VEOZAH- fezolinetant tablet, film coated
VEOZAH by
Drug Labeling and Warnings
VEOZAH by is a Prescription medication manufactured, distributed, or labeled by Astellas Pharma US, Inc., AndersonBrecon Inc., Fujimoto Chemicals Co., Ltd., Juzen Chemical Corporation, Astellas Pharma Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VEOZAH safely and effectively. See full prescribing information for VEOZAH.
VEOZAH® (fezolinetant) tablets, for oral use
Initial U.S. Approval: 2023WARNING: RISKS OF HEPATOTOXICITY
See full prescribing information for complete boxed warning.
Hepatotoxicity has occurred with the use of VEOZAH in the postmarketing setting (5.1).
- Perform hepatic laboratory tests prior to initiation of treatment to evaluate for hepatic function and injury. Do not start VEOZAH if either aminotransferase is ≥ 2 x ULN or if the total bilirubin is ≥ 2 x ULN for the evaluating laboratory.
- Perform follow-up hepatic laboratory testing monthly for the first 3 months, at 6 months, and 9 months of treatment (2.1, 5.1).
- Advise patients to discontinue VEOZAH immediately and seek medical attention including hepatic laboratory tests if they experience signs or symptoms that may suggest liver injury (new onset fatigue, decreased appetite, nausea, vomiting, pruritus, jaundice, pale feces, dark urine, or abdominal pain) (2.1, 5.1).
- Discontinue VEOZAH if transaminase elevations are > 5 x ULN, or if transaminase elevations are > 3 x ULN and the total bilirubin level is > 2 x ULN.
- If transaminase elevations > 3 x ULN occur, perform more frequent follow-up hepatic laboratory tests until resolution (5.1).
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
VEOZAH is a neurokinin 3 (NK3) receptor antagonist indicated for the treatment of moderate to severe vasomotor symptoms due to menopause. (1)
DOSAGE AND ADMINISTRATION
One 45 mg tablet orally once daily with or without food.
Perform baseline hepatic laboratory tests to evaluate for hepatic function and injury before beginning VEOZAH. While using VEOZAH, perform follow-up hepatic laboratory tests monthly for the first 3 months, at 6 months, and 9 months after initiation of therapy or when signs or symptoms suggest liver injury. (2.1)
DOSAGE FORMS AND STRENGTHS
Tablets: 45 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
Hepatotoxicity: Cases of hepatotoxicity and jaundice have been reported in the postmarketing setting. Perform hepatic laboratory tests prior to initiation of VEOZAH to evaluate for hepatic function and injury. Perform follow-up hepatic laboratory tests monthly for the first 3 months, at 6 months, and 9 months after initiation of therapy.
Advise patients to discontinue VEOZAH immediately and seek medical attention including hepatic laboratory tests if they experience signs or symptoms that may suggest liver injury (new onset fatigue, decreased appetite, nausea, vomiting, pruritus, jaundice, pale feces, dark urine, or abdominal pain). (5.1)
ADVERSE REACTIONS
The most common adverse reactions with VEOZAH [at least 2% in VEOZAH 45 mg and greater than placebo] are: abdominal pain, diarrhea, insomnia, back pain, hot flush, and hepatic transaminase elevation. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Astellas Pharma US, Inc. at 1-800-727-7003 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: RISKS OF HEPATOTOXICITY
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on VEOZAH
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Effects on Vasomotor Symptoms in Postmenopausal Women
14.2 Effects on Endometrium in Postmenopausal Women
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: RISKS OF HEPATOTOXICITY
Hepatotoxicity has occurred with the use of VEOZAH in the postmarketing setting (5.1).
- Perform hepatic laboratory tests prior to initiation of treatment to evaluate for hepatic function and injury. Do not start VEOZAH if either aminotransferase is ≥ 2 x the upper limit of normal (ULN) or if the total bilirubin is ≥ 2 x ULN for the evaluating laboratory.
- Perform follow-up hepatic laboratory testing monthly for the first 3 months, at 6 months, and 9 months of treatment (2.1, 5.1).
- Advise patients to discontinue VEOZAH immediately and seek medical attention including hepatic laboratory tests if they experience signs or symptoms that may suggest liver injury (new onset fatigue, decreased appetite, nausea, vomiting, pruritus, jaundice, pale feces, dark urine, or abdominal pain) (2.1, 5.1).
- Discontinue VEOZAH if transaminase elevations are > 5 x ULN, or if transaminase elevations are > 3 x ULN and the total bilirubin level is > 2 x ULN.
- If transaminase elevations > 3 x ULN occur, perform more frequent follow-up hepatic laboratory tests until resolution (5.1).
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Take a single 45 mg VEOZAH tablet orally once daily with or without food.
Take VEOZAH with liquids and swallow whole. Do not cut, crush, or chew tablets.
