CYRAMZA- ramucirumab solution
CYRAMZA by
Drug Labeling and Warnings
CYRAMZA by is a Prescription medication manufactured, distributed, or labeled by Eli Lilly and Company. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use CYRAMZA safely and effectively. See full prescribing information for CYRAMZA.
CYRAMZA (ramucirumab) injection, for intravenous use
Initial U.S. Approval: 2014RECENT MAJOR CHANGES
INDICATIONS AND USAGE
CYRAMZA® is a human vascular endothelial growth factor receptor 2 (VEGFR2) antagonist indicated:
- as a single agent or in combination with paclitaxel, for treatment of advanced or metastatic gastric or gastro-esophageal junction adenocarcinoma with disease progression on or after prior fluoropyrimidine- or platinum-containing chemotherapy. (1.1)
- in combination with docetaxel, for treatment of metastatic non-small cell lung cancer with disease progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving CYRAMZA. (1.2)
- in combination with FOLFIRI, for the treatment of metastatic colorectal cancer with disease progression on or after prior therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine. (1.3)
- as a single agent, for the treatment of hepatocellular carcinoma in patients who have an alpha fetoprotein of ≥400 ng/mL and have been treated with sorafenib. (1.4)
DOSAGE AND ADMINISTRATION
- For intravenous infusion only. Do not administer as an intravenous push or bolus. (2.7)
- Premedicate before each infusion. (2.1)
- Gastric Cancer: Administer CYRAMZA 8 mg/kg every 2 weeks as a single agent or in combination with weekly paclitaxel. (2.2)
- Non-Small Cell Lung Cancer: Administer CYRAMZA 10 mg/kg on Day 1 of a 21-day cycle prior to docetaxel. (2.3)
- Colorectal Cancer: Administer CYRAMZA 8 mg/kg every 2 weeks prior to FOLFIRI. (2.4)
- Hepatocellular Carcinoma: Administer CYRAMZA 8 mg/kg every 2 weeks. (2.5)
DOSAGE FORMS AND STRENGTHS
Injection: 100 mg/10 mL (10 mg/mL) or 500 mg/50 mL (10 mg/mL) solution in a single-dose vial (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Hemorrhage: CYRAMZA increases the risk of hemorrhage and gastrointestinal hemorrhage, including severe and fatal events. Permanently discontinue CYRAMZA in patients who experience severe bleeding. (5.1)
- Gastrointestinal Perforations: CYRAMZA increases the risk of gastrointestinal perforation, which can be fatal. Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation. (5.2)
- Impaired Wound Healing: Withhold CYRAMZA for 28 days prior to elective surgery. Do not administer CYRAMZA for at least 2 weeks following a major surgical procedure and until adequate wound healing. The safety of resumption of CYRAMZA after resolution of wound healing complications has not been established. (5.3)
- Arterial Thromboembolic Events (ATEs): Serious and sometimes fatal ATEs can occur with CYRAMZA. Permanently discontinue CYRAMZA in patients who experience an ATE. (5.4)
- Hypertension: Monitor blood pressure and treat hypertension. Withhold CYRAMZA for severe hypertension. Permanently discontinue CYRAMZA for hypertension that cannot be controlled with antihypertensive therapy and for hypertensive crisis or hypertensive encephalopathy. (5.5)
- Infusion-Related Reactions (IRR): Monitor for signs and symptoms during infusion. Reduce the infusion rate for Grade 1 or 2 IRR and permanently discontinue for Grade 3 or 4 IRR. (5.6)
- Worsening of Pre-existing Hepatic Impairment: New onset or worsening encephalopathy, ascites or hepatorenal syndrome can occur in patients with Child-Pugh B or C cirrhosis. (5.7)
- Posterior Reversible Encephalopathy Syndrome: Permanently discontinue CYRAMZA. (5.8)
- Proteinuria Including Nephrotic Syndrome: Monitor for proteinuria. Withhold CYRAMZA for urine protein levels greater than or equal to 2 g per 24 hours. Permanently discontinue CYRAMZA for urine protein levels greater than 3 g per 24 hours or nephrotic syndrome. (5.9)
- Thyroid Dysfunction: Monitor thyroid function during treatment. (5.10)
- Embryo-Fetal Risk: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception (5.11, 8.1, 8.3)
ADVERSE REACTIONS
- The most common adverse reactions observed in single agent CYRAMZA-treated gastric cancer patients at a rate of ≥10% and ≥2% higher than placebo were hypertension and diarrhea. (6.1)
- The most common adverse reactions observed in patients treated with CYRAMZA with paclitaxel at a rate of ≥30% and ≥2% higher than placebo with paclitaxel were fatigue/asthenia, neutropenia, diarrhea, and epistaxis. (6.1)
- The most common adverse reactions observed in patients treated with CYRAMZA with docetaxel at a rate of ≥30% and ≥2% higher than placebo with docetaxel were neutropenia, fatigue/asthenia, and stomatitis/mucosal inflammation. (6.1)
- The most common adverse reactions observed in patients treated with CYRAMZA with FOLFIRI at a rate of ≥30% and ≥2% higher than placebo with FOLFIRI were diarrhea, neutropenia, decreased appetite, epistaxis, and stomatitis. (6.1)
- The most common adverse reactions observed in single agent CYRAMZA-treated HCC patients at a rate of ≥15% and ≥2% higher than placebo were fatigue, peripheral edema, hypertension, abdominal pain, decreased appetite, proteinuria, nausea, and ascites. The most common laboratory abnormalities at a rate of ≥30% and a ≥2% difference in incidence between arms were thrombocytopenia, hypoalbuminemia, and hyponatremia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eli Lilly and Company at 1-800-LillyRx (1-800-545-5979) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Gastric Cancer
1.2 Non-Small Cell Lung Cancer
1.3 Colorectal Cancer
1.4 Hepatocellular Carcinoma
2 DOSAGE AND ADMINISTRATION
2.1 Premedication
2.2 Recommended Dosage for Gastric Cancer
2.3 Recommended Dosage for Non-Small Cell Lung Cancer
2.4 Recommended Dosage for Colorectal Cancer
2.5 Recommended Dosage for Hepatocellular Carcinoma
2.6 Dosage Modifications for Adverse Reactions
2.7 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hemorrhage
5.2 Gastrointestinal Perforations
5.3 Impaired Wound Healing
5.4 Arterial Thromboembolic Events
5.5 Hypertension
5.6 Infusion-Related Reactions
5.7 Worsening of Pre-existing Hepatic Impairment
5.8 Posterior Reversible Encephalopathy Syndrome
5.9 Proteinuria Including Nephrotic Syndrome
5.10 Thyroid Dysfunction
5.11 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Gastric Cancer
14.2 Non-Small Cell Lung Cancer
14.3 Colorectal Cancer
14.4 Hepatocellular Carcinoma
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Gastric Cancer
CYRAMZA®, as a single agent or in combination with paclitaxel, is indicated for the treatment of patients with advanced or metastatic, gastric or gastro-esophageal junction (GEJ) adenocarcinoma with disease progression on or after prior fluoropyrimidine- or platinum-containing chemotherapy.
1.2 Non-Small Cell Lung Cancer
CYRAMZA, in combination with docetaxel, is indicated for the treatment of patients with metastatic non-small cell lung cancer (NSCLC) with disease progression on or after platinum-based chemotherapy. Patients with epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving CYRAMZA.
-
2 DOSAGE AND ADMINISTRATION
2.1 Premedication
- Prior to each CYRAMZA infusion, premedicate all patients with an intravenous histamine-1 receptor antagonist (e.g., diphenhydramine hydrochloride) [see Warnings and Precautions (5.6)].
- For patients who have experienced a Grade 1 or 2 IRR, premedicate with a histamine-1 receptor antagonist, dexamethasone (or equivalent), and acetaminophen prior to each CYRAMZA infusion [see Dosage and Administration (2.6)].
2.2 Recommended Dosage for Gastric Cancer
- The recommended dosage of CYRAMZA, either as a single agent or in combination with weekly paclitaxel, is 8 mg/kg every 2 weeks administered by intravenous infusion over 60 minutes. If the first infusion is tolerated, all subsequent CYRAMZA infusions may be administered over 30 minutes. Continue CYRAMZA until disease progression or unacceptable toxicity.
- When given in combination with paclitaxel, administer CYRAMZA prior to administration of paclitaxel.
- Refer to the prescribing information for paclitaxel for dosage information.
2.3 Recommended Dosage for Non-Small Cell Lung Cancer
- The recommended dosage of CYRAMZA is 10 mg/kg administered by intravenous infusion over 60 minutes on Day 1 of a 21-day cycle prior to docetaxel infusion. If the first infusion is tolerated, all subsequent CYRAMZA infusions may be administered over 30 minutes. Continue CYRAMZA until disease progression or unacceptable toxicity.
- Refer to the prescribing information for docetaxel for dosage information.
