ADREVIEW- iobenguane i-123 injection
AdreView by
Drug Labeling and Warnings
AdreView by is a Prescription medication manufactured, distributed, or labeled by Medi-Physics Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use AdreView safely and effectively. See full prescribing information for AdreView.
AdreView (Iobenguane I 123 Injection) for Intravenous Use
Initial U.S. Approval: 2008INDICATIONS AND USAGE
AdreView is a radiopharmaceutical agent for gamma-scintigraphy indicated for:
- use in the detection of primary or metastatic pheochromocytoma or neuroblastoma as an adjunct to other diagnostic tests (1.1)
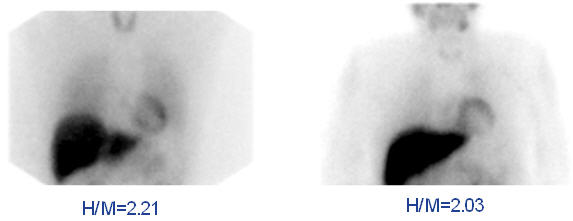
- scintigraphic assessment of sympathetic innervation of the myocardium by measurement of the heart to mediastinum (H/M) ratio of radioactivity uptake in patients with New York Heart Association (NYHA) class II or class III heart failure and left ventricular ejection fraction (LVEF) ≤ 35%. Among these patients, AdreView may be used to help identify patients with lower one and two year mortality risks, as indicated by an H/M ratio ≥ 1.6. (1.2)
Limitations of Use: In patients with congestive heart failure, AdreView utility has not been established for:
- selecting a therapeutic intervention or for monitoring the response to therapy;
- using the H/M ratio to identify a patient with a high risk for death.
DOSAGE AND ADMINISTRATION
- AdreView emits radiation and must be handled with appropriate safety measures. (2.1, 2.6)
- Administer thyroid blockade medications to patients at risk for thyroid accumulation of AdreView. (2.2, 5.6)
- Measure patient dose by a suitable radioactivity calibration system immediately prior to administration. (2.4)
- For patients ≥ 16 years of age or < 16 years of age and ≥ 70 kg: administer 10 mCi (370 MBq). (2.4, 2.5)
- For patients < 16 years of age and < 70 kg: amount scaled to the adult reference activity based on weight. (2.5)
DOSAGE FORMS AND STRENGTHS
5 mL of sterile solution for intravenous injection in a single use vial (2 mCi/mL at calibration time) (3)
CONTRAINDICATIONS
Known hypersensitivity to iobenguane or iobenguane sulfate. (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity reactions have followed AdreView administration. Have anaphylactic and hypersensitivity treatment measures available prior to AdreView administration. (5.1)
- Drugs which block norepinephrine uptake or deplete norepinephrine stores may decrease AdreView uptake. When medically feasible, stop these drugs before AdreView administration and monitor patients for withdrawal signs and symptoms. (5.2)
- AdreView contains benzyl alcohol (10.3 mg/mL) which may cause serious reactions in premature or low birth-weight infants. (5.3)
- Patients with severe renal impairment may have increased radiation exposure and decreased quality of AdreView images. (5.4)
- Failure to block thyroid iodine uptake may result in iodine 123 accumulation in the thyroid. (5.6)
ADVERSE REACTIONS
Serious hypersensitivity reactions have been reported following AdreView administration. The most common adverse reactions, dizziness, rash, pruritis, flushing, headache, and injection site hemorrhage occurred in < 1.3% of patients. (6.1, 6.2)
To report SUSPECTED ADVERSE REACTIONS, contact GE Healthcare at 1-800-654-0118 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Amitryptiline and derivatives, imipramine and derivatives, other antidepressants that inhibit norepinephrine transporter, antihypertensives that deplete norepinephrine stores or inhibit reuptake, sympathomimetic amines and cocaine: Discontinue for 5 biological half-lives prior to AdreView administration (7)
USE IN SPECIFIC POPULATIONS
- Pregnancy: May cause fetal harm. (8.1)
- Lactation: Advise a lactating woman to interrupt breastfeeding and pump and discard breastmilk for at least 6 days after AdreView administration. (8.2)
- Pediatrics: safety and effectiveness have not been established in pediatric patients < 1 month of age or in any pediatric patient with heart failure. (8.4)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Pheochromocytoma and Neuroblastoma
1.2 Congestive Heart Failure
2 DOSAGE AND ADMINISTRATION
2.1 Radiation Safety
2.2 Thyroid Blockade
2.3 Preparation and Administration
2.4 Recommended Dose for Adults
2.5 Recommended Dose for Pediatric Patients
2.6 Radiation Dosimetry
2.7 Imaging Guidelines
2.8 Estimation of the H/M Ratio among Patients with Congestive Heart Failure
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 Imaging Errors due to Concomitant Medications and Risks Associated with Withdrawal of Medications
5.3 Risk of Serious Adverse Reactions in Infants due to Benzyl Alcohol Preservative
5.4 Increased Radiation Exposure in Patients with Severe Renal Impairment
5.5 Imaging Errors due to Conditions that Affect the Sympathetic Nervous System
5.6 Thyroid Accumulation
5.7 Hypertension
6 ADVERSE REACTIONS
6.1 Clinical Study Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
11.1 Physical Characteristics
11.2 External Radiation
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Pheochromocytoma and Neuroblastoma
14.2 Congestive Heart Failure
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Pheochromocytoma and Neuroblastoma
AdreView is a radiopharmaceutical indicated for use in the detection of primary or metastatic pheochromocytoma or neuroblastoma as an adjunct to other diagnostic tests.
