SORIATANE- acitretin capsule
SORIATANE by
Drug Labeling and Warnings
SORIATANE by is a Prescription medication manufactured, distributed, or labeled by Stiefel Laboratories Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- SPL UNCLASSIFIED SECTION
-
BOXED WARNING
(What is this?)
CONTRAINDICATIONS AND WARNINGS: Pregnancy
SORIATANE must not be used by females who are pregnant, or who intend to become pregnant during therapy or at any time for at least 3 years following discontinuation of therapy. SORIATANE also must not be used by females who may not use reliable contraception while undergoing treatment and for at least 3 years following discontinuation of treatment. Acitretin is a metabolite of etretinate (TEGISON), and major human fetal abnormalities have been reported with the administration of acitretin and etretinate. Potentially, any fetus exposed can be affected.
Clinical evidence has shown that concurrent ingestion of acitretin and ethanol has been associated with the formation of etretinate, which has a significantly longer elimination half-life than acitretin. Because the longer elimination half-life of etretinate would increase the duration of teratogenic potential for female patients, ethanol must not be ingested by female patients of childbearing potential either during treatment with SORIATANE or for 2 months after cessation of therapy. This allows for elimination of acitretin, thus removing the substrate for transesterification to etretinate. The mechanism of the metabolic process for conversion of acitretin to etretinate has not been fully defined. It is not known whether substances other than ethanol are associated with transesterification.
Acitretin has been shown to be embryotoxic and/or teratogenic in rabbits, mice, and rats at oral doses of 0.6, 3, and 15 mg per kg, respectively. These doses are approximately 0.2, 0.3, and 3 times the maximum recommended therapeutic dose, respectively, based on a mg-per-m2 comparison.
Major human fetal abnormalities associated with acitretin and/or etretinate administration have been reported including meningomyelocele; meningoencephalocele; multiple synostoses; facial dysmorphia; syndactyly; absence of terminal phalanges; malformations of hip, ankle, and forearm; low-set ears; high palate; decreased cranial volume; cardiovascular malformation; and alterations of the skull and cervical vertebrae.
SORIATANE should be prescribed only by those who have special competence in the diagnosis and treatment of severe psoriasis, are experienced in the use of systemic retinoids, and understand the risk of teratogenicity.
Because of the teratogenicity of SORIATANE, a program called the Do Your P.A.R.T. program, Pregnancy Prevention Actively Required During and After Treatment, has been developed to educate women of childbearing potential and their healthcare providers about the serious risks associated with acitretin and to help prevent pregnancies from occurring with the use of this drug and for 3 years after its discontinuation. The Do Your P.A.R.T. program requirements are described below and program materials are available at www.soriatane.com or may be requested by calling 1-888-784-3335 (1-888-STIEFEL) (see also PRECAUTIONS section).
Important Information for Women of Childbearing Potential:
SORIATANE should be considered only for women with severe psoriasis unresponsive to other therapies or whose clinical condition contraindicates the use of other treatments.
Females of reproductive potential must not be given a prescription for SORIATANE until pregnancy is excluded. SORIATANE is contraindicated in females of reproductive potential unless the patient meets ALL of the following conditions:
- Must have had 2 negative urine or serum pregnancy tests with a sensitivity of at least 25 mIU per mL before receiving the initial prescription for SORIATANE. The first test (a screening test) is obtained by the prescriber when the decision is made to pursue therapy with SORIATANE. The second pregnancy test (a confirmation test) should be done during the first 5 days of the menstrual period immediately preceding the beginning of therapy with SORIATANE. For patients with amenorrhea, the second test should be done at least 11 days after the last act of unprotected sexual intercourse (without using 2 effective forms of contraception [birth control] simultaneously). If the second pregnancy test is negative, initiation of treatment with SORIATANE should begin within 7 days of the specimen collection. SORIATANE should be limited to a monthly supply.
- Must have a pregnancy test with a sensitivity of at least 25 mIU per mL repeated every month during treatment with SORIATANE. The patient must have a negative result from a urine or serum pregnancy test before receiving a prescription for SORIATANE. To encourage compliance with this recommendation, a monthly supply of the drug should be prescribed. For at least 3 years after discontinuing therapy with SORIATANE, a pregnancy test must be repeated every 3 months.
- Must have selected and have committed to use 2 effective forms of contraception (birth control) simultaneously, at least 1 of which must be a primary form, unless absolute abstinence is the chosen method, or the patient has undergone a hysterectomy or is clearly postmenopausal.