2.4 Recommended Dosage for Colorectal Cancer
- The recommended dosage of CYRAMZA is 8 mg/kg every 2 weeks administered by intravenous infusion over 60 minutes prior to FOLFIRI administration. If the first infusion is tolerated, all subsequent CYRAMZA infusions may be administered over 30 minutes. Continue CYRAMZA until disease progression or unacceptable toxicity.
- Refer to the prescribing information for fluorouracil, leucovorin, and irinotecan for dosage information.
2.5 Recommended Dosage for Hepatocellular Carcinoma
- The recommended dosage of CYRAMZA is 8 mg/kg every 2 weeks administered by intravenous infusion over 60 minutes. If the first infusion is tolerated, all subsequent CYRAMZA infusions may be administered over 30 minutes. Continue CYRAMZA until disease progression or unacceptable toxicity.
2.6 Dosage Modifications for Adverse Reactions
Reduce dose, withhold dose, or discontinue CYRAMZA to manage adverse reactions as described in Table 1.
Table 1: Dosage Modifications for CYRAMZA Adverse Reaction Severitya Dosage Modification a National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI CTCAE) version 4.0 used to identify adverse reactions
Hemorrhage
[see Warnings and Precautions (5.1)]Grade 3 or 4 Permanently discontinue CYRAMZA Gastrointestinal Perforation
[see Warnings and Precautions (5.2)]All Grades Permanently discontinue CYRAMZA Wound Healing Complications
[see Warnings and Precautions (5.3)]All Grades - Withhold CYRAMZA for 28 days prior to elective surgery. Resume CYRAMZA no sooner than 2 weeks after surgery and until adequate wound healing.
- The safety of resumption of CYRAMZA after resolution of wound healing complications has not been established.
Arterial Thromboembolic Events
[see Warnings and Precautions (5.4)]All Grades Permanently discontinue CYRAMZA Hypertension
[see Warnings and Precautions (5.5)]Severe hypertension Withhold CYRAMZA until controlled with medical management Severe hypertension that cannot be controlled with antihypertensive therapy Permanently discontinue CYRAMZA Infusion-Related Reaction (IRR)
[see Dosage and Administration (2.1), Warnings and Precautions (5.6)]Grade 1 or 2 IRR Reduce the infusion rate of CYRAMZA by 50% Grade 3 or 4 IRR Permanently discontinue CYRAMZA Posterior Reversible Encephalopathy Syndrome (PRES) [see Warnings and Precautions (5.8)] All Grades Permanently discontinue CYRAMZA Proteinuria
[see Warnings and Precautions (5.9)]First occurrence of increased urine protein levels greater than or equal to 2 g per 24 hours - Withhold CYRAMZA until urine protein level is less than 2 g per 24 hours
- Resume CYRAMZA at a reduced dose:
- Reduce 8 mg dose to 6 mg
- Reduce 10 mg dose to 8 mg
Reoccurrence of urine protein level greater than 2 g per 24 hours following initial dose reduction - Withhold CYRAMZA until urine protein level is less than 2 g per 24 hours
- Resume CYRAMZA at a reduced dose:
- Reduce 6 mg dose to 5 mg
- Reduce 8 mg dose to 6 mg
Urine protein level greater than 3 g per 24 hours or in the setting of nephrotic syndrome Permanently discontinue CYRAMZA 2.7 Preparation and Administration
Preparation
- Visually inspect vials for particulate matter and discoloration. Discard if particulate matter or discolorations are identified.
- Calculate the dose and the required volume of CYRAMZA needed for the calculated dose.
- Withdraw the required volume of CYRAMZA and further dilute with only 0.9% Sodium Chloride Injection in an intravenous infusion container to a final volume of 250 mL. Do not use dextrose containing solutions.
- Do not shake. Gently invert the container to ensure adequate mixing.
- Do not dilute with other solutions or co-infuse with other electrolytes or medications.
- Do not freeze. Store diluted solution for no more than 24 hours at 2°C to 8°C (36°F to 46°F) or 4 hours at room temperature (below 25°C [77°F]).
- Discard any unused portion of CYRAMZA.
Administration
- Visually inspect the diluted solution for particulate matter and discoloration prior to administration. Discard if particulate matter or discolorations are identified.
- Do not administer CYRAMZA as an intravenous push or bolus.
- Administer diluted CYRAMZA solution via infusion pump through a separate infusion line. Use of a protein sparing 0.22 micron filter is recommended.
- Flush the line with sterile 0.9% Sodium Chloride Injection at the end of the infusion.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hemorrhage
CYRAMZA increased the risk of hemorrhage and gastrointestinal hemorrhage, including Grade ≥3 hemorrhagic events. Across five clinical studies in 1916 patients with various cancers treated with CYRAMZA, the incidence of all Grade hemorrhage occurred between 13-44%. Grade 3-5 hemorrhage incidence ranged from 2-5% [see Adverse Reactions (6.1)].
Patients with gastric cancer receiving nonsteroidal anti-inflammatory drugs (NSAIDs) were excluded from enrollment in REGARD and RAINBOW; therefore, the risk of gastric hemorrhage in CYRAMZA-treated patients with gastric tumors receiving NSAIDs is unknown.
Patients with NSCLC receiving therapeutic anticoagulation or chronic therapy with NSAIDs or other anti-platelet therapy other than once daily aspirin or with radiographic evidence of major airway or blood vessel invasion or intratumor cavitation were excluded from REVEL; therefore, the risk of pulmonary hemorrhage in these groups of patients is unknown.
Permanently discontinue CYRAMZA in patients who experience severe (Grade 3 or 4) bleeding [see Dosage and Administration (2.6)].
5.2 Gastrointestinal Perforations
CYRAMZA can increase the risk of gastrointestinal perforation, a potentially fatal event. Across five clinical studies in 1916 patients with various cancers treated with CYRAMZA, the incidence of all Grade and Grade 3-5 gastrointestinal perforations ranged from <1-2% [see Adverse Reactions (6.1)].
Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation [see Dosage and Administration (2.6)].
5.3 Impaired Wound Healing
Impaired wound healing can occur in patients who receive drugs that inhibit the VEGF or VEGFR pathway. CYRAMZA, a VEGFR2 antagonist, has the potential to adversely affect wound healing. CYRAMZA has not been studied in patients with serious or non-healing wounds.
Withhold CYRAMZA for 28 days prior to elective surgery. Do not administer CYRAMZA for at least 2 weeks following a major surgical procedure and until adequate wound healing. The safety of resumption of CYRAMZA after resolution of wound healing complications has not been established [see Dosage and Administration (2.6)].
5.4 Arterial Thromboembolic Events
Serious, sometimes fatal, arterial thromboembolic events (ATEs), including myocardial infarction, cardiac arrest, cerebrovascular accident, and cerebral ischemia, occurred across clinical trials. Across five clinical studies in 1916 patients with various cancers treated with CYRAMZA, the incidence of all Grade ATE was 2-3%. Grade 3-5 ATE incidence was 1-2% [see Adverse Reactions (6.1)].
Permanently discontinue CYRAMZA in patients who experience an ATE [see Dosage and Administration (2.6)].
5.5 Hypertension
An increased incidence of severe hypertension occurred in patients receiving CYRAMZA. Across five clinical studies in 1916 patients with various cancers treated with CYRAMZA, the incidence of all Grade hypertension occurred between 11-26%. Grade 3-5 hypertension incidence ranged from 6-15% [see Adverse Reactions (6.1)].
Control hypertension prior to initiating treatment with CYRAMZA. Monitor blood pressure every two weeks or more frequently as indicated during treatment. Withhold CYRAMZA for severe hypertension until medically controlled. Permanently discontinue CYRAMZA for medically significant hypertension that cannot be controlled with antihypertensive therapy or in patients with hypertensive crisis or hypertensive encephalopathy [see Dosage and Administration (2.6)].
5.6 Infusion-Related Reactions
Infusion-related reactions (IRR), including severe and life threatening IRR, occurred in CYRAMZA clinical trials. The majority of IRR across trials occurred during or following a first or second CYRAMZA infusion. Symptoms of IRR included rigors/tremors, back pain/spasms, chest pain and/or tightness, chills, flushing, dyspnea, wheezing, hypoxia, and paresthesia. In severe cases, symptoms included bronchospasm, supraventricular tachycardia, and hypotension. Across five clinical studies in 1916 patients with various cancers treated with CYRAMZA in which premedication was recommended or required, the incidence of all Grade IRR occurred between <1-9%. Grade 3-5 IRR incidence was <1% [see Adverse Reactions (6.1)].
Premedicate prior to each CYRAMZA infusion [see Dosage and Administration (2.1)]. Monitor patients during the infusion for signs and symptoms of IRR in a setting with available resuscitation equipment. Premedicate prior to each CYRAMZA infusion [see Dosage and Administration (2.1)]. Reduce the infusion rate by 50% for Grade 1-2 IRR. Permanently discontinue CYRAMZA for Grade 3-4 IRR [see Dosage and Administration (2.6)].
