and impact on activities of daily living and mood associated with RLS. The range of scores is 0 to 40, with 0 being absence of RLS symptoms and 40 the most severe symptoms. Three of the controlled trials utilized the change from baseline in the IRLS scale at the Week 12 endpoint as the primary efficacy outcome.
Three hundred eighty patients were randomized to receive ropinirole (n = 187) or placebo (n = 193) in a US trial (RLS-1); 284 were randomized to receive either ropinirole (n = 146) or placebo (n = 138) in a multinational trial (excluding US) (RLS-2); and 267 patients were randomized to ropinirole (n = 131) or placebo (n = 136) in a multinational trial (including US) (RLS-3). Across the three trials, the mean duration of RLS was 16 to 22 years (range: 0 to 65 years), mean age was approximately 54 years (range: 18 to 79 years), and approximately 61% were women. The mean dose at Week 12 was approximately 2 mg/day for the three trials.
At baseline, mean total IRLS score was 22 for ropinirole and 21.6 for placebo in RLS-1, was 24.4 for ropinirole and 25.2 for placebo in RLS-2, and was 23.6 for ropinirole and 24.8 for placebo in RLS-3. In all three trials, a statistically significant difference between the treatment group receiving ropinirole and the treatment group receiving placebo was observed at Week 12 for both the mean change from baseline in the IRLS scale total score and the percentage of patients rated as responders (much improved or very much improved) on the CGI-I (see Table 9).
Table 9 Mean Change in Total IRLS Score and Percent Responders on CGI-I Ropinirole
Placebo
Difference from Placebo
Mean change in total IRLS score at Week 12
RLS-1
-13.5
-9.8
-3.7
RLS-2
-11
-8
-3
RLS-3
-11.2
-8.7
-2.5
Percent responders on CGI-I at Week 12
RLS-1
73.3%
56.5%
16.8%
RLS-2
53.4%
40.9%
12.5%
RLS-3
59.5%
39.6%
19.9%
Long-term maintenance of efficacy in the treatment of RLS was demonstrated in a 36-week trial. Following a 24-week, single-blind treatment phase (flexible dosages of ropinirole of 0.25 to 4 mg once daily), patients who were responders (defined as a decrease of > 6 points on the IRLS scale total score relative to baseline) were randomized in double-blind fashion to placebo or continuation of ropinirole for an additional 12 weeks. Relapse was defined as an increase of at least 6 points on the IRLS scale total score to a total score of at least 15, or withdrawal due to lack of efficacy. For patients who were responders at Week 24, the mean dose of ropinirole was 2 mg (range: 0.25 to 4 mg). Patients continued on ropinirole demonstrated a significantly lower relapse rate compared with patients randomized to placebo (32.6% versus 57.8%, P = 0.0156).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Ropinirole Hydrochloride Tablets equivalent to 0.25 mg of ropinirole are white-colored, round-shaped, film-coated tablets debossed with "ZF22" on one side and plain on other side and are supplied as follows:
NDC: 68382-338-01 in bottle of 100 tablets
NDC: 68382-338-10 in bottle of 1000 tablets
Ropinirole Hydrochloride Tablets equivalent to 0.5 mg of ropinirole are yellow-colored, round-shaped, film-coated tablets, debossed with "ZF23" on one side and plain on the other side and are supplied as follows:
NDC: 68382-339-01 in bottle of 100 tablets
NDC: 68382-339-10 in bottle of 1000 tablets
Ropinirole Hydrochloride Tablets equivalent to 1 mg of ropinirole are green-colored, round-shaped, film-coated tablets, debossed with "ZF24" on one side and plain on the other side and are supplied as follows:
NDC: 68382-340-01 in bottle of 100 tablets
NDC: 68382-340-10 in bottle of 1000 tablets
Ropinirole Hydrochloride Tablets equivalent to 2 mg of ropinirole are pink-colored, round-shaped, film-coated tablets, debossed with "ZF25" on one side and plain on the other side and are supplied as follows:
NDC: 68382-341-01 in bottle of 100 tablets
NDC: 68382-341-10 in bottle of 1000 tablets
Ropinirole Hydrochloride Tablets equivalent to 3 mg of ropinirole are purple-colored, round-shaped, film-coated tablets, debossed with "ZF42" on one side and plain on the other side and are supplied as follows:
NDC: 68382-342-01 in bottle of 100 tablets
NDC: 68382-342-10 in bottle of 1000 tablets
Ropinirole Hydrochloride Tablets equivalent to 4 mg of ropinirole are brown-colored, round-shaped, film-coated tablets, debossed with "ZF43" on one side and plain on the other side and are supplied as follows:
NDC: 68382-343-01 in bottle of 100 tablets
NDC: 68382-343-10 in bottle of 1000 tablets
Ropinirole Hydrochloride Tablets equivalent to 5 mg of ropinirole are blue-colored, round-shaped, film-coated tablets, debossed with "ZF26" on one side and plain on the other side and are supplied as follows:
NDC: 68382-344-01 in bottle of 100 tablets
NDC: 68382-344-10 in bottle of 1000 tablets
Storage
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature]. Protect from light and moisture. Close container tightly after each use.
