DEXRAZOXANE by AuroMedics Pharma LLC / GLAND PHARMA LIMITED DEXRAZOXANE kit
DEXRAZOXANE by
Drug Labeling and Warnings
DEXRAZOXANE by is a Prescription medication manufactured, distributed, or labeled by AuroMedics Pharma LLC, GLAND PHARMA LIMITED. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DEXRAZOXANE FOR INJECTION safely and effectively. See full prescribing information for DEXRAZOXANE FOR INJECTION.
DEXRAZOXANE for injection, for intravenous use
Initial U.S. Approval: 1995INDICATIONS AND USAGE
Dexrazoxane for injection is a cytoprotective agent indicated for reducing the incidence and severity of cardiomyopathy associated with doxorubicin administration in women with metastatic breast cancer who have received a cumulative doxorubicin dose of 300 mg/m2 and who will continue to receive doxorubicin therapy to maintain tumor control. Do not use dexrazoxane for injection with doxorubicin initiation. (1)
DOSAGE AND ADMINISTRATION
- Reconstitute vial contents and dilute before use. (2.3)
- Administer dexrazoxane for injection by slow I.V. push or rapid drip intravenous infusion from a bag. (2.1, 2.3)
- The recommended dosage ratio of dexrazoxane for injection to doxorubicin is 10:1 (e.g., 500 mg/m2 dexrazoxane for injection to 50 mg/m2 doxorubicin). Do not administer doxorubicin before dexrazoxane for injection. (2.1)
- Reduce dose by 50% for patients with creatinine clearance < 40 mL/min. (2.2, 8.7)
DOSAGE FORMS AND STRENGTHS
250 mg single-dose vial as sterile, pyrogen-free lyophilizates. (3)
CONTRAINDICATIONS
Dexrazoxane for injection should not be used with non-anthracycline chemotherapy regimens. (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
In clinical studies, dexrazoxane was administered to patients also receiving chemotherapeutic agents for cancer. Pain on injection was observed more frequently in patients receiving dexrazoxane versus placebo. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AuroMedics Pharma LLC at 1-866-850-2876 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.USE IN SPECIFIC POPULATIONS
- Nursing Mothers: Discontinue drug or nursing. (8.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2021
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
2.2 Dose Modifications
2.3 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
5.2 Concomitant Chemotherapy
5.3 Cardiac Toxicity
5.4 Secondary Malignancies
5.5 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Females of Reproductive Potential
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Myelosuppression
17.2 Embryo-Fetal Toxicity
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Dexrazoxane for injection is indicated for reducing the incidence and severity of cardiomyopathy associated with doxorubicin administration in women with metastatic breast cancer who have received a cumulative doxorubicin dose of 300 mg/m2 and who will continue to receive doxorubicin therapy to maintain tumor control. Do not use with the initiation of doxorubicin therapy [see Warnings and Precautions (5.2)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
Administer dexrazoxane for injection by slow I.V. push or rapid drip intravenous infusion from a bag.
The recommended dosage ratio of dexrazoxane for injection to doxorubicin is 10:1 (e.g., 500 mg/m2 dexrazoxane for injection to 50 mg/m2 doxorubicin). Do not administer doxorubicin before dexrazoxane for injection. Administer doxorubicin within 30 minutes after the completion of dexrazoxane for injection infusion.
2.2 Dose Modifications
Dosing in Patients with Renal Impairment: Reduce dexrazoxane for injection dosage in patients with moderate to severe renal impairment (creatinine clearance values less than 40 mL/min) by 50% (dexrazoxane for injection to doxorubicin ratio reduced to 5:1; such as 250 mg/m2 dexrazoxane for injection to 50 mg/m2 doxorubicin) [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Dosing in Patients with Hepatic Impairment: Since a doxorubicin dose reduction is recommended in the presence of hyperbilirubinemia, reduce the dexrazoxane for injection dosage proportionately (maintaining the 10:1 ratio) in patients with hepatic impairment.
2.3 Preparation and Administration
Preparation and Handling of Infusion Solution: Dexrazoxane for injection must be reconstituted with 0.167 Molar (M/6) sodium lactate injection, USP to give a concentration of 10 mg dexrazoxane for injection for each mL of sodium lactate. The reconstituted solution should be given by slow I.V. push or rapid drip intravenous infusion from a bag. After completing the infusion of dexrazoxane for injection, and prior to a total elapsed time of 30 minutes (from the beginning of the dexrazoxane for injection infusion), the intravenous injection of doxorubicin should be given.