5.7 Worsening of Pre-existing Hepatic Impairment
Clinical deterioration, manifested by new onset or worsening encephalopathy, ascites, or hepatorenal syndrome, was reported in patients with Child-Pugh B or C cirrhosis who received single agent CYRAMZA. Use CYRAMZA in patients with Child-Pugh B or C cirrhosis only if the potential benefits of treatment are judged to outweigh the risks of clinical deterioration.
Based on safety data from REACH-2, in patients with Child-Pugh A liver cirrhosis, the pooled incidence of hepatic encephalopathy and hepatorenal syndrome was higher for patients who received CYRAMZA (6%) compared to patients who received placebo (0%) [see Adverse Reactions (6.1)].
5.8 Posterior Reversible Encephalopathy Syndrome
Posterior Reversible Encephalopathy Syndrome (PRES) (also known as Reversible Posterior Leukoencephalopathy Syndrome [RPLS]) has been reported in <0.1% of 1916 patients enrolled in five clinical studies with CYRAMZA. Symptoms of PRES include seizure, headache, nausea/vomiting, blindness, or altered consciousness, with or without associated hypertension.
Confirm the diagnosis of PRES with magnetic resonance imaging and permanently discontinue CYRAMZA in patients who develop PRES. Symptoms may resolve or improve within days, although some patients with PRES can experience ongoing neurologic sequelae or death [see Dosage and Administration (2.6)].
5.9 Proteinuria Including Nephrotic Syndrome
Across five clinical studies in 1916 patients with various cancers treated with CYRAMZA, the incidence of all Grade proteinuria ranged from 3-20%. Grade ≥3 proteinuria (including 4 patients with nephrotic syndrome) incidence ranged from <1-3% [see Adverse Reactions (6.1)].
Monitor proteinuria by urine dipstick and/or urinary protein creatinine ratio. If the result of the urine dipstick is 2+ or greater, perform a 24-hour urine collection for protein measurement. Withhold CYRAMZA for urine protein levels that are 2 or more grams over 24 hours. Reinitiate CYRAMZA at a reduced dose once the urine protein level returns to less than 2 grams over 24 hours. Permanently discontinue CYRAMZA for urine protein levels greater than 3 grams over 24 hours or in the setting of nephrotic syndrome [see Dosage and Administration (2.6)].
5.10 Thyroid Dysfunction
Across five clinical studies in 1916 patients with various cancers treated with CYRAMZA, the incidence of Grade 1-2 hypothyroidism ranged from <1-3%; there were no reports of Grade 3-5 hypothyroidism [see Adverse Reactions (6.1)]. Monitor thyroid function during treatment with CYRAMZA.
5.11 Embryo-Fetal Toxicity
Based on its mechanism of action, CYRAMZA can cause fetal harm when administered to pregnant women. Animal models link angiogenesis, VEGF and VEGFR2 to critical aspects of female reproduction, embryo-fetal development, and postnatal development. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with CYRAMZA and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
- Hemorrhage [see Warnings and Precautions (5.1)].
- Gastrointestinal Perforations [see Warnings and Precautions (5.2)].
- Impaired Wound Healing [see Warnings and Precautions (5.3)].
- Arterial Thromboembolic Events [see Warnings and Precautions (5.4)].
- Hypertension [see Warnings and Precautions (5.5)].
- Infusion-Related Reactions [see Warnings and Precautions (5.6)].
- Worsening of Pre-existing Hepatic Impairment [see Warnings and Precautions (5.7)].
- Posterior Reversible Encephalopathy Syndrome [see Warnings and Precautions (5.8)].
- Proteinuria Including Nephrotic Syndrome [see Warnings and Precautions (5.9)].
- Thyroid Dysfunction [see Warnings and Precautions (5.10)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the Warnings and Precautions section reflect exposure to CYRAMZA in 1916 patients from five studies: REGARD, RAINBOW, RAISE, REVEL, and REACH-2.
Gastric Cancer
The safety of CYRAMZA was evaluated in REGARD and RAINBOW [see Clinical Studies (14.1)]. Patients in both trials had locally advanced or metastatic gastric cancer (including GEJ adenocarcinoma) and had previously received platinum- or fluoropyrimidine-containing chemotherapy. Patients had Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1. Both trials excluded patients with uncontrolled hypertension, major surgery within 28 days, or patients receiving chronic anti-platelet therapy other than once daily aspirin. REGARD excluded patients with bilirubin ≥1.5 mg/dL and RAINBOW excluded patients with bilirubin >1.5 times the upper limit of normal (ULN).
CYRAMZA Administered as a Single Agent (REGARD)
Patients received either CYRAMZA 8 mg/kg or placebo intravenously every two weeks. Patients randomized to CYRAMZA received a median of 4 doses; the median duration of exposure was 8 weeks and 32 (14% of 236) patients received CYRAMZA for at least six months.
The most common serious adverse reactions with CYRAMZA were anemia (3.8%) and intestinal obstruction (2.1%). Red blood cell transfusions were given to 11% of CYRAMZA-treated patients versus 8.7% of patients who received placebo.
The most common adverse reactions (all grades) observed in CYRAMZA-treated patients at a rate of ≥10% and ≥2% higher than placebo were hypertension and diarrhea. Table 2 provides the frequency and severity of adverse reactions (CTCAE, version 4.0) in REGARD.
Table 2: Adverse Reactions Occurring in ≥5% of Patients with a ≥2% Difference Between Arms in REGARD a Hypertension is a consolidated term.
Adverse Reactions CYRAMZA
(N=236)Placebo
(N=115)All Grades
(%)Grade 3-4
(%)All Grades
(%)Grade 3-4
(%)Vascular Hypertensiona 16 8 8 3 Gastrointestinal Diarrhea 14 1 9 2 Nervous System Headache 9 0 3 0 Metabolism and Nutrition Hyponatremia 6 3 2 1 Clinically relevant adverse reactions reported in ≥1% and <5% of CYRAMZA-treated patients in REGARD were:
- Neutropenia (4.7%)
- Epistaxis (4.7%)
- Rash (4.2%)
- Intestinal obstruction (2.1%)
- Arterial thromboembolic events (1.7%)
Across clinical trials of CYRAMZA administered as a single agent, clinically relevant adverse reactions (including Grade ≥3) reported in CYRAMZA-treated patients included proteinuria, gastrointestinal perforation, and IRR. In REGARD, according to laboratory assessment, 8% of CYRAMZA-treated patients developed proteinuria versus 3% of placebo-treated patients. Two patients discontinued CYRAMZA due to proteinuria. The rate of gastrointestinal perforation in REGARD was 0.8% and the rate of IRR was 0.4%.
CYRAMZA Administered in Combination with Paclitaxel (RAINBOW)
Patients received paclitaxel 80 mg/m2 on Days 1, 8, and 15 of each 28-day cycle with either CYRAMZA 8 mg/kg or placebo intravenously every two weeks. Patients randomized to CYRAMZA received a median of 9 doses; the median duration of exposure was 18 weeks, and 93 (28% of 327) patients received CYRAMZA for at least six months.
The most common serious adverse reactions in patients who received CYRAMZA with paclitaxel were neutropenia (3.7%) and febrile neutropenia (2.4%); 19% of patients who received CYRAMZA with paclitaxel received granulocyte colony-stimulating factors.
Adverse reactions resulting in discontinuation of any component of the CYRAMZA with paclitaxel combination in ≥2% of patients in RAINBOW were neutropenia (4%) and thrombocytopenia (3%).
The most common adverse reactions (all grades) observed in patients who received CYRAMZA with paclitaxel at a rate of ≥30% and ≥2% higher than placebo with paclitaxel were fatigue/asthenia, neutropenia, diarrhea, and epistaxis. Table 3 provides the frequency and severity of adverse reactions (CTCAE, version 4.0) in RAINBOW.
Table 3: Adverse Reactions Occurring in ≥5% of Patients with a ≥2% Difference Between Arms in RAINBOW a Neutropenia, gastrointestinal hemorrhage events, hypertension, proteinuria, and hypoalbuminemia are consolidated terms.
b Includes 1 fatal event in the CYRAMZA arm.