Dispense in a tight, light-resistant container.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Dosing Instructions
Instruct patients to take ropinirole only as prescribed. If a dose is missed, advise patients not to double their next dose. Ropinirole can be taken with or without food [see Dosage and Administration (2.1)].
Ropinirole is the active ingredient in ropinirole hydrochloride tablets (the immediate-release formulation). Ask your patients if they are taking another medication containing ropinirole.
Hypersensitivity/Allergic Reactions
Advise patients about the potential for developing a hypersensitivity/allergic reaction including manifestations such as urticaria, angioedema, rash, and pruritus when taking any ropinirole product. Inform patients who experience these or similar reactions to immediately contact their healthcare professional [see Contraindications (4)].
Falling Asleep during Activities of Daily Living and Somnolence
Alert patients to the potential sedating effects caused by ropinirole, including somnolence and the possibility of falling asleep while engaged in activities of daily living. Because somnolence is a frequent adverse reaction with potentially serious consequences, patients should not drive a car, operate machinery, or engage in other potentially dangerous activities until they have gained sufficient experience with ropinirole to gauge whether or not it adversely affects their mental and/or motor performance. Advise patients that if increased somnolence or episodes of falling asleep during activities of daily living (e.g., conversations, eating, driving a motor vehicle, etc.) are experienced at any time during treatment, they should not drive or participate in potentially dangerous activities until they have contacted their physician.
Advise patients of possible additive effects when patients are taking other sedating medications, alcohol, or other central nervous system depressants (e.g., benzodiazepines, antipsychotics, antidepressants, etc.) in combination with ropinirole or when taking a concomitant medication (e.g., ciprofloxacin) that increases plasma levels of ropinirole [see Warnings and Precautions (5.1)].
Syncope and Hypotension/Orthostatic Hypotension
Advise patients that they may experience syncope and may develop hypotension with or without symptoms such as dizziness, nausea, syncope, and sometimes sweating while taking ropinirole, especially if they are elderly. Hypotension and/or orthostatic symptoms may occur more frequently during initial therapy or with an increase in dose at any time (cases have been seen after weeks of treatment). Postural/orthostatic symptoms may be related to sitting up or standing. Accordingly, caution patients against standing rapidly after sitting or lying down, especially if they have been doing so for prolonged periods and especially at the initiation of treatment with ropinirole [see Warnings and Precautions (5.2, 5.3)].
Hallucinations/Psychotic-like Behavior
Inform patients that they may experience hallucinations (unreal visions, sounds, or sensations), and that other psychotic-like behavior can occur while taking ropinirole. The elderly are at greater risk than younger patients with Parkinson's disease. This risk is greater in patients who are taking ropinirole with L-dopa or taking higher doses of ropinirole and may also be further increased in patients taking any other drugs that increase dopaminergic tone. Tell patients to report hallucinations or psychotic-like behavior to their healthcare provider promptly should they develop [see Warnings and Precautions (5.4)].
Dyskinesia
Inform patients that ropinirole may cause and/or exacerbate pre-existing dyskinesias [see Warnings and Precautions (5.5)].