Reconstituted dexrazoxane for injection, when transferred to an empty infusion bag, is stable for 6 hours from the time of reconstitution when stored at controlled room temperature, 20° to 25°C (68° to 77°F) or under refrigeration, 2° to 8°C (36° to 46°F). Discard unused solutions.
The reconstituted dexrazoxane for injection solution may be diluted with either 0.9% sodium chloride injection, USP or 5% dextrose injection, USP to a concentration range of 1.3 to 5 mg/mL in intravenous infusion bags. The resultant solutions are stable for 6 hours when stored at controlled room temperature, 20° to 25°C (68° to 77°F) or under refrigeration, 2° to 8°C (36° to 46°F). Discard unused solutions.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Solutions containing a precipitate should be discarded.
Use caution when handling and preparing the reconstituted solution. The use of gloves is recommended. If dexrazoxane for injection powder or solutions contact the skin or mucosae, wash exposed area immediately and thoroughly with soap and water. Follow special handling and disposal procedures.1
Administration: Do not mix dexrazoxane for injection with other drugs. The reconstituted solution should be given by slow I.V. push or rapid drip intravenous infusion from a bag. After completing the infusion of dexrazoxane for injection, and prior to a total elapsed time of 30 minutes (from the beginning of the dexrazoxane for injection infusion), the intravenous injection of doxorubicin should be given.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
Dexrazoxane may add to the myelosuppression caused by chemotherapeutic agents. Obtain a complete blood count prior to and during each course of therapy, and administer dexrazoxane and chemotherapy only when adequate hematologic parameters are met.
5.2 Concomitant Chemotherapy
Only use dexrazoxane in those patients who have received a cumulative doxorubicin dose of 300 mg/m2 and are continuing with doxorubicin therapy. Do not use with chemotherapy initiation as dexrazoxane may interfere with the antitumor activity of the chemotherapy regimen. In a trial conducted in patients with metastatic breast cancer who were treated with fluorouracil, doxorubicin, and cyclophosphamide (FAC) with or without dexrazoxane starting with their first cycle of FAC therapy, patients who were randomized to receive dexrazoxane had a lower response rate (48% vs. 63%) and shorter time to progression than patients who were randomized to receive placebo.
5.3 Cardiac Toxicity
Treatment with dexrazoxane does not completely eliminate the risk of anthracycline-induced cardiac toxicity. Monitor cardiac function before and periodically during therapy to assess left ventricular ejection fraction (LVEF). In general, if test results indicate deterioration in cardiac function associated with doxorubicin, the benefit of continued therapy should be carefully evaluated against the risk of producing irreversible cardiac damage.
5.4 Secondary Malignancies
Secondary malignancies such as acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) have been reported in studies of pediatric patients who have received dexrazoxane in combination with chemotherapy. Dexrazoxane for injection is not indicated for use in pediatric patients. Some adult patients who received dexrazoxane in combination with anti-cancer agents known to be carcinogenic have also developed secondary malignancies, including AML and MDS.
Razoxane is the racemic mixture, of which dexrazoxane is the S(+)-enantiomer. Secondary malignancies (primarily acute myeloid leukemia) have been reported in patients treated chronically with oral razoxane. In these patients, the total cumulative dose of razoxane ranged from 26 grams to 480 grams and the duration of treatment was from 42 to 319 weeks. One case of T-cell lymphoma, one case of B-cell lymphoma, and six to eight cases of cutaneous basal cell or squamous cell carcinoma have also been reported in patients treated with razoxane. Long-term administration of razoxane to rodents was associated with the development of malignancies [see Nonclinical Toxicology (13.1)].
5.5 Embryo-Fetal Toxicity
Dexrazoxane can cause fetal harm when administered to pregnant women. Dexrazoxane administration during the period of organogenesis resulted in maternal toxicity, embryotoxicity and teratogenicity in rats and rabbits at doses significantly lower than the clinically recommended dose [see Use in Specific Populations (8.1)]. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus.
Advise female patients of reproductive potential to avoid becoming pregnant and to use highly effective contraception during treatment [see Use in Specific Populations (8.6)].
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other trials and may not reflect the rates observed in clinical practice.
The adverse reaction profile described in this section was identified from randomized, placebo-controlled, double-blind studies in patients with metastatic breast cancer who received the combination of the FAC chemotherapy regimen with or without dexrazoxane. The dose of doxorubicin was 50 mg/m2 in each of these trials. Treatment was administered every three weeks until disease progression or cardiac toxicity.