1.2 Congestive Heart Failure
AdreView is indicated for scintigraphic assessment of sympathetic innervation of the myocardium by measurement of the heart to mediastinum (H/M) ratio of radioactivity uptake in patients with New York Heart Association (NYHA) class II or class III heart failure and left ventricular ejection fraction (LVEF) ≤ 35%. Among these patients, AdreView may be used to help identify patients with lower one and two year mortality risks, as indicated by an H/M ratio ≥ 1.6.
Limitations of Use: In patients with congestive heart failure, AdreView utility has not been established for:
- selecting a therapeutic intervention or for monitoring the response to therapy;
- using the H/M ratio to identify a patient with a high risk for death.
-
2 DOSAGE AND ADMINISTRATION
2.1 Radiation Safety
AdreView emits radiation and must be handled with appropriate safety measures to minimize radiation exposure to clinical personnel and patients. Radiopharmaceuticals should be used by or under the control of physicians who are qualified by specific training and experience in the safe use and handling of radionuclides, and whose experience and training have been approved by the appropriate government agency authorized to license the use of radionuclides. AdreView dosing is based upon the radioactivity determined using a suitable calibration system immediately prior to administration.
To minimize radiation dose to the bladder, prior to and following AdreView administration, encourage hydration to permit frequent voiding. Encourage the patient to void frequently for the first 48 hours following AdreView administration [see Clinical Pharmacology (12.2)].
2.2 Thyroid Blockade
Before administration of AdreView to patients at risk for thyroid accumulation of the drug, administer Potassium Iodide Oral Solution or Lugol's Solution (equivalent to 100 mg iodide for adults, body-weight adjusted for children) or potassium perchlorate (400 mg for adults, body-weight adjusted for children) to block uptake of iodine 123 by the patient's thyroid. Administer the blocking agent at least one hour before the dose of AdreView [see Warnings and Precautions (5.6)]. Individualize thyroid blockade; for example, the blockade may not be necessary for patients who have undergone thyroidectomy or those with a very limited life expectancy.
2.3 Preparation and Administration
- Assess pregnancy status before administering AdreView to a female of reproductive potential.
- Inspect the AdreView vial for particulate matter and discoloration prior to administration. Use aseptic procedures and a radiation shielding syringe during administration. Administer the dose as an intravenous injection over 1 to 2 minutes. A subsequent injection of 0.9% sodium chloride may be used to ensure full delivery of the dose.
2.4 Recommended Dose for Adults
For adults (≥ 16 years of age), the recommended dose is 10 mCi (370 MBq) [see Clinical Studies (14.1, 14.2)].
2.5 Recommended Dose for Pediatric Patients
For pediatric patients < 16 years of age weighing ≥ 70 kg, the recommended dose is 10 mCi (370 MBq) [see Clinical Studies (14.1)].
For pediatric patients < 16 years of age weighing < 70 kg, the recommended dose should be calculated according to patient body weight as shown in Table 1 [see Clinical Studies (14.1)]. The benzyl alcohol in AdreView may cause serious adverse reactions in premature or low birth-weight infants [see Warnings and Precautions (5.3) and Use in Specific Populations (8.4)].
Table 1. AdreView Dose Preparation for Pediatric Patients* Weight (kg) Fraction of adult activity AdreView (mCi) pediatric dose AdreView (MBq) pediatric dose - * Based on a reference activity for an adult scaled to body weight according to the schedule proposed by the European Association of Nuclear Medicine Paediatric Task Group.
3 0.1 1 37 4 0.14 1.4 52 6 0.19 1.9 70 8 0.23 2.3 85.1 10 0.27 2.7 99.9 12 0.32 3.2 118.4 14 0.36 3.6 133.2 16 0.4 4 148 18 0.44 4.4 162.8 20 0.46 4.6 170.2 22 0.5 5 185 24 0.53 5.3 196.1 26 0.56 5.6 207.2 28 0.58 5.8 214.6 30 0.62 6.2 229.4 32 0.65 6.5 240.5 34 0.68 6.8 251.6 36 0.71 7.1 262.7 38 0.73 7.3 270.1 40 0.76 7.6 281.2 42 0.78 7.8 288.6 44 0.8 8 296 46 0.82 8.2 303.4 48 0.85 8.5 314.5 50 0.88 8.8 325.6 52 0.9 9 333 54 0.9 9 333 56 0.92 9.2 340.4 58 0.92 9.2 340.4 60 0.96 9.6 355.2 62 0.96 9.6 355.2 64 0.98 9.8 362.6 66 0.98 9.8 362.6 68 0.99 9.9 366.3 2.6 Radiation Dosimetry
The estimated absorbed radiation doses to adults and children from intravenous administration of AdreView are as shown in Table 2:
Table 2. Estimated Absorbed Radiation Dose from AdreView ORGAN / TISSUE ABSORBED DOSE PER UNIT ADMINISTERED ACTIVITY ADULT 15-YEAR OLD 10-YEAR OLD 5-YEAR OLD 1-YEAR OLD NEONATES μGy/
MBqrad/mCi μGy/
MBqrad/mCi μGy/
MBqrad/mCi μGy/
MBqrad/mCi μGy/
MBqrad/mCi μGy/
