NPLATE- romiplostim injection, powder, lyophilized, for solution
Nplate by
Drug Labeling and Warnings
Nplate by is a Prescription medication manufactured, distributed, or labeled by Amgen Inc, Amgen Manufacturing Ltd, Amgen Technology (ireland) Unlimited Company, Amgen, Inc, Immunex Rhode Island Corporation, Patheon Italia S.p.A.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NPLATE safely and effectively. See full prescribing information for NPLATE.
NPLATE® (romiplostim) for injection, for subcutaneous use
Initial U.S. Approval: 2008
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
Nplate is a thrombopoietin receptor agonist indicated for the treatment of thrombocytopenia in:
- Adult patients with immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
- Pediatric patients 1 year of age and older with ITP for at least 6 months who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Limitations of Use:
- Nplate is not indicated for the treatment of thrombocytopenia due to myelodysplastic syndrome (MDS) or any cause of thrombocytopenia other than ITP.
- Nplate should be used only in patients with ITP whose degree of thrombocytopenia and clinical condition increases the risk for bleeding.
- Nplate should not be used in an attempt to normalize platelet counts. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
- For injection: 125 mcg, 250 mcg or 500 mcg of deliverable romiplostim as a lyophilized powder in single-dose vials. (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- In some patients with MDS, Nplate increases blast cell counts and increases the risk of progression to acute myelogenous leukemia. (5.1)
- Thrombotic/thromboembolic complications may result from increases in platelet counts with Nplate use. Portal vein thrombosis has been reported in patients with chronic liver disease receiving Nplate. (5.2)
- If severe thrombocytopenia develops during Nplate treatment, assess patients for the formation of neutralizing antibodies. (5.3)
ADVERSE REACTIONS
- In adult patients, the most common adverse reactions (≥ 5% higher patient incidence in Nplate versus placebo) are arthralgia, dizziness, insomnia, myalgia, pain in extremity, abdominal pain, shoulder pain, dyspepsia, and paresthesia. Headache was the most commonly reported adverse reaction that did not occur at ≥ 5% higher patient incidence in Nplate versus placebo. (6.1)
- In pediatric patients, the most common adverse reactions (≥ 25%) are: contusion, upper respiratory tract infection, and oropharyngeal pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Inc. at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 10/2019
- Adult patients with immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage Regimen
2.2 Preparation and Administration
2.3 Monitoring to Assess Effectiveness and Safety
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Progression of Myelodysplastic Syndromes to Acute Myelogenous Leukemia
5.2 Thrombotic/Thromboembolic Complications
5.3 Loss of Response to Nplate
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
6.3 Immunogenicity
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Adults with ITP
14.2 Pediatric Patients with ITP
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1
INDICATIONS AND USAGE
Nplate is indicated for the treatment of thrombocytopenia in:
Adult patients with immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Pediatric patients 1 year of age and older with ITP for at least 6 months who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Limitations of Use:
Nplate is not indicated for the treatment of thrombocytopenia due to myelodysplastic syndrome (MDS) or any cause of thrombocytopenia other than ITP [see Warnings and Precautions (5.1)].
Nplate should be used only in patients with ITP whose degree of thrombocytopenia and clinical condition increases the risk for bleeding.
Nplate should not be used in an attempt to normalize platelet counts [see Warnings and Precautions (5.2)].
-
2
DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage Regimen
For All Patients
Use the lowest dose of Nplate to achieve and maintain a platelet count ≥ 50 × 109/L as necessary to reduce the risk for bleeding. Administer Nplate as a weekly subcutaneous injection with dose adjustments based upon the platelet count response.
The prescribed Nplate dose may consist of a very small volume (e.g., 0.15 mL). Administer Nplate only with a syringe that contains 0.01 mL graduations.
See Section 2.3 for monitoring recommendations during Nplate therapy.
Discontinue Nplate if the platelet count does not increase to a level sufficient to avoid clinically important bleeding after 4 weeks of Nplate therapy at the maximum weekly dose of 10 mcg/kg [see Warnings and Precautions (5.3)].
For Adult Patients with ITP
The initial dose of Nplate is 1 mcg/kg. Actual body weight at initiation of treatment should always be used when calculating the initial dose. In adults, future dose adjustments are based on changes in platelet counts only.
Adjust the weekly dose of Nplate by increments of 1 mcg/kg until the patient achieves a platelet count ≥ 50 × 109/L as necessary to reduce the risk for bleeding; do not exceed a maximum weekly dose of 10 mcg/kg. In clinical studies, most adult patients who responded to Nplate achieved and maintained platelet counts ≥ 50 × 109/L with a median dose of 2 mcg/kg.
Adjust the dose as follows for adult patients:
If the platelet count is < 50 × 109/L, increase the dose by 1 mcg/kg.
If platelet count is > 200 × 109/L and ≤ 400 × 109/L for 2 consecutive weeks, reduce the dose by 1 mcg/kg.
If platelet count is > 400 × 109/L, do not dose. Continue to assess the platelet count weekly. After the platelet count has fallen to < 200 × 109/L, resume Nplate at a dose reduced by 1 mcg/kg.
For Pediatric Patients with ITP
The initial dose of Nplate is 1 mcg/kg. Actual body weight at initiation of treatment should always be used when calculating initial dose. In pediatric patients, future dose adjustments are based on changes in platelet counts and changes in body weight. Reassessment of body weight is recommended every 12 weeks.
Adjust the weekly dose of Nplate by increments of 1 mcg/kg until the patient achieves a platelet count ≥ 50 × 109/L as necessary to reduce the risk for bleeding; do not exceed a maximum weekly dose of 10 mcg/kg. In a pediatric placebo-controlled clinical study, the median of the most frequent dose of Nplate received by patients during weeks 17 through 24 was 5.5 mcg/kg.
Adjust the dose as follows for pediatric patients:
If the platelet count is < 50 × 109/L, increase the dose by 1 mcg/kg.
If platelet count is > 200 × 109/L and ≤ 400 × 109/L for 2 consecutive weeks, reduce the dose by 1 mcg/kg.
If platelet count is > 400 × 109/L, do not dose. Continue to assess the platelet count weekly. After the platelet count has fallen to < 200 × 109/L, resume Nplate at a dose reduced by 1 mcg/kg.
2.2 Preparation and Administration
To mitigate against medication errors (both overdose and underdose), ensure that these preparation and administration instructions are followed. Use aseptic technique. Only administer subcutaneously [see Overdosage (10)].