Patients in clinical trials who received FAC with dexrazoxane experienced more severe leukopenia, granulocytopenia, and thrombocytopenia than patients receiving FAC without dexrazoxane [see Warnings and Precautions (5.1)].
Table 1 below lists the incidence of adverse reactions for patients receiving FAC with either dexrazoxane or placebo in the breast cancer studies. Adverse experiences occurring during courses 1 through 6 are displayed for patients receiving dexrazoxane or placebo with FAC beginning with their first course of therapy (columns 1 and 3, respectively). Adverse experiences occurring at course 7 and beyond for patients who received placebo with FAC during the first six courses and who then received either dexrazoxane or placebo with FAC are also displayed (columns 2 and 4, respectively).
The adverse reactions listed below in Table 1 demonstrate that the frequency of adverse reaction “Pain on Injection” has been greater for dexrazoxane arm, as compared to placebo.
Table 1: Adverse Reaction
Percentage (%) of Breast Cancer Patients with Adverse Reaction
FAC + Dexrazoxane
FAC + Placebo
Courses 1 to 6
N = 413
Courses ≥ 7
N = 102
Courses 1 to 6
N = 458
Courses ≥ 7
N = 99
Alopecia
94
100
97
98
Nausea
77
51
84
60
Vomiting
59
42
72
49
Fatigue/Malaise
61
48
58
55
Anorexia
42
27
47
38
Stomatitis
34
26
41
28
Fever
34
22
29
18
Infection
23
19
18
21
Diarrhea
21
14
24
7
Pain on Injection
12
13
3
0
Sepsis
17
12
14
9
Neurotoxicity
17
10
13
5
Streaking/Erythema
5
4
4
2
Phlebitis
6
3
3
5
Esophagitis
6
3
7
4
Dysphagia
8
0
10
5
Hemorrhage
2
3
2
1
Extravasation
1
3
1
2
Urticaria
2
2
2
0
Recall Skin Reaction
1
1
2
0
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Dexrazoxane can cause fetal harm when administered to pregnant women. Dexrazoxane administration resulted in maternal toxicity, embryotoxicity and teratogenicity in rats and rabbits at doses significantly lower than the clinically recommended dose. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus [see Warnings and Precautions (5.5)].
Animal Data
Dexrazoxane resulted in maternal toxicity in rats at doses of ≥ 2 mg/kg (1/40 the human dose on a mg/m2 basis) and embryotoxicity and teratogenicity at 8 mg/kg (approximately 1/10 the human dose on a mg/m2 basis) when given daily to pregnant rats during the period of organogenesis. Teratogenic effects in the rat included imperforate anus, microphthalmia, and anophthalmia. In offspring allowed to develop to maturity, fertility was impaired in the male and female rats treated in utero during organogenesis at 8 mg/kg. In rabbits, doses of ≥ 5 mg/kg (approximately 1/10 the human dose on a mg/m2 basis) daily during the period of organogenesis caused maternal toxicity and doses of 20 mg/kg (1/2 the human dose on a mg/m2 basis) were embryotoxic and teratogenic. Teratogenic effects in the rabbit included several skeletal malformations such as short tail, rib and thoracic malformations, and soft tissue variations including subcutaneous, eye and cardiac hemorrhagic areas, as well as agenesis of the gallbladder and of the intermediate lobe of the lung.
8.3 Nursing Mothers
It is not known whether dexrazoxane or its metabolites are excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from dexrazoxane, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and effectiveness of dexrazoxane in pediatric patients have not been established [see Warnings and Precautions (5.4)].
8.5 Geriatric Use
Clinical studies of dexrazoxane did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently than younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Females of Reproductive Potential
Contraception: Dexrazoxane can cause fetal harm when administered during pregnancy. Advise female patients of reproductive potential to use highly effective contraception during treatment [see Use in Specific Populations (8.1)].
8.7 Renal Impairment
Greater exposure to dexrazoxane may occur in patients with compromised renal function. Reduce the dexrazoxane dose by 50% in patients with creatinine clearance values < 40 mL/min [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There are no data on overdosage in the cardioprotective trials; the maximum dose administered during the cardioprotective trials was 1000 mg/m2 every three weeks.
