FLOXURIDINE- floxuridine injection, powder, lyophilized, for solution
Floxuridine by
Drug Labeling and Warnings
Floxuridine by is a Prescription medication manufactured, distributed, or labeled by Fresenius Kabi USA, LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
SPL UNCLASSIFIED SECTION
FOR INTRA-ARTERIAL INFUSION ONLY
WARNING
It is recommended that Floxuridine for Injection, USP be given only by or under the supervision of a qualified physician who is experienced in cancer chemotherapy and intra-arterial drug therapy and is well versed in the use of potent antimetabolites.
Because of the possibility of severe toxic reactions, all patients should be hospitalized for initiation of the first course of therapy.
-
DESCRIPTION:
Floxuridine for Injection, USP, an antineoplastic antimetabolite, is available as a sterile, nonpyrogenic, lyophilized powder for reconstitution. Each vial contains 500 mg of floxuridine which is to be reconstituted with 5 mL of sterile water for injection. An appropriate amount of reconstituted solution is then diluted with a parenteral solution for intra-arterial infusion (see DOSAGE AND ADMINISTRATION).
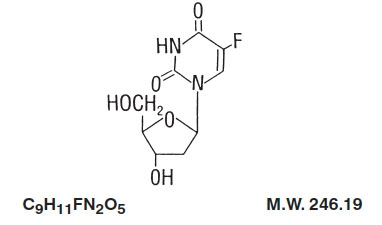
Floxuridine is a fluorinated pyrimidine. Chemically, floxuridine is 2’-deoxy-5-fluorouridine. It is a white to off-white odorless solid which is freely soluble in water. The 2% aqueous solution has a pH of between 4.0 and 5.5.
The structural formula is:
-
CLINICAL PHARMACOLOGY:
When floxuridine is given by rapid intra-arterial injection it is apparently rapidly catabolized to 5-fluorouracil. Thus, rapid injection of floxuridine produces the same toxic and antimetabolic effects as does 5-fluorouracil. The primary effect is to interfere with the synthesis of deoxyribonucleic acid (DNA) and to a lesser extent inhibit the formation of ribonucleic acid (RNA). However, when floxuridine is given by continuous intra-arterial infusion its direct anabolism to floxuridine-monophosphate is enhanced, thus increasing the inhibition of DNA.
Floxuridine is metabolized in the liver. The drug is excreted intact and as urea, fluorouracil, α-fluoro-β-ureidopropionic acid, dihydrofluorouracil, α-fluoro-β-guanidopropionic acid and α-fluoro-β-alanine in the urine; it is also expired as respiratory carbon dioxide. Pharmacokinetic data on intra-arterial infusion of floxuridine are not available.
-
INDICATIONS AND USAGE:
Floxuridine for Injection, USP is effective in the palliative management of gastrointestinal adenocarcinoma metastatic to the liver, when given by continuous regional intra-arterial infusion in carefully selected patients who are considered incurable by surgery or other means. Patients with known disease extending beyond an area capable of infusion via a single artery should, except in unusual circumstances, be considered for systemic therapy with other chemotherapeutic agents.
- CONTRAINDICATIONS:
-
WARNINGS:
BECAUSE OF THE POSSIBILITY OF SEVERE TOXIC REACTIONS, ALL PATIENTS SHOULD BE HOSPITALIZED FOR THE FIRST COURSE OF THERAPY.
Floxuridine should be used with extreme caution in poor risk patients with impaired hepatic or renal function or a history of high-dose pelvic irradiation or previous use of alkylating agents. The drug is not intended as an adjuvant to surgery.
Floxuridine may cause fetal harm when administered to a pregnant woman. It has been shown to be teratogenic in the chick embryo, mouse (at doses of 2.5 to 100 mg/kg) and rat (at doses of 75 to 150 mg/kg). Malformations included cleft palates; skeletal defects; and deformed appendages, paws and tails. The dosages which were teratogenic in animals are 4.2 to 125 times the recommended human therapeutic dose.
There are no adequate and well-controlled studies with floxuridine in pregnant women. If this drug is used during pregnancy or if the patient becomes pregnant while taking (receiving) this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
Combination Therapy
Any form of therapy which adds to the stress of the patient, interferes with nutrition or depresses bone marrow function will increase the toxicity of floxuridine.
-
PRECAUTIONS:
General
Floxuridine is a highly toxic drug with a narrow margin of safety. Therefore, patients should be carefully supervised since therapeutic response is unlikely to occur without some evidence of toxicity. Severe hematological toxicity, gastrointestinal hemorrhage and even death may result from the use of floxuridine despite meticulous selection of patients and careful adjustment of dosage. Although severe toxicity is more likely in poor risk patients, fatalities may be encountered occasionally even in patients in relatively good condition.