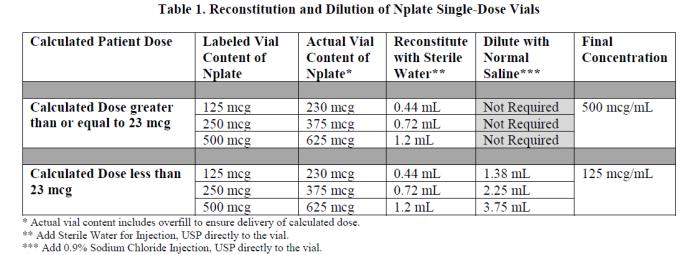
Nplate is supplied in single-dose vials as a sterile, preservative-free, white lyophilized powder that must be reconstituted as outlined in Table 1 and administered using a syringe with 0.01 mL graduations.
Calculation of Patient Dose
Multiply the patient’s weight (kg) by the prescribed dose to obtain the Calculated Patient Dose.
Calculated Patient Dose (mcg) = Weight (kg) × Prescribed dose (mcg/kg) Reconstitution and Dilution of Nplate Single-Dose Vials
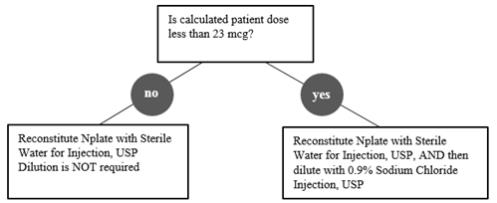
Reconstitute Nplate with Sterile Water for Injection, USP. If the Calculated Patient Dose is less than 23 mcg, dilution with 0.9% Sodium Chloride Injection, USP is required. Follow instructions in Table 1.
Gently swirl and invert the vial to reconstitute. Avoid excess or vigorous agitation: DO NOT SHAKE. Generally, dissolution of Nplate takes less than 2 minutes. The reconstituted Nplate solution should be clear and colorless. Visually inspect the reconstituted solution for particulate matter and/or discoloration. Do not administer Nplate if particulate matter and/or discoloration is observed.
Initial reconstitution of Nplate with designated volumes of Sterile Water for Injection, USP results in a concentration of 500 mcg/mL in all vial sizes. Do not reconstitute or dilute with Bacteriostatic Water for Injection, USP or dilute with Bacteriostatic Sodium Chloride Injection, USP.
If a patient’s dose is less than 23 mcg, then additional dilution with 0.9% Sodium Chloride Injection, USP is required. Dilution per reconstitution instructions results in reducing the concentration of Nplate from 500 mcg/mL to 125 mcg/mL in all vial sizes (see Table 1). This reduced concentration allows for low-doses to be accurately calculated, and consistently measured with a 0.01 mL graduated syringe.
Administration of Prepared Nplate Solution
Calculate Volume to Administer by dividing the Calculated Patient Dose (mcg) by the final concentration. See Table 2 for final concentrations.
Table 2. Administration of Prepared Nplate Solution Calculated Patient Dose Final Concentration Volume to Administer (mL) Calculated Dose greater than or equal to 23 mcg 500 mcg/mL = Calculated Patient Dose / 500 mcg/mL Calculated Dose less than 23 mcg 125 mcg/mL = Calculated Patient Dose / 125 mcg/mL Administer Nplate only using a syringe with 0.01 mL graduations for accurate dosage. Round volume to the nearest hundredth mL. Verify that the syringe contains the correct dosage.
Discard any unused portion. Do not pool unused portions from the vials. Do not administer more than one dose from a vial.
Storage of Reconstituted Solution
Reconstituted product with Sterile Water for Injection, USP that has not been further diluted can remain in the original vial at room temperature 25°C (77°F) or be refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours following reconstitution. Reconstituted product with Sterile Water for Injection, USP may be held in a syringe at room temperature 25°C (77°F) for a maximum of 4 hours following reconstitution. Protect product from light. Do not shake.
Storage of Diluted solution (after initial reconstitution)
Reconstituted and further diluted product with 0.9% Sodium Chloride Injection, USP can be held in a syringe at room temperature 25°C (77°F) or in the original vial refrigerated at 2°C to 8°C (36°F to 46°F) for no longer than 4 hours prior to administration. Protect product from light. Do not shake.
2.3 Monitoring to Assess Effectiveness and Safety
Obtain CBCs, including platelet counts, weekly during the dose adjustment phase of Nplate therapy and then monthly following establishment of a stable Nplate dose. Obtain CBCs, including platelet counts, weekly for at least 2 weeks following discontinuation of Nplate [see Dosage and Administration (2.1)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5
WARNINGS AND PRECAUTIONS
5.1 Risk of Progression of Myelodysplastic Syndromes to Acute Myelogenous Leukemia
Progression from myelodysplastic syndromes (MDS) to acute myelogenous leukemia (AML) has been observed in adult clinical trials with Nplate.
A randomized, double-blind, placebo-controlled trial enrolling adult patients with severe thrombocytopenia and International Prognostic Scoring System (IPSS) low or intermediate-1 risk MDS was terminated due to more cases of AML observed in the Nplate arm. This trial consisted of a 58-week study period with a 5-year long-term follow-up phase. The patients were randomized 2:1 to treatment with Nplate or placebo (167 Nplate, 83 placebo). During the 58-week study period, progression to AML occurred in 10 (6.0%) patients in the Nplate arm and 4 (4.8%) patients in the placebo arm (hazard ratio [95%CI] = 1.20 [0.38, 3.84]). Of the 250 patients, 210 (84.0%) entered the long-term follow-up phase of this study. With 5-years of follow-up, 29 (11.6%) patients showed progression to AML, including 20/168 (11.9%) patients in the Nplate arm versus 9/82 (11.0%) patients in the placebo arm (HR [95% CI] = 1.06 [0.48, 2.33]). The incidence of death (overall survival) was 55.7% (93/167) in the Nplate arm versus 54.2% (45/83) in the placebo arm (HR [95% CI] = 1.03 [0.72, 1.47]). In the baseline low IPSS group, there was a higher incidence of death in the Nplate arm [41.3% (19/46)] compared to the placebo arm [30.4% (7/23)] (HR [95% CI] = 1.59 [0.67, 3.80]).
In a single-arm trial of Nplate given to 72 patients with thrombocytopenia-related MDS, 8 (11.1%) patients were reported as having possible disease progression, of which 3 (4.2%) had confirmation of AML during follow-up. In addition, in 3 (4.2%) patients, increased peripheral blood blast cell counts decreased to baseline after discontinuation of Nplate.
Nplate is not indicated for the treatment of thrombocytopenia due to MDS or any cause of thrombocytopenia other than ITP.
5.2 Thrombotic/Thromboembolic Complications
Thrombotic/thromboembolic complications may result from increases in platelet counts with Nplate use. Portal vein thrombosis has been reported in patients with chronic liver disease receiving Nplate.