Disposition studies with dexrazoxane have not been conducted in cancer patients undergoing dialysis, but retention of a significant dose fraction (> 0.4) of the unchanged drug in the plasma pool, minimal tissue partitioning or binding, and availability of greater than 90% of the systemic drug levels in the unbound form suggest that it could be removed using conventional peritoneal or hemodialysis.
There is no known antidote for dexrazoxane. Instances of suspected overdose should be managed with good supportive care until resolution of myelosuppression and related conditions is complete. Management of overdose should include treatment of infections, fluid regulation, and maintenance of nutritional requirements.
-
11 DESCRIPTION
Dexrazoxane for injection, a cardioprotective agent for use in conjunction with doxorubicin, is a sterile, pyrogen-free lyophilizate intended for intravenous administration.
Chemically, dexrazoxane is (S)-4,4’-(1-methyl-1,2-ethanediyl)bis-2,6- piperazinedione. The structural formula is as follows:
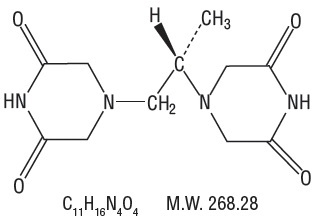
Dexrazoxane, an intracellular chelating agent, is a derivative of EDTA. Dexrazoxane for injection is a white to off-white or pale pink lyophilized powder or cake that melts at 191° to 197°C. It is sparingly soluble in water and 0.1 N HCl, slightly soluble in ethanol and methanol, and practically insoluble in nonpolar organic solvents. The pKa is 2.1. Dexrazoxane has an octanol/water partition coefficient of 0.025 and degrades rapidly above a pH of 7.0.
Each 250 mg vial contains dexrazoxane hydrochloride equivalent to 250 mg dexrazoxane. Hydrochloric Acid, NF is added for pH adjustment. When reconstituted as directed with the 25 mL vial of 0.167 Molar (M/6) sodium lactate injection, USP diluent provided, each mL contains: 10 mg dexrazoxane. The pH of the resultant solution is 3.5 to 5.5.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism by which dexrazoxane exerts its cytoprotective activity is not fully understood. Dexrazoxane is a cyclic derivative of EDTA that penetrates cell membranes. Results of laboratory studies suggest that dexrazoxane is converted intracellularly to a ring-opened chelating agent that interferes with iron-mediated free radical generation thought to be responsible, in part, for anthracycline-induced cardiomyopathy.
12.3 Pharmacokinetics
The pharmacokinetics of dexrazoxane have been studied in advanced cancer patients with normal renal and hepatic function. The pharmacokinetics of dexrazoxane can be adequately described by a two-compartment open model with first-order elimination. Dexrazoxane has been administered as a 15 minute infusion over a dose range of 60 to 900 mg/m2 with 60 mg/m2 of doxorubicin, and at a fixed dose of 500 mg/m2 with 50 mg/m2 doxorubicin. The disposition kinetics of dexrazoxane are dose-independent, as shown by linear relationship between the area under plasma concentration-time curves and administered doses ranging from 60 to 900 mg/m2. The mean peak plasma concentration of dexrazoxane was 36.5 mcg/mL at 15 minute after intravenous administration of 500 mg/m2 dose of dexrazoxane for injection over 15 to 30 minutes prior to the 50 mg/m2 doxorubicin dose.
The important pharmacokinetic parameters of dexrazoxane are summarized in Table 2:
Table 2: Summary of Mean (%CVa) Dexrazoxane Pharmacokinetic Parameters at a Dosage Ratio of 10:1 of Dexrazoxane: Doxorubicin a Coefficient of variation
b Steady-state volume of distribution
Dose Doxorubicin (mg/m2)
Dose
Dexrazoxane (mg/m2)
Number of Subjects
Elimination Half-Life (h)
Plasma Clearance (L/h/m2)
Renal Clearance (L/h/m2)
bVolume of Distribution (L/m2)
50
500
10
2.5 (16)
7.88 (18)
3.35 (36)
22.4 (22)
60
600
5
2.1 (29)
6.25 (31)
---
22.0 (55)
Distribution: Following a rapid distributive phase (0.2 to 0.3 hours), dexrazoxane reaches post-distributive equilibrium within 2 to 4 hours. The estimated mean steady-state volume of distribution of dexrazoxane is 22.4 L/m2 after 500 mg/m2 of dexrazoxane dose followed by 50 mg/m2 of doxorubicin, suggesting distribution throughout total body water (25 L/m2).
