ILUMYA- tildrakizumab-asmn injection, solution
ILUMYA by
Drug Labeling and Warnings
ILUMYA by is a Prescription medication manufactured, distributed, or labeled by Sun Pharmaceutical Industries, Inc., Vetter Pharma Fertigung GmbH & Co. KG (Ravensburg Schuetzenstrasse), Vetter Pharma Fertigung GmbH & Co. KG (Ravensburg Mooswiesen), Samsung Biologics Co., Ltd.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ILUMYA safely and effectively. See full prescribing information for ILUMYA.
ILUMYA™ (tildrakizumab-asmn) injection, for subcutaneous use Initial U.S. Approval: 2018INDICATIONS AND USAGE
ILUMYA is an interleukin-23 antagonist indicated for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. (1) (1)
DOSAGE AND ADMINISTRATION
- Administer by subcutaneous injection. (2.1)
- Recommended dose is 100 mg at Weeks 0, 4, and every twelve weeks thereafter. (2.1)
DOSAGE FORMS AND STRENGTHS
Injection: 100 mg/mL solution in a single-dose prefilled syringe. (3) (3)
CONTRAINDICATIONS
Serious hypersensitivity reaction to tildrakizumab or to any of the excipients. (4) (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity: If a serious allergic reaction occurs, discontinue ILUMYA immediately and initiate appropriate therapy. (5.1)
- Infections: ILUMYA may increase the risk of infection. Instruct patients to seek medical advice if signs or symptoms of clinically important chronic or acute infection occur. If a serious infection develops, consider discontinuing ILUMYA until the infection resolves. (5.2)
- Tuberculosis (TB): Evaluate for TB prior to initiating treatment. (5.3)
ADVERSE REACTIONS
Most common (≥1%) adverse reactions associated with ILUMYA treatment are upper respiratory infections, injection site reactions, and diarrhea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Sun Pharmaceutical Industries, Inc. at 1-800-818-4555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Live Vaccines: Avoid use of live vaccines in patients treated with ILUMYA. (7.1) (7)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 9/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
2.2 Tuberculosis Assessment Prior to Initiation of ILUMYA
2.3 Important Administration Instructions
2.4 Preparation and Administration of ILUMYA
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
5.2 Infections
5.3 Pretreatment Evaluation for Tuberculosis
5.4 Immunizations
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
7 DRUG INTERACTIONS
7.1 Live Vaccinations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
ILUMYA is administered by subcutaneous injection. The recommended dose is 100 mg at Weeks 0, 4, and every twelve weeks thereafter. Each syringe contains 1 mL of 100 mg/mL tildrakizumab-asmn.
2.2 Tuberculosis Assessment Prior to Initiation of ILUMYA
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with ILUMYA [see Warnings and Precautions (5.3)].
2.3 Important Administration Instructions
ILUMYA should only be administered by a healthcare provider. Administer ILUMYA subcutaneously. Each pre-filled syringe is for single-dose only. Inject the full amount (1 mL), which provides 100 mg of tildrakizumab per syringe. If a dose is missed, administer the dose as soon as possible. Thereafter, resume dosing at the regularly scheduled interval.
2.4 Preparation and Administration of ILUMYA
Before injection, remove ILUMYA carton from the refrigerator, and let the prefilled syringe (in the ILUMYA carton with the lid closed) sit at room temperature for 30 minutes.
Follow the instructions on the ILUMYA carton to remove the prefilled syringe correctly, and remove only when ready to inject. Do not pull off the needle cover until you are ready to inject.
Inspect ILUMYA visually for particulate matter and discoloration prior to administration. ILUMYA is a clear to slightly opalescent, colorless to slightly yellow solution. Do not use if the liquid contains visible particles or the syringe is damaged. Air bubbles may be present; there is no need to remove them.