Administer VEOZAH orally at about the same time each day. If a dose of VEOZAH is missed or not taken at the usual time, administer the missed dose as soon as possible, unless there is less than 12 hours before the next scheduled dose. Return to the regular schedule the following day.
Perform baseline hepatic laboratory tests to evaluate for hepatic function and injury [including serum alanine aminotransferase (ALT), serum aspartate aminotransferase (AST), serum alkaline phosphatase (ALP), and serum bilirubin (total and direct)] before initiating treatment with VEOZAH. Do not start VEOZAH if ALT or AST is ≥ 2 x ULN or if the total bilirubin is ≥ 2 x ULN for the evaluating laboratory.
While using VEOZAH, perform follow-up hepatic laboratory tests monthly for the first 3 months, at 6 months, and 9 months after initiation of therapy.
Advise patients to discontinue VEOZAH immediately and seek medical attention including hepatic laboratory tests if they experience signs or symptoms that may suggest liver injury [see Warnings and Precautions (5.1)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
VEOZAH is contraindicated in women with any of the following conditions:
- Known cirrhosis [see Warnings and Precautions (5.1), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
- Severe renal impairment or end-stage renal disease [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
- Concomitant use with CYP1A2 inhibitors [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
In three clinical trials, elevations in serum transaminase [alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST)] levels > 3 x the upper limit of normal (ULN) occurred in 2.3% [exposure adjusted incidence rate (EAIR) of 2.7 per 100 person-years] of women receiving VEOZAH and 0.9% (EAIR of 1.5 per 100 person-years) of women receiving placebo. No elevations in serum total bilirubin (> 2 x ULN) occurred. Women with ALT or AST elevations were generally asymptomatic. Transaminase levels returned to pretreatment levels (or close to these) without sequelae with dose continuation, and upon dose interruption, or discontinuation. Women with cirrhosis were not studied [see Adverse Reactions (6.1)].
In the postmarketing setting, cases of drug-induced liver injury with elevations of ALT, AST, alkaline phosphatase (ALP), and total bilirubin occurred within 40 days of starting VEOZAH. Patients reported a general sense of feeling unwell and symptoms of fatigue, nausea, pruritus, jaundice, pale feces, and dark urine. The patients’ signs and symptoms gradually resolved after discontinuation of VEOZAH [see Adverse Reactions (6.2)].
Perform baseline hepatic laboratory tests to evaluate for hepatic function and injury [including serum ALT, serum AST, serum ALP, and serum bilirubin (total and direct)] prior to VEOZAH initiation. Do not start VEOZAH if ALT or AST is ≥ 2 x ULN or if the total bilirubin is ≥ 2 x ULN for the evaluating laboratory.
Perform follow-up hepatic laboratory tests monthly for the first 3 months, at 6 months, and 9 months after initiation of therapy.
Advise patients to discontinue VEOZAH immediately and seek medical attention including hepatic laboratory tests if they experience signs or symptoms that may suggest liver injury:
- new onset fatigue, decreased appetite, nausea, vomiting, pruritus, jaundice, pale feces, dark urine, or abdominal pain.
Discontinue VEOZAH if:
- transaminase elevations are > 5 x ULN.
- transaminase elevations are > 3 x ULN and total bilirubin is > 2 x ULN.
If transaminase elevations > 3 x ULN occur, perform more frequent follow-up hepatic laboratory tests until resolution.
Exclude alternative causes of hepatic laboratory test elevations.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in the labeling:
- Hepatic Transaminase Elevation and Hepatotoxicity [see Warnings and Precautions (5.1)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of VEOZAH was evaluated in three 52-week clinical trials [see Clinical Studies (14)]. Across the three clinical trials, a total of 1100 women received VEOZAH. Trials 1 and 2 were placebo-controlled for the first 12 weeks, followed by re-randomization of women previously receiving placebo to VEOZAH (women on VEOZAH remained on VEOZAH) for an additional 40 weeks of uncontrolled treatment. Trial 3 was a randomized, placebo-controlled, double-blind safety study evaluating the safety of VEOZAH for 52 weeks. The adverse reactions reported in at least 2% in VEOZAH 45 mg and greater than placebo in Trial 3 are presented in Table 1.
Table 1: Adverse Reactions Reported in at Least 2% in VEOZAH 45 mg and Greater Than Placebo in a Placebo‑Controlled, Double-Blind 52-Week Trial (Trial 3) - * EAIR = Number of individuals experiencing an adverse event divided by exposure time (total person-years) x 100.
- † Abdominal pain (including Abdominal pain, Abdominal pain lower, Abdominal pain upper).
- ‡ Hepatic transaminase elevations (including Alanine aminotransferase abnormal, Alanine aminotransferase increased, Aspartate aminotransferase abnormal, Aspartate aminotransferase increased).