Impulse Control/Compulsive Behaviors
Advise patients that they may experience impulse control and/or compulsive behaviors while taking 1 or more of the medications (including ropinirole) that increase central dopaminergic tone, that are generally used for the treatment of Parkinson's disease. Advise patients to inform their physician or healthcare provider if they develop new or increased gambling urges, sexual urges, uncontrolled spending, binge or compulsive eating, or other urges while being treated with ropinirole. Physicians should consider dose reduction or stopping the medication if a patient develops such urges while taking ropinirole [see Warnings and Precautions (5.6)].
Withdrawal-Emergent Hyperpyrexia and Confusion
Advise patients to contact their healthcare provider if they wish to discontinue ropinirole or decrease the dose of ropinirole [see Warnings and Precautions (5.7)].
Melanoma
Advise patients with Parkinson's disease that they have a higher risk of developing melanoma. Advise patients to have their skin examined on a regular basis by a qualified healthcare provider (e.g., dermatologist) when using ropinirole for any indication [see Warnings and Precautions (5.8)].
Augmentation and Rebound
Inform patients with RLS that augmentation and/or rebound may occur after starting treatment with ropinirole [see Warnings and Precautions (5.9)].
Nursing Mothers
Because of the possibility that ropinirole may be excreted in breast milk, discuss the developmental and health benefits of breastfeeding along with the mother's clinical need for ropinirole and any potential adverse effects on the breastfed child from ropinirole or from the underlying maternal condition [see Use in Specific Populations (8.2)]. Advise patients that ropinirole could inhibit lactation because ropinirole inhibits prolactin secretion.
Pregnancy
Because experience with ropinirole in pregnant women is limited and ropinirole has been shown to have adverse effects on embryofetal development in animals, including teratogenic effects, advise patients of this potential risk. Advise patients to notify their physician if they become pregnant or intend to become pregnant during therapy [see Use in Specific Populations (8.1)].
- SPL UNCLASSIFIED SECTION
-
SPL PATIENT PACKAGE INSERT
PHARMACIST--DETACH HERE AND GIVE INSTRUCTIONS TO PATIENT
Patient Information
Ropinirole Hydrochloride
(roe-PIN-i-ROLE HYE-droe-KLOR-ide)
Tablets
If you have Parkinson's disease, read this side.
What is the most important information I should know about ropinirole hydrochloride tablets?
Ropinirole hydrochloride tablets can cause serious side effects, including:
- Falling asleep during normal activities. You may fall asleep while doing normal activities such as driving a car, doing physical tasks, or using hazardous machinery while taking ropinirole hydrochloride tablets. You may suddenly fall asleep without being drowsy or without warning. This may result in having accidents. Your chances of falling asleep while doing normal activities while taking ropinirole hydrochloride tablets are greater if you take other medicines that cause drowsiness. Tell your healthcare provider right away if this happens. Before starting ropinirole hydrochloride tablets, be sure to tell your healthcare provider if you take any medicines that make you drowsy.
- Fainting. Fainting can happen, and sometimes your heart rate may be decreased. This can happen especially when you start taking ropinirole hydrochloride tablets or your dose is increased. Tell your healthcare provider if you faint, feel dizzy, or feel light-headed.
- Decrease in blood pressure. Ropinirole hydrochloride tablets can decrease your blood pressure (hypotension), especially when you start taking ropinirole hydrochloride tablets or when your dose is changed. If you faint or feel dizzy, nauseated, or sweaty when you stand up from sitting or lying down (orthostatic hypotension), this may mean that your blood pressure is decreased. When you change position from lying down or sitting to standing up, you should do it carefully and slowly. Call your healthcare provider if you have any of the symptoms of decreased blood pressure listed above.
- Increase in blood pressure. Ropinirole hydrochloride tablets may increase your blood pressure.
- Changes in heart rate (decrease or increase). Ropinirole hydrochloride tablets can decrease or increase your heart rate.
- Hallucinations and other psychotic-like behavior. Ropinirole hydrochloride tablets can cause or worsen psychotic-like behavior including hallucinations (seeing or hearing things that are not real), confusion, excessive suspicion, aggressive behavior, agitation, delusional beliefs (believing things that are not real), and disorganized thinking. The chances of having hallucinations or these other psychotic-like changes are higher in people with Parkinson's disease who are taking ropinirole hydrochloride tablets or taking higher doses of these drugs. If you have hallucinations or any of these other psychotic-like changes, talk with your healthcare provider.