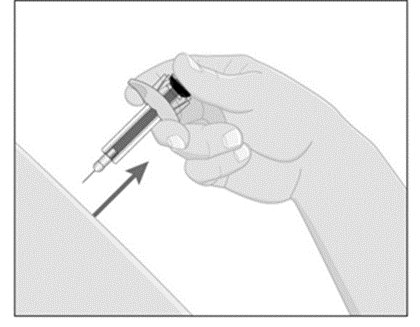
Choose an injection site with clear skin and easy access (such as abdomen, thighs, or upper arm). Do not administer 2 inches around the navel or where the skin is tender, bruised, erythematous, indurated, or affected by psoriasis. Also do not inject into scars, stretch marks, or blood vessels.
- While holding the body of the syringe, pull the needle cover straight off (do not twist) and discard.
- Inject ILUMYA subcutaneously as recommended [see Dosage and Administration (2.3)].
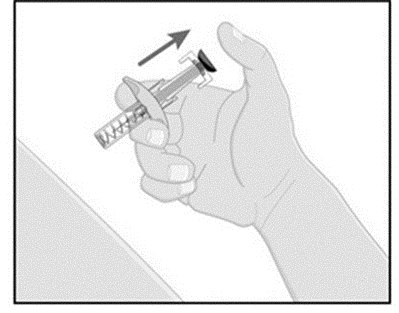
- Press down the blue plunger until it can go no further. This activates the safety mechanism that will ensure full retraction of the needle after the injection is given.
- Remove the needle from the skin entirely before letting go of the blue plunger. After the blue plunger is released, the safely lock will draw the needle inside the needle guard.
- Discard of any unused portion. Dispose of used syringe.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
Cases of angioedema and urticaria occurred in ILUMYA treated subjects in clinical trials. If a serious hypersensitivity reaction occurs, discontinue ILUMYA immediately and initiate appropriate therapy [see Adverse Reactions (6.1)].
5.2 Infections
ILUMYA may increase the risk of infection. Although infections were slightly more common in the ILUMYA group (23%), the difference in frequency of infections between the ILUMYA group and the placebo group was less than 1% during the placebo-controlled period. However, subjects with active infections or a history of recurrent infections were not included in clinical trials. Upper respiratory infections occurred more frequently in the ILUMYA group than in the placebo group [see Adverse Reactions (6.1)].
The rates of serious infections for the ILUMYA group and the placebo group were ≤0.3%. Treatment with ILUMYA should not be initiated in patients with any clinically important active infection until the infection resolves or is adequately treated.
In patients with a chronic infection or a history of recurrent infection, consider the risks and benefits prior to prescribing ILUMYA. Instruct patients to seek medical help if signs or symptoms of clinically important chronic or acute infection occur. If a patient develops a clinically important or serious infection or is not responding to standard therapy, monitor the patient closely and consider discontinuation of ILUMYA until the infection resolves [see Adverse Reactions (6.1)].
5.3 Pretreatment Evaluation for Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with ILUMYA. Initiate treatment of latent TB prior to administering ILUMYA. In clinical trials, of 55 subjects with latent TB who were concurrently treated with ILUMYA and appropriate TB prophylaxis, no subjects developed active TB (during the mean follow-up of 56.5 weeks). One other subject developed TB while receiving ILUMYA. Monitor patients for signs and symptoms of active TB during and after ILUMYA treatment. Consider anti-TB therapy prior to initiation of ILUMYA in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Do not administer ILUMYA to patients with active TB infection.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in the labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Infections [see Warnings and Precautions(5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In clinical trials, a total of 1994 subjects with plaque psoriasis were treated with ILUMYA, of which 1083 subjects were treated with ILUMYA 100 mg. Of these, 672 subjects were exposed for at least 12 months, 587 for 18 months, and 469 for 24 months.
Data from three placebo-controlled trials (Trials 1, 2, and 3) in 705 subjects (mean age 46 years, 71% males, 81% white) were pooled to evaluate the safety of ILUMYA (100 mg administered subcutaneously at Weeks 0 and 4, followed by every 12 weeks [Q12W]) [see Clinical Studies (14)].