In vitro studies have shown that dexrazoxane is not bound to plasma proteins.
Metabolism: Qualitative metabolism studies with dexrazoxane have confirmed the presence of unchanged drug, a diacid-diamide cleavage product, and two monoacid-monoamide ring products in the urine of animals and man. The metabolite levels were not measured in the pharmacokinetic studies.
Excretion: Urinary excretion plays an important role in the elimination of dexrazoxane. Forty-two percent of a 500 mg/m2 dose of dexrazoxane was excreted in the urine. Renal clearance averages 3.35 L/h/m2 after the 500 mg/m2 dexrazoxane for injection dose followed by 50 mg/m2 of doxorubicin.
Specific Populations:
Pediatric: Pharmacokinetics following dexrazoxane administration have not been evaluated in pediatric patients.
Effect of Renal Impairment: The pharmacokinetics of dexrazoxane were assessed following a single 15 minute IV infusion of 150 mg/m2 of dexrazoxane. Dexrazoxane clearance was reduced in subjects with renal dysfunction. Compared with controls, the mean AUC0-inf value was 2-fold greater in subjects with moderate (CLCR 30 to 50 mL/min) to severe (CLCR < 30 mL/min) renal dysfunction. Modeling demonstrated that equivalent exposure (AUC-inf) could be achieved if dosing were reduced by 50% in subjects with creatinine clearance values < 40 mL/min compared with control subjects (CLCR > 80 mL/min) [see Use in Specific Populations (8.7) and Dosage and Administration (2.2)].
Effect of Hepatic Impairment: Pharmacokinetics following dexrazoxane administration have not been evaluated in patients with hepatic impairment. The dexrazoxane dose is dependent upon the dose of doxorubicin [see Dosage and Administration (2.2)].
Drug Interactions: There was no significant change in the pharmacokinetics of doxorubicin (50 mg/m2) and its predominant metabolite, doxorubicinol, in the presence of dexrazoxane (500 mg/m2) in a crossover study in cancer patients.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term carcinogenicity studies have been carried out with dexrazoxane in animals. Nevertheless, a study by the National Cancer Institute has reported that long-term dosing with razoxane (the racemic mixture of dexrazoxane, ICRF-187, and its enantiomer ICRF-186) is associated with the development of malignancies in rats and possibly in mice [see Warnings and Precautions (5.4)].
Dexrazoxane was not mutagenic in the bacterial reverse mutation (Ames) test, but was found to be clastogenic to human lymphocytes in vitro and to mouse bone marrow erythrocytes in vivo (micronucleus test).
Dexrazoxane has the potential to impair fertility in male patients based on effects in repeat-dose toxicology studies. Testicular atrophy was seen with dexrazoxane administration at doses as low as 30 mg/kg weekly for 6 weeks in rats (1/3 the human dose on a mg/m2 basis) and as low as 20 mg/kg weekly for 13 weeks in dogs (approximately equal to the human dose on a mg/m2 basis).
-
14 CLINICAL STUDIES
The ability of dexrazoxane to prevent/reduce the incidence and severity of doxorubicin-induced cardiomyopathy was evaluated in three prospectively randomized placebo-controlled studies. In these studies, patients were treated with a doxorubicin-containing regimen and either dexrazoxane or placebo starting with the first course of chemotherapy. There was no restriction on the cumulative dose of doxorubicin. Cardiac function was assessed by measurement of the LVEF, utilizing resting multigated nuclear medicine (MUGA) scans, and by clinical evaluations. Patients receiving dexrazoxane had significantly smaller mean decreases from baseline in LVEF and lower incidences of congestive heart failure than the control group; however, in the largest study, patients with advanced breast cancer receiving FAC with dexrazoxane had a lower response rate (48% vs. 63%) and a shorter time to progression than patients who received FAC versus placebo.
In the clinical trials, patients who were initially randomized to receive placebo were allowed to receive dexrazoxane after a cumulative dose of doxorubicin above
300 mg/m2. Retrospective historical analyses showed that the risk of experiencing a cardiac event (see Table 3 for definition) at a cumulative dose of doxorubicin above 300 mg/m2 was greater in the patients who did not receive dexrazoxane beginning with their seventh course of FAC than in the patients who did receive dexrazoxane (HR=13.08; 95% CI: 3.72, 46.03; p < 0.001). Overall, 3% of patients treated with dexrazoxane developed CHF compared with 22% of patients not receiving dexrazoxane.