Adverse Reaction
VEOZAH 45 mg
(n=609)
Total Person-Years=504.2
n (%, EAIR*)
Placebo
(n=610)
Total Person-Years=475.0
n (%, EAIR*)
Abdominal pain†
26 (4.3%, 5.2)
13 (2.1%, 2.7)
Diarrhea
24 (3.9%, 4.8)
16 (2.6%, 3.4)
Insomnia
24 (3.9%, 4.8)
11 (1.8%, 2.3)
Back pain
18 (3.0%, 3.6)
13 (2.1%, 2.7)
Hot flush
15 (2.5%, 3.0)
10 (1.6%, 2.1)
Hepatic transaminase elevation‡
14 (2.3%, 2.8)
5 (0.8%, 1.1)
In the pooled laboratory data of Trials 1, 2, and 3, elevated hepatic transaminases (> 3 x ULN) occurred in 25 women (2.3%, 2.7 EAIR) exposed to VEOZAH 45 mg (n=1100, 912.1 total person-years) as compared to 8 women (0.9%, 1.5 EAIR) exposed to placebo (n=952, 549.1 total person-years).
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of VEOZAH. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hepatic: Cases of serious drug-induced hepatotoxicity occurred within 40 days of starting VEOZAH. Patients experienced elevated transaminases (up to 50 x ULN at peak elevation), elevated alkaline phosphatase (up to 4 x ULN at peak elevation), and bilirubin (up to 5 x ULN at peak elevation) coupled with symptoms of fatigue, nausea, pruritus, jaundice, pale feces, and dark urine. After discontinuation of VEOZAH, these abnormalities gradually resolved.
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on VEOZAH
CYP1A2 Inhibitors
VEOZAH is a substrate of CYP1A2. Concomitant use of VEOZAH with drugs that are weak, moderate, or strong CYP1A2 inhibitors, increase the plasma Cmax and AUC of VEOZAH [see Clinical Pharmacology (12.3)].
VEOZAH is contraindicated in individuals using CYP1A2 inhibitors.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no data on VEOZAH use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes.
In embryo-fetal toxicity animal studies with fezolinetant, embryo-lethality occurred at high doses above the human therapeutic dose in rats and rabbits, but no teratogenicity was observed. In the pre- and post-natal development animal study, delayed parturition and embryo-lethality occurred at high doses above the human therapeutic dose in rats. Additionally, in the male offspring delayed male reproductive maturation was observed, characterized by incomplete preputial separation, which affected male fertility at doses above the human therapeutic dose in rats [see Data].
In the U.S. general population, the estimated background risk of major birth defects or miscarriage in clinically recognized pregnancies are 2-4% and 15-20%, respectively.
Animal Data
In embryo-fetal development toxicity studies in rats and rabbits, embryo-lethality was noted at the highest doses (128- and 174-fold the human AUC24 at the human therapeutic dose for rats and rabbits, respectively). The no observed adverse effect level (NOAEL) for embryo-fetal development was 50 mg/kg/day in rats and 45 mg/kg/day in rabbits (62- and 16‑fold the human AUC24 at the human therapeutic dose for rats and rabbits, respectively). Fezolinetant showed no effects on fertility and early embryonic development in rats [see Nonclinical Toxicology (13.1)].
In the pre- and post-natal development study in rats, the NOAEL for maternal and fetal toxicity was 30 mg/kg/day (36‑fold the human AUC24 at the human therapeutic dose) based on delayed parturition and embryo-lethality at 100 mg/kg/day. The NOAEL for F1 generation development was determined to be 100 mg/kg/day for females (204-fold the human AUC24 at the human therapeutic dose) and 10 mg/kg/day for males (11-fold the human AUC24 at the human therapeutic dose). The F1 male showed delayed male reproductive maturation, characterized as incomplete balanopreputial separation at time of mating, at doses of greater than or equal to 30 mg/kg/day (36-fold the human AUC24 at the human therapeutic dose), which affected male fertility [see Nonclinical Toxicology (13.1)].
8.2 Lactation
Risk Summary
There are no data on the presence of fezolinetant in human milk, the effects on the breastfed child, or the effects on milk production. It is not known if fezolinetant is present in human milk.
Data
Animal Data
Following administration of radiolabeled fezolinetant to lactating rats, the radioactivity concentration in milk was higher than that in the plasma at all time points, indicating that fezolinetant-derived components transferred to the tissues in infant rats via breast milk.
8.4 Pediatric Use
The efficacy and safety of VEOZAH in individuals less than 18 years of age have not been established.
8.5 Geriatric Use
There have not been sufficient numbers of geriatric women involved in clinical trials utilizing VEOZAH to determine whether those over 65 years of age differ from younger women in their response to VEOZAH.