Placebo-Controlled Period (Weeks 0-16 of Trial 1 and Weeks 0-12 of Trials 2 and 3)
In the placebo-controlled period of Trials 1, 2, and 3 in the 100 mg group, adverse events occurred in 48.2% of subjects in the ILUMYA group compared to 53.8% of subjects in the placebo group. The rates of serious adverse events were 1.4% in the ILUMYA group and 1.7% in the placebo group.
Table 1 summarizes the adverse reactions that occurred at a rate of at least 1% and at a higher rate in the ILUMYA group than in the placebo group.
Table 1:Adverse Reactions Occurring in ≥1% of Subjects in the ILUMYA Group and More Frequently than in the Placebo Group in the Plaque Psoriasis Trials 1, 2, and 3 Adverse Reaction
ILUMYA
100 mg
(N=705)
N (%)
Placebo
(N=355)
N (%)
Upper respiratory infections*
98 (14)
41 (12)
Injection site reactions†
24 (3)
7 (2)
Diarrhea
13 (2)
5 (1)
- * Upper respiratory infections include nasopharyngitis, upper respiratory tract infection, viral upper respiratory tract infection, and pharyngitis.
- † Injection site reactions include injection site urticaria, pruritus, pain, reaction, erythema, inflammation, edema, swelling, bruising, hematoma, and hemorrhage.
During the placebo-controlled period of Trials 1, 2, and 3, adverse reactions that occurred at rates less than 1% but greater than 0.1% in the ILUMYA group and at a higher rate than in the placebo group included dizziness and pain in extremity.
Specific Adverse Reactions
Hypersensitivity Reactions
Cases of angioedema and urticaria occurred in ILUMYA-treated subjects in clinical trials [see Warnings and Precautions (5.1)].
Infections
Infections were slightly more common in the ILUMYA group. The difference in frequency of infections between the ILUMYA group (23%) and the placebo group was less than 1% during the placebo-controlled period. The most common (≥1%) infections were upper respiratory infections. The rates of severe infections for the ILUMYA group and the placebo group were ≤0.3%.
Safety Through Week 52/64
Through Week 52 (Trials 1 and 3) and Week 64 (Trial 2), no new adverse reactions were identified with ILUMYA use and the frequency of the adverse reactions was similar to that observed during the placebo-controlled period.
6.2 Immunogenicity
As with all therapeutic proteins there is the potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to tildrakizumab in the studies described below with the incidences of antibodies in other studies or to other products may be misleading.
Up to Week 64, approximately 6.5% of subjects treated with ILUMYA 100 mg developed antibodies to tildrakizumab. Of the subjects who developed antibodies to tildrakizumab, approximately 40% (2.5% of all subjects receiving ILUMYA) had antibodies that were classified as neutralizing. Development of neutralizing antibodies to tildrakizumab was associated with lower serum tildrakizumab concentrations and reduced efficacy.
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited available data with ILUMYA use in pregnant women are insufficient to inform a drug associated risk of adverse developmental outcomes. Human IgG is known to cross the placental barrier; therefore, ILUMYA may be transferred from the mother to the fetus. An embryofetal developmental study conducted with tildrakizumab in pregnant monkeys revealed no treatment-related effects to the developing fetus when tildrakizumab was administered subcutaneously during organogenesis to near parturition at doses up to 159 times the maximum recommended human dose (MRHD). When dosing was continued until parturition, a small increase in neonatal death was observed at 59 times the MRHD [see Data].The clinical significance of this nonclinical finding is unknown.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In an embryofetal developmental study, subcutaneous doses up to 300 mg/kg tildrakizumab were administered to pregnant cynomolgus monkeys once every two weeks during organogenesis to gestation day 118 (22 days from parturition). No maternal or embryofetal toxicities were observed at doses up to 300 mg/kg (159 times the MRHD of 100 mg, based on AUC comparison). Tildrakizumab crossed the placenta in monkeys.
In a pre- and postnatal developmental study, subcutaneous doses up to 100 mg/kg tildrakizumab were administered to pregnant cynomolgus monkeys once every two weeks from gestation day 50 to parturition. Neonatal deaths occurred in the offspring of one control monkey, two monkeys at 10 mg/kg dose (6 times the MRHD based on AUC comparison), and four monkeys at 100 mg/kg dose (59 times the MRHD based on AUC comparison). The clinical significance of these nonclinical findings is unknown. No tildrakizumab-related adverse effects were noted in the remaining infants from birth through 6 months of age.