To minimize the risk for thrombotic/thromboembolic complications, do not use Nplate in an attempt to normalize platelet counts. Follow the dose adjustment guidelines [see Dosage and Administration (2.1)].
5.3 Loss of Response to Nplate
Hyporesponsiveness or failure to maintain a platelet response with Nplate should prompt a search for causative factors, including neutralizing antibodies to Nplate [see Adverse Reactions (6.3)]. To detect antibody formation, submit blood samples to Amgen (1-800-772-6436). Amgen will assay these samples for antibodies to Nplate and thrombopoietin (TPO). Discontinue Nplate if the platelet count does not increase to a level sufficient to avoid clinically important bleeding after 4 weeks at the highest weekly dose of 10 mcg/kg.
-
6
ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections:
- Progression of Myelodysplastic Syndromes [see Warnings and Precautions (5.1)]
- Thrombotic/Thromboembolic Complications [see Warnings and Precautions (5.2)]
- Loss of Response to Nplate [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adults
The data described below reflect Nplate exposure to 271 adult patients with ITP, aged 18 to 88, of whom 62% were female. Nplate was studied in two randomized, placebo-controlled, double-blind studies that were identical in design, with the exception that Study 1 evaluated nonsplenectomized patients with ITP and Study 2 evaluated splenectomized patients with ITP. Data are also reported from an open-label, single-arm study in which patients received Nplate over an extended period of time. Overall, Nplate was administered to 114 patients for at least 52 weeks and 53 patients for at least 96 weeks.
In the placebo-controlled studies, headache was the most commonly reported adverse drug reaction, occurring in 35% of patients receiving Nplate and 32% of patients receiving placebo. For those patients receiving Nplate, 14 (48%) of headaches were mild, 9 (31%) were moderate, and 6 (21%) were severe. Table 3 presents adverse drug reactions from Studies 1 and 2 with a ≥ 5% higher patient incidence in Nplate versus placebo.
Table 3. Adverse Reactions Identified in Two Placebo-Controlled Studies Adverse Reactions by Body System Nplate (%)
(n=84)Placebo (%)
(n=41)Musculoskeletal and Connective Tissue Disorders Arthralgia 22 (26%) 8 (20%) Myalgia 12 (14%) 1 (2%) Pain in Extremity 11 (13%) 2 (5%) Shoulder Pain 7 (8%) 0 Nervous System Disorders Dizziness 14 (17%) 0 Paresthesia 5 (6%) 0 Psychiatric Disorders Insomnia 13 (16%) 3 (7%) Gastrointestinal Disorders Abdominal pain 9 (11%) 0 Dyspepsia 6 (7%) 0 MedDRA version 9 is used. Among 291 adult patients with ITP who received Nplate in the single-arm extension study, the incidence rates of the adverse reactions occurred in a pattern similar to those reported in the placebo-controlled clinical studies.
The safety profile of Nplate was similar across patients, regardless of ITP duration. The following adverse reactions (at least 5% incidence and at least 5% more frequent with Nplate compared with placebo or standard of care) occurred in Nplate patients with ITP duration up to 12 months: bronchitis, sinusitis, vomiting, arthralgia, myalgia, headache, dizziness, diarrhea, upper respiratory tract infection, cough, nausea and oropharyngeal pain. The adverse reaction of thrombocytosis occurred with an incidence of 2% in adults with ITP duration up to 12 months.
Bone Marrow Reticulin Formation and Collagen Fibrosis
Nplate administration may increase the risk for development or progression of reticulin fiber formation within the bone marrow. This formation may improve upon discontinuation of Nplate. In a clinical trial, one patient with ITP and hemolytic anemia developed marrow fibrosis with collagen during Nplate therapy.
An open-label clinical trial prospectively evaluated changes in bone marrow reticulin formation and collagen fibrosis in adult patients with ITP treated with Nplate or a non-US approved romiplostim product. Patients were administered romiplostim by SC injection once weekly for up to 3 years. Based on cohort assignment at time of study enrollment, patients were evaluated for bone marrow reticulin and collagen at year 1 (cohort 1), year 2 (cohort 2), or year 3 (cohort 3) in comparison to the baseline bone marrow at start of the trial. Patients were evaluated for bone marrow reticulin formation and collagen fibrosis using the modified Bauermeister grading scale. From the total of 169 patients enrolled in the 3 cohorts, 132 (78%) patients were evaluable for bone marrow collagen fibrosis and 131 (78%) patients were evaluable for bone marrow reticulin formation. Two percent (2/132) of patients (both from cohort 3) developed Grade 4 findings (presence of collagen). There was no detectable bone marrow collagen in one patient on repeat testing 12 weeks after discontinuation of romiplostim. Progression of bone marrow reticulin formation (increase greater than or equal to 2 grades or more) or an increase to Grade 4 (presence of collagen) was reported in 7% (9/131) of patients.
Pediatric Patients
The data described below reflect median exposure to Nplate of 168 days for 59 pediatric patients (aged 1 to 17 years) with ITP for at least 6 months, of whom 47.5% were female, across the randomized phase of two placebo-controlled trials. Table 4 presents the most common adverse reactions experienced by at least 5% of the pediatric patients (1 year and older) receiving Nplate across the two placebo-controlled trials with at least a 5% higher incidence in patients who received Nplate compared to those who received placebo.
Table 4. Common Adverse Reactions (≥ 5% Incidence and ≥ 5% More Frequent on the Nplate Arm) from Two Placebo-Controlled Trials in Pediatric Patients with ITP for at least 6 months Adverse Reactions by Body System Nplate (%)
(N = 59)Placebo (%)
(N = 24)Infections and Infestations Upper Respiratory Tract Infection 18 (31%) 6 (25%) Ear Infection 3 (5%) 0 Gastroenteritis 3 (5%) 0 Sinusitis 3 (5%) 0 Respiratory, Thoracic and Mediastinal Disorders Oropharyngeal Pain 15 (25%) 1 (4%) Gastrointestinal Disorders Diarrhea 12 (20%) 3 (13%) Abdominal Pain Upper 8 (14%) 1 (4%) Skin and Subcutaneous Tissue Disorders Rash 9 (15%) 2 (8%) Purpura 4 (7%) 0 Urticaria 3 (5%) 0 General Disorders and Administration Site Conditions Pyrexia 14 (24%) 2 (8%) Peripheral Swelling 4 (7%) 0 Injury, Poisoning and Procedural Complications Contusion 24 (41%) 8 (33%) MedDRA version 20.1 is used.