- Patients must use 2 effective forms of contraception (birth control) simultaneously for at least 1 month prior to initiation of therapy with SORIATANE, during therapy with SORIATANE, and for at least 3 years after discontinuing therapy with SORIATANE. A Contraception Counseling Referral Form is available so that patients can receive an initial free contraception counseling session and pregnancy testing. Counseling about contraception and behaviors associated with an increased risk of pregnancy must be repeated on a monthly basis by the prescriber during therapy with SORIATANE and every 3 months for at least 3 years following discontinuation of SORIATANE.
-
Effective forms of contraception include both primary and secondary forms of contraception. Primary forms of contraception include: tubal ligation, partner’s vasectomy, intrauterine devices, birth control pills, and injectable/implantable/insertable/topical hormonal birth control products. Secondary forms of contraception include condoms (with or without spermicide), diaphragms and cervical caps (which must be used with a spermicide), and vaginal sponges (contains spermicide).
Any birth control method can fail. Therefore, it is critically important that women of childbearing potential use 2 effective forms of contraception (birth control) simultaneously. It has not been established if there is a pharmacokinetic interaction between acitretin and combined oral contraceptives. However, it has been established that acitretin interferes with the contraceptive effect of microdosed progestin preparations.1 Microdosed “minipill” progestin preparations are not recommended for use with SORIATANE. It is not known whether other progestin-only contraceptives, such as implants and injectables, are adequate methods of contraception during acitretin therapy. Prescribers are advised to consult the package insert of any medication administered concomitantly with hormonal contraceptives, since some medications may decrease the effectiveness of these birth control products. Patients should be prospectively cautioned not to self-medicate with the herbal supplement St. John’s wort because a possible interaction has been suggested with hormonal contraceptives based on reports of breakthrough bleeding on oral contraceptives shortly after starting St. John’s wort. Pregnancies have been reported by users of combined hormonal contraceptives who also used some form of St. John’s wort (see PRECAUTIONS). -
Must have signed a Patient Agreement/Informed Consent for Female Patients that contains warnings about the risk of potential birth defects if the fetus is exposed to SORIATANE, about contraceptive failure, about the fact that they must not ingest beverages or products containing ethanol while taking SORIATANE and for 2 months after treatment with SORIATANE has been discontinued, and about preventing pregnancy while taking SORIATANE and for at least 3 years after discontinuing SORIATANE.
If pregnancy does occur during therapy with SORIATANE or at any time for at least 3 years following discontinuation of SORIATANE, the prescriber and patient should discuss the possible effects on the pregnancy. The available information is as follows:
Acitretin, the active metabolite of etretinate, is teratogenic and is contraindicated during pregnancy. The risk of severe fetal malformations is well established when systemic retinoids are taken during pregnancy. Pregnancy must also be prevented after stopping acitretin therapy, while the drug is being eliminated to below a threshold blood concentration that would be associated with an increased incidence of birth defects. Because this threshold has not been established for acitretin in humans and because elimination rates vary among patients, the duration of posttherapy contraception to achieve adequate elimination cannot be calculated precisely. It is strongly recommended that contraception be continued for at least 3 years after stopping treatment with acitretin, based on the following considerations:- In the absence of transesterification to form etretinate, greater than 98% of the acitretin would be eliminated within 2 months, assuming a mean elimination half-life of 49 hours.
-
In cases where etretinate is formed, as has been demonstrated with concomitant administration of acitretin and ethanol,
- greater than 98% of the etretinate formed would be eliminated in 2 years, assuming a mean elimination half-life of 120 days.
-
greater than 98% of the etretinate formed would be eliminated in 3 years, based on the longest demonstrated elimination half-life of 168 days.
However, etretinate was found in plasma and subcutaneous fat in one patient reported to have had sporadic alcohol intake, 52 months after she stopped acitretin therapy.2
-
Severe birth defects have been reported where conception occurred during the time interval when the patient was being treated with acitretin and/or etretinate. In addition, severe birth defects have also been reported when conception occurred after the mother completed therapy. These cases have been reported both prospectively (before the outcome was known) and retrospectively (after the outcome was known). The events below are listed without distinction as to whether the reported birth defects are consistent with retinoid-induced embryopathy or not.