8.2 Lactation
Risk Summary
There are no data on the presence of tildrakizumab in human milk, the effects on the breastfed infant, or the effects on milk production. Human IgG is known to be present in human milk. Tildrakizumab was detected in the milk of monkeys [see Data].
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ILUMYA and any potential adverse effects on the breastfed child from ILUMYA or from the underlying maternal condition.
Data
Animal Data
Very low levels of tildrakizumab were detected in breast milk of monkeys in the pre- and postnatal developmental study described in 8.1. The mean tildrakizumab concentrations in milk were approximately 0.09 – 0.2% of that in serum on postpartum days 28 and 91.
8.4 Pediatric Use
Safety and effectiveness of ILUMYA in pediatric patients (<18 years of age) have not been established.
8.5 Geriatric Use
A total of 1083 subjects were exposed to ILUMYA 100 mg during Phase 2 and 3 trials. A total of 92 subjects were 65 years or older, and 17 subjects were 75 years or older. Although no differences in safety or efficacy were observed between older and younger subjects, the number of subjects aged 65 and over is not sufficient to determine whether they respond differently from younger subjects [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
Tildrakizumab-asmn is a humanized IgG1/k antibody that specifically binds to the p19 subunit of interleukin-23 (IL-23).
Tildrakizumab-asmn is produced in a recombinant Chinese hamster ovary (CHO) cell line and has an approximate molecular mass of 147 kilodaltons.
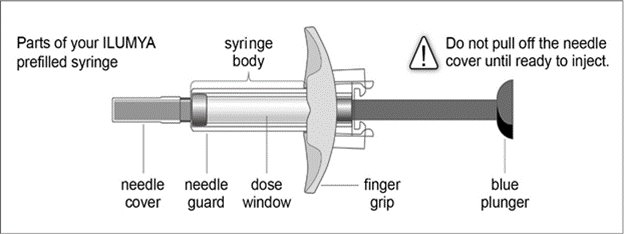
ILUMYA (tildrakizumab-asmn) injection, for subcutaneous use, is a sterile, clear to slightly opalescent, colorless to slightly yellow solution. ILUMYA is supplied in a single-dose prefilled syringe with a glass barrel and 29-gauge fixed, 1/2-inch needle.
The syringe is fitted with a passive needle guard and a needle cover.
Each 1 mL single-dose prefilled syringe contains 100 mg of tildrakizumab-asmn formulated in: L-histidine (0.495 mg), L-histidine hydrochloride monohydrate (1.42 mg), polysorbate 80 (0.5 mg), sucrose (70.0 mg), and Water for Injection, USP with a pH of 5.7-6.3.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tildrakizumab is a humanized IgG1/k monoclonal antibody that selectively binds to the p19 subunit of IL-23 and inhibits its interaction with the IL-23 receptor. IL-23 is a naturally occurring cytokine that is involved in inflammatory and immune responses. Tildrakizumab inhibits the release of proinflammatory cytokines and chemokines.
12.3 Pharmacokinetics
Tildrakizumab pharmacokinetics increases proportionally over a dose range from 50 mg to 200 mg (0.5 to 2 times the approved recommended dosage) following subcutaneous administration in subjects with plaque psoriasis. Steady-state concentrations were achieved by Week 16 following subcutaneous administration of tildrakizumab at Weeks 0, 4, and every 12 weeks thereafter. At the 100 mg dose at Week 16, the mean (± SD) steady-state trough concentrations ranged from 1.22 ± 0.94 mcg/mL to 1.47 ± 1.12 mcg/mL. The geometric mean (CV%) steady-state Cmax was 8.1 mcg/mL (34%).
Absorption
The absolute bioavailability of tildrakizumab was estimated to be 73-80% following subcutaneous injection. The peak concentration (Cmax) was reached by approximately 6 days.