8.6 Renal Impairment
VEOZAH is contraindicated in individuals with severe (eGFR 15 to less than 30 mL/min/1.73 m2) renal impairment or end-stage renal disease (eGFR less than 15 mL/min/1.73 m2) [see Clinical Pharmacology (12.3)]. No dose adjustment of VEOZAH is recommended for individuals with mild (eGFR 60 to less than 90 mL/min/1.73 m2) or moderate (eGFR 30 to less than 60 mL/min/1.73 m2) renal impairment.
8.7 Hepatic Impairment
Child-Pugh Class A or B hepatic impairment increased the exposure of VEOZAH [see Clinical Pharmacology (12.3)]. VEOZAH has not been studied in individuals with Child-Pugh Class C hepatic impairment.
VEOZAH is contraindicated in individuals with cirrhosis [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
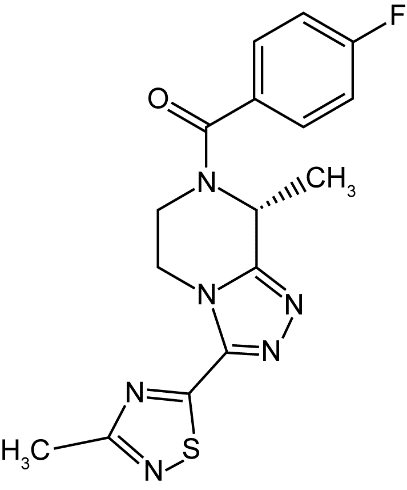
11 DESCRIPTION
VEOZAH (fezolinetant) is a small-molecule NK3 receptor antagonist.
The chemical name of fezolinetant is (4-Fluorophenyl)[(8R)-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]methanone having a molecular formula of C16H15FN6OS and a molecular weight of 358.39. The structural formula of fezolinetant is:
Fezolinetant is a white powder. It is very slightly soluble in water (0.29 mg/mL).
Each VEOZAH (fezolinetant) tablet for oral use contains 45 mg of fezolinetant and the following inactive ingredients: ferric oxide, hydroxypropyl cellulose, hypromellose, low-substituted hydroxypropyl cellulose, magnesium stearate, mannitol, microcrystalline cellulose, polyethylene glycol, talc, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
VEOZAH is a neurokinin 3 (NK3) receptor antagonist that blocks neurokinin B (NKB) binding on the kisspeptin/neurokinin B/dynorphin (KNDy) neuron to modulate neuronal activity in the thermoregulatory center. Fezolinetant has high affinity for the NK3 receptor (Ki value of 19.9 to 22.1 nmol/L), which is more than 450-fold higher than binding affinity to NK1 or NK2 receptors.
12.2 Pharmacodynamics
Treatment with fezolinetant did not show any clear trends in sex hormones measured (follicle-stimulating hormone, testosterone, estrogen, and dehydroepiandrosterone sulfate) in menopausal women. Transient decrease of luteinizing hormone (LH) levels was observed at peak concentrations of fezolinetant.
Cardiac Electrophysiology
At a dose 20 times the maximum approved recommended dose, fezolinetant does not prolong the QT interval to any clinically relevant extent.
12.3 Pharmacokinetics
In healthy women, fezolinetant Cmax and AUC increased proportionally over a dosage range from 20 to 60 mg once daily (0.44 to 1.33 times the approved recommended dosage).
Steady-state plasma concentrations of fezolinetant were reached after two once daily doses, with minimal fezolinetant accumulation.
Absorption
The median (range) time to reach fezolinetant Cmax is 1.5 (1 to 4) hours in healthy women.
Effect of Food
No clinically significant differences in fezolinetant pharmacokinetics were observed following administration with a high‑calorie, high-fat meal containing approximately 1000 calories (500-600 calories from fat, 250 calories from carbohydrates, and 150 calories from protein).
Distribution
The mean apparent volume of distribution (Vz/F) of fezolinetant is 189 L. The plasma protein binding of fezolinetant is 51%. The blood-to-plasma ratio is 0.9.
Elimination
The effective half-life (t1/2) of fezolinetant is 9.6 hours in women with vasomotor symptoms. The apparent clearance at steady-state of fezolinetant is 10.8 L/h.
Metabolism
Fezolinetant is primarily metabolized by CYP1A2 and to a lesser extent by CYP2C9 and CYP2C19. A major metabolite of fezolinetant, ES259564, was identified in plasma. ES259564 is approximately 20-fold less potent than the parent. The metabolite-to-parent ratio ranges from 0.7 to 1.8.
Excretion
Following oral administration of fezolinetant, 76.9% of the dose was excreted in urine (1.1% unchanged) and 14.7% in feces (0.1% unchanged).
Specific Populations
There were no substantive differences in the pharmacokinetics of VEOZAH based on race and body weight (93 to 278 pounds).