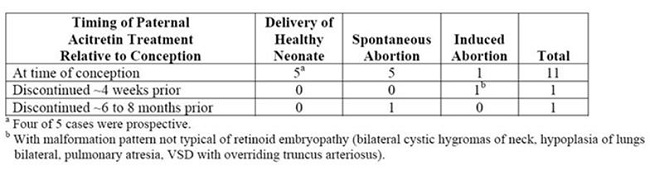
- There have been 318 prospectively reported cases involving pregnancies and the use of etretinate, acitretin, or both. In 238 of these cases, the conception occurred after the last dose of etretinate (103 cases), acitretin (126), or both (9). Fetal outcome remained unknown in approximately one-half of these cases, of which 62 were terminated and 14 were spontaneous abortions. Fetal outcome is known for the other 118 cases and 15 of the outcomes were abnormal (including cases of absent hand/wrist, clubfoot, GI malformation, hypocalcemia, hypotonia, limb malformation, neonatal apnea/anemia, neonatal ichthyosis, placental disorder/death, undescended testicle, and 5 cases of premature birth). In the 126 prospectively reported cases where conception occurred after the last dose of acitretin only, 43 cases involved conception at least 1 year but less than 2 years after the last dose. There were 3 reports of abnormal outcomes out of these 43 cases (involving limb malformation, GI tract malformations, and premature birth). There were only 4 cases where conception occurred at least 2 years after the last dose but there were no reports of birth defects in these cases.
- There is also a total of 35 retrospectively reported cases where conception occurred at least 1 year after the last dose of etretinate, acitretin, or both. From these cases there are 3 reports of birth defects when the conception occurred at least 1 year but less than 2 years after the last dose of acitretin (including heart malformations, Turner’s Syndrome, and unspecified congenital malformations) and 4 reports of birth defects when conception occurred 2 or more years after the last dose of acitretin (including foot malformation, cardiac malformations [2 cases], and unspecified neonatal and infancy disorder). There were 3 additional abnormal outcomes in cases where conception occurred 2 or more years after the last dose of etretinate (including chromosome disorder, forearm aplasia, and stillbirth).
- Females who have taken TEGISON (etretinate) must continue to follow the contraceptive recommendations for TEGISON. TEGISON is no longer marketed in the US; for information, call Stiefel at 1-888-784-3335 (1-888-STIEFEL).
- Patients should not donate blood during and for at least 3 years following the completion of therapy with SORIATANE because women of childbearing potential must not receive blood from patients being treated with SORIATANE.
- Important Information for Males Taking SORIATANE:
- Patients should not donate blood during and for at least 3 years following therapy with SORIATANE because women of childbearing potential must not receive blood from patients being treated with SORIATANE.
- Samples of seminal fluid from 3 male patients treated with acitretin and 6 male patients treated with etretinate have been assayed for the presence of acitretin. The maximum concentration of acitretin observed in the seminal fluid of these men was 12.5 ng per mL. Assuming an ejaculate volume of 10 mL, the amount of drug transferred in semen would be 125 ng, which is 1/200,000 of a single 25 mg capsule. Thus, although it appears that residual acitretin in seminal fluid poses little, if any, risk to a fetus while a male patient is taking the drug or after it is discontinued, the no-effect limit for teratogenicity is unknown and there is no registry for birth defects associated with acitretin. The available data are as follows:
There have been 25 cases of reported conception when the male partner was taking acitretin. The pregnancy outcome is known in 13 of these 25 cases. Of these, 9 reports were retrospective and 4 were prospective (meaning the pregnancy was reported prior to knowledge of the outcome)3.
For All Patients: A SORIATANE MEDICATION GUIDE MUST BE GIVEN TO THE PATIENT EACH TIME SORIATANE IS DISPENSED, AS REQUIRED BY LAW.
-
DESCRIPTION
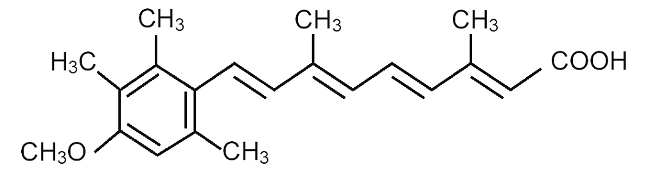
SORIATANE (acitretin), a retinoid, is available in 10-mg and 25-mg gelatin capsules for oral administration. Chemically, acitretin is all-trans-9-(4-methoxy-2,3,6-trimethylphenyl)-3,7-dimethyl-2,4,6,8-nonatetraenoic acid. It is a metabolite of etretinate and is related to both retinoic acid and retinol (vitamin A). It is a yellow to greenish-yellow powder with a molecular weight of 326.44. The structural formula is:
Each capsule contains acitretin, black monogramming ink, gelatin, maltodextrin (a mixture of polysaccharides), microcrystalline cellulose, and sodium ascorbate.