Adverse Reactions CYRAMZA with Paclitaxel
(N=327)Placebo with Paclitaxel
(N=329)All Grades
(%)≥ Grade 3
(%)All Grades
(%)≥ Grade 3
(%)General Fatigue/Asthenia 57 12 44 6 Peripheral edema 25 2 14 1 Hematology Neutropeniaa 54 41 31 19 Thrombocytopenia 13 2 6 2 Gastrointestinal Diarrhea 32 4 23 2 Stomatitis 20 1 7 1 Gastrointestinal hemorrhage eventsa,b 10 4 6 2 Respiratory, Thoracic, and Mediastinal Epistaxis 31 0 7 0 Vascular Hypertensiona 25 15 6 3 Renal and Urinary Proteinuriaa 17 1 6 0 Metabolism and Nutrition Hypoalbuminemiaa 11 1 5 1 Clinically relevant adverse reactions reported in ≥1% and <5% of patients receiving CYRAMZA with paclitaxel were:
- Sepsis (3.1%), including 5 fatal events
- Gastrointestinal perforations (1.2%), including 1 fatal event
Non-Small Cell Lung Cancer
The safety of CYRAMZA was evaluated in REVEL [see Clinical Studies (14.2)]. Patients had NSCLC with disease progression on or after one platinum-based therapy for locally advanced or metastatic disease and ECOG PS 0 or 1. REVEL excluded patients with bilirubin greater than the ULN, uncontrolled hypertension, major surgery within 28 days, radiographic evidence of major airway or blood vessel invasion by cancer, radiographic evidence of intra-tumor cavitation, or gross hemoptysis within the preceding 2 months, and patients receiving therapeutic anticoagulation or chronic anti-platelet therapy other than once daily aspirin.
Patients received either CYRAMZA 10 mg/kg or placebo intravenously in combination with docetaxel 75 mg/m2 intravenously every 21 days. Due to an increased incidence of neutropenia and febrile neutropenia in patients enrolled in East Asian sites, REVEL was amended and 24 patients (11 patients receiving CYRAMZA with docetaxel, 13 patients receiving placebo with docetaxel) at East Asian sites received a starting dose of docetaxel at 60 mg/m2 every three weeks. Patients randomized to CYRAMZA received a median of 4.5 doses; the median duration of exposure was 3.5 months, and 195 (31% of 627) patients received CYRAMZA for at least six months.
The most common serious adverse reactions in patients who received CYRAMZA with docetaxel were febrile neutropenia (14%), pneumonia (6%), and neutropenia (5%). The use of granulocyte colony-stimulating factors was 42% in CYRAMZA with docetaxel-treated patients versus 37% in patients who received placebo with docetaxel.
The most common adverse reactions leading to treatment discontinuation of CYRAMZA were IRR (0.5%) and epistaxis (0.3%). For patients with non-squamous histology, the overall incidence of pulmonary hemorrhage was 7% and the incidence of Grade ≥3 pulmonary hemorrhage was 1% for CYRAMZA with docetaxel compared to 6% overall incidence and 1% for Grade ≥3 pulmonary hemorrhage for placebo with docetaxel. For patients with squamous histology, the overall incidence of pulmonary hemorrhage was 10% and the incidence of Grade ≥3 pulmonary hemorrhage was 2% for CYRAMZA with docetaxel compared to 12% overall incidence and 2% for Grade ≥3 pulmonary hemorrhage for placebo with docetaxel. Treatment discontinuation due to adverse reactions occurred more frequently in CYRAMZA with docetaxel-treated patients (9%) than in placebo with docetaxel-treated patients (5%).
The most common adverse reactions (all grades) observed in CYRAMZA with docetaxel-treated patients at a rate of ≥30% and ≥2% higher than placebo with docetaxel were neutropenia, fatigue/asthenia, and stomatitis/mucosal inflammation. Table 4 provides the frequency and severity of adverse reactions (NCI CTCAE, version 4.0) in REVEL.
Table 4: Adverse Reactions Occurring in ≥5% of Patients with a ≥2% Difference Between Arms in REVEL a Neutropenia, thrombocytopenia, and hypertension are consolidated terms.
Adverse Reactions CYRAMZA with Docetaxel
(N=627)Placebo with Docetaxel
(N=618)All Grades
(%)Grade 3-4
(%)All Grades
(%)Grade 3-4
(%)Hematology Neutropeniaa 55 49 46 40 Febrile neutropenia 16 16 10 10 Thrombocytopeniaa 13 3 5 <1 General Fatigue/Asthenia 55 14 50 11 Peripheral edema 16 0 9 <1 Gastrointestinal Stomatitis/Mucosal inflammation 37 7 19 2 Respiratory, Thoracic, and Mediastinal Epistaxis 19 <1 7 <1 Eye Lacrimation increased 13 <1 5 0 Vascular Hypertensiona 11 6 5 2 Clinically relevant adverse drug reactions reported in ≥1% and <5% of CYRAMZA with docetaxel-treated patients in REVEL were:
- Hyponatremia (4.8%)
- Proteinuria (3.3%)
Colorectal Cancer
The safety of CYRAMZA was evaluated in RAISE [see Clinical Studies (14.3)]. Patients had mCRC with disease progression on or after therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine and ECOG PS 0 or 1. RAISE excluded patients with uncontrolled hypertension, major surgery within 28 days, and those who experienced any of the following during first-line therapy with a bevacizumab-containing regimen: an arterial thrombotic/thromboembolic event; Grade 4 hypertension; Grade 3 proteinuria; a Grade 3-4 bleeding event; or bowel perforation.
Patients received either CYRAMZA 8 mg/kg or placebo intravenously in combination with FOLFIRI intravenously every two weeks. Patients randomized to CYRAMZA received a median of 8 doses (range 1-68); the median duration of exposure was 4.4 months, and 169 (32% of 529) patients received CYRAMZA for at least six months.
The most common serious adverse reactions with CYRAMZA with FOLFIRI were diarrhea (3.6%), intestinal obstruction (3.0%), and febrile neutropenia (2.8%).
Treatment discontinuation of any study drug due to adverse reactions occurred more frequently in CYRAMZA with FOLFIRI-treated patients (29%) than in placebo with FOLFIRI-treated patients (13%). The most common adverse reactions leading to discontinuation of any component of CYRAMZA with FOLFIRI as compared to placebo with FOLFIRI were neutropenia (12.5% versus 5.3%) and thrombocytopenia (4.2% versus 0.8%). The most common adverse reactions leading to treatment discontinuation of CYRAMZA were proteinuria (1.5%) and gastrointestinal perforation (1.7%).
The most common adverse reactions (all grades) observed in CYRAMZA with FOLFIRI-treated patients at a rate of ≥30% and ≥2% higher than placebo with FOLFIRI were diarrhea, neutropenia, decreased appetite, epistaxis, and stomatitis. Twenty percent of patients treated with CYRAMZA with FOLFIRI received granulocyte colony-stimulating factors. Table 5 provides the frequency and severity of adverse reactions (CTCAE, version 4.0) in RAISE.
Table 5: Adverse Reactions Occurring in ≥5% of Patients with a ≥2% Difference Between Arms in RAISE a Gastrointestinal hemorrhage events, neutropenia, thrombocytopenia, hypertension, proteinuria, and hypoalbuminemia, are consolidated terms.
b Includes 3 fatal events in the CYRAMZA arm.
c Includes 3 patients with nephrotic syndrome in the CYRAMZA arm.
Adverse Reactions CYRAMZA with FOLFIRI
(N=529)Placebo with FOLFIRI
(N=528)All Grades
(%)≥ Grade 3
(%)All Grades
(%)≥ Grade 3
(%)Gastrointestinal Diarrhea 60 11 51 10 Decreased appetite 37 2 27 2 Stomatitis 31 4 21 2 Gastrointestinal hemorrhage eventsa,b 12 2 7 1 Hematology Neutropeniaa 59 38 46 23 Thrombocytopeniaa 28 3 14 <1 Respiratory, Thoracic, and Mediastinal Epistaxis 33 0 15 0 Vascular Hypertensiona 26 11 9 3 General Peripheral edema 20 <1 9 0 Renal and Urinary Proteinuriaa,c 17 3 5 <1 Skin and Subcutaneous Tissue Palmar-plantar erythrodysesthesia syndrome 13 1 5 <1 Metabolism and Nutrition Hypoalbuminemiaa 6 1 2 0 Clinically relevant adverse reactions reported in ≥1% and <5% of patients receiving CYRAMZA with FOLFIRI were:
- Gastrointestinal perforation (1.7%) including 4 fatal events
Thyroid stimulating hormone (TSH) levels were evaluated in 224 patients (115 CYRAMZA with FOLFIRI-treated patients and 109 placebo with FOLFIRI-treated patients) with normal baseline TSH levels. Patients underwent periodic TSH laboratory assessments until 30 days after the last dose of study treatment. Increased TSH levels were observed in 53 (46%) patients treated with CYRAMZA with FOLFIRI compared with 4 (4%) patients treated with placebo with FOLFIRI.
Hepatocellular Carcinoma
The safety of CYRAMZA was evaluated in REACH-2 [see Clinical Studies (14.4)]. Patients had Barcelona Clinic Liver Cancer (BCLC) stage B HCC who were no longer amenable to locoregional therapy, or BCLC stage C HCC, Child-Pugh score A, and baseline AFP ≥400 ng/mL. Patients had ECOG PS 0 or 1. REACH-2 excluded patients with clinically meaningful ascites, history of or current hepatic encephalopathy, uncontrolled hypertension, major surgery within 28 days, bilirubin >1.5 times ULN, severe variceal bleeding in the 3 months prior to treatment or with varices at high risk of bleeding, and patients receiving therapeutic anticoagulation or chronic anti-platelet therapy other than once daily aspirin.