Women with Renal Impairment
Following single-dose administration of 30 mg fezolinetant, there was no effect on VEOZAH exposure (Cmax and AUC) in women with mild (eGFR 60 to less than 90 mL/min/1.73 m2) to severe (eGFR 15 to less than 30 mL/min/1.73 m2) renal impairment. The AUC of ES259564 (a major metabolite of fezolinetant) in women with moderate (eGFR 30 to less than 60 mL/min/1.73 m2) and severe (eGFR 15 to less than 30 mL/min/1.73 m2) renal impairment increased by approximately 75% and 380%, respectively. VEOZAH has not been studied in individuals with end-stage renal disease (eGFR less than 15 mL/min/1.73 m2).
Women with Hepatic Impairment
Following single-dose administration of 30 mg fezolinetant in women with mild Child-Pugh Class A cirrhosis, the mean Cmax increased by 23% and AUCinf increased by 56%, relative to women with normal hepatic function. In women with moderate Child-Pugh Class B cirrhosis, the mean Cmax of fezolinetant decreased by 15% and AUCinf increased by 96%. The Cmax of ES259564 decreased in both mild and moderate cirrhosis while AUCinf and AUClast increased less than 15%. VEOZAH has not been studied in individuals with severe Child-Pugh Class C cirrhosis.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Strong CYP1A2 Inhibitors: Fezolinetant Cmax increased by 80% and AUC increased by 840% following concomitant use with fluvoxamine (strong CYP1A2 inhibitor).
Moderate CYP1A2 Inhibitors: Fezolinetant Cmax increased by 40% and AUC increased by 360% following concomitant use with mexiletine (moderate CYP1A2 inhibitor) 400 mg every 8 hours.
Weak CYP1A2 Inhibitors: Fezolinetant Cmax increased by 30% and AUC increased by 100% following concomitant use with cimetidine (weak CYP1A2 inhibitor) 300 mg every 6 hours.
No clinically significant differences in fezolinetant exposure were observed in smokers (moderate CYP1A2 inducer).
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Fezolinetant and ES259564 are not inhibitors of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Fezolinetant and ES259564 are not inducers of CYP1A2, CYP2B6, and CYP3A4.
Transporter Systems
Fezolinetant is not a substrate nor an inhibitor of P-glycoprotein (P-gp). ES259564 is a substrate of P-gp, but not an inhibitor of P-gp.
Both fezolinetant and ES259564 are not a substrate of BCRP, OATP1B1, and OATP1B3. In addition, ES259564 is not a substrate of OAT1, OAT3, OCT2, MATE1, and MATE2-K.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 2-year female rat carcinogenicity study and a 26-week carcinogenicity study in rasH2 transgenic mice, there was no evidence of drug-related carcinogenicity at 186-fold and 47-fold the human AUC24 at the human therapeutic dose of 45 mg, respectively.
Mutagenesis
Fezolinetant showed no genotoxic potential by the bacterial reverse mutation test, chromosomal aberration test, or in vivo micronucleus test.
Impairment of Fertility
Fezolinetant had no effect on female fertility or early embryonic development up to 100 mg/kg/day in rats (143-fold the human AUC24 at the human therapeutic dose). In the pre- and post-natal development study in rats, the F1 male showed incomplete balanopreputial separation at doses greater than or equal to 30 mg/kg/day (36-fold the human AUC24 at the human therapeutic dose), which delayed male reproductive maturation and affected fertility. These effects were not observed following dosing at 10 mg/kg/day (11-fold the human AUC24 at the human therapeutic dose) [see Use in Specific Populations (8.1)].
13.2 Animal Toxicology and/or Pharmacology
Repeat dose toxicity studies were conducted in intact female rats and cynomolgus monkeys. In female rats, daily administration of fezolinetant for 26 weeks at doses equal to or greater than 30 mg/kg/day (56-fold the human AUC24 at the human therapeutic dose) showed uterine atrophy and epithelial mucification of the vagina and cervix. In female cynomolgus monkeys, daily administration for 39 weeks at doses equal to or greater than 10 mg/kg/day (19-fold the human AUC24 at the human therapeutic dose) showed reduced ovarian activity.
-
14 CLINICAL STUDIES
14.1 Effects on Vasomotor Symptoms in Postmenopausal Women
The efficacy of VEOZAH for the treatment of moderate to severe vasomotor symptoms due to menopause was evaluated in the first 12-week, randomized, placebo-controlled, double-blind portion of each of two phase 3 clinical trials. In each of these two trials, after the first 12 weeks, women on placebo were then re-randomized to VEOZAH for a 40-week extension to evaluate safety for up to 52 weeks total exposure.
In Trials 1 (NCT04003155) and 2 (NCT04003142), 1022 women (522 in Trial 1 and 500 in Trial 2) who had a minimum average of 7 moderate to severe vasomotor symptoms per day were randomized to one of two doses of fezolinetant (including the 45 mg dosage strength) or placebo. Randomization was stratified by smoking status.