MBqrad/mCi *OLINDA/EXM calculation based on biodistribution data from Swanson et al. and Publication 53 of the ICRP (International Commission on Radiological Protection) [Annals of the ICRP 1987; 18 (1-4): 329-331] Adrenals 16 0.059 21 0.078 31 0.115 42 0.155 67 0.248 111 0.411 Brain 3.9 0.014 4.9 0.018 8.1 0.030 13 0.048 24 0.089 55.9 0.207 Breast 4.7 0.017 5.9 0.022 9.4 0.035 15 0.056 28 0.104 65.3 0.242 Gallbladder 20 0.074 24 0.089 34 0.126 51 0.189 95 0.352 200 0.740 GI Tract Stomach Wall 7.6 0.028 10 0.037 17 0.063 27 0.100 51 0.189 114 0.422 Small Intestine Wall 7.7 0.028 9.8 0.036 16 0.059 25 0.093 46 0.170 104 0.385 Colon Wall 8.1 0.030 10 0.037 16 0.059 26 0.096 46 0.170 104.3 0.386 Upper Large Intestine Wall 8.4 0.031 11 0.041 18 0.067 30 0.111 53 0.196 119 0.440 Lower Large Intestine Wall 7.7 0.028 9.6 0.036 15 0.056 21 0.078 38 0.141 84.9 0.314 Heart Wall 18 0.067 23 0.085 35 0.130 53 0.196 94 0.348 182 0.673 Kidneys 13 0.048 16 0.059 24 0.089 35 0.130 59 0.218 132 0.488 Liver 67 0.248 87 0.322 130 0.481 180 0.666 330 1.221 720 2.664 Lungs 16 0.059 23 0.085 32 0.118 48 0.178 89 0.329 215 0.796 Muscles 6 0.022 7.6 0.028 12 0.044 17 0.063 33 0.122 75.1 0.278 Esophagus 6 0.022 7.6 0.028 11 0.041 18 0.067 32 0.118 72.2 0.267 Osteogenic Cells 16 0.059 21 0.078 31 0.115 47 0.174 100 0.370 254 0.940 Ovaries 7.9 0.029 10 0.037 15 0.056 22 0.081 41 0.152 92.3 0.342 Pancreas 12 0.044 15 0.056 25 0.093 39 0.144 68 0.252 143 0.529 Red marrow 5.6 0.021 6.8 0.025 10 0.037 15 0.056 30 0.111 89.5 0.331 Skin 3.7 0.014 4.4 0.016 7.1 0.026 11 0.041 21 0.078 53.1 0.196 Spleen 20 0.074 27 0.100 42 0.155 64 0.237 110 0.407 282 1.043 Testes 5.4 0.020 7.1 0.026 11 0.041 16 0.059 30 0.111 69.9 0.259 Thymus 6 0.022 7.6 0.028 11 0.041 18 0.067 32 0.118 72.2 0.267 Thyroid 4.7 0.017 6.1 0.023 9.9 0.037 16 0.059 30 0.111 69.4 0.257 Urinary Bladder Wall 66 0.244 84 0.311 110 0.407 110 0.407 200 0.740 478.0 1.769 Uterus 11 0.041 14 0.052 21 0.078 28 0.104 51 0.189 110.0 0.407 Whole Body 8.1 0.030 10 0.037 16 0.059 24 0.089 44 0.163 104.0 0.385 EFFECTIVE DOSE µSv/MBq 13.7 18.1 26.7 37.6 68 162 mSv/mCi 0.507 0.670 0.988 1.39 2.52 6 The effective dose resulting from an administered activity amount of 10 mCi is 5.07 mSv in an adult.
2.7 Imaging Guidelines
Pheochromocytoma and Neuroblastoma
Begin whole body planar scintigraphy imaging 24 ± 6 hours following administration of AdreView. Single photon emission computed tomography (SPECT) may be performed following planar scintigraphy, as appropriate [see Clinical Studies (14.1)].
Congestive Heart Failure
Begin anterior planar imaging of the chest at 4 hours (± 10 minutes) following administration of AdreView. Single photon emission computed tomography (SPECT) can then be performed. The recommended collimator for all imaging is a low-energy high-resolution. The recommended matrix for planar images is 128×128. The camera should be positioned to include the entire heart and as much of the upper chest as possible within the field of view [see Clinical Studies (14.2)].
2.8 Estimation of the H/M Ratio among Patients with Congestive Heart Failure
Initial evaluation of cardiac AdreView images involves visual examination of the location, pattern and intensity of cardiac radioactivity uptake to guide quantitative assessment (see Step 1 below). Perform quantitative assessment of radioactivity uptake in terms of the heart/mediastinum ratio (H/M) on anterior planar images of the chest (see Step 2 below).
Step 1. Visual Guidelines for AdreView Cardiac Uptake on Anterior Planar Chest Images
-
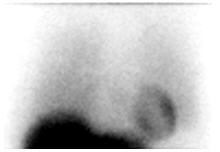
a. Normal:
Distinct visualization of the left ventricular myocardium in the left lower chest, with greater uptake in the heart than in the adjacent lungs and mediastinum (Figure 1). -
Figure 1. Normal anterior planar AdreView image of the chest
-
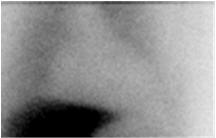
b. Abnormal:
Homogeneously or heterogeneously decreased cardiac uptake, with indistinct or absent visualization of the left ventricular myocardium. Cardiac activity is usually less than or equal to that of the adjacent left lung (Figure 2a). In extreme cases, little or no AdreView uptake is seen in the left lower chest (Figure 2b). -
Figure 2. Abnormal anterior planar AdreView images of the chest: a) Heterogeneously reduced cardiac uptake; b) Absent cardiac uptake
Figure 2a
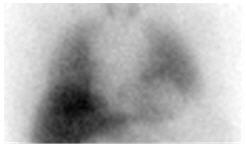
Figure 2b
Step 2. Quantitate AdreView Cardiac Uptake
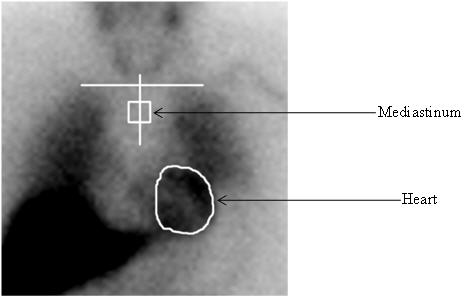
The AdreView H/M ratio is determined from the activity in heart (H) and mediastinum (M) regions of interest (ROIs) drawn on the anterior planar chest image (Figure 3) using the following procedure:
- (1) Draw an irregular ROI defining the epicardial border of the heart. If the epicardial border cannot be defined because all or the majority of the myocardium is not visualized, draw the ROI based upon the presumed location of the heart, using the medial aspects of the left and right lower lung for anatomical guidance.