- Uncontrolled sudden movements. Ropinirole hydrochloride tablets may cause uncontrolled sudden movements or make such movements you already have worse or more frequent. Tell your healthcare provider if this happens. The doses of your anti-Parkinson's medicine may need to be changed.
- Unusual urges. Some patients taking ropinirole hydrochloride tablets get urges to behave in a way unusual for them. Examples of this are an unusual urge to gamble, increased sexual urges and behaviors, or an uncontrollable urge to shop, spend money, or eat. If you notice or your family notices that you are developing any unusual behaviors, talk to your healthcare provider.
- Increased chance of skin cancer (melanoma). People with Parkinson's disease may have a higher chance of getting melanoma. It is not known if ropinirole hydrochloride tablets increase your chances of getting melanoma. You and your healthcare provider should check your skin on a regular basis. Tell your healthcare provider right away if you notice any changes in your skin such as a change in the size, shape, or color of moles on your skin.
What are ropinirole hydrochloride tablets?
- Ropinirole hydrochloride tablets are a short-acting prescription medicine containing ropinirole (usually taken 3 times a day) that is used to treat Parkinson's disease. It is also used to treat a condition called Restless Legs Syndrome (RLS).
Having one of these conditions does not mean you have or will develop the other condition.
You should not be taking more than 1 medicine containing ropinirole. Tell your healthcare provider if you are taking any other medicine containing ropinirole.
It is not known if ropinirole hydrochloride tablets are safe and effective for use in children younger than 18 years of age.
Do not take ropinirole hydrochloride tablets if you:
- are allergic to ropinirole or any of the ingredients in ropinirole hydrochloride tablets. See the end of this page for a complete list of the ingredients in ropinirole hydrochloride tablets.
Get help right away if any of the symptoms of an allergic reaction cause problems swallowing or breathing. Call your healthcare provider if you have any of the symptoms of an allergic reaction.
Symptoms of an allergic reaction may include:
- hives
- rash
- swelling of the face, lips, mouth, tongue, or throat
- itching
Before you take ropinirole hydrochloride tablets, tell your healthcare provider about all of your medical conditions, including if you:
- have daytime sleepiness from a sleep disorder or have unexpected or unpredictable sleepiness or periods of sleep.
- start or stop taking other medicines while you are taking ropinirole hydrochloride tablets. This may increase your chances of getting side effects.
- start or stop smoking while you are taking ropinirole hydrochloride tablets. Smoking may decrease the treatment effect of ropinirole hydrochloride tablets.
- feel dizzy, nauseated, sweaty, or faint when you stand up from sitting or lying down.
- drink alcoholic beverages. This may increase your chances of becoming drowsy or sleepy while taking ropinirole hydrochloride tablets.
- have high or low blood pressure.
- have or have had heart problems.
- are pregnant or plan to become pregnant. It is not known if ropinirole hydrochloride tablets can harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if ropinirole hydrochloride tablets passes into your breast milk. The amount of breast milk you make may be decreased while taking ropinirole hydrochloride tablets. Talk to your healthcare provider to decide if you should breastfeed while taking ropinirole hydrochloride tablets.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Some of these medicines may increase your chances of getting side effects while taking ropinirole hydrochloride tablets.
How should I take ropinirole hydrochloride tablets?
- Take ropinirole hydrochloride tablets exactly as directed by your healthcare provider.
- Take ropinirole hydrochloride tablets with or without food.
- Do not suddenly stop taking ropinirole hydrochloride tablets without talking to your healthcare provider. If you stop this medicine suddenly, you may develop fever, confusion, or severe muscle stiffness.
- Before starting ropinirole hydrochloride tablets, you should talk to your healthcare provider about what to do if you miss a dose. If you have missed the previous dose and it is time for your next dose, do not double the dose.
- Your healthcare provider will start you on a low dose of ropinirole hydrochloride tablets. Your healthcare provider will change the dose until you are taking the right amount of medicine to control your symptoms. It may take several weeks before you reach a dose that controls your symptoms.