The mean age of the postmenopausal women was 54 years. Women self-identified as Caucasian (81%), African American (17%), Asian (1%), and Hispanic/Latina ethnicity (24%). The study population included menopausal women with one or more of the following: prior hysterectomy (32.1%), prior oophorectomy (21.6%), or prior hormone therapy use (19.9%). Those who were on prior hormone therapy underwent a wash-out period prior to trial participation.
The co-primary efficacy endpoints for both trials were the mean change from baseline in moderate to severe vasomotor symptoms frequency and severity to Weeks 4 and 12. Data from each trial demonstrated statistically significant and clinically meaningful (≥ 2 hot flashes over 24 hours) reduction from baseline in the frequency of moderate to severe vasomotor symptoms for VEOZAH 45 mg compared to placebo at Weeks 4 and 12. Data from each trial also demonstrated a statistically significant reduction from baseline in the severity of moderate to severe vasomotor symptoms (over 24 hours) at Weeks 4 and 12 for VEOZAH 45 mg compared to placebo.
Results of the co-primary endpoint for change from baseline to Weeks 4 and 12 in mean frequency of moderate to severe vasomotor symptoms over 24 hours from Trials 1 and 2 are shown in Table 2.
Table 2: Mean Baseline and Change from Baseline to Weeks 4 and 12 for Mean Frequency of Moderate to Severe Vasomotor Symptoms over 24 Hours in Women Treated with VEOZAH in Trials 1 and 2 - * Statistically significantly superior compared to placebo at the 0.05 level with multiplicity adjustment.
LS Mean: Least Squares Mean estimated from a mixed model for repeated measures analysis of covariance; SD: Standard Deviation;
SE: Standard Error.Parameter
Trial 1
Trial 2
VEOZAH
45 mg
(n=174)
Placebo
(n=175)
VEOZAH
45 mg
(n=167)
Placebo
(n=167)
Baseline
Mean (SD)
10.4 (3.92)
10.5 (3.79)
11.8 (8.26)
11.6 (5.02)Change from Baseline to Week 4
LS Mean (SE)
Difference vs Placebo (95% CI)
P-value
-5.4 (0.30)-2.1 (-2.9, -1.3)
< 0.001*
-3.3 (0.29)--
--
-6.3 (0.33)-2.6 (-3.5, -1.6)
< 0.001*
-3.7 (0.33)--
--
Change from Baseline to Week 12
LS Mean (SE)
Difference vs Placebo (95% CI)
P-value
-6.4 (0.31)-2.6 (-3.4, -1.7)
< 0.001*
-3.9 (0.31)--
--
-7.5 (0.39)-2.5 (-3.6, -1.5)
< 0.001*
-5.0 (0.39)--
--
Results of the co-primary endpoint for change from baseline to Weeks 4 and 12 in mean severity of moderate to severe vasomotor symptoms over 24 hours from Trials 1 and 2 are shown in Table 3.
Table 3: Mean Baseline and Change from Baseline to Weeks 4 and 12 for Mean Severity of Moderate to Severe Vasomotor Symptoms over 24 Hours in Women Treated with VEOZAH in Trials 1 and 2 Parameter Trial 1 Trial 2 VEOZAH
45 mg
(n=174)Placebo
(n=175)VEOZAH
45 mg
(n=167)Placebo
(n=167)- * Statistically significantly superior compared to placebo at the 0.05 level with multiplicity adjustment.
LS Mean: Least Squares Mean estimated from a mixed model for repeated measures analysis of covariance; SD: Standard Deviation;
SE: Standard Error.Baseline
Mean (SD)
2.4 (0.35)
2.4 (0.35)
2.4 (0.34)
2.4 (0.32)Change from Baseline to Week 4
LS Mean (SE)
Difference vs Placebo (95% CI)
P-value
-0.5 (0.04)-0.2 (-0.3, -0.1)
0.002*
-0.3 (0.04)--
--
-0.6 (0.05)-0.3 (-0.4, -0.2)
< 0.001*
-0.3 (0.05)--
--
Change from Baseline to Week 12
LS Mean (SE)
Difference vs Placebo (95% CI)
P-value
-0.6 (0.05)-0.2 (-0.4, -0.1)
0.007*
-0.4 (0.05)--
--
-0.8 (0.06)-0.3 (-0.5, -0.1)
< 0.001*
-0.5 (0.06)--
--
14.2 Effects on Endometrium in Postmenopausal Women
In the VEOZAH 45 mg dose group across Trials 1, 2, and 3, endometrial biopsy assessments identified one case of endometrial hyperplasia and one case of endometrial malignancy. The rate of these events in the VEOZAH 45 mg dose group was ≤ 1% with the upper bound of the one-sided 95% confidence limit being ≤ 4%.