- (2) Draw a horizontal line to mark the estimated location of the lung apices. If the most superior aspect of the image does not include the lung apices (because of limited field of view for a small gamma camera), draw this line at the top of the image display.
- (3) Draw a vertical line approximately equidistant from the medial aspects of the right and left lung.
- (4) Examine the counts for the 12 pixels along the vertical line starting 4 pixels below the intersection point with the horizontal line determined in step 2, and identify the pixel with the lowest counts. If more than one pixel has this same number of counts, choose the most superiorly located pixel.
- (5) Using the pixel defined in step 4 as the center, draw a square ROI of 7×7 dimensions.
- (6) Calculate the H/M ratio by dividing the counts/pixel in the total myocardium ROI determined in step 1 by the counts/pixel in the 7×7 pixel mediastinal ROI determined in step 5.
Figure 3. Illustration of creation of regions of interest for determination of the H/M ratio
Step 3. Interpretation of AdreView H/M Ratio
The expected range for AdreView H/M ratio is 1.0 to 2.4 [see Clinical Studies (14.2)].
Figure 4. Examples of AdreView Images in Subjects with Lower One and Two Year Mortality Risk
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions have been reported following AdreView administration. Prior to administration, question the patient for a history of prior reactions to iodine, an iodine-containing contrast agent or other products containing iodine. If the patient is known or strongly suspected to have hypersensitivity to iodine, an iodine-containing contrast agent or other products containing iodine, the decision to administer AdreView should be based upon an assessment of the expected benefits compared to the potential hypersensitivity risks. Have anaphylactic and hypersensitivity treatment measures available prior to AdreView administration [see Adverse Reactions (6.2)].
5.2 Imaging Errors due to Concomitant Medications and Risks Associated with Withdrawal of Medications
Many medications have the potential to interfere with AdreView imaging and review of the patient's medications is required prior to AdreView dosing due to the risk for unreliable imaging results. If the AdreView imaging information is essential for clinical care, consider the withdrawal of the following categories of medications if the withdrawal can be accomplished safely: antihypertensives that deplete norepinephrine stores or inhibit reuptake (e.g., reserpine, labetalol), antidepressants that inhibit norepinephrine transporter function (e.g., amitriptyline and derivatives, imipramine and derivatives, selective serotonin reuptake inhibitors), and sympathomimetic amines (e.g., phenylephrine, phenylpropanolamine, pseudoephedrine and ephedrine). The period of time necessary to discontinue any specific medication prior to AdreView dosing has not been established [see Drug Interactions (7)].
Pheochromocytoma and Neuroblastoma
Drugs which interfere with norepinephrine uptake in neuroendocrine tumors may lead to false negative imaging results. When medically feasible, stop these drugs before AdreView administration and monitor patients for the occurrence of clinically significant withdrawal symptoms, especially patients with elevated levels of circulating catecholamines and their metabolites.
Congestive Heart Failure
Many commonly used cardiovascular, pulmonary, and neuropsychiatric medications interfere with AdreView imaging (see above). AdreView imaging should not be performed if discontinuation of these medications would involve risks which outweigh the value of AdreView imaging. In clinical trials, patients were not eligible for AdreView imaging if they were receiving medications in the above categories and the risks for medication withdrawal were unacceptable or if they were not clinically stable (e.g., experiencing continuing chest pain, hemodynamic instability, or clinically significant arrhythmia).
5.3 Risk of Serious Adverse Reactions in Infants due to Benzyl Alcohol Preservative
Serious and fatal adverse reactions including "gasping syndrome" can occur in neonates and infants treated with benzyl alcohol-preserved drugs, including AdreView. The "gasping syndrome" is characterized by central nervous system depression, metabolic acidosis, and gasping respirations. When administering AdreView in infants consider the combined daily metabolic load of benzyl alcohol from all sources including AdreView (contains 10.3 mg of benzyl alcohol per mL) and other drugs containing benzyl alcohol. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known [see Use in Specific Populations (8.4)].
5.4 Increased Radiation Exposure in Patients with Severe Renal Impairment
AdreView is cleared by glomerular filtration and is not dialyzable. The radiation dose to patients with severe renal impairment may be increased due to the delayed elimination of the drug. Delayed AdreView clearance may also reduce the target to background ratios and decrease the quality of scintigraphic images. These risks importantly may limit the role of AdreView in the diagnostic evaluation of patients with severe renal impairment. AdreView safety and efficacy have not been established in these patients [see Clinical Pharmacology (12.2)].