- Contact your healthcare provider if you stop taking ropinirole hydrochloride tablets for any reason. Do not restart without talking with your healthcare provider.
- Your healthcare provider may prescribe ropinirole hydrochloride tablets alone, or add ropinirole hydrochloride tablets to medicine that you are already taking for Parkinson's disease.
- You should not substitute ropinirole hydrochloride tablets for ropinirole hydrochloride extended-release tablets or ropinirole hydrochloride extended-release tablets for ropinirole hydrochloride tablets without talking with your healthcare provider.
What are the possible side effects of ropinirole hydrochloride tablets?
Ropinirole hydrochloride tablets can cause serious side effects, including:
- See "What is the most important information I should know about ropinirole hydrochloride tablets?"
The most common side effects of ropinirole hydrochloride tablets include:
- fainting
- sleepiness or drowsiness
- hallucinations (seeing or hearing things that are not real)
- dizziness
- nausea or vomiting
- uncontrolled sudden movements
- leg swelling
- fatigue, tiredness, or weakness
- confusion
- headache
- upset stomach, abdominal pain or discomfort
- increased sweating
- constipation
- suddenly falling asleep
- high blood pressure (hypertension)
Tell your healthcare provider about any side effect that bothers you or does not go away.
These are not all of the possible side effects with ropinirole hydrochloride tablets.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store ropinirole hydrochloride tablets?
- Store ropinirole hydrochloride tablets at room temperature between 20° to 25°C (68° to 77°F).
- Keep ropinirole hydrochloride tablets in a tightly closed container and out of direct sunlight.
Keep ropinirole hydrochloride tablets and all medicines out of the reach of children.
General information about the safe and effective use of ropinirole hydrochloride tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet.
Do not use ropinirole hydrochloride tablets for a condition for which it was not prescribed. Do not give ropinirole hydrochloride tablets to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your pharmacist or healthcare provider for information about ropinirole hydrochloride tablets that is written for health professionals.
Please address medical inquiries to, MedicalAffairs@zydususa.com Tel.: 1-877-993-8779.
What are the ingredients in ropinirole hydrochloride tablets?
The following ingredients are in ropinirole hydrochloride tablets:
Active ingredient: ropinirole (as ropinirole hydrochloride)
Inactive ingredients: croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol and titanium dioxide. Additionally, each 0.5 mg tablet contains: FD&C blue# 2, iron oxide red and iron oxide yellow; each 1 mg tablet contains: FD&C blue#2 and iron oxide yellow; each 2 mg tablet contains: iron oxide red and iron oxide yellow; each 3 mg tablet contains carmine, FD&C blue # 2 and FD&C yellow # 6; each 4 mg tablet contains: iron oxide black, iron oxide red and iron oxide yellow and each 5 mg tablet contains: FD&C blue# 2.
Cadila Healthcare Ltd.
Ahmedabad, India
Distributed by:
Zydus Pharmaceuticals (USA) Inc.
Pennington, NJ 08534
Rev.: 12/17
Patient Information Leaflet
Ropinirole Hydrochloride
(roe-PIN-i-ROLE HYE-droe-KLOR-ide)
Tablets
If you have Restless Legs Syndrome (RLS), read this side.
An immediate-release form of ropinirole is approved for the treatment of moderate to severe primary RLS (see other side of this leaflet).
People with RLS should take ropinirole hydrochloride tablets differently than people with Parkinson's disease (see "How should I take ropinirole hydrochloride tablets for RLS?" for the recommended dosing for RLS). A lower dose is generally needed for people with RLS, and is taken once daily before bedtime.
What is the most important information I should know about ropinirole hydrochloride tablets?
Ropinirole hydrochloride tablets can cause serious side effects, including:
- Falling asleep during normal activities. You may fall asleep while doing normal activities such as driving a car, doing physical tasks, or using hazardous machinery while taking ropinirole hydrochloride tablets. You may suddenly fall asleep without being drowsy or without warning. This may result in having accidents. Your chances of falling asleep while doing normal activities while taking ropinirole hydrochloride tablets are greater if you take other medicines that cause drowsiness. Tell your healthcare provider right away if this happens. Before starting ropinirole hydrochloride tablets, be sure to tell your healthcare provider if you take any medicines that make you drowsy.