Table 4: Incidence of Endometrial Hyperplasia or Carcinoma after 12 Months of Treatment in Trials 1, 2, and 3 Final Diagnosis
VEOZAH
45 mg
(n=350)
Placebo
(n=186)
Endometrial Hyperplasia and Carcinoma, n (%)
One-sided upper limit of 95% CI
- Simple hyperplasia without atypia
- Endometrial adenocarcinoma
2 (0.6%)
1.8%
1
1
0
1.6%
0
0
Five cases of disordered proliferative endometrium were reported in women receiving VEOZAH 45 mg and four cases were reported in women receiving placebo across the three clinical trials. The EAIR was 1.4 per 100 person-years in VEOZAH 45 mg versus 2.0 per 100 person-years in the placebo group.
- * Statistically significantly superior compared to placebo at the 0.05 level with multiplicity adjustment.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
VEOZAH (fezolinetant) 45 mg tablets are supplied as round, light red, film-coated tablets, debossed with the Astellas logo and ‘645’ on the same side. VEOZAH tablets are available in the following package sizes:
- Bottles of 30 tablets with child resistant closure, (NDC: 0469-2660-30)
- Bottles of 90 tablets with child resistant closure, (NDC: 0469-2660-90)
-
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information).
Evaluation of Hepatic Injury During Treatment with VEOZAH
Inform patients that they will have to have a blood test to evaluate their liver function before beginning VEOZAH and while using VEOZAH monthly for the first 3 months, at 6 months, and 9 months after initialization of therapy. Advise patients to discontinue VEOZAH immediately and seek medical attention including hepatic laboratory tests if they experience signs or symptoms that may suggest liver abnormalities such as new onset fatigue, decreased appetite, nausea, vomiting, pruritus, jaundice, pale feces, dark urine, or abdominal pain [see Warnings and Precautions (5.1)].
Serious Adverse Reactions with VEOZAH
Inform patients of possible serious adverse reactions of VEOZAH including hepatic transaminase elevation and liver injury [see Warnings and Precautions (5.1)].
Common Adverse Reactions with VEOZAH
Inform patients of possible less serious but common adverse reactions of VEOZAH including abdominal pain, diarrhea, insomnia, back pain, and hot flush [see Adverse Reactions (6.1)].
Drug Interactions
Advise patients to report their use of any other prescription or nonprescription medications or dietary supplements [see Drug Interactions (7.1)].
Distributed by:
Astellas Pharma US, Inc.
Northbrook, IL 60062
© 2024 Astellas Pharma US, Inc. or its affiliates
432286-VEO-USA
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
VEOZAH® (vee-O-zah)
(fezolinetant)
tablets, for oral use
What is the most important information I should know about VEOZAH?
VEOZAH can cause serious side effects, including:
- Liver Problems. Your healthcare provider will do a blood test to check your liver before you start taking VEOZAH. Your healthcare provider will also do this blood test monthly for the first 3 months, at month 6, and month 9 after you start taking VEOZAH or if you have signs or symptoms that suggest liver problems. If your liver blood test values are elevated, your healthcare provider may advise you to stop treatment or request additional liver blood tests.
Stop VEOZAH right away and call your healthcare provider if you have the following signs or symptoms of liver problems:
- feeling more tired than you do usually
- decreased appetite
- nausea
- vomiting
- itching
- yellowing of the eyes or skin (jaundice)
- pale feces
- dark urine
- pain in the stomach (abdomen)
See “What are the possible side effects of VEOZAH?” for more information about side effects.
What is VEOZAH?
VEOZAH is a prescription medicine used to reduce moderate to severe vasomotor symptoms due to menopause. VEOZAH is not a hormone. Vasomotor symptoms are the feelings of warmth in the face, neck, and chest, or sudden intense feelings of heat and sweating (“hot flashes” or “hot flushes”).
Do not use VEOZAH if you:
- have cirrhosis.
- have severe kidney problems or kidney failure.
- are taking certain medicines called CYP1A2 inhibitors. Ask your healthcare provider if you are not sure.
Before you use VEOZAH, tell your healthcare provider about all of your medical conditions, including if you:
- have liver disease or liver problems.
- have kidney problems.
- have any medical conditions that may become worse while you are using VEOZAH.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. VEOZAH may affect the way other medicines work, and other medicines may affect how VEOZAH works. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take VEOZAH?
- Take VEOZAH exactly as your healthcare provider tells you to take it.
- Take 1 VEOZAH tablet by mouth with or without food at about the same time each day.
- Swallow the VEOZAH tablet whole with liquid. Do not cut, crush, or chew the tablet.
- If you miss a dose of VEOZAH, take the missed dose as soon as possible on the same day, with at least 12 hours before the next scheduled dose. Return to your normal schedule the following day.
What are the possible side effects of VEOZAH?
VEOZAH can cause serious side effects, including:
- See “What is the most important information I should know about VEOZAH?”
Common side effects of VEOZAH include:
- stomach (abdominal) pain
- diarrhea
- difficulty sleeping (insomnia)
- back pain
- hot flashes or hot flushes
Tell your healthcare provider if you have any side effect that bothers you or does not go away.