5.5 Imaging Errors due to Conditions that Affect the Sympathetic Nervous System
Individuals with conditions that affect the sympathetic nervous system, e.g., Parkinsonian syndromes such as Parkinson's disease or multiple system atrophy, may show decreased cardiac uptake of AdreView independent of heart disease.
5.6 Thyroid Accumulation
Failure to block thyroid uptake of iodine 123 may result in an increased long term risk for thyroid neoplasia [see Dosage and Administration (2.2)].
5.7 Hypertension
Assess the patient's pulse and blood pressure before and intermittently for 30 minutes after AdreView administration. AdreView may increase release of norepinephrine from chromaffin granules and produce a transient episode of hypertension, although this was not observed in the clinical studies. Prior to AdreView administration, ensure emergency cardiac and anti-hypertensive treatments are readily available.
-
6 ADVERSE REACTIONS
6.1 Clinical Study Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
During clinical development 1346 patients were exposed to AdreView, 251 patients with known or suspected pheochromocytoma or neuroblastoma, 985 patients with heart failure, and 110 control patients. All patients were monitored for adverse reactions over a 24 hour period following AdreView administration.
Pheochromocytoma and Neuroblastoma
Serious adverse reactions were not observed in the AdreView clinical study. Adverse reactions were all mild to moderate in severity and were predominantly isolated occurrences (≤ 2 patients) of one of the following reactions: dizziness, rash, pruritus, flushing or injection site hemorrhage.
Congestive Heart Failure
No serious adverse reactions to AdreView were observed in clinical studies. Adverse reactions that occurred with a frequency > 1% were associated with the injection site (1.3%), problems such as hematoma and bruising. The other most common reactions were flushing (0.3%) and headache (0.4%). The adverse reactions were predominantly of mild to moderate intensity.
6.2 Postmarketing Experience
Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hypersensitivity reactions have uncommonly been reported during the postmarketing use of AdreView [see Warnings and Precautions (5.1)].
-
7 DRUG INTERACTIONS
The following drugs have the potential to decrease the uptake of norepinephrine and cause false negative imaging results: antihypertensives that deplete norepinephrine stores or inhibit reuptake (e.g., reserpine, labetalol), antidepressants that inhibit norepinephrine transporter function (e.g., amitriptyline and derivatives, imipramine and derivatives, selective serotonin reuptake inhibitors), sympathomimetic amines (e.g., phenylephrine, phenylpropanolamine, pseudoephedrine and ephedrine), and cocaine. Clinical studies have not determined which specific drugs may cause false negative imaging results nor whether all drugs in any specific pharmacologic class have the same potential to produce the negative imaging results. Increasing the dose of AdreView will not overcome any potential uptake limiting effect of these drugs. Before AdreView administration, discontinue (for at least 5 biological half-lives) drugs known or expected to reduce norepinephrine uptake, as clinically tolerated.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Radioactive iodine products cross the placenta and can permanently impair fetal thyroid function. Administration of an appropriate thyroid blocking agent is recommended before use of AdreView in a pregnant woman to protect the woman and fetus from accumulation of I 123 [see Dosage and Administration (2.2) ].
There are no available data on AdreView use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Animal reproduction studies have not been conducted with iobenguane I 123. All radiopharmaceuticals have the potential to cause fetal harm depending on the fetal stage of development and the magnitude of the radiation dose. Advise pregnant women of the potential risks of fetal exposure to radiation doses with administration of AdreView.
AdreView contains 10.3 mg/mL of benzyl alcohol. Because benzyl alcohol is rapidly metabolized by a pregnant woman, benzyl alcohol exposure in the fetus is unlikely. However, adverse reactions have occurred in premature neonates and low birth weight infants who received intravenously administered benzyl alcohol-containing drugs [see Warnings and Precautions (5.3) and Use in Specific Populations (8.4)].
The estimated background risk of major birth defects and miscarriage for the indicated population(s) is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
8.2 Lactation
Risk Summary
Iodine 123 (I 123), the radionuclide in AdreView, is present in human milk. There is no information on the effects on the breastfed infant or on milk production. Advise a lactating woman to interrupt breastfeeding and pump and discard breastmilk for at least 6 days (>10 physical half-lives) after AdreView administration in order to minimize radiation exposure to a breastfed infant.
8.4 Pediatric Use
The safety and effectiveness of AdreView have been established in the age groups 1 month to 16 years in patients with known or suspected neuroblastoma [see Clinical Studies (14.1)]. Safety and effectiveness in pediatric patients below the age of 1 month or in any pediatric patient with congestive heart failure have not been established.
Serious adverse reactions including fatal reactions and the "gasping syndrome" occurred in premature neonates and infants in the neonatal intensive care unit who received drugs containing benzyl alcohol as a preservative. In these cases, benzyl alcohol dosages of 99 to 234 mg/kg/day produced high levels of benzyl alcohol and its metabolites in the blood and urine (blood levels of benzyl alcohol were 0.61 to 1.378 mmol/L). Additional adverse reactions included gradual neurological deterioration, seizures, intracranial hemorrhage, hematologic abnormalities, skin breakdown, hepatic and renal failure, hypotension, bradycardia, and cardiovascular collapse. Preterm, low-birth weight infants may be more likely to develop these reactions because they may be less able to metabolize benzyl alcohol. When administering AdreView in infants consider the combined daily metabolic load of benzyl alcohol from all sources including AdreView (contains 10.3 mg of benzyl alcohol per mL) and other drugs containing benzyl alcohol. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known [see Warnings and Precautions (5.3)].