Distribution
The geometric mean (CV%) volume of distribution is 10.8 L (24%).
Elimination
The geometric mean (CV%) systemic clearance was 0.32 L/day (38%) and the half-life was approximately 23 days (23%).
Metabolism
The metabolic pathway of tildrakizumab has not been characterized. As a humanized IgG1/k monoclonal antibody, tildrakizumab is expected to be degraded into small peptides and amino acids via catabolic pathways in a manner similar to endogenous IgG.
Specific Populations
No clinically significant differences in the pharmacokinetics of tildrakizumab were observed based on age (≥18 years). No specific studies have been conducted to determine the effect of renal or hepatic impairment on the pharmacokinetics of tildrakizumab.
Body Weight
Tildrakizumab concentrations were lower in subjects with higher body weight.
Drug Interaction Studies
Cytochrome P450 Substrates
The AUCinf of dextromethorphan (CYP2D6 substrate) increased by 20% when used concomitantly with tildrakizumab 200 mg (two times the approved recommended dose) administered subcutaneously at Weeks 0 and 4 in subjects with plaque psoriasis. No clinically significant changes in AUCinf of caffeine (CYP1A2 substrate), warfarin (CYP2C9 substrate), omeprazole (CYP2C19 substrate), and midazolam (CYP3A4 substrate) were observed.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Animal studies have not been conducted to evaluate the carcinogenic or mutagenic potential of ILUMYA.
No effects on fertility parameters were observed in male or female cynomolgus monkeys that were administered tildrakizumab at subcutaneous or intravenous doses up to 140 mg/kg once every two weeks for 3 months (133 or 155 times the MRHD, respectively, based on AUC comparison). The monkeys were not mated to evaluate fertility.
-
14 CLINICAL STUDIES
In two multicenter, randomized, double-blind, placebo-controlled trials (Trial 2 [NCT01722331] and Trial 3 [NCT01729754]), 926 subjects were treated with ILUMYA 100 mg (N=616) or placebo (N=310). Subjects had a Physician Global Assessment (PGA) score of ≥3 (moderate) on a 5-point scale of overall disease severity, Psoriasis Area and Severity Index (PASI) score ≥12, and a minimum body surface area (BSA) involvement of 10%. Subjects with guttate, erythrodermic, or pustular psoriasis were excluded.
In both trials, subjects were randomized to either placebo or ILUMYA (100 mg at Week 0, Week 4, and every twelve weeks thereafter [Q12W]) up to 64 weeks.
Trials 2 and 3 assessed the changes from baseline to Week 12 in the two co-primary endpoints:
- PASI 75, the proportion of subjects who achieved at least a 75% reduction in the PASI composite score.
- PGA of 0 (“cleared”) or 1 (“minimal”), the proportion of subjects with a PGA of 0 or 1 and at least a 2-point improvement.
Other evaluated outcomes in Trials 2 and 3 included the proportion of subjects who achieved a reduction from baseline in PASI score of at least 90% (PASI 90) and a reduction of 100% in PASI score (PASI 100) at Week 12 and maintenance of efficacy up to Week 64.
In both trials, subjects in the ILUMYA 100 mg and placebo treatment groups were predominantly men (69%) and White (80%), with a mean age of 46 years. At baseline, these subjects had a median affected BSA of 27%, a median PASI score of 17.8, and approximately 33% had a PGA score of 4 (“marked”) or 5 (“severe”). Approximately 34% had received prior phototherapy, 39% had received prior conventional systemic therapy, and 18% had received prior biologic therapy for the treatment of psoriasis. Approximately 16% of subjects had a history of psoriatic arthritis.
Clinical Response at Week 12
The results of Trials 2 and 3 are presented in Table 2.