Patients received either CYRAMZA 8 mg/kg or placebo intravenously every two weeks. Patients received a median of 6 doses (range 1-51) of CYRAMZA; the median duration of exposure was 12 weeks (range 2-107 weeks) and 48 patients (24% of 197) received CYRAMZA for at least six months.
The most common serious adverse reactions with CYRAMZA were ascites (3%) and pneumonia (3%).
Treatment discontinuations due to adverse reactions occurred in 18% of CYRAMZA-treated patients, with proteinuria being the most frequent (2%).
The most common adverse reactions reported in ≥15% of patients and ≥2% higher than placebo were fatigue, peripheral edema, hypertension, abdominal pain, decreased appetite, proteinuria, nausea, and ascites. The most common laboratory abnormalities ≥30% and ≥2% higher than placebo were thrombocytopenia, hypoalbuminemia, and hyponatremia. Table 6 provides the frequency and severity of adverse reactions (CTCAE, version 4.0) and Table 7 provides the incidence and severity of laboratory abnormalities in REACH-2.
Table 6: Adverse Reactions Occurring in ≥10% of Patients with a ≥2% Difference Between Arms in REACH-2 a Fatigue, hypertension, abdominal pain, and proteinuria are consolidated terms.
b Includes 1 fatal event in the CYRAMZA arm.
c Includes 1 patient with nephrotic syndrome in the CYRAMZA arm.
Adverse Reactions CYRAMZA
(N=197)Placebo
(N=95)All Grades
(%)≥ Grade 3
(%)All Grades
(%)≥ Grade 3
(%)General Fatiguea 36 5 20 3 Peripheral edema 25 2 14 0 Decreased appetite 23 2 20 1 Insomnia 11 0 6 1 Pyrexia 10 0 3 0 Vascular Hypertensiona 25 13 13 5 Gastrointestinal Abdominal Paina 25 2 16 2 Nausea 19 0 12 0 Ascitesb 18 4 7 1 Vomiting 10 0 7 0 Renal and Urinary Proteinuriaa, c 20 2 4 0 Nervous System Headache 14 0 5 1 Respiratory, Thoracic, and Mediastinal Epistaxis 14 <1 3 0 Musculoskeletal Back Pain 10 <1 7 1 Clinically relevant adverse drug reactions reported in ≥1% and <10% of CYRAMZA treated patients in REACH-2 were:
- IRR (9%)
- Hepatic encephalopathy (5%) including 1 fatal event
- Hepatorenal syndrome (2%) including 1 fatal event
Table 7: Laboratory Abnormalities Worsening from Baseline in ≥15% (All Grades) of Patients Receiving CYRAMZA with a Difference Between Arms of ≥2% in REACH-2 a Laboratory abnormalities were not included if the ≥ Grade 3 percentage was less than placebo-treated patients.
b The denominator used to calculate the incidence varied based on the number of patients with a baseline and at least one on study laboratory measurement: CYRAMZA-treated patients (range 179-193 patients) and placebo-treated patients (range 84-92 patients).
Laboratory Abnormalitya CYRAMZAb Placebob All Grades
(%)≥ Grade 3
(%)All Grades
(%)≥ Grade 3
(%)Hematology Thrombocytopenia 46 8 15 1 Neutropenia 24 8 12 3 Chemistry Hypoalbuminemia 33 <1 16 0 Hyponatremia 32 16 25 5 Hypocalcemia 16 2 5 0 6.2 Immunogenicity
As with all therapeutic proteins, there is the potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to CYRAMZA with the incidences of antibodies to other products may be misleading.
In clinical trials, 86/2890 (3%) of CYRAMZA-treated patients tested positive for treatment-emergent anti-ramucirumab antibodies by an enzyme-linked immunosorbent assay (ELISA). Neutralizing antibodies were detected in 14 of the 86 patients who tested positive for treatment-emergent anti-ramucirumab antibodies.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of CYRAMZA. Because such reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Blood and lymphatic system: Thrombotic microangiopathy
- Neoplasms benign, malignant and unspecified: Hemangioma
- Respiratory, thoracic, and mediastinal: Dysphonia
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action [see Clinical Pharmacology (12.1)], CYRAMZA can cause fetal harm when administered to a pregnant woman. There are no available data on CYRAMZA use in pregnant women. Animal models link angiogenesis, VEGF and VEGFR2 to critical aspects of female reproduction, embryo-fetal development, and postnatal development. No animal studies have been conducted to evaluate the effect of ramucirumab on reproduction and fetal development. Advise a pregnant woman of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
No animal studies have been specifically conducted to evaluate the effect of ramucirumab on reproduction and fetal development. In mice, loss of the VEGFR2 gene resulted in embryo-fetal death and these fetuses lacked organized blood vessels and blood islands in the yolk sac. In other models, VEGFR2 signaling was associated with development and maintenance of endometrial and placental vascular function, successful blastocyst implantation, maternal and feto-placental vascular differentiation, and development during early pregnancy in rodents and non-human primates. Disruption of VEGF signaling has also been associated with developmental anomalies including poor development of the cranial region, forelimbs, forebrain, heart, and blood vessels.
8.2 Lactation
Risk Summary
There is no information on the presence of ramucirumab in human milk or its effects on the breastfed child or on milk production. Human IgG is present in human milk, but published data suggest that breast milk antibodies do not enter the neonatal and infant circulation in substantial amounts. Because of the potential risk for serious adverse reactions in breastfed children from ramucirumab, advise women not to breastfeed during treatment with CYRAMZA and for 2 months after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating CYRAMZA [see Use in Specific Populations (8.1)].
Contraception
Based on its mechanism of action, CYRAMZA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Infertility
Females
Advise females of reproductive potential that based on animal data CYRAMZA may impair fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of CYRAMZA in pediatric patients have not been established.
Juvenile Animal Toxicity Data
In animal studies, effects on epiphyseal growth plates were identified. In cynomolgus monkeys, anatomical pathology revealed adverse effects on the epiphyseal growth plate (thickening and osteochondropathy) at all doses tested (5-50 mg/kg). Ramucirumab exposure at the lowest weekly dose tested in the cynomolgus monkey was 0.2 times the exposure in humans at the recommended dose of ramucirumab as a single agent.
8.5 Geriatric Use
Of the 563 CYRAMZA-treated patients in REGARD and RAINBOW, 205 (36%) were 65 and over, while 41 (7%) were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects.
Of the 1253 patients in REVEL, 455 (36%) were 65 and over and 84 (7%) were 75 and over. Of the 627 patients who received CYRAMZA with docetaxel in REVEL, 237 (38%) were 65 and over, while 45 (7%) were 75 and over. In an exploratory subgroup analysis of REVEL, the hazard ratio for overall survival in patients less than 65 years old was 0.74 (95% CI: 0.62, 0.87) and in patients 65 years and over was 1.10 (95% CI: 0.89, 1.36).
Of the 529 patients who received CYRAMZA with FOLFIRI in RAISE, 209 (40%) were 65 and over, while 51 (10%) were 75 and over. Overall, no differences in safety or effectiveness were observed between these subjects and younger subjects.
Of the 197 patients who received CYRAMZA in REACH-2, 95 (48%) were 65 years and over, while 37 (19%) were 75 years and over. Overall, no differences in efficacy were observed between these subjects and younger subjects.
8.6 Hepatic Impairment
No dose adjustment is recommended for patients with mild (total bilirubin within ULN and aspartate aminotransferase [AST] >ULN or total bilirubin >1 to 1.5 times ULN and any AST) or moderate (total bilirubin >1.5 to 3 times ULN and any AST) hepatic impairment. Clinical deterioration was reported in patients with Child-Pugh B or C cirrhosis who received single agent CYRAMZA [see Warnings and Precautions (5.7), Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
Ramucirumab is a human VEGFR2 antagonist. It is a recombinant human IgG1 monoclonal antibody. Ramucirumab has an approximate molecular weight of 147 kDa. Ramucirumab is produced in genetically engineered mammalian NS0 cells.
CYRAMZA (ramucirumab) injection for intravenous use is a sterile, preservative-free, clear to slightly opalescent and colorless to slightly yellow solution. CYRAMZA is supplied at a concentration of 10 mg/mL in either 100 mg (10 mL) or 500 mg (50 mL) single-dose vials. CYRAMZA is formulated in glycine (9.98 mg/mL), histidine (0.65 mg/mL), histidine monohydrochloride (1.22 mg/mL), polysorbate 80 (0.1 mg/mL), sodium chloride (4.383 mg/mL), and Water for Injection, USP, pH 6.0.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ramucirumab is a VEGFR2 antagonist that specifically binds VEGFR2 and blocks binding of VEGFR ligands, VEGF-A, VEGF-C, and VEGF-D. As a result, ramucirumab inhibits ligand-stimulated activation of VEGFR2, thereby inhibiting ligand-induced proliferation, and migration of human endothelial cells. Ramucirumab inhibited angiogenesis in an in vivo animal model.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of ramucirumab were studied in patients with various cancers over a dose range of 6-12 mg/kg administered every two or three weeks. The PK characteristics of ramucirumab are similar for patients across cancer types based on a population PK analysis. Ramucirumab systemic exposure increased dose proportionally at doses of 8 mg/kg and above and steady state concentrations were achieved at approximately 12 weeks.