In pediatric patients of age ≥ 1 year receiving Nplate for ITP, adverse reactions with an incidence of ≥ 25% in the two randomized trials were: contusion (41%), upper respiratory tract infection (31%), and oropharyngeal pain (25%).6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of Nplate. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Erythromelalgia
- Hypersensitivity
- Angioedema
6.3 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to Nplate in the studies described below with the incidence of antibodies in other studies or to other products may be misleading. Patients were screened for immunogenicity to romiplostim using a BIAcore-based biosensor immunoassay. This assay is capable of detecting both high- and low-affinity binding antibodies that bind to romiplostim and cross-react with TPO. The samples from patients that tested positive for binding antibodies were further evaluated for neutralizing capacity using a cell-based bioassay.
In adult clinical studies in adult patients with ITP, the incidence of pre-existing antibodies to romiplostim was 3.3% (35/1046) and the incidence of binding antibody development during treatment with Nplate or a non-US approved romiplostim product was 5.7% (60/1046). The incidence of pre-existing antibodies to endogenous TPO was 3% (31/1046) and the incidence of binding antibody development to endogenous TPO during treatment was 3.2% (33/1046). Of the patients with positive binding antibodies that developed to romiplostim or to TPO, four patients had neutralizing activity to romiplostim and none had neutralizing activity to TPO. No apparent correlation was observed between antibody activity and clinical effectiveness or safety.
In pediatric studies, the incidence of binding antibodies to Nplate at any time was 7.8% (22/282). Of the 22 patients, 2 patients had pre-existing binding non-neutralizing Nplate antibodies at baseline. Additionally, 2.5% (7/282) developed neutralizing antibodies to Nplate. A total of 3.2% (9/282) patients had binding antibodies to TPO at any time during Nplate treatment. Of these 9 patients, 2 patients had pre-existing binding non-neutralizing antibodies to TPO. All patients were negative for neutralizing activity to TPO.
A postmarketing registry study involving patients with thrombocytopenia on Nplate or a non-US approved romiplostim product was conducted to assess the long-term consequences of the anti-romiplostim antibodies. Adult patients who lacked response or lost response to Nplate or a non-US approved romiplostim product were enrolled. The incidence of new binding antibody development was 3.8% (7/184) to romiplostim and 2.2% (4/184) were positive for binding, non-neutralizing antibodies to TPO; two patients were positive for binding antibodies to both romiplostim and TPO. Of the seven patients with positive binding antibodies to romiplostim, one patient (0.5%; 1/184) was positive for neutralizing antibodies to romiplostim only.
Nineteen confirmed pediatric patients were included in the postmarketing registry study. The incidence of binding antibody post treatment was 16% (3/19) to romiplostim, of which 5.3% (1/19) were positive for neutralizing antibodies to romiplostim. There were no antibodies detected to TPO.
Immunogenicity assay results are highly dependent on the sensitivity and specificity of the assay used in detection and may be influenced by several factors, including sample handling, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to romiplostim with the incidence of antibodies to other products may be misleading.
- Progression of Myelodysplastic Syndromes [see Warnings and Precautions (5.1)]
-
7
DRUG INTERACTIONS
Nplate may be used with other medical ITP therapies, such as corticosteroids, danazol, azathioprine, intravenous immunoglobulin (IVIG), and anti-D immunoglobulin [see Clinical Studies (14.1)].
-
8
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal reproduction studies, Nplate may cause fetal harm when administered to a pregnant woman. Available data with Nplate use in pregnant women are insufficient to draw conclusions about any drug-associated risk for major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction and developmental toxicity studies, romiplostim crossed the placenta, and adverse fetal effects included thrombocytosis, postimplantation loss, and an increase in pup mortality (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In rat and rabbit embryo-fetal development toxicity studies, no evidence of fetal harm was observed at romiplostim doses up to 11 times (rats) and 82 times (rabbits) the maximum human dose (MHD) based on systemic exposure (AUC). In mice at doses 5 times the MHD, reductions in maternal body weight and increased postimplantation loss occurred.
In a prenatal and postnatal development study in rats, at doses 11 times the MHD, there was an increase in perinatal pup mortality. Romiplostim crossed the placental barrier in rats and increased fetal platelet counts at clinically equivalent and higher doses.
8.2 Lactation
Risk Summary
There is no information regarding the presence of romiplostim in human milk, the effects on the breastfed child, or the effects on milk production. Maternal IgG is known to be present in human milk. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed child to romiplostim are unknown. Due to the potential for serious adverse reactions in a breastfed child from Nplate, advise women not to breastfeed during treatment with Nplate.
8.4 Pediatric Use
Safety and effectiveness have been established in pediatric patients age 1 year and older with ITP for at least 6 months evaluated in two randomized, placebo-controlled studies [see Adverse Reactions (6.1), Clinical Studies (14.2)]. The pharmacokinetics of romiplostim have been evaluated in pediatric patients 1 year and older with ITP [see Clinical Pharmacology (12.3)]. See Dosage and Administration (2.1) for dosing recommendations for pediatric patients 1 year and older. The safety and efficacy of Nplate in pediatric patients younger than 1 year with ITP have not been established. Serum concentrations of romiplostim in pediatric patients with ITP were within the range observed in adult patients with ITP receiving the same dose range of romiplostim.
8.5 Geriatric Use
Of the 271 patients who received Nplate in ITP clinical studies, 55 (20%) were age 65 and over, and 27 (10%) were 75 and over. No overall differences in safety or efficacy have been observed between older and younger patients in the placebo-controlled studies, but greater sensitivity of some older individuals cannot be ruled out. In general, dose adjustment for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
10
OVERDOSAGE
Overdoses due to medication errors have been reported in patients receiving Nplate. In the event of overdose, platelet counts may increase excessively and result in thrombotic/thromboembolic complications. In this case, discontinue Nplate and monitor platelet counts. Reinitiate treatment with Nplate in accordance with dosing and administration recommendations [see Dosage and Administration (2.1, 2.2)].
-
11
DESCRIPTION
Romiplostim is a thrombopoietin receptor agonist (TPO-RA). Romiplostim, a member of the TPO mimetic class, is an Fc-peptide fusion protein (peptibody). The peptibody molecule contains two identical single-chain subunits, each consisting of human immunoglobulin IgG1 Fc domain, covalently linked at the C-terminus to a peptide containing two thrombopoietin receptor-binding domains. Romiplostim has no amino acid sequence homology to endogenous TPO. Romiplostim is produced by recombinant DNA technology in Escherichia coli (E. coli).