These are not all the possible side effects of VEOZAH.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store VEOZAH?
- Store VEOZAH at room temperature between 68°F to 77°F (20°C to 25°C).
- Dispose of the unused medicine through a take-back option, if available. See www.fda.gov/drugdisposal for more information.
- Keep VEOZAH and all medicines out of the reach of children.
General information about the safe and effective use of VEOZAH.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use VEOZAH for a condition for which it was not prescribed. Do not give VEOZAH to other people, even if they have the same symptoms you have. It may harm them.
This Patient Information leaflet summarizes the most important information about VEOZAH. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about VEOZAH that is written for healthcare professionals.
What are the ingredients in VEOZAH?
Active ingredient: fezolinetant
Inactive ingredients: ferric oxide, hydroxypropyl cellulose, hypromellose, low-substituted hydroxypropyl cellulose, magnesium stearate, mannitol, microcrystalline cellulose, polyethylene glycol, talc, and titanium dioxide
Distributed by:
Astellas Pharma US, Inc.
Northbrook, IL 60062
© 2024 Astellas Pharma US, Inc. or its affiliates
432286-VEO-USA
For more information, go to www.VEOZAH.com or call 1-800-727-7003.
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 02/2026
- Package/Label Display Panel – VEOZAH Bottle Carton Label
-
Package/Label Display Panel – VEOZAH Sample Bottle Carton Label
NDC: 0469-2460-07
PROFESSIONAL SAMPLE-NOT FOR SALE
Rx Only
VEOZAH®
(fezolinetant) tablets
45 mg7 tablets
-
Package/Label Display Panel- VEOZAH Sample Blister Carton Label
NDC: 0469-2760-07
PROFESSIONAL SAMPLE-NOT FOR SALE
Rx Only
VEOZAH®
(fezolinetant) tablets
45 mg7 tablets
-
INGREDIENTS AND APPEARANCE
VEOZAH
fezolinetant tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0469-2660 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FEZOLINETANT (UNII: 83VNE45KXX) (FEZOLINETANT - UNII:83VNE45KXX) FEZOLINETANT 45 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TALC (UNII: 7SEV7J4R1U) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color RED (LIGHT RED) Score no score Shape ROUND Size 7mm Flavor Imprint Code Astellas;logo;645 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0469-2660-30 1 in 1 CARTON 05/12/2023 1 30 in 1 BOTTLE; Type 0: Not a Combination Product 2 NDC: 0469-2660-90 1 in 1 CARTON 05/12/2023 2 90 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216578 05/12/2023 VEOZAH
fezolinetant tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0469-2460 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FEZOLINETANT (UNII: 83VNE45KXX) (FEZOLINETANT - UNII:83VNE45KXX) FEZOLINETANT 45 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TALC (UNII: 7SEV7J4R1U) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color RED (LIGHT RED) Score no score Shape ROUND Size 7mm Flavor Imprint Code Astellas;logo;645 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0469-2460-28 4 in 1 TRAY 05/16/2023 1 NDC: 0469-2460-07 1 in 1 CARTON 1 7 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216578 05/16/2023 VEOZAH
fezolinetant tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0469-2760 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FEZOLINETANT (UNII: 83VNE45KXX) (FEZOLINETANT - UNII:83VNE45KXX) FEZOLINETANT 45 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TALC (UNII: 7SEV7J4R1U) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color RED (LIGHT RED) Score no score Shape ROUND Size 7mm Flavor Imprint Code Astellas;logo;645 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0469-2760-28 4 in 1 TRAY 05/12/2023 1 NDC: 0469-2760-07 1 in 1 CARTON 1 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216578 05/12/2023 Labeler - Astellas Pharma US, Inc. (605764828) Registrant - Astellas Pharma US, Inc. (605764828) Establishment Name Address ID/FEI Business Operations AndersonBrecon Inc. 053217022 LABEL(0469-2660, 0469-2460, 0469-2760) , PACK(0469-2660, 0469-2460, 0469-2760) Establishment Name Address ID/FEI Business Operations Fujimoto Chemicals Co., Ltd. 717836717 API MANUFACTURE(0469-2660, 0469-2460, 0469-2760) Establishment Name Address ID/FEI Business Operations Juzen Chemical Corporation 691036974 API MANUFACTURE(0469-2660, 0469-2460, 0469-2760) , ANALYSIS(0469-2660, 0469-2460, 0469-2760) Establishment Name Address ID/FEI Business Operations Astellas Pharma Inc. 695086702 MANUFACTURE(0469-2660, 0469-2460, 0469-2760) , ANALYSIS(0469-2660, 0469-2460, 0469-2760)


Trademark Results [VEOZAH]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VEOZAH 97614529 not registered Live/Pending |
Astellas US LLC 2022-09-30 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.