Table 3: Definition of Cardiac Events
- Development of congestive heart failure, defined as having two or more of the following:
a. Cardiomegaly by X-ray
b. Basilar Rales
c. S3 Gallop
d. Paroxysmal nocturnal dyspnea and/or orthopnea and/ or significant dyspnea on exertion. - Decline from baseline in LVEF by ≥ 10% and to below the lower limit of normal for the institution.
- Decline in LVEF by ≥ 20% from baseline value.
- Decline in LVEF to ≥ 5% below lower limit of normal for the institution.
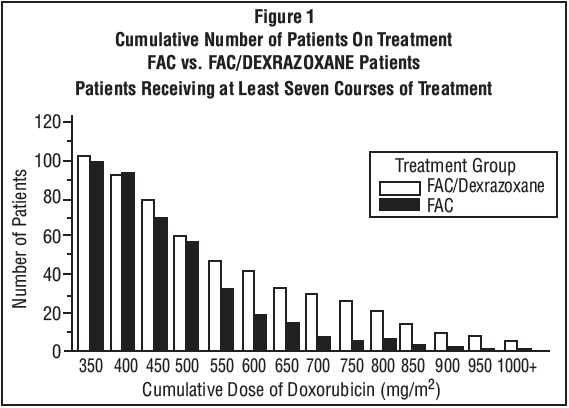
Figure 1 shows the number of patients still on treatment at increasing cumulative doses.
- Development of congestive heart failure, defined as having two or more of the following:
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Dexrazoxane for injection is available in the following strengths as sterile, pyrogen-free lyophilizates.
NDC: 55150-434-02
250 mg single-dose vial with a green flip-top seal, packaged in single vial packs. (This package also contains a 25 mL vial of 0.167 Molar (M/6) sodium lactate injection, USP.)Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature]. Reconstituted solutions of dexrazoxane for injection are stable for 6 hours at controlled room temperature or under refrigeration, 2° to 8°C (36° to 46°F).
Discard unused solutions.
Follow special handling and disposal procedures.
The vial stopper is not made with natural rubber latex.
-
17 PATIENT COUNSELING INFORMATION
17.1 Myelosuppression
Treatment with dexrazoxane is associated with leukopenia, neutropenia, and thrombocytopenia. Perform hematological monitoring [see Warnings and Precautions (5.1)].
17.2 Embryo-Fetal Toxicity
Counsel patients on pregnancy planning and prevention. Advise female patients of reproductive potential that dexrazoxane can cause fetal harm and to use highly effective contraception during treatment [see Warnings and Precautions (5.5) and Use in Specific Populations (8.1, 8.6)].
Distributed by:
AuroMedics Pharma LLC
279 Princeton-Hightstown Rd.
E. Windsor, NJ 08520Manufactured by:
Gland Pharma Limited
D.P. Pally, Dundigal Post
Hyderabad-500 043, India

Novaplus is a registered trademark of Vizient Inc. - PACKAGE LABEL.PRINCIPAL DISPLAY PANEL 250 mg per vial - Container Label
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL 25 mL - Container Label Diluent
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL 250 mg per vial - Container-Carton
-
INGREDIENTS AND APPEARANCE
DEXRAZOXANE
dexrazoxane kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55150-434 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55150-434-02 01 in 1 PACKAGE; Type 0: Not a Combination Product 05/12/2016 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-DOSE 25 mL Part 2 1 VIAL 25 mL Part 1 of 2 DEXRAZOXANE HYDROCHLORIDE
dexrazoxane injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC: 55150-432 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEXRAZOXANE HYDROCHLORIDE (UNII: 5346058Q7S) (DEXRAZOXANE - UNII:048L81261F) DEXRAZOXANE 250 mg in 25 mL Inactive Ingredients Ingredient Name Strength HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55150-432-02 25 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA200752 05/12/2016 Part 2 of 2 SODIUM LACTATE
sodium lactate injection, solutionProduct Information Item Code (Source) NDC: 55150-433 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength SODIUM LACTATE (UNII: TU7HW0W0QT) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55150-433-02 25 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA200752 05/12/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA200752 05/12/2016 Labeler - AuroMedics Pharma LLC (968961354) Establishment Name Address ID/FEI Business Operations GLAND PHARMA LIMITED 918601238 ANALYSIS(55150-434) , API MANUFACTURE(55150-434) , MANUFACTURE(55150-434) , PACK(55150-434)



© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.