Nplate (romiplostim) for injection is supplied as a sterile, preservative-free, lyophilized, solid white powder for subcutaneous injection. Three vial presentations are available, which contain a sufficient amount of active ingredient to provide either 125 mcg, 250 mcg or 500 mcg of deliverable romiplostim. Each single-dose 125 mcg vial of Nplate contains the following: 230 mcg romiplostim, 0.7 mg L-histidine, 18 mg mannitol, 0.02 mg polysorbate 20, 9 mg sucrose, and sufficient HCL to adjust the pH to a target of 5.0. Each single-dose 250 mcg vial of Nplate contains the following: 375 mcg romiplostim, 1.2 mg L-histidine, 30 mg mannitol, 0.03 mg polysorbate 20, 15 mg sucrose, and sufficient HCl to adjust the pH to a target of 5.0. Each single-dose 500 mcg vial of Nplate contains the following: 625 mcg romiplostim, 1.9 mg L-histidine, 50 mg mannitol, 0.05 mg polysorbate 20, 25 mg sucrose, and sufficient HCl to adjust the pH to a target of 5.0 [see Dosage and Administration (2.2)].
-
12
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Nplate increases platelet production through binding and activation of the TPO receptor, a mechanism analogous to endogenous TPO.
12.2 Pharmacodynamics
In clinical studies, treatment with Nplate resulted in dose-dependent increases in platelet counts. After a single subcutaneous dose of 1 to 10 mcg/kg Nplate in patients with ITP, the peak platelet count was 1.3 to 14.9 times greater than the baseline platelet count over a 2- to 3-week period. The platelet counts were above 50 × 109/L for seven out of eight patients with ITP who received six weekly doses of Nplate at 1 mcg/kg.
12.3 Pharmacokinetics
In the long-term extension study in adult patients with ITP receiving weekly treatment of Nplate subcutaneously, the pharmacokinetics of romiplostim over the dose range of 3 to 15 mcg/kg indicated that peak serum concentrations of romiplostim were observed about 7 to 50 hours post dose (median: 14 hours) with half-life values ranging from 1 to 34 days (median: 3.5 days). The serum concentrations varied among patients and did not correlate with the dose administered. The elimination of serum romiplostim is in part dependent on the TPO receptor on platelets. As a result, for a given dose, patients with high platelet counts are associated with low serum concentrations and vice versa. In another ITP clinical study, no accumulation in serum concentrations was observed (n = 4) after six weekly doses of Nplate (3 mcg/kg). The accumulation at higher doses of romiplostim is unknown.
Serum concentrations of romiplostim in pediatrics with ITP were within the range observed in adult ITP patients receiving the same dose range of romiplostim. Similar to adults with ITP, romiplostim pharmacokinetics are highly variable in pediatric patients with ITP.
-
13
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of romiplostim has not been evaluated. The mutagenic potential of romiplostim has not been evaluated. Romiplostim had no effect on the fertility of rats at doses up to 37 times the MHD based on systemic exposure.
13.2 Animal Toxicology and/or Pharmacology
In a 4-week repeat-dose toxicity study in which rats were dosed subcutaneously three times per week, romiplostim caused extramedullary hematopoiesis, bone hyperostosis, and marrow fibrosis at clinically equivalent and higher doses. In this study, these findings were not observed in animals after a 4-week post treatment recovery period. Studies of long-term treatment with romiplostim in rats have not been conducted; therefore, it is not known if the fibrosis of the bone marrow is reversible in rats after long-term treatment.
-
14
CLINICAL STUDIES
14.1 Adults with ITP
The safety and efficacy of Nplate in adults with ITP were assessed in two double-blind, placebo-controlled clinical studies, an open-label single-arm study, and in an open-label extension study.
Studies 1 (NCT00102336) and 2 (NCT00102323)
In Studies 1 and 2, patients with ITP who had completed at least one prior treatment and had a platelet count of ≤ 30 × 109/L prior to study entry were randomized (2:1) to 24 weeks of Nplate (1 mcg/kg subcutaneous [SC]) or placebo. The median time since ITP diagnosis for Studies 1 and 2 was 2.1 years (range 0.1 to 31.6) and 8 years (range 0.6 to 44.8), respectively. Prior ITP treatments in both study groups included corticosteroids, immunoglobulins, rituximab, cytotoxic therapies, danazol, and azathioprine. Patients already receiving ITP medical therapies at a constant dosing schedule were allowed to continue receiving these medical treatments throughout the studies. Rescue therapies (i.e., corticosteroids, IVIG, platelet transfusions, and anti-D immunoglobulin) were permitted for bleeding, wet purpura, or if the patient was at immediate risk for hemorrhage. Patients received single weekly SC injections of Nplate, with individual dose adjustments to maintain platelet counts (50 × 109/L to 200 × 109/L).
Study 1 evaluated patients who had not undergone a splenectomy. The patients had been diagnosed with ITP for approximately 2 years and had received a median of three prior ITP treatments. Overall, the median platelet count was 19 × 109/L at study entry. During the study, the median weekly Nplate dose was 2 mcg/kg (25th–75th percentile: 1–3 mcg/kg).
Study 2 evaluated patients who had undergone a splenectomy. The patients had been diagnosed with ITP for approximately 8 years and had received a median of six prior ITP treatments. Overall, the median platelet count was 14 × 109/L at study entry. During the study, the median weekly Nplate dose was 3 mcg/kg (25th–75th percentile: 2-7 mcg/kg).
Study 1 and 2 outcomes are shown in Table 5. A durable platelet response was the achievement of a weekly platelet count ≥ 50 × 109/L for any 6 of the last 8 weeks of the 24-week treatment period in the absence of rescue medication at any time. A transient platelet response was the achievement of any weekly platelet counts ≥ 50 × 109/L for any 4 weeks during the treatment period without a durable platelet response. An overall platelet response was the achievement of either a durable or a transient platelet response. Platelet responses were excluded for 8 weeks after receiving rescue medications.
Table 5. Results from Placebo-Controlled Studiesa Outcomes Study 1
Nonsplenectomized PatientsStudy 2
Splenectomized PatientsNplate
(n = 41)Placebo
(n = 21)Nplate
(n = 42)Placebo
(n = 21)Platelet Responses and Rescue Therapy Durable Platelet Response, n (%) 25 (61%) 1 (5%) 16 (38%) 0 (0%) Overall Platelet Response, n (%) 36 (88%) 3 (14%) 33 (79%) 0 (0%) Number of Weeks with Platelet Counts ≥ 50 × 109/L, average 15 1 12 0 Requiring Rescue Therapy, n (%) 8 (20%) 13 (62%) 11 (26%) 12 (57%) Reduction/Discontinuation of Baseline Concurrent ITP Medical Therapy Receiving Therapy at Baseline (n = 11) (n = 10) (n = 12) (n = 6) Patients Who Had > 25% Dose Reduction in Concurrent Therapy, n (%) 4/11
(36%)2/10
(20%)4/12
(33%)1/6
(17%)Patients Who Discontinued Baseline Therapy, n (%)b 4/11
(36%)3/10
(30%)8/12
(67%)0/6
(0%)a All p values < 0.05 for platelet response and rescue therapy comparisons between Nplate and placebo.
b For multiple concomitant baseline therapies, all therapies were discontinued.In Studies 1 and 2, nine patients reported a serious bleeding event [five (6%) Nplate, four (10%) placebo]. Bleeding events that were Grade 2 severity or higher occurred in 15% of patients treated with Nplate and 34% of patients treated with placebo.