- Table 2: Efficacy Results at Week 12 in Adults with Plaque Psoriasis in Trials 2 and 3 (NRI*)
Trial 2 (NCT01722331)
Trial 3 (NCT01729754)
ILUMYA 100 mg
(N=309)
n (%)
Placebo
(N=154)
n (%)
ILUMYA 100 mg
(N=307)
n (%)
Placebo
(N=156)
n (%)
PGA of 0 or 1†,‡
179 (58)
11 (7)
168 (55)
7 (4)
PASI 75†
197 (64)
9 (6)
188 (61)
9 (6)
PASI 90
107 (35)
4 (3)
119 (39)
2 (1)
PASI 100
43 (14)
2 (1)
38 (12)
0 (0)
* NRI = Non-Responder Imputation
† Co-Primary Endpoints
‡ PGA score of 0 (“cleared”) or 1 (“minimal”)
Examination of age, gender, race, and previous treatment with a biologic did not identify differences in response to ILUMYA among these subgroups at Week 12.
Maintenance of Response and Durability of Response
In Trial 2, subjects originally randomized to ILUMYA and who were responders at Week 28 (i.e., PASI 75) were re-randomized to an additional 36 weeks of either maintaining the same dose of ILUMYA Q12W (every twelve weeks) or placebo.
At Week 28, 229 (74%) subjects treated with ILUMYA 100 mg were PASI 75 responders. At Week 64, 84% of subjects who continued on ILUMYA 100 mg Q12W maintained PASI 75 compared to 22% of subjects who were re-randomized to placebo. In addition, for subjects who were re-randomized and also had a PGA score of 0 or 1 at Week 28, 69% of subjects who continued on ILUMYA 100 mg Q12W maintained this response (PGA 0 or 1) at Week 64 compared to 14% of subjects who were re-randomized to placebo.
For PASI 75 responders at Week 28 who were re-randomized to treatment withdrawal (i.e., placebo), the median time to loss of PASI 75 was approximately 20 weeks.
In addition, for subjects who were re-randomized to placebo and also had a PGA score of 0 or 1 at Week 28, the median time to loss of PGA score of 0 or 1 was approximately 16 weeks.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
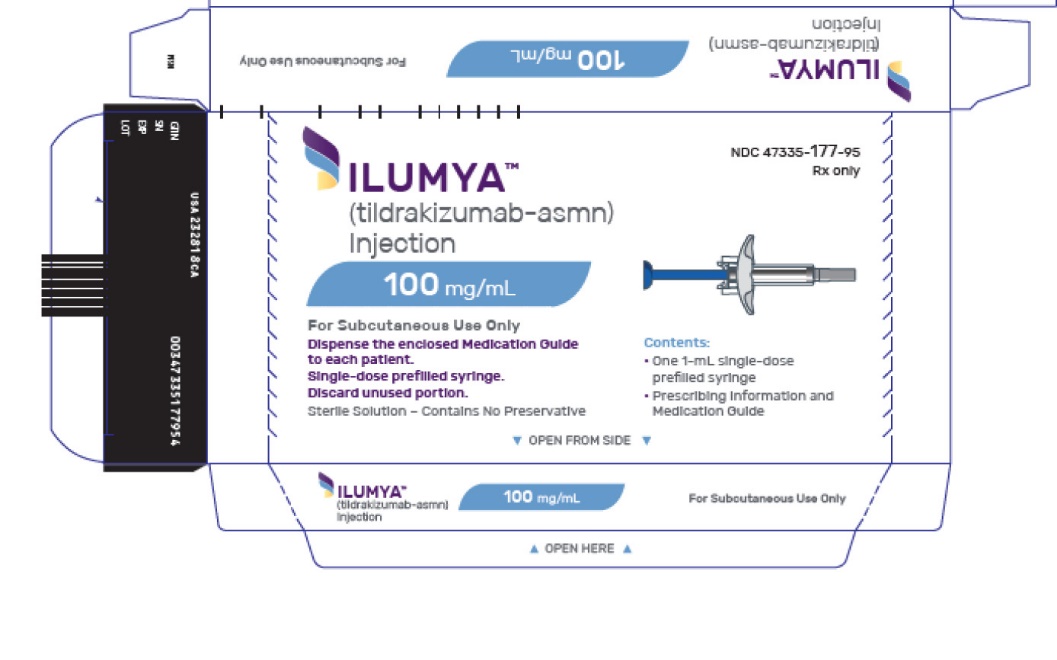
ILUMYA (tildrakizumab-asmn) Injection is a sterile, preservative-free, clear to slightly opalescent, colorless to slightly yellow solution. ILUMYA is supplied as one single-dose prefilled syringe per carton that delivers 1 mL of a 100 mg/mL solution.