Therapy is to be discontinued promptly whenever one of the following signs of toxicity appears:
Myocardial ischemia
Stomatitis or esophagopharyngitis, at the first visible sign
Leukopenia (WBC under 3,500) or a rapidly falling white blood count
Vomiting, intractable
Diarrhea, frequent bowel movements or watery stools
Gastrointestinal ulceration and bleeding
Thrombocytopenia (platelets under 100,000)
Hemorrhage from any site
Information for Patients
Patients should be informed of expected toxic effects, particularly oral manifestations. Patients should be alerted to the possibility of alopecia as a result of therapy and should be informed that it is usually a transient effect.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term studies in animals to evaluate the carcinogenic potential of floxuridine have not been conducted. On the basis of the available data, no evaluation can be made of the carcinogenic risk of floxuridine to humans.
Mutagenesis
Oncogenic transformation of fibroblasts from mouse embryo has been induced in vitro by floxuridine, but the relationship between oncogenicity and mutagenicity is not clear. Floxuridine has also been shown to be mutagenic in human leukocytes in vitro and in the Drosophila test system. In addition, 5-fluorouracil, to which floxuridine is catabolized when given by intraarterial injection, has been shown to be mutagenic in in vitro tests.
Impairment of Fertility
The effects of floxuridine on fertility and general reproductive performance have not been studied in animals. However, because floxuridine is catabolized to 5-fluorouracil, it should be noted that 5-fluorouracil has been shown to induce chromosomal aberrations and changes in chromosome organization of spermatogonia in rats at doses of 125 or 250 mg/kg, administered intraperitoneally.
Spermatogonial differentiation was also inhibited by fluorouracil, resulting in transient infertility. In female rats, fluorouracil, administered intraperitoneally at doses of 25 or 50 mg/kg during the preovulatory phase of oogenesis, significantly reduced the incidence of fertile matings, delayed the development of pre- and post-implantation embryos, increased the incidence of preimplantation lethality and induced chromosomal anomalies in these embryos. Compounds such as floxuridine, which interfere with DNA, RNA and protein synthesis, might be expected to have adverse effects on gametogenesis.
Pregnancy
Teratogenic Effects: Pregnancy category D.
See WARNINGS. Floxuridine has been shown to be teratogenic in the chick embryo, mouse (at doses of 2.5 to 100 mg/kg) and rat (at doses of 75 to 150 mg/kg). Malformations included cleft palates, skeletal defects and deformed appendages, paws and tails. The dosages which were teratogenic in animals are 3.2 to 125 times the recommended human therapeutic dose.
There are no adequate and well-controlled studies with floxuridine in pregnant women. While there is no evidence of teratogenicity in humans due to floxuridine, it should be kept in mind that other drugs which inhibit DNA synthesis (eg, methotrexate and aminopterin) have been reported to be teratogenic in humans. Floxuridine should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nonteratogenic Effects
Floxuridine has not been studied in animals for its effects on peri- and postnatal development. However, compounds which inhibit DNA, RNA and protein synthesis might be expected to have adverse effects on peri- and postnatal development.
-
ADVERSE REACTIONS:
Adverse reactions to the arterial infusion of floxuridine are generally related to the procedural complications of regional arterial infusion.
The more common adverse reactions to the drug are nausea, vomiting, diarrhea, enteritis, stomatitis and localized erythema. The more common laboratory abnormalities are anemia, leukopenia, thrombocytopenia and elevations of alkaline phosphatase, serum transaminase, serum bilirubin and lactic dehydrogenase.
Other adverse reactions are:
Gastrointestinal: duodenal ulcer, duodenitis, gastritis, bleeding, gastroenteritis, glossitis, pharyngitis, anorexia, cramps, abdominal pain; possible intra- and extrahepatic biliary sclerosis, as well as acalculous cholecystitis.
Dermatologic: alopecia, dermatitis, nonspecific skin toxicity, rash.
Cardiovascular: myocardial ischemia.
Miscellaneous Clinical Reactions: fever, lethargy, malaise, weakness.
Laboratory Abnormalities: BSP, prothrombin, total proteins, sedimentation rate and thrombopenia.
Procedural Complications of Regional Arterial Infusion: arterial aneurysm; arterial ischemia; arterial thrombosis; embolism; fibromyositis; thrombophlebitis; hepatic necrosis; abscesses; infection at catheter site; bleeding at catheter site; catheter blocked, displaced or leaking.
The following adverse reactions have not been reported with floxuridine but have been noted following the administration of 5-fluorouracil. While the possibility of these occurring following floxuridine therapy is remote because of its regional administration, one should be alert for these reactions following the administration of floxuridine because of the pharmacological similarity of these two drugs: pancytopenia, agranulocytosis, myocardial ischemia, angina, anaphylaxis, generalized allergic reactions, acute cerebellar syndrome, nystagmus, headache, dry skin, fissuring, photosensitivity, pruritic maculopapular rash, increased pigmentation of the skin, vein pigmentation, lacrimal duct stenosis, visual changes, lacrimation, photophobia, disorientation, confusion, euphoria, epistaxis and nail changes, including loss of nails.