8.5 Geriatric Use
In clinical studies of AdreView in heart disease, 27% of subjects were 65-74 years of age and 17% of subjects were 75 years of age or over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
AdreView is excreted by the kidneys, and the risks of adverse reactions, increased radiation dose, and occurrence of falsely negative imaging results, may be greater in patients with severely impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection and image interpretation. Consider assessment of renal function in elderly patients prior to AdreView administration.
- 10 OVERDOSAGE
-
11 DESCRIPTION
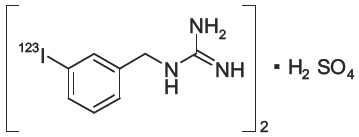
AdreView (Iobenguane I 123 Injection) is a sterile, pyrogen-free radiopharmaceutical for intravenous injection. Each mL contains 0.08 mg iobenguane sulfate, 74 MBq (2 mCi) of I 123 (as iobenguane sulfate I 123) at calibration date and time on the label, 23 mg sodium dihydrogen phosphate dihydrate, 2.8 mg disodium hydrogen phosphate dihydrate and 10.3 mg (1% v/v) benzyl alcohol with a pH of 5.0 – 6.5. Iobenguane sulfate I 123 is also known as I 123 meta-iodobenzlyguanidine sulfate and has the following structural formula:
11.1 Physical Characteristics
Iodine 123 is a cyclotron-produced radionuclide that decays to Te 123 by electron capture and has a physical half-life of 13.2 hours.
Table 3. Principal Radiation Emission Data – Iodine 123 Radiation Energy Level (keV) Abundance (%) Gamma 159 83 11.2 External Radiation
The specific gamma ray constant for iodine 123 is 1.6 R/mCi-hr at 1 cm. The first half value thickness of lead (Pb) for I 123 is 0.04 cm. The relative transmission of radiation emitted by the radionuclide that results from interposition of various thicknesses of Pb is shown in Table 4 (e.g., the use of 2.16 cm Pb will decrease the external radiation exposure by a factor of about 1,000).
Table 4. Reduction in In-air Collision Kerma Caused by Lead Shielding* Shield Thickness cm of lead (Pb) Reduction in In-air Collision Kerma - * Calculation based on attenuation and energy-transfer coefficients obtained from National Institute of Standards & Technology Report NISTIR 5632.
0.04 0.5 0.13 10-1 0.77 10-2 2.16 10-3 3.67 10-4 -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Iobenguane is similar in structure to the antihypertensive drug guanethedine and to the neurotransmitter norepinephrine (NE). Iobenguane is, therefore, largely subject to the same uptake and accumulation pathways as NE. Iobenguane is taken up by the NE transporter in adrenergic nerve terminals and stored in the presynaptic storage vesicles. Iobenguane accumulates in adrenergically innervated tissues such as the adrenal medulla, salivary glands, heart, liver, spleen and lungs as well as tumors derived from the neural crest. By labeling iobenguane with the isotope iodine 123, it is possible to obtain scintigraphic images of the organs and tissues in which the radiopharmaceutical accumulates.
12.2 Pharmacodynamics
AdreView is a diagnostic radiopharmaceutical which contains a small quantity of iobenguane that is not expected to produce a pharmacodynamic effect [see Description (11)]. To minimize radiation dose to the thyroid gland, this organ should be blocked before dosing [see Dosage and Administration (2.2)]. Since iobenguane is excreted mainly via the kidneys, patients with severe renal insufficiency may experience increased radiation exposure and impaired imaging results. Frequent voiding should be encouraged after administration to minimize the radiation dose to the bladder [see Warnings and Precautions (5.4)]. The calculation of the estimated radiation dose is shown in Table 2 [see Dosage and Administration (2.6)].
12.3 Pharmacokinetics
Iobenguane is rapidly cleared from the blood and accumulates in adrenergically innervated tissues [see Clinical Pharmacology (12.1)]. Retention is especially prolonged in highly adrenergically innervated tissues (e.g., the adrenal medulla, heart, and salivary glands).
The majority of the iobenguane dose is excreted unaltered by the kidneys via glomerular filtration. A rapid initial clearance of circulating iobenguane is observed, followed by a slow clearance as iobenguane is released from other compartments. In patients with normal renal function, 70 to 90% of the administered dose is recovered unaltered in urine within 4 days. Iobenguane is not cleared by dialysis [see Warnings and Precautions (5.4)]. Most of the remaining radioactivity recovered in the urine is in the form of the radioiodinated metabolite m-iodohippuric acid (MIHA) (typically ≤ 10%) and free radioiodide (typically ≤ 6%). The enzymatic process responsible for metabolism has not been well characterized and the pharmacologic activity of these metabolites has not been studied. Only a small amount (< 1%) of the injected dose is eliminated via the feces.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Iobenguane hemisulfate was not mutagenic in vitro in the Ames bacterial mutation assay and in the in vitro mouse lymphoma test, and was negative in the in vivo micronucleus test in rats.
Long-term animal studies have not been conducted to evaluate AdreView's carcinogenic potential or potential effects on fertility.
13.2 Animal Toxicology and/or Pharmacology
Iobenguane sulfate testing in dogs revealed electrocardiographic (ECG) changes after administration of 202 times the mg/m2 conversion of the maximum human dose for a 60 kg adult; the no observable effect level (NOEL) was not determined. When iobenguane was tested in a cell system stably expressing hERG-1 potassium channels, inhibition of potassium channels was not observed at an 80 μM iobenguane concentration and the IC50 was 487 μM.