Study 3 (NCT01143038)
Study 3 was a single-arm, open-label study designed to assess the safety and efficacy of Nplate in adult patients who had an insufficient response (platelet count ≤ 30 x 109/L) to first-line therapy. The study enrolled 75 patients of whom the median age was 39 years (range 19 to 85) and 59% were female.
The median time from ITP diagnosis to study enrollment was 2.2 months (range 0.1 to 6.6). Sixty percent of patients had ITP duration < 3 months and 40% had ITP duration ≥ 3 months. The median platelet count at screening was 20 x 109/L. Prior ITP treatments included corticosteroids, immunoglobulins and anti-D immunoglobulins. Patients already receiving ITP medical therapies at a constant dosing schedule were allowed to continue receiving these medical treatments throughout the studies. Rescue therapies (i.e., corticosteroids, IVIG, platelet transfusions, anti-D immunoglobulin, dapsone, danazol, and azathioprine) were permitted.
Patients received single weekly SC injections of Nplate over a 12-month treatment period, with individual dose adjustments to maintain platelet counts (50 x 109/L to 200 x 109/L). During the study, the median weekly Nplate dose was 3 mcg/kg (25th-75th percentile: 2-4 mcg/kg).
Of the 75 patients enrolled in Study 3, 70 (93%) had a platelet response ≥ 50 x 109/L during the 12-month treatment period. The mean number of months with platelet response during the 12-month treatment period was 9.2 (95% CI: 8.3, 10.1) months; the median was 11 (95% CI: 10, 11) months. The Kaplan-Meier estimate of the median time to first platelet response was 2.1 weeks (95% CI: 1.1, 3.0). Twenty-four (32%) patients maintained every platelet count ≥ 50 x 109/L for at least 6 months in the absence of Nplate and any medication for ITP (concomitant or rescue); the median time to onset of maintaining every platelet count ≥ 50 x 109/L for at least 6 months was 27 weeks (range 6 to 57).
Study 4 (NCT00116688) Extension Study
Patients who had completed a prior Nplate study (including Study 1 and Study 2) were allowed to enroll in a long-term open-label extension study. Following Nplate discontinuation in Studies 1 and 2, seven patients maintained platelet counts of ≥ 50 × 109/L. Among 291 patients who subsequently entered the extension study and received Nplate, platelet counts were increased and sustained regardless of whether they had received Nplate or placebo in the prior placebo-controlled studies. The majority of patients reached a median platelet count of 50 × 109/L after receiving one to three doses of Nplate, and these platelet counts were maintained throughout the remainder of the study with a median duration of Nplate treatment of 78 weeks and a maximum duration of 277 weeks.
14.2 Pediatric Patients with ITP
The safety and efficacy of Nplate in pediatric patients 1 year and older with ITP for at least 6 months were assessed in two double-blind, placebo-controlled clinical trials.
Study 5 (NCT01444417)
In Study 5, patients refractory or relapsed after at least one prior ITP therapy with a platelet count ≤ 30 x 109/L were stratified by age and randomized (2:1) to receive Nplate (n = 42) or placebo (n = 20). The starting dose for all ages was 1 mcg/kg weekly. Over a 24-week treatment period, dose was titrated up to a maximum of 10 mcg/kg weekly of either Nplate or placebo in an effort to maintain a target platelet count of ≥ 50 × 109/L to 200 × 109/L.
The median age of the patients was 9.5 years (range 3 to 17) and 57% were female. Approximately 58% of patients had a baseline count ≤ 20 x 109/L, which was similar between treatment arms. The percentage of patients with at least 2 prior ITP therapies (predominantly immunoglobulins and corticosteroids) was 81% in the group treated with Nplate and 70% in the group treated with placebo. One patient in each group had undergone splenectomy.
Study 5 results are shown in Table 6. The efficacy of Nplate in this trial was measured by the proportion of patients receiving Nplate achieving a durable platelet response and the proportion of patient achieving an overall platelet response. A durable platelet response was defined as achieving at least 6 weekly platelet counts ≥ 50 × 109/L during weeks 18 through 25 of treatment. A transient platelet response was defined as a weekly platelet count ≥ 50 × 109/L for 4 or more times during weeks 2 through 25, but without durable platelet response. An overall platelet response was defined as a durable or a transient platelet response. Platelet responses were excluded for 4 weeks after receiving rescue medications.
Table 6. Results from Pediatric Placebo-Controlled Studiesa Outcomes Study 5 Nplate
(n = 42)Placebo
(n = 20)Platelet Responses and Rescue Therapy Durable Platelet Responsea, n (%) 22 (52%) 2 (10%) Overall Platelet Responsea, n (%) 30 (71%) 4 (20%) Number of Weeks with Platelet Counts ≥ 50 x 109/L, mediana 12 1 a All p values < 0.05 for platelet response between Nplate and placebo. Study 6 (NCT00515203)
In study 6, patients diagnosed with ITP at least 6 months prior to enrollment with a platelet count ≤ 30 x 109/L were stratified by age and randomized (3:1) to receive Nplate (n = 17) or placebo (n = 5). The starting dose for all ages was 1 mcg/kg weekly. Over a 12-week treatment period dose was titrated up to a maximum of 10 mcg/kg weekly of either Nplate or placebo in an effort to maintain a target platelet count of ≥ 50 × 109/L to 250 × 109/L.
The median age of the patients was 10 years (range 1 to 17 years) and 27.3% of patients were female. Approximately 82% of patients had a baseline count ≤ 20 x 109/L, which was similar between treatment arms. The percentage of patients with at least 2 prior ITP therapies (predominantly IVIG and corticosteroids) was 88% in the group treated with Nplate and 100% in the group treated with placebo. Six patients in the Nplate group and 2 patients in the placebo group had undergone splenectomy.
The efficacy of Nplate in this trial was measured by the proportion of patients who achieved a platelet count of ≥ 50 × 109/L for 2 consecutive weeks and by the proportion of patients who achieved an increase in platelet count of ≥ 20 × 109/L above baseline for 2 consecutive weeks. Platelet responses within 4 weeks following rescue medications use were excluded. Of the 17 patients who received romiplostim, 15 achieved a platelet count of ≥ 50 × 109/L for 2 consecutive weeks (88.2%, 95% CI: 63.6%, 98.5%).