- Fainting. Fainting can happen, and sometimes your heart rate may be decreased. This can happen especially when you start taking ropinirole hydrochloride tablets or your dose is increased. Tell your healthcare provider if you faint or feel dizzy or light-headed.
- Decrease in blood pressure. Ropinirole hydrochloride tablets can decrease your blood pressure (hypotension), especially when you start taking ropinirole hydrochloride tablets or when your dose is changed. If you feel faint or feel dizzy, nauseated, or sweaty when you stand up from sitting or lying down (orthostatic hypotension), this may mean that your blood pressure is decreased. When you change position from lying down or sitting to standing up, you should do it carefully and slowly. Call your healthcare provider if you have any of the symptoms of decreased blood pressure listed above.
- Hallucinations and other psychotic-like behavior. Ropinirole hydrochloride tablets can cause or worsen psychotic-like behavior including hallucinations (seeing or hearing things that are not real), confusion, excessive suspicion, aggressive behavior, agitation, delusional beliefs (believing things that are not real), and disorganized thinking. If you have hallucinations or any of these other psychotic-like changes, talk with your healthcare provider.
- Unusual urges. Some patients taking ropinirole hydrochloride tablets get urges to behave in a way unusual for them. Examples of this are an unusual urge to gamble, increased sexual urges and behaviors, or an uncontrollable urge to shop, spend money, or eat. If you notice or your family notices that you are developing any unusual behaviors, talk to your healthcare provider.
- Increased chance of skin cancer (melanoma). It is not known if ropinirole hydrochloride tablets increase your chance of getting melanoma. You and your healthcare provider should check your skin on a regular basis. Tell your healthcare provider right away if you notice any changes in your skin such as a change in the size, shape, or color of moles on your skin.
- Changes in Restless Legs Syndrome symptoms. Ropinirole hydrochloride tablets may cause Restless Legs symptoms to come back in the morning (rebound), happen earlier in the evening, or even happen in the afternoon.
What are ropinirole hydrochloride tablets?
Ropinirole hydrochloride tablets are a prescription medicine containing ropinirole used to treat moderate-to-severe primary Restless Legs Syndrome. It is also used to treat Parkinson's disease.
Having one of these conditions does not mean you have or will develop the other condition.
You should not be taking more than 1 medicine containing ropinirole. Tell your healthcare provider if you are taking any other medicine containing ropinirole.
It is not known if ropinirole hydrochloride tablets are safe and effective for use in children younger than 18 years of age.
Do not take ropinirole hydrochloride tablets if you:
- are allergic to ropinirole or any of the ingredients in ropinirole hydrochloride tablets. See the end of this leaflet for a complete list of the ingredients in ropinirole hydrochloride tablets.
Get help right away if any of the symptoms of an allergic reaction cause problems swallowing or breathing. Call your healthcare provider if you have any of the symptoms of an allergic reaction. Symptoms of an allergic reaction may include:
- hives
- rash
- swelling of the face, lips, mouth, tongue, or throat
- itching
Before you take ropinirole hydrochloride tablets, tell your healthcare provider all about your medical conditions, including if you:
- have daytime sleepiness from a sleep disorder or have unexpected or unpredictable sleepiness or periods of sleep.
- start or stop taking other medicines while you are taking ropinirole hydrochloride tablets. This may increase your chances of getting side effects.
- start or stop smoking while you are taking ropinirole hydrochloride tablets. Smoking may decrease the treatment effect of ropinirole hydrochloride tablets.
- feel dizzy, nauseated, sweaty, or faint when you stand up from sitting or lying down.
- drink alcoholic beverages. This may increase your chances of becoming drowsy or sleepy while taking ropinirole hydrochloride tablets.
- have high or low blood pressure.
- have or have had heart problems.
- are pregnant or plan to become pregnant. It is not known if ropinirole hydrochloride tablets can harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if ropinirole hydrochloride tablets passes into your breast milk. The amount of breast milk you make may be decreased while taking ropinirole hydrochloride tablets.
Talk to your healthcare provider to decide if you should breastfeed while taking ropinirole hydrochloride tablets. Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Some of these medicines may increase your chances of getting side effects while taking ropinirole hydrochloride tablets.