Distribution
The mean (% coefficient of variation [CV%]) volume of distribution of ramucirumab at steady-state (Vss) was 5.4 L (15%).
Elimination
The mean (CV%) clearance of ramucirumab was 0.015 L/hour (30%) and the mean elimination half-life was 14 days (20%).
Specific Populations
Age (19-88 years), sex (68% male), race (70% White, 24% Asian), renal impairment (creatinine clearance [CLcr] calculated by Cockcroft-Gault, 15-89 mL/min, mild hepatic impairment (total bilirubin within ULN and AST>ULN or total bilirubin >1 to 1.5 times ULN and any AST), or moderate hepatic impairment (total bilirubin >1.5 to 3 times ULN) had no clinically meaningful effect on the PK of ramucirumab. The effect of severe hepatic impairment (total bilirubin >3 times ULN and any AST) on the PK of ramucirumab is unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No animal studies have been performed to test ramucirumab for potential carcinogenicity or genotoxicity.
Inhibition of VEGFR2 signaling in animal models was shown to result in changes to hormone levels critical for pregnancy, and, in monkeys, an increased duration of the follicular cycle. In a 39 week animal study, female monkeys treated with ramucirumab showed dose dependent increases in follicular mineralization of the ovary.
13.2 Animal Toxicology and/or Pharmacology
Adverse effects in the kidney (glomerulonephritis) occurred in monkeys at doses of 16-50 mg/kg (0.7-5.5 times the exposure in humans at the recommended dose of ramucirumab as a single agent).
A single dose of ramucirumab resulting in an exposure approximately 10 times the human exposure at the recommended dose of ramucirumab as a single agent did not significantly impair wound healing in monkeys using a full-thickness incisional model.
-
14 CLINICAL STUDIES
14.1 Gastric Cancer
REGARD
The efficacy of CYRAMZA was evaluated in REGARD (NCT00917384), a multinational, randomized, double-blind, multicenter study in patients with locally advanced or metastatic gastric cancer (including adenocarcinoma of the GEJ) who previously received platinum- or fluoropyrimidine-containing chemotherapy. Patients were required to have experienced disease progression either within 4 months after the last dose of first-line therapy for locally advanced or metastatic disease or within 6 months after the last dose of adjuvant therapy. Patients were also required to have ECOG PS of 0 or 1. Patients were randomized (2:1) to receive either an intravenous infusion of CYRAMZA 8 mg/kg or placebo every 2 weeks. Randomization was stratified by weight loss over the prior 3 months (≥10% versus <10%), geographic region, and location of the primary tumor (gastric versus GEJ). The major efficacy outcome measure was overall survival (OS). An additional efficacy outcome measure was progression-free survival (PFS).
A total of 355 patients were randomized, 238 to the CYRAMZA-treatment group and 117 to the placebo-treatment group. Baseline demographic and disease characteristics were similar between treatment arms. The median age was 60 years (range 24-87); 70% were men; 77% were White, 16% Asian; 28% had ECOG PS 0 and 72% had ECOG PS 1; 91% had measurable disease; 75% had gastric cancer; and 25% had adenocarcinoma of the GEJ. The majority of patients (85%) experienced disease progression during or following first-line therapy for metastatic disease. Prior chemotherapy for gastric cancer consisted of platinum/fluoropyrimidine combination therapy (81%), fluoropyrimidine-containing regimens without platinum (15%), and platinum-containing regimens without fluoropyrimidine (4%). Patients received a median of 4 doses (range 1-34) of CYRAMZA or a median of 3 doses (range 1-30) of placebo.
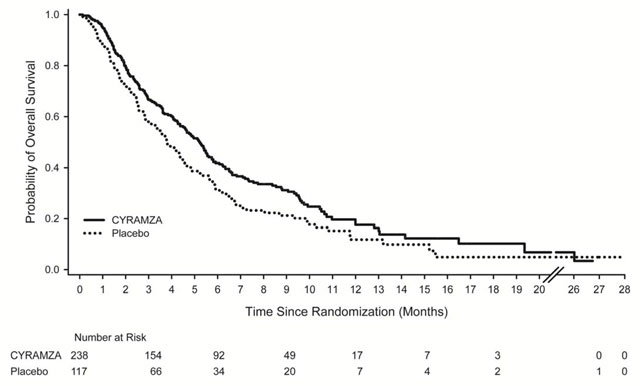
Efficacy results are shown in Table 8 and Figure 1.
Table 8: Efficacy Results in REGARD Abbreviations: BSC = best supportive care; CI = confidence interval
a 65 of 199 events in CYRAMZA-treated patients and 31 of 108 events in placebo-treated patients were deaths.
CYRAMZA + BSC
N=238Placebo + BSC
N=117Overall Survival Number of deaths (%) 179 (75%) 99 (85%) Median – months (95% CI) 5.2 (4.4, 5.7) 3.8 (2.8, 4.7) Hazard Ratio (95% CI) 0.78 (0.60, 0.998) Stratified Log-rank p-value 0.047 Progression-free Survival Number of events (%)a 199 (84%) 108 (92%) Median – months (95% CI) 2.1 (1.5, 2.7) 1.3 (1.3, 1.4) Hazard Ratio (95% CI) 0.48 (0.38, 0.62) Stratified Log-rank p-value <0.001 Figure 1: Kaplan-Meier Curves for Overall Survival in REGARD
RAINBOW
The efficacy of CYRAMZA was evaluated in RAINBOW (NCT01170663), a multinational, randomized, double-blind study in patients with locally advanced or metastatic gastric cancer (including adenocarcinoma of the GEJ) who previously received platinum- and fluoropyrimidine-containing chemotherapy. Patients were required to have experienced disease progression during, or within 4 months after the last dose of first-line therapy. Patients were also required to have ECOG PS of 0 or 1. Patients were randomized (1:1) to receive either CYRAMZA 8 mg/kg or placebo as an intravenous infusion every 2 weeks (on Days 1 and 15) of each 28-day cycle. Patients in both arms received paclitaxel 80 mg/m2 by intravenous infusion on Days 1, 8, and 15 of each 28-day cycle. Prior to administration of each dose of paclitaxel, patients were required to have adequate hematopoietic and hepatic function. The paclitaxel dose was permanently reduced, in increments of 10 mg/m2, for a maximum of two dose reductions for Grade 4 hematologic toxicity or Grade 3 paclitaxel-related non-hematologic toxicity. Randomization was stratified by geographic region, time to progression from the start of first-line therapy (<6 months versus ≥6 months), and disease measurability. The major efficacy outcome measure was OS. Additional efficacy outcome measures were PFS and overall response rate (ORR).
A total of 665 patients were randomized, 330 to the CYRAMZA-treatment group and 335 to the placebo-treatment group. Baseline demographics and disease characteristics were similar between treatment arms. The median age was 61 years (range: 24-84); 71% were men; 61% were White, 35% Asian; 39% had ECOG PS 0 and 61% had ECOG PS 1; 78% had measurable disease; 79% had gastric cancer; and 21% had adenocarcinoma of the GEJ. Two-thirds of the patients experienced disease progression while on first-line therapy (67%) and 25% of patients received an anthracycline in combination with platinum/fluoropyrimidine combination therapy.
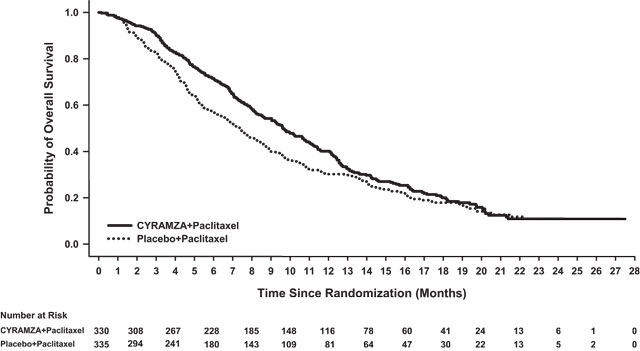
Efficacy results are shown in Table 9 and Figure 2.
Table 9: Efficacy Results in RAINBOW Abbreviations: CI = confidence interval, CMH = Cochran-Mantel-Haenszel
a 56 of 279 events in CYRAMZA-treated patients and 55 of 296 events in placebo-treated patients were deaths.
b 2 complete responses in CYRAMZA-treated patients and 1 complete response in placebo-treated patients.