- NDC: 47335-177-95
Each prefilled syringe is equipped with a passive needle guard and a needle cover.
16.2 Storage and Handling
Store refrigerated at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light until the time of use. Do not freeze. Do not shake. ILUMYA can be kept at room temperature at 25°C (77°F) for up to 30 days in the original carton to protect from light. Once stored at room temperature, do not place back in the refrigerator. If not used within 30 days, discard ILUMYA. Do not store ILUMYA above 25°C (77°F).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide).
Instruct patients and/or caregivers to read the Medication Guide before starting ILUMYA therapy and to reread the Medication Guide each time the prescription is renewed. Advise patients of the potential benefits and risks of ILUMYA.
Hypersensitivity
Advise patients to seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions [see Warnings and Precautions (5.1)].
Infections
Instruct patients of the importance of communicating any history of infections to the doctor and contacting their doctor if they develop any symptoms of infection [see Warnings and Precautions (5.2)].
Manufactured by: Sun Pharma Global FZE
Sharjah, U.A.E.
U.S. License No. 2092
In: Germany
U.S. Patent No. 8,404,813, 8,293,883 and 9,809,648
Tildrakizumab-asmn (active ingred.) Product of The Netherlands.
Distributed by: Sun Pharmaceutical Industries, Inc.,
Cranbury, NJ 08512
ILUMYA is a trademark of Sun Pharma Global FZE
Copyright © 2019 Sun Pharma Global FZE
All rights reserved.
uspi-tildrakizumab-pfs-00002 -
Medication Guide
ILUMYA™ (“e-loom’-me-a”)
(tildrakizumab-asmn)
injection, for subcutaneous use
What is the most important information I should know about ILUMYA?
ILUMYA may cause serious side effects, including:
Serious allergic reactions. Get emergency medical help right away if you get any of the following symptoms of a serious allergic reaction:
- feel faint
- swelling of your face, eyelids, lips, mouth, tongue or throat
- skin rash
- trouble breathing or throat tightness
- chest tightness
Infections. ILUMYA is a medicine that may lower the ability of your immune system to fight infections and may increase your risk of infections. Your healthcare provider should check you for infections and tuberculosis (TB) before starting treatment with ILUMYA and may treat you for TB before you begin treatment with ILUMYA if you have a history of TB or have active TB. Your healthcare provider should watch you closely for signs and symptoms of TB during and after treatment with ILUMYA.
Tell your healthcare provider right away if you have an infection or have symptoms of an infection, including:
- fever, sweats, or chills
- cough
- shortness of breath
- blood in your phlegm (mucus)
- muscle aches
- warm, red, or painful skin or sores on your body different from your psoriasis
- weight loss
- diarrhea or stomach pain
- burning when you urinate or urinating more often than normal
See “What are the possible side effects of ILUMYA?” for more information about side effects.
What is ILUMYA?
ILUMYA is a prescription medicine used to treat adults with moderate to severe plaque psoriasis who may benefit from taking injections, pills (systemic therapy) or treatment using ultraviolet or UV light (phototherapy).
It is not known if ILUMYA is safe and effective in children under 18 years of age.
Do not use ILUMYA if you have had a severe allergic reaction to tildrakizumab or any of the other ingredients in ILUMYA. See the end of this Medication Guide for a complete list of ingredients in ILUMYA.
Before receiving ILUMYA, tell your healthcare provider about all of your medical conditions, including if you:
- have any of the conditions or symptoms listed in the section “What is the most important information I should know about ILUMYA?”
- have an infection that does not go away or that keeps coming back.
- have TB or have been in close contact with someone with TB.