How should I take ropinirole hydrochloride tablets?
- Take ropinirole hydrochloride tablets exactly as directed by your healthcare provider.
- Ropinirole hydrochloride tablets are usually taken once in the evening, 1 to 3 hours before bedtime.
- Take ropinirole hydrochloride tablets with or without food.
- Do not suddenly stop taking ropinirole hydrochloride tablets without talking to your healthcare provider. If you stop this medicine suddenly, you may develop fever, confusion, or severe muscle stiffness.
- Your healthcare provider will start you on a low dose of ropinirole hydrochloride tablets. Your healthcare provider may change the dose until you are taking the right amount of medicine to control your symptoms.
- If you miss your dose, do not double your next dose. Take only your usual dose 1 to 3 hours before your next bedtime.
- Contact your healthcare provider if you stop taking ropinirole hydrochloride tablets for any reason. Do not restart without talking with your healthcare provider.
What are the possible side effects of ropinirole hydrochloride tablets?
Ropinirole hydrochloride tablets can cause serious side effects, including:
- See "What is the most important information I should know about ropinirole hydrochloride tablets?"
The most common side effects of ropinirole hydrochloride tablets include:
- nausea or vomiting
- drowsiness or sleepiness
- dizziness
- fatigue, tiredness, or weakness
Tell your healthcare provider about any side effect that bothers you or that does not go away.
These are not all of the possible side effects with ropinirole hydrochloride tablets. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store ropinirole hydrochloride tablets?
- Store ropinirole hydrochloride tablets at room temperature between 20° to 25°C (68° to 77°F).
- Keep ropinirole hydrochloride tablets in a tightly closed container and out of direct sunlight.
Keep ropinirole hydrochloride tablets and all medicines out of the reach of children.
General information about the safe and effective use of ropinirole hydrochloride tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ropinirole hydrochloride tablets for a condition for which it was not prescribed. Do not give ropinirole hydrochloride tablets to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your pharmacist or healthcare provider for information about ropinirole hydrochloride tablets that is written for health professionals.
Please address medical inquiries to, MedicalAffairs@zydususa.com Tel.: 1-877-993-8779.
What are the ingredients in ropinirole hydrochloride tablets?
Active ingredient: ropinirole (as ropinirole hydrochloride)
Inactive ingredients: croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol and titanium dioxide. Additionally, each 0.5 mg tablet contains: FD&C blue# 2, iron oxide red and iron oxide yellow; each 1 mg tablet contains: FD&C blue#2 and iron oxide yellow; each 2 mg tablet contains: iron oxide red and iron oxide yellow; each 3 mg tablet contains carmine, FD&C blue # 2 and FD&C yellow # 6; each 4 mg tablet contains: iron oxide black, iron oxide red and iron oxide yellow and each 5 mg tablet contains: FD&C blue# 2.
This Patient Information has been approved by the U.S. Food and Drug Administration.