The same 15 patients also achieved an increase in platelet count of ≥ 20 × 109/L above baseline for 2 consecutive weeks during the treatment period (88.2%, 95% CI: 63.6%, 98.5%). None of the patients treated with placebo achieved either endpoint.
16 HOW SUPPLIED/STORAGE AND HANDLING
Nplate (romiplostim) for injection is supplied as a sterile, preservative-free, solid white lyophilized powder in single-dose vials that deliver 125 mcg (NDC-55513-223-01), 250 mcg (NDC: 55513-221-01) and 500 mcg (NDC: 55513-222-01) of romiplostim.
Store Nplate vials in the refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Do not freeze.
If needed, unopened Nplate vials may be stored in the original carton at room temperature up to a maximum of 25°C (77°F) for a single period of up to 30 days. The new expiration date must be written in the space provided on the carton. Once stored at room temperature, do not place back in the refrigerator. If not used within the 30 days, discard Nplate.
-
17
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Inform patients of the following risks and considerations for Nplate:
- Nplate therapy is administered to achieve and maintain a platelet count ≥ 50 × 109/L as necessary to reduce the risk for bleeding; Nplate is not used to normalize platelet counts.
- Following discontinuation of Nplate, thrombocytopenia and risk of bleeding may develop that is worse than that experienced prior to the Nplate therapy.
- Nplate therapy may increase the risk of reticulin fiber formation within the bone marrow. This formation may improve upon discontinuation. Detection of peripheral blood cell abnormalities may necessitate a bone marrow examination.
- Too much Nplate may result in excessive platelet counts and a risk for thrombotic/thromboembolic complications.
- Nplate stimulates certain bone marrow cells to make platelets and increases the risk of progression to acute myelogenous leukemia in patients with myelodysplastic syndromes.
- Platelet counts and CBCs must be performed weekly until a stable Nplate dose has been achieved; thereafter, platelet counts and CBCs must be performed monthly while taking Nplate.
- Patients must be closely monitored with weekly platelet counts and CBCs for at least 2 weeks following Nplate discontinuation.
- Even with Nplate therapy, patients should continue to avoid situations or medications that may increase the risk for bleeding.
Pregnancy:
- Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their prescriber of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Lactation:
- Advise women not to breastfeed during treatment with Nplate [see Use in Specific Populations (8.2)].
Nplate® (romiplostim)
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799
U.S. License No. 1080Patent: http://pat.amgen.com/nplate/
© 2008-2019 Amgen Inc. All rights reserved.
www.nplate.com
1xxxxx
v15
- Nplate therapy is administered to achieve and maintain a platelet count ≥ 50 × 109/L as necessary to reduce the risk for bleeding; Nplate is not used to normalize platelet counts.
-
MEDICATION GUIDE
MEDICATION GUIDE
Nplate® (N-plāt)
(romiplostim)
for injectionWhat is the most important information I should know about Nplate?
Nplate can cause serious side effects, including:
-
Worsening of a precancerous blood condition to a blood cancer (leukemia). Nplate is not for use in people with a precancerous condition called myelodysplastic syndromes (MDS), or for any condition other than immune thrombocytopenia (ITP). If you have MDS and receive Nplate, your MDS condition may worsen and become an acute leukemia. If MDS worsens to become acute leukemia you may die sooner from the acute leukemia.
-
Higher risk for blood clots.
○ You may have a higher risk of getting a blood clot if your platelet count becomes high during treatment with Nplate. You may have severe complications or die from some forms of blood clots, such as clots that spread to the lungs or that cause heart attacks or strokes.
○ If you have a chronic liver disease, you may get blood clots in the veins of your liver. This may affect your liver function.
- Injection of too much Nplate may cause a dangerous increase in your blood platelet count and serious side effects. Your healthcare provider may change your dose or stop Nplate depending upon the change in your blood platelet count. You must have blood platelet counts done before you start, during, and after Nplate therapy is stopped (see “How will I receive Nplate?”).
What is Nplate?
Nplate is a prescription medicine used to treat low blood platelet counts (thrombocytopenia) in:
adults with immune thrombocytopenia (ITP) when certain medicines or surgery to remove your spleen have not worked well enough.
children 1 year of age and older with ITP for at least 6 months when certain medicines or surgery to remove your spleen have not worked well enough.- Nplate is not for use in people with a precancerous condition called myelodysplastic syndrome (MDS), or low platelet count caused by any condition other than ITP.
- Nplate is only used if your low platelet count and medical condition increase your risk of bleeding.
- Nplate is used to try to keep your platelet count about 50,000 per microliter in order to lower the risk for bleeding. Nplate is not used to make your platelet count normal.
- It is not known if Nplate is safe and effective in children under the age of 1.
Before receiving Nplate, first speak to your healthcare provider and understand the benefits and risks of Nplate. Be sure to tell your healthcare provider about all of your medical conditions, including if you:
- have had surgery to remove your spleen (splenectomy)
- have a bone marrow problem, including a blood cancer or MDS
- have or had a blood clot
- have chronic liver disease
- have bleeding problems
- are pregnant or plan to become pregnant. Nplate may harm your unborn baby. Tell your healthcare provider if you become pregnant or think you may be pregnant during treatment with Nplate.
- are breastfeeding or plan to breastfeed. Nplate may pass into your breast milk and harm your baby. Do not breastfeed during treatment with Nplate.
Know the medicines you take. Keep a list of them and show it to your healthcare provider or pharmacist when you get a new medicine.How will I receive Nplate? - Nplate is given as an injection under the skin (subcutaneous) one time each week by your healthcare provider.
- During treatment, your healthcare provider will closely monitor your Nplate dose and platelet counts.
○ Your healthcare provider will check your platelet count every week and change your dose of Nplate as needed. This will continue until your healthcare provider decides that your dose of Nplate can stay the same. After that, you will need to get blood tests every month. When you stop receiving Nplate, you will need blood tests for at least 2 weeks to check if your platelet count drops too low.
○ Tell your healthcare provider about any bruising or bleeding that occurs during treatment with Nplate.
- If you miss a scheduled dose of Nplate, call your healthcare provider to schedule your next dose as soon as possible.
What should I avoid while receiving Nplate?
Avoid situations or medicines that may increase your risk of bleeding.What are the possible side effects of Nplate?
Nplate may cause serious side effects. See “What is the most important information I should know about Nplate?”