- recently received or are scheduled to receive a vaccine (immunization). You should avoid receiving live vaccines during treatment with ILUMYA.
- are pregnant or plan to become pregnant. It is not known if ILUMYA can harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if ILUMYA passes into your breast milk.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How will I receive ILUMYA?
- ILUMYA should only be given to you by a healthcare provider.
- ILUMYA is given as an injection under your skin (subcutaneous injection) in areas of your body such as your thighs, stomach area (abdomen), or upper arm.
- If you miss a follow-up appointment and do not receive your dose of ILUMYA, schedule another appointment as soon as possible.
What are the possible side effects of ILUMYA?
ILUMYA may cause serious side effects. See “What is the most important information I should know about ILUMYA?”
The most common side effects of ILUMYA include:
- upper respiratory infections
- injection site reactions
- diarrhea
These are not all of the possible side effects of ILUMYA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of ILUMYA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your healthcare provider for information about ILUMYA that is written for health professionals.
What are the ingredients in ILUMYA?
Active ingredient: tildrakizumab-asmn
Inactive ingredients: L-histidine, L-histidine hydrochloride monohydrate, polysorbate 80, sucrose, and Water for Injection, USP.
Manufactured by: Sun Pharma Global FZE
Sharjah, U.A.E.
U.S. License No. 2092
In: Germany
U.S. Patent No. 8,404,813, 8,293,883 and 9,809,648
Tildrakizumab-asmn (active ingred.) Product of The Netherlands.
Distributed by: Sun Pharmaceutical Industries, Inc.
Cranbury, NJ 08512
ILUMYA is a trademark of Sun Pharma Global FZE
Copyright © 2019 Sun Pharma Global FZE
All rights reserved.
usmg-tildrakizumab-pfs-00002- This Medication Guide has been approved by the U.S. Food and Drug Administration. Issued: 09/2019
- Package/Label Display Panel
-
INGREDIENTS AND APPEARANCE
ILUMYA
tildrakizumab-asmn injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 47335-177 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TILDRAKIZUMAB (UNII: DEW6X41BEK) (TILDRAKIZUMAB - UNII:DEW6X41BEK) TILDRAKIZUMAB 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 0.495 mg in 1 mL HISTIDINE MONOHYDROCHLORIDE (UNII: 1D5Q932XM6) 1.42 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.5 mL in 1 mL SUCROSE (UNII: C151H8M554) 70 mg in 1 mL WATER (UNII: 059QF0KO0R) 1 mL in 1 mL Product Characteristics Color YELLOW (clear to slightly opalescent, colorless to slightly yellow solution) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 47335-177-10 1 mL in 1 CARTON; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 08/06/2018 2 NDC: 47335-177-01 1 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 08/06/2018 3 NDC: 47335-177-95 1 mL in 1 CARTON; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 08/06/2018 4 NDC: 47335-177-96 1 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 08/06/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761067 08/06/2018 Labeler - Sun Pharmaceutical Industries, Inc. (146974886) Registrant - Sun Pharmaceutical Industries, Inc. (146974886) Establishment Name Address ID/FEI Business Operations Sun Pharmaceutical Industries, Inc. 146974886 MANUFACTURE(47335-177) Establishment Name Address ID/FEI Business Operations Vetter Pharma Fertigung GmbH & Co. KG (Ravensburg Schuetzenstrasse) 316126754 manufacture(47335-177) Establishment Name Address ID/FEI Business Operations Vetter Pharma Fertigung GmbH & Co. KG (Ravensburg Mooswiesen) 312670654 pack(47335-177)
Trademark Results [ILUMYA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ILUMYA 90644697 not registered Live/Pending |
Sun Pharma Global FZE 2021-04-14 |
 ILUMYA 87835715 5657826 Live/Registered |
Sun Pharma Global FZE 2018-03-15 |
 ILUMYA 87459018 not registered Dead/Abandoned |
Sun Pharma Global FZE 2017-05-22 |
 ILUMYA 87358465 not registered Live/Pending |
Sun Pharma Global FZE 2017-03-04 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.