-
14 CLINICAL STUDIES
14.1 Pheochromocytoma and Neuroblastoma
The safety and efficacy of AdreView were assessed in an open-label, multicenter, multinational trial of 251 subjects with known or suspected neuroblastoma or pheochromocytoma. Diagnostic efficacy for the detection of metabolically active neuroblastoma or pheochromocytoma was determined by comparison of focal increased radionuclide uptake on planar scintigraphy at 24 ± 6 hours post-administration of AdreView against the definitive diagnosis (standard of truth). Anterior and posterior planar whole-body images, or alternatively whole-body overlapping spot images, were acquired from the head to below the knees. Additional spot images were performed as deemed appropriate at the discretion of the clinical image reviewer. SPECT imaging of the thorax and abdomen was then obtained when possible.
Of the 251 subjects dosed with AdreView, 100 had known or suspected neuroblastoma and 151 had known or suspected pheochromocytoma. The population included 154 adults and 97 pediatric patients; the majority of adults were female (59%), the majority of pediatric subjects were male (58%). The adult subjects had a mean age of 49 years (range 17 to 88 years). The pediatric patients (56 males and 41 females) consisted of 32 infants (1 month up to 2 years of age), 62 children (2 years up to 12 years) and three adolescents (12 years up to 16 years).
The definitive diagnosis (standard of truth) for the presence or absence of metabolically active pheochromocytoma or neuroblastoma was determined by histopathology or, when histopathology was unavailable, a composite of imaging (i.e., CT, MRI, [131I]-mIBG scintigraphy), plasma/urine catecholamine and/or catecholamine metabolite measurements, and clinical follow-up.
A standard of truth was available for 211 subjects (127 with pheochromocytoma, 84 with neuroblastoma) and this group comprised the diagnostic efficacy population. For 93 of these subjects, the standard of truth was based solely upon histopathology. Of 211 subjects in the efficacy population, all had planar scintigraphy and 167 subjects had SPECT in addition to planar imaging. All images were assessed independently by three readers blinded to all clinical data. Table 5 summarizes the AdreView performance characteristics, by reader.
Table 5. AdreView Planar Imaging: Sensitivity and Specificity Outcome Reader A Reader B Reader C Sensitivity (n = 159) Point estimate 0.80 0.77 0.79 95% confidence interval 0.73 - 0.86 0.70 - 0.84 0.71 - 0.85 Specificity (n = 52) Point estimate 0.77 0.73 0.69 95% confidence interval 0.63 - 0.87 0.59 - 0.84 0.55 - 0.81 Performance characteristics (sensitivity and specificity) of AdreView planar imaging in patients with known or suspected neuroblastoma were similar to those in patients with known or suspected pheochromocytoma. Among the selected patients who also underwent SPECT imaging, similar performance characteristics of AdreView scintigraphy were observed when SPECT plus planar imaging was compared to planar imaging alone.
14.2 Congestive Heart Failure
The safety and efficacy of AdreView were evaluated in two open label, multicenter trials in patients with New York Heart Association (NYHA) class II or III heart failure and left ventricular ejection fraction ≤ 35%. The trials excluded subjects with an acute myocardial infarction within the prior thirty days, subjects with a functioning ventricular pacemaker as well as subjects who had received defibrillation to treat a previous arrhythmic event. Subjects underwent AdreView myocardial imaging (planar and SPECT) and continued standard clinical care; AdreView results were not used in a patient's clinical care. Mortality was assessed for up to two years after AdreView imaging and the results from the trials were analyzed using a pre-specified data integration plan.
AdreView images in each trial were reviewed by three independent readers who assessed the H/M ratio on 3 hour 50 minute post-injection planar scintigraphy. Readers were masked to clinical information and the majority read value was used in analyses. The prognostic performance of the H/M ratio in estimating mortality was analyzed using the pre-specified 1.6 ratio cut-point to distinguish patients with higher risk from those with lower risk; other cut-points were also analyzed.
Within the two trials, 964 patients were enrolled; 80% were men, 83% were categorized as NYHA class II and 17% as class III. The average age was 62 years (range 20 - 90 years of age). Most patients had ischemic heart disease (66%) and a history of smoking (74%). The patients were on a stable regimen of cardiovascular medications, including angiotensin converting enzyme (ACE) inhibitors and/or angiotensin receptor blockers (ARBs) (93%) and beta-blockers (92%). The range of AdreView H/M ratios in these subjects was 1.0-2.4 [mean 1.4 (± 0.2 standard deviation)].
Within the two trials, 94 age-matched control subjects without heart disease were enrolled, 64% were men, average age was 59 years (range 29 - 82 years of age). The range of AdreView H/M ratios in these subjects was 1.1-2.4 [mean 1.8 (± 0.2 standard deviation)].
One Year Results: By 12 months following enrollment, 50 (5%) patients had died, 61 (6%) had missing follow-up information and three patients had missing H/M ratios.
Two Year Results: By 23 months following enrollment (the requirement for designation of two-year follow-up), 96 (10%) patients had died, 201 (21%) patients had missing follow-up information and three patients had missing H/M ratio data.
Table 6 summarizes the mortality results by categories of H/M ratio.