The most common side effects of Nplate in adults include:headache tingling or numbness in hands and feet joint pain bronchitis dizziness inflammation of the sinuses (sinusitis) trouble sleeping vomiting muscle tenderness or weakness
pain in the arms and legs
stomach (abdomen) pain
shoulder pain
indigestiondiarrhea
upper respiratory tract infection
cough
nausea
pain in mouth and throat (oropharyngeal pain)The most common side effects of Nplate in children 1 year of age and older include: bruising
upper respiratory tract infectionpain in mouth and throat (oropharyngeal pain)
People who take Nplate may have an increased risk of developing new or worsening changes in the bone marrow called “increased reticulin”. These changes may improve if you stop taking Nplate. Your healthcare provider may need to check your bone marrow for this problem during treatment with Nplate.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to Amgen at 1-800-77-AMGEN (1-800-772-6436).General information about the safe and effective use of Nplate.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about Nplate that is written for health professionals.What are the ingredients in Nplate?
Active ingredient: romiplostim
Inactive ingredients: L-histidine, mannitol, polysorbate 20, sucrose, and hydrochloric acid
Nplate® (romiplostim)
Manufactured by: Amgen Inc., One Amgen Center Drive, Thousand Oaks, California 91320-1799
US License No. 1080.
Patent: http://pat.amgen.com/nplate/
© 2008-2019 Amgen Inc. All rights reserved. For more information go to www.nplate.com.
1xxxxxx v8This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 10/2019 -
Worsening of a precancerous blood condition to a blood cancer (leukemia). Nplate is not for use in people with a precancerous condition called myelodysplastic syndromes (MDS), or for any condition other than immune thrombocytopenia (ITP). If you have MDS and receive Nplate, your MDS condition may worsen and become an acute leukemia. If MDS worsens to become acute leukemia you may die sooner from the acute leukemia.
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel
Single Dose Vial
Rx Only
NDC: 55513-221-01
Amgen®
Nplate®
(romiplostim)
250 mcg*
250 mcg*
*Reconstitute with 0.72 mL Sterile Water for Injection, USP.
Delivers 250 mcg in 0.5 mL
For Subcutaneous Use Only
Single Dose Vial; Discard unused portion
Dispense the enclosed Medication Guide to each patient.
Store at 2º to 8ºC (36º to 46ºF).
Protect from light. Do not freeze.
DO NOT SHAKE reconstituted solution
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel
Single Dose Vial
Rx Only
NDC: 55513-222-01
Amgen®
Nplate®
(romiplostim)
500 mcg*
500 mcg*
*Reconstitute with 1.2 mL Sterile Water for Injection, USP.
Delivers 500 mcg in 1 mL
For Subcutaneous Use Only
Single Dose Vial; Discard unused portion
Dispense the enclosed Medication Guide to each patient.
Store at 2º to 8ºC (36º to 46ºF).
Protect from light. Do not freeze.
DO NOT SHAKE reconstituted solution

-
PRINCIPAL DISPLAY PANEL
Principal Display Panel
Single Dose Vial
Rx Only
NDC: 55513-223-01
Amgen®
Nplate®
(romiplostim)
125 mcg*
125 mcg*
*Reconstitute with 0.44 mL Sterile Water for Injection, USP.
Delivers 125 mcg in 0.25 mL
For Subcutaneous Use Only
Single Dose Vial; Discard unused portion
Dispense the enclosed Medication Guide to each patient.
Store at 2º to 8ºC (36º to 46ºF).
Protect from light. Do not freeze.
DO NOT SHAKE reconstituted solution

-
INGREDIENTS AND APPEARANCE
NPLATE
romiplostim injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55513-222 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROMIPLOSTIM (UNII: GN5XU2DXKV) (ROMIPLOSTIM - UNII:GN5XU2DXKV) ROMIPLOSTIM 500 ug in 1 mL Inactive Ingredients Ingredient Name Strength HYDROCHLORIC ACID (UNII: QTT17582CB) HISTIDINE (UNII: 4QD397987E) 10 mmol in 1 mL MANNITOL (UNII: 3OWL53L36A) 40 mg in 1 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.04 mg in 1 mL SUCROSE (UNII: C151H8M554) 20 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-222-01 1 in 1 CARTON 08/25/2008 1 1 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125268 08/25/2008 NPLATE
romiplostim injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55513-223 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROMIPLOSTIM (UNII: GN5XU2DXKV) (ROMIPLOSTIM - UNII:GN5XU2DXKV) ROMIPLOSTIM 125 ug in 0.25 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 0.7 mg in 0.25 mL MANNITOL (UNII: 3OWL53L36A) 18 mg in 0.25 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.02 mg in 0.25 mL SUCROSE (UNII: C151H8M554) 9 mg in 0.25 mL HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-223-01 1 in 1 CARTON 08/02/2019 1 0.25 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125268 08/02/2019 NPLATE
romiplostim injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55513-221 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROMIPLOSTIM (UNII: GN5XU2DXKV) (ROMIPLOSTIM - UNII:GN5XU2DXKV) ROMIPLOSTIM 250 ug in 0.5 mL Inactive Ingredients Ingredient Name Strength HYDROCHLORIC ACID (UNII: QTT17582CB) HISTIDINE (UNII: 4QD397987E) 5 mmol in 0.5 mL MANNITOL (UNII: 3OWL53L36A) 20 mg in 0.5 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.02 mg in 0.5 mL SUCROSE (UNII: C151H8M554) 10 mg in 0.5 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-221-01 1 in 1 CARTON 08/25/2008 1 0.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125268 08/25/2008 Labeler - Amgen Inc (039976196) Establishment Name Address ID/FEI Business Operations Amgen Manufacturing Ltd 785800020 ANALYSIS(55513-221, 55513-222, 55513-223) , LABEL(55513-221, 55513-222, 55513-223) , PACK(55513-221, 55513-222, 55513-223) Establishment Name Address ID/FEI Business Operations Amgen Technology (ireland) Unlimited Company 896293920 ANALYSIS(55513-221, 55513-222, 55513-223) , LABEL(55513-221, 55513-222, 55513-223) , MANUFACTURE(55513-221, 55513-222, 55513-223) , PACK(55513-221, 55513-222, 55513-223) Establishment Name Address ID/FEI Business Operations Amgen, Inc 039976196 ANALYSIS(55513-221, 55513-222, 55513-223) Establishment Name Address ID/FEI Business Operations Immunex Rhode Island Corporation 968084785 ANALYSIS(55513-221, 55513-222, 55513-223) Establishment Name Address ID/FEI Business Operations Patheon Italia S.p.A. 338336589 ANALYSIS(55513-221, 55513-222, 55513-223) , MANUFACTURE(55513-221, 55513-222, 55513-223)
Trademark Results [Nplate]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 NPLATE 78732010 3574008 Live/Registered |
Amgen Inc. 2005-10-12 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.