-
OVERDOSAGE:
The possibility of overdosage with floxuridine is unlikely in view of the mode of administration. Nevertheless, the anticipated manifestations would be nausea, vomiting, diarrhea, gastrointestinal ulceration and bleeding, bone marrow depression (including thrombocytopenia, leukopenia and agranulocytosis). No specific antidotal therapy exists. Patients who have been exposed to an overdosage of floxuridine should be monitored hematologically for at least 4 weeks. Should abnormalities appear, appropriate therapy should be utilized.
The acute intravenous toxicity of floxuridine is as follows:
Species
LD 50
(mg/kg ± S.E.)
Mouse
880 ± 51
Rat
670 ± 73
Rabbit
94 ± 19.6
Dog
157 ± 46
-
DOSAGE AND ADMINISTRATION:
Each vial must be reconstituted with 5 mL of sterile water for injection to yield a solution containing approximately 100 mg of floxuridine/mL. The calculated daily dose(s) of the drug is then diluted with 5% dextrose or 0.9% sodium chloride injection to a volume appropriate for the infusion apparatus to be used. The administration of floxuridine is best achieved with the use of an appropriate pump to overcome pressure in large arteries and to ensure a uniform rate of infusion.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit.
The recommended therapeutic dosage schedule of floxuridine by continuous arterial infusion is 0.1 to 0.6 mg/kg/day. The higher dosage ranges (0.4 to 0.6 mg) are usually employed for hepatic artery infusion because the liver metabolizes the drug, thus reducing the potential for systemic toxicity. Therapy can be given until adverse reactions appear. (See PRECAUTIONS). When these side effects have subsided, therapy may be resumed. The patients should be maintained on therapy as long as response to floxuridine continues.
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published. 1-7 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
-
HOW SUPPLIED:
Product
No.
NDC
No.
104507
63323-145-07
Floxuridine for Injection, USP, 500 mg floxuridine powder in a 5 mL vial, packaged individually. This is to be reconstituted with 5 mL sterile water for injection.
The container closure is not made with natural rubber latex.
The sterile powder should be stored at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature]. Reconstituted vials should be stored under refrigeration 2° to 8°C (36° to 46°F) for not more than 2 weeks.
-
REFERENCES:
- Recommendations for the safe handling of parenteral antineoplastic drugs. Washington, DC, US Government Printing Office NIH publication 83-2621.
- AMA Council Report. Guidelines for handling parenteral antineoplastics. JAMA. Mar 15, 1985, 253:1590-1592.
- National Study Commission on Cytotoxic Exposure: Recommendations for handling cytotoxic agents. Available from Louis P. Jeffrey, ScD, Director of Pharmacy Services, Rhode Island Hospital, 593 Eddy Street, Providence, Rhode Island 02902.
- Clinical Oncological Society of Australia: Guidelines and recommendations for safe handling of antineoplastic agents. Med J Aust. Apr 30, 1983, 1:426-428.
- Jones, RB, Frank R, Mass T: Safe handling of chemotherapeutic agents: a report from the Mount Sinai Medical Center. CA. Sept-Oct, 1983, 33:258-263.
- ASHP technical assistance bulletin on handling cytotoxic drugs in hospitals. AM J Hosp Pharm. Jan, 1985, 42:131-137.
- OSHA Work-Practice Guidelines for Personnel Dealing with Cytotoxic (Antineoplastic) Drugs. Am J Hosp Pharm. 1986; 43:1193-1204.
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL
PACKAGE LABEL - PRINCIPAL DISPLAY - Floxuridine 500 mg Vial Label
Floxuridine for Injection, USP
500 mg per vial
For intra-arterial use only.
Powder for reconstitution of parenteral solutions.
Rx only

PACKAGE LABEL - PRINCIPAL DISPLAY - Floxuridine 500 mg Vial Carton Panel
Floxuridine for Injection, USP
500 mg per vial
For intra-arterial use only.
Powder for reconstitution of parenteral solutions.
Rx only
-
INGREDIENTS AND APPEARANCE
FLOXURIDINE
floxuridine injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 63323-145 Route of Administration INTRA-ARTERIAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FLOXURIDINE (UNII: 039LU44I5M) (FLOXURIDINE - UNII:039LU44I5M) FLOXURIDINE 500 mg in 5 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 63323-145-07 1 in 1 CARTON 03/15/2001 1 5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA075837 03/15/2001 Labeler - Fresenius Kabi USA, LLC (608775388) Establishment Name Address ID/FEI Business Operations Fresenius Kabi USA, LLC 023648251 manufacture(63323-145)

© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.