This product's package insert may have been updated. For current package insert, please visit www.zydususa.com
- SPL UNCLASSIFIED SECTION
-
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
NDC: 68382-338-01 in bottle of 100 tablets
Ropinirole Hydrochloride Tablets, 0.25 mg
100 tablets
ZYDUS

NDC: 68382-339-01 in bottle of 100 tablets
Ropinirole Hydrochloride Tablets, 0.5 mg
100 tablets
ZYDUS

NDC: 68382-340-01 in bottle of 100 tablets
Ropinirole Hydrochloride Tablets, 1 mg
100 tablets
ZYDUS

NDC: 68382-341-01 in bottle of 100 tablets
Ropinirole Hydrochloride Tablets, 2 mg
100 tablets
ZYDUS

NDC: 68382-342-01 in bottle of 100 tablets
Ropinirole Hydrochloride Tablets, 3 mg
100 tablets
ZYDUS

NDC: 68382-343-01 in bottle of 100 tablets
Ropinirole Hydrochloride Tablets, 4 mg
100 tablets
ZYDUS

NDC: 68382-344-01 in bottle of 100 tablets
Ropinirole Hydrochloride Tablets, 5 mg
100 tablets
ZYDUS

-
INGREDIENTS AND APPEARANCE
ROPINIROLE HYDROCHLORIDE
ropinirole hydrochloride tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68382-338 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROPINIROLE HYDROCHLORIDE (UNII: D7ZD41RZI9) (ROPINIROLE - UNII:030PYR8953) ROPINIROLE 0.25 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) Product Characteristics Color WHITE (WHITE) Score no score Shape ROUND (ROUND) Size 7mm Flavor Imprint Code ZF22 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68382-338-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 2 NDC: 68382-338-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090411 09/23/2009 ROPINIROLE HYDROCHLORIDE
ropinirole hydrochloride tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68382-339 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROPINIROLE HYDROCHLORIDE (UNII: D7ZD41RZI9) (ROPINIROLE - UNII:030PYR8953) ROPINIROLE 0.5 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) HYPROMELLOSES (UNII: 3NXW29V3WO) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) Product Characteristics Color YELLOW (YELLOW) Score no score Shape ROUND (ROUND) Size 7mm Flavor Imprint Code ZF23 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68382-339-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 2 NDC: 68382-339-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090411 09/23/2009 ROPINIROLE HYDROCHLORIDE
ropinirole hydrochloride tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68382-340 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROPINIROLE HYDROCHLORIDE (UNII: D7ZD41RZI9) (ROPINIROLE - UNII:030PYR8953) ROPINIROLE 1 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) Product Characteristics Color GREEN (GREEN) Score no score Shape ROUND (ROUND) Size 7mm Flavor Imprint Code ZF24 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68382-340-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 2 NDC: 68382-340-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090411 09/23/2009 ROPINIROLE HYDROCHLORIDE
ropinirole hydrochloride tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68382-341 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROPINIROLE HYDROCHLORIDE (UNII: D7ZD41RZI9) (ROPINIROLE - UNII:030PYR8953) ROPINIROLE 2 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) Product Characteristics Color PINK (PINK) Score no score Shape ROUND (ROUND) Size 7mm Flavor Imprint Code ZF25 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68382-341-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 2 NDC: 68382-341-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090411 09/23/2009 ROPINIROLE HYDROCHLORIDE
ropinirole hydrochloride tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68382-342 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROPINIROLE HYDROCHLORIDE (UNII: D7ZD41RZI9) (ROPINIROLE - UNII:030PYR8953) ROPINIROLE 3 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) CARMINIC ACID (UNII: CID8Z8N95N) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) Product Characteristics Color PURPLE (PURPLE) Score no score Shape ROUND (ROUND) Size 7mm Flavor Imprint Code ZF42 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68382-342-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 2 NDC: 68382-342-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090411 09/23/2009 ROPINIROLE HYDROCHLORIDE
ropinirole hydrochloride tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68382-343 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROPINIROLE HYDROCHLORIDE (UNII: D7ZD41RZI9) (ROPINIROLE - UNII:030PYR8953) ROPINIROLE 4 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) Product Characteristics Color BROWN (BROWN) Score no score Shape ROUND (ROUND) Size 7mm Flavor Imprint Code ZF43 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68382-343-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 2 NDC: 68382-343-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090411 09/23/2009 ROPINIROLE HYDROCHLORIDE
ropinirole hydrochloride tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68382-344 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROPINIROLE HYDROCHLORIDE (UNII: D7ZD41RZI9) (ROPINIROLE - UNII:030PYR8953) ROPINIROLE 5 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) Product Characteristics Color BLUE (BLUE) Score no score Shape ROUND (ROUND) Size 7mm Flavor Imprint Code ZF26 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68382-344-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 2 NDC: 68382-344-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 09/23/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090411 09/23/2009 Labeler - Zydus Pharmaceuticals (USA) Inc. (156861945) Registrant - Zydus Pharmaceuticals (USA) Inc. (156861945) Establishment Name Address ID/FEI Business Operations Cadila Healthcare Limited 918596198 ANALYSIS(68382-338, 68382-339, 68382-340, 68382-341, 68382-342, 68382-343, 68382-344) , MANUFACTURE(68382-338, 68382-339, 68382-340, 68382-341, 68382-342, 68382-343, 68382-344)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.