Table 6. AdreView H/M Ratios and One and Two Year* Mortality Rates Range of H/M Ratio Values
(number of patients)One-year Mortality Rate (%)
(95% confidence interval)Two-year* Mortality Rate (%)
(95% confidence interval)- * Calculated at 23 months based upon protocol definition for 2-year follow-up
≥ 1.6 (201) 1.0 (0.0, 2.4) 3.3 (0.7, 5.9) 1.2 – 1.6 (668) 5.5 (3.8, 7.3) 11.5 (9.0, 14.0) < 1.2 (92) 13.4 (6.4, 20.5) 22.0 (13.2, 30.8) H/M Ratio Prognostic Performance Characteristics:
Follow-up mortality results were used to estimate the baseline H/M ratio prognostic performance characteristics. In these estimates, various H/M ratio "cut points" were used to group patients into those with higher versus lower H/M values, such as < 1.6 versus ≥ 1.6. The group of patients who died was examined to determine the probability of these patients having had a lower baseline H/M ratio (sensitivity). The group of patients who survived was examined to determine the probability of these patients having had a higher baseline H/M ratio (specificity). Based upon these results, the prognostic usefulness of any given H/M ratio in a patient was estimated by the positive predictive value (PPV) and the negative predictive value (NPV). The PPV is the probability of death given a lower H/M ratio; the NPV is the probability of survival given a higher H/M ratio.
Table 7 summarizes the performance characteristics by various H/M ratio categories for one year, the time point with the most complete data. Results were similar for the two year follow-up time point analyses.
Table 7. One Year Mortality Outcomes and AdreView Prognostic Performance Characteristics H/M Group* Subjects
(n = 961)Death† Survival Sensitivity
(%)Specificity
(%)PPV
(%)NPV
(%)- * subjects grouped by H/M ratio cut-off values;
- † 6% discontinued patients are counted as survived (non-events)
< 1.2 92 12 80 24 91 13 96 ≥ 1.2 869 38 831 < 1.4 429 33 396 66 57 8 97 ≥ 1.4 532 17 515 < 1.6 760 48 712 96 22 6 99 ≥ 1.6 201 2 199 < 1.8 914 50 864 100 5 5 100 ≥ 1.8 47 0 47 Cox Proportional Hazards Analyses:
The association of potential risk factors with mortality for up to two years was analyzed in Cox multivariate proportional hazard modeling that included such variables as demographics, hypertension, dyslipidemia, diabetes, cardiovascular medications, smoking, and NYHA classification. The initial model included all pre-specified variables except for H/M ratio and used backward selection of variables found to be significant risk factors of all-cause mortality (p < 0.05) for inclusion in the final model. The final model consisted of the significant variables from the initial model plus the H/M ratio. In addition to age, in the final model, the H/M ratio was found to be a significant risk factor for mortality (Hazard Ratio < 0.08, p < 0.001). Left ventricular ejection fraction and brain natriuretic peptide (BNP) were not included in these models, and AdreView SPECT defect scores were not tested in these models.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
AdreView is supplied in 10 mL glass vials containing a total volume of 5 mL of solution with a total radioactivity of 370 MBq (10 mCi) at calibration time. Each vial is enclosed in a lead container of appropriate thickness.
NDC: 17156-235-01
Storage
Store AdreView at 20°-25°C (68°-77°F); excursions permitted to 15°-30°C (59°-86°F) [see USP]. This product does not contain a preservative. Store within the original lead container or equivalent radiation shielding.
In accordance with USP recommendations Iobenguane I 123 Injection preparations should not be used after the expiration date and time stated on the label.
-
17 PATIENT COUNSELING INFORMATION
Instruct patients to inform their physician or healthcare provider if they:
- 1. are pregnant. Advise a pregnant woman of the potential risks of fetal exposure to radiation doses with AdreView [see Use in Specific Populations (8.1)].
- 2. are breast feeding. Advise a lactating woman to interrupt breastfeeding and pump and discard breastmilk for at least 6 days (>10 physical half-lives) after AdreView administration in order to minimize radiation exposure to a breastfed infant [see Use in Specific Populations (8.2].
- 3. are sensitive to iodine, an iodine-containing contrast agent or other products that contain iodine.
- 4. are sensitive to Potassium Iodide Oral Solution, or Lugol's Solution.
- 5. have reduced renal function.
Instruct patients to increase their level of hydration prior to receiving AdreView and to void frequently for the first 48 hours following AdreView administration.
-
SPL UNCLASSIFIED SECTION
Manufactured and Distributed by GE Healthcare, Medi-Physics, Inc., Arlington Heights, IL 60004 U.S.A.
AdreView is a trademark of GE Healthcare or one of its subsidiaries.
GE and the GE Monogram are trademarks of General Electric Company.© 2020 General Electric Company – All rights reserved.
43-2035D
- PRINCIPAL DISPLAY PANEL - 5 mL Vial Label
-
INGREDIENTS AND APPEARANCE
ADREVIEW
iobenguane i-123 injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 17156-235 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Iobenguane I-123 (UNII: P2TH1XYZ84) (Iobenguane I-123 - UNII:P2TH1XYZ84) Iobenguane I-123 2 mCi in 1 mL Inactive Ingredients Ingredient Name Strength Sodium phosphate, monobasic, dihydrate (UNII: 5QWK665956) 23 mg in 1 mL Sodium phosphate, dibasic, dihydrate (UNII: 94255I6E2T) 2.8 mg in 1 mL Benzyl alcohol (UNII: LKG8494WBH) 10.3 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 17156-235-01 1 in 1 CONTAINER 09/19/2008 1 5 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022290 09/19/2008 Labeler - Medi-Physics Inc. (095263729) Establishment Name Address ID/FEI Business Operations Medi-Physics Inc. 095263729 MANUFACTURE(17156-235) , RELABEL(17156-235) , REPACK(17156-235) , ANALYSIS(17156-235)

Trademark Results [AdreView]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ADREVIEW 78318112 3014913 Live/Registered |
GE HEALTHCARE LIMITED 2003-10-24 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.