CYRAMZA + paclitaxel
N=330Placebo + paclitaxel
N=335Overall Survival Number of deaths (%) 256 (78%) 260 (78%) Median – months (95% CI) 9.6 (8.5, 10.8) 7.4 (6.3, 8.4) Hazard Ratio (95% CI) 0.81 (0.68, 0.96) Stratified Log-rank p-value 0.017 Progression-free Survival Number of events (%)a 279 (85%) 296 (88%) Median – months (95% CI) 4.4 (4.2, 5.3) 2.9 (2.8, 3.0) Hazard Ratio (95% CI) 0.64 (0.54, 0.75) Stratified Log-rank p-value <0.001 Overall Response Rateb Rate – percent (95% CI) 28 (23, 33) 16 (13, 20) Stratified CMH p-value <0.001 Figure 2: Kaplan-Meier Curves for Overall Survival in RAINBOW
14.2 Non-Small Cell Lung Cancer
The efficacy of CYRAMZA was evaluated in REVEL (NCT01168973), a multinational, randomized, double-blind study in patients with NSCLC with disease progression on or after one platinum-based therapy for locally advanced or metastatic disease. Patients in REVEL were also required to have ECOG PS 0 or 1. Patients were randomized (1:1) to receive either CYRAMZA at 10 mg/kg or placebo by intravenous infusion, in combination with docetaxel at 75 mg/m2, every 21 days. Sites in East Asia administered a reduced dose of docetaxel at 60 mg/m2 every 21 days. Patients who discontinued combination therapy because of an adverse reaction attributed to either CYRAMZA/placebo or docetaxel were permitted to continue monotherapy with the other treatment component until disease progression or intolerable toxicity. Randomization was stratified by geographic region, sex, prior maintenance therapy, and ECOG PS. The major efficacy outcome measure was OS. Additional efficacy outcome measures included PFS and ORR.
A total of 1253 patients were randomized, 628 to the CYRAMZA-treatment group and 625 to the placebo-treatment group. Baseline demographics and disease characteristics were similar between treatment arms. The median age was 62 years (range 21-86); 67% were men; 82% were White and 13% were Asian; 32% had ECOG PS 0; 73% had nonsquamous histology and 26% had squamous histology. In addition to platinum chemotherapy (99%), the most common prior therapies were pemetrexed (38%), gemcitabine (25%), taxane (24%), and bevacizumab (14%). Twenty-two percent of patients received prior maintenance therapy. Tumor EGFR status was unknown for the majority of patients (65%). Where tumor EGFR status was known (n=445), 7.4% were positive for EGFR mutation (n=33). No data were collected regarding tumor ALK rearrangement status.
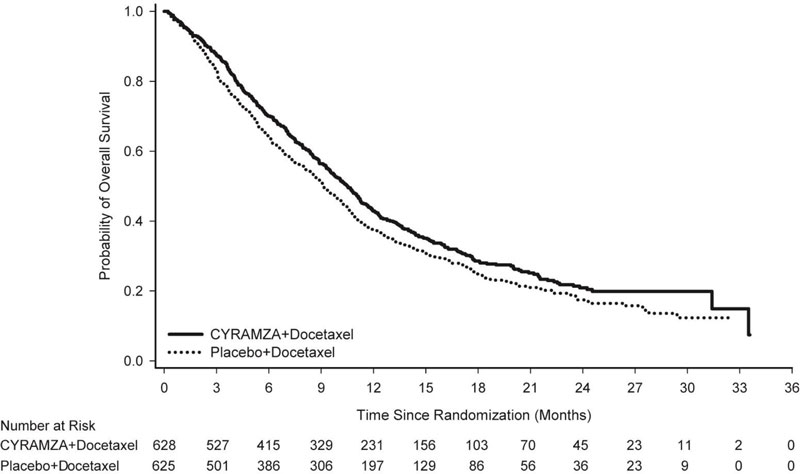
Overall response rate (complete response + partial response) was 23% (95% CI: 20, 26) for CYRAMZA with docetaxel and 14% (95% CI: 11, 17) for placebo with docetaxel, p-value of <0.001. Efficacy results are shown in Table 10 and Figure 3.
Table 10: Efficacy Results in REVEL Abbreviations: CI = confidence interval
a 126 of 558 events in CYRAMZA-treated patients and 109 of 583 events in placebo-treated patients were deaths.
CYRAMZA + docetaxel
N=628Placebo + docetaxel
N=625Overall Survival Number of deaths (%) 428 (68%) 456 (73%) Median – months (95% CI) 10.5 (9.5, 11.2) 9.1 (8.4, 10.0) Hazard Ratio (95% CI) 0.86 (0.75, 0.98) Stratified Log-rank p-value 0.024 Progression-free Survival Number of events (%)a 558 (89%) 583 (93%) Median – months (95% CI) 4.5 (4.2, 5.4) 3.0 (2.8, 3.9) Hazard Ratio (95% CI) 0.76 (0.68, 0.86) Stratified Log-rank p-value <0.001 14.3 Colorectal Cancer
The efficacy of CYRAMZA was evaluated in RAISE (NCT01183780), a multinational, randomized, double-blind study in patients with mCRC, who had disease progression on or after prior therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine. Patients in RAISE were required to have ECOG PS 0 or 1 and to have disease progression within 6 months of the last dose of first-line therapy. Patients were randomized (1:1) to receive either CYRAMZA at 8 mg/kg as an intravenous infusion or placebo, in combination with FOLFIRI: irinotecan 180 mg/m2 administered intravenously over 90 minutes and folinic acid 400 mg/m2 administered intravenously simultaneously over 120 minutes; followed by fluorouracil 400 mg/m2 intravenous bolus over 2 to 4 minutes; followed by fluorouracil 2400 mg/m2 administered intravenously by continuous infusion over 46 to 48 hours. Treatment cycles on both arms were repeated every 2 weeks. Patients who discontinued one or more components of treatment because of an adverse reaction were permitted to continue therapy with the other treatment component(s) until disease progression or unacceptable toxicity. Randomization was stratified by geographic region, tumor KRAS status, and time to disease progression after beginning first-line treatment (<6 months versus ≥6 months). The major efficacy outcome measure was OS. An additional efficacy outcome measure was PFS.
A total of 1072 patients were randomized, 536 to the CYRAMZA-treatment group and 536 to the placebo-treatment group. Baseline demographics and disease characteristics were similar between treatment arms. The median age was 62 years (range 21-87); 57% were men; 76% were White and 20% were Asian; 49% had ECOG PS 0; 49% had KRAS mutant tumors; and 24% had <6 months from time to disease progression after beginning first-line treatment. The treatment effect was consistent across the pre-specified stratification factors.
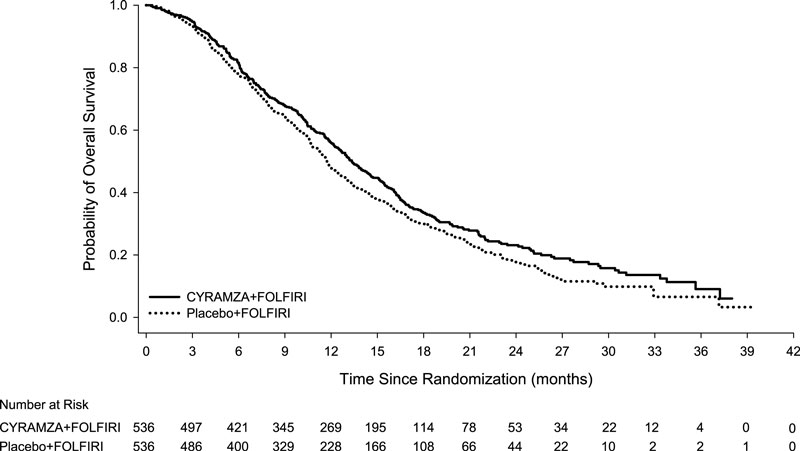
Efficacy results are shown in Table 11 and Figure 4.
Table 11: Efficacy Results in RAISE Abbreviations: CI = confidence interval.
a 73 of 476 events in CYRAMZA-treated patients and 64 of 494 events in placebo-treated patients were deaths.
CYRAMZA + FOLFIRI
N=536Placebo + FOLFIRI
N=536Overall Survival Number of deaths (%) 372 (69%) 397 (74%) Median – months (95% CI) 13.3 (12.4, 14.5) 11.7 (10.8, 12.7) Hazard Ratio (95% CI) 0.85 (0.73, 0.98) Stratified Log-rank p-value 0.023 Progression-free Survival Number of events (%)a 476 (89%) 494 (92%) Median – months (95% CI) 5.7 (5.5, 6.2) 4.5 (4.2, 5.4) Hazard Ratio (95% CI) 0.79 (0.70, 0.90) Stratified Log-rank p-value <0.001 14.4 Hepatocellular Carcinoma
The efficacy of CYRAMZA was evaluated in REACH-2 (NCT02435433), a multinational, randomized, double-blind, placebo-controlled, multicenter study in patients with advanced HCC with AFP ≥400 ng/mL who had disease progression on or after prior sorafenib therapy or who were intolerant to sorafenib. Patients in REACH-2 were required to have ECOG PS of 0 or 1, Child-Pugh A, BCLC stage B and no longer amenable to locoregional therapy, or BCLC stage C. Patients were randomized (2:1) to receive CYRAMZA 8 mg/kg or placebo every 2 weeks as an intravenous infusion until disease progression or unacceptable toxicity. Randomization was stratified by geographic region, macrovascular invasion (yes versus no), and ECOG PS (0 versus 1). The major efficacy outcome measure was OS. Additional efficacy outcome measures included PFS and ORR based on investigator assessment.
A total of 292 patients were randomized, 197 to the CYRAMZA-treatment group and 95 to the placebo-treatment group. Baseline demographics and disease characteristics were similar between the arms. The median age was 64 years (range 26-88); 80% were men; 50% were Asian; 58% had ECOG PS 0; 35% had macrovascular invasion; 72% had extrahepatic spread; 17% were sorafenib intolerant, 37% had hepatitis B, 26% had hepatitis C, 24% had significant prior alcohol use, and 64% had prior locoregional therapy.
Efficacy results are shown in Table 12 and Figure 5.
Table 12: Efficacy Results in REACH-2 Abbreviations: BSC = best supportive care; CI = confidence interval
a 26 of 172 events in CYRAMZA-treated patients and 9 of 86 events in placebo-treated patients were deaths.
b all responses were partial
CYRAMZA + BSC
N=197Placebo + BSC
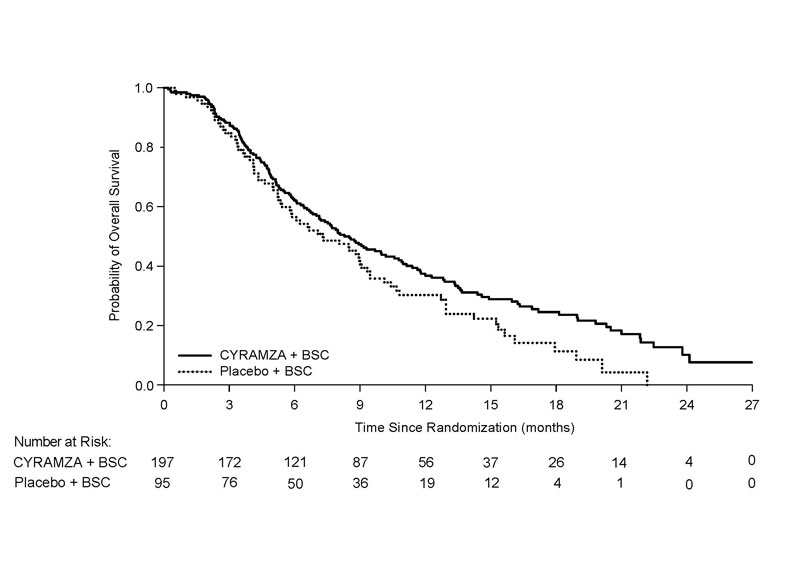
N=95Overall Survival Number of deaths (%) 147 (75%) 74 (78%) Median – months (95% CI) 8.5 (7.0, 10.6) 7.3 (5.4, 9.1) Hazard Ratio (95% CI) 0.71 (0.53, 0.95) Stratified Log-rank p-value 0.020 Progression-free Survival Number of events (%)a 172 (87%) 86 (91%) Median – months (95% CI) 2.8 (2.8, 4.1) 1.6 (1.5, 2.7) Hazard Ratio (95% CI) 0.45 (0.34, 0.60) Stratified Log-rank p-value <0.0001 Overall Response Rateb Rate – percent (95% CI) 4.6% (1.7, 7.5) 1.1% (0, 3.1) Figure 5: Kaplan-Meier Curves for Overall Survival in REACH-2
-
16 HOW SUPPLIED/STORAGE AND HANDLING
CYRAMZA (ramucirumab) injection is a clear to slightly opalescent and colorless to slightly yellow, preservative-free solution supplied in single-dose vials.
- NDC: 0002-7669-01
100 mg/10 mL (10 mg/mL), individually packaged in a carton - NDC: 0002-7678-01
500 mg/50 mL (10 mg/mL), individually packaged in a carton
Store vials in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light until time of use. Do not freeze or shake the vial.
- NDC: 0002-7669-01
-
17 PATIENT COUNSELING INFORMATION
Hemorrhage
Advise patients that CYRAMZA can cause severe bleeding. Advise patients to contact their health care provider for bleeding or symptoms of bleeding including lightheadedness [see Warnings and Precautions (5.1)].
Gastrointestinal Perforations
Advise patients to notify their health care provider for severe diarrhea, vomiting, or severe abdominal pain [see Warnings and Precautions (5.2)].
Impaired Wound Healing
Advise patients that CYRAMZA has the potential to impair wound healing. Instruct patients not to undergo surgery without first discussing this potential risk with their health care provider [see Warnings and Precautions (5.3)].
Arterial Thromboembolic Events
Advise patients of an increased risk of an arterial thromboembolic event [see Warnings and Precautions (5.4)].
Hypertension
Advise patients to undergo routine blood pressure monitoring and to contact their health care provider if blood pressure is elevated or if symptoms from hypertension occur including severe headache, lightheadedness, or neurologic symptoms [see Warnings and Precautions (5.5)].
Posterior Reversible Encephalopathy Syndrome
Advise patients to inform their health care provider if they develop neurological symptoms consistent with PRES (seizure, headache, nausea/vomiting, blindness, or altered consciousness) [see Warnings and Precautions (5.8)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus. Advise females to inform their health care provider of a known or suspected pregnancy and to use effective contraception during CYRAMZA treatment and for 3 months after the last dose [see Warnings and Precautions (5.11), Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during CYRAMZA treatment and for 2 months after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise females of reproductive potential that CYRAMZA may impair fertility [see Use in Specific Populations (8.3)].
-
PRINCIPAL DISPLAY PANEL
PACKAGE LABELING
This section contains a representative sample of product package labeling. Product may be manufactured at other manufacturing sites.
PACKAGE CARTON –CYRAMZA 100 mg/10 mL single-use vial.
NDC: 0002-7669-01
Cyramza®
(ramucirumab)
Injection
100 mg/10 mL
(10 mg/mL)
For Intravenous Infusion Only
Must Dilute Prior to Use
Single-Dose Vial
Discard Unused Portion
Keep Refrigerated
Rx only
www.cyramza.com
Lilly
CARTON FOR US ORIGIN
CARTON FOR IRELAND ORIGIN
-
PRINCIPAL DISPLAY PANEL
PACKAGE CARTON – CYRAMZA 500 mg/50mL single-use vial.
NDC: 0002-7678-01
Cyramza®
(ramucirumab)
Injection
500 mg/50 mL
(10 mg/mL)
For Intravenous Infusion Only
Must Dilute Prior to Use
Single-Dose Vial
Discard Unused Portion
Keep Refrigerated
Rx only
www.cyramza.com
Lilly
CARTON FOR US ORIGIN
CARTON FOR IRELAND ORIGIN
-
INGREDIENTS AND APPEARANCE
CYRAMZA
ramucirumab solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0002-7669 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ramucirumab (UNII: D99YVK4L0X) (ramucirumab - UNII:D99YVK4L0X) ramucirumab 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength histidine (UNII: 4QD397987E) 0.65 mg in 1 mL histidine monohydrochloride (UNII: 1D5Q932XM6) 1.22 mg in 1 mL sodium chloride (UNII: 451W47IQ8X) 4.38 mg in 1 mL glycine (UNII: TE7660XO1C) 9.98 mg in 1 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.1 mg in 1 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0002-7669-01 1 in 1 CARTON 04/21/2014 1 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125477 04/21/2014 CYRAMZA
ramucirumab solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0002-7678 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ramucirumab (UNII: D99YVK4L0X) (ramucirumab - UNII:D99YVK4L0X) ramucirumab 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength histidine (UNII: 4QD397987E) 0.65 mg in 1 mL histidine monohydrochloride (UNII: 1D5Q932XM6) 1.22 mg in 1 mL sodium chloride (UNII: 451W47IQ8X) 4.38 mg in 1 mL glycine (UNII: TE7660XO1C) 9.98 mg in 1 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.1 mg in 1 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0002-7678-01 1 in 1 CARTON 04/21/2014 1 50 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125477 04/21/2014 Labeler - Eli Lilly and Company (006421325)




Trademark Results [CYRAMZA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 CYRAMZA 86315717 4899164 Live/Registered |
ImClone LLC 2014-06-20 |
 CYRAMZA 86241873 not registered Dead/Abandoned |
ImClone LLC 2014-04-03 |
 CYRAMZA 85447460 4580501 Live/Registered |
ImClone LLC 2011-10-14 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.