Gelatin capsule shells contain gelatin, iron oxide (yellow, black, and red), and titanium dioxide. They may also contain benzyl alcohol, carboxymethylcellulose sodium, edetate calcium disodium.
-
CLINICAL PHARMACOLOGY
The mechanism of action of SORIATANE is unknown.
Pharmacokinetics:
Absorption:
Oral absorption of acitretin is optimal when given with food. For this reason, acitretin was given with food in all of the following trials. After administration of a single 50-mg oral dose of acitretin to 18 healthy subjects, maximum plasma concentrations ranged from 196 to 728 ng per mL (mean: 416 ng per mL) and were achieved in 2 to 5 hours (mean: 2.7 hours). The oral absorption of acitretin is linear and proportional with increasing doses from 25 to 100 mg. Approximately 72% (range: 47% to 109%) of the administered dose was absorbed after a single 50-mg dose of acitretin was given to 12 healthy subjects.
Metabolism:
(See Pharmacokinetic Drug Interactions: Ethanol.)
Following oral absorption, acitretin undergoes extensive metabolism and interconversion by simple isomerization to its 13-cis form (cis-acitretin). The formation of cis-acitretin relative to parent compound is not altered by dose or fed/fast conditions of oral administration of acitretin. Both parent compound and isomer are further metabolized into chain-shortened breakdown products and conjugates, which are excreted. Following multiple-dose administration of acitretin, steady-state concentrations of acitretin and cis-acitretin in plasma are achieved within approximately 3 weeks.
Elimination:
The chain-shortened metabolites and conjugates of acitretin and cis-acitretin are ultimately excreted in the feces (34% to 54%) and urine (16% to 53%). The terminal elimination half-life of acitretin following multiple-dose administration is 49 hours (range: 33 to 96 hours), and that of cis-acitretin under the same conditions is 63 hours (range: 28 to 157 hours). The accumulation ratio of the parent compound is 1.2; that of cis-acitretin is 6.6.
Special Populations:
Psoriasis:
In an 8-week trial of acitretin pharmacokinetics in subjects with psoriasis, mean steady-state trough concentrations of acitretin increased in a dose-proportional manner with dosages ranging from 10 to 50 mg daily. Acitretin plasma concentrations were nonmeasurable (<4 ng per mL) in all subjects 3 weeks after cessation of therapy.
Pharmacokinetic Drug Interactions
(see also boxed CONTRAINDICATIONS AND WARNINGS and PRECAUTIONS: Drug Interactions): In studies of in vivo pharmacokinetic drug interactions, no interaction was seen between acitretin and cimetidine, digoxin, phenprocoumon, or glyburide.
Ethanol:
Clinical evidence has shown that etretinate (a retinoid with a much longer half-life, see below) can be formed with concurrent ingestion of acitretin and ethanol. In a 2-way crossover trial, all 10 subjects formed etretinate with concurrent ingestion of a single 100-mg oral dose of acitretin during a 3-hour period of ethanol ingestion (total ethanol, approximately 1.4 g per kg body weight). A mean peak etretinate concentration of 59 ng per mL (range: 22 to 105 ng per mL) was observed, and extrapolation of AUC values indicated that the formation of etretinate in this trial was comparable to a single 5-mg oral dose of etretinate. There was no detectable formation of etretinate when a single 100-mg oral dose of acitretin was administered without concurrent ethanol ingestion, although the formation of etretinate without concurrent ethanol ingestion cannot be excluded (see boxed CONTRAINDICATIONS AND WARNINGS). Of 93 evaluable psoriatic subjects on acitretin therapy in several foreign trials (10 to 80 mg per day), 16% had measurable etretinate levels (>5 ng per mL).
Etretinate has a much longer elimination half-life compared with that of acitretin. In one trial the apparent mean terminal half-life after 6 months of therapy was approximately 120 days (range: 84 to 168 days). In another trial of 47 subjects treated chronically with etretinate, 5 had detectable serum drug levels (in the range of 0.5 to 12 ng per mL) 2.1 to 2.9 years after therapy was discontinued. The long half-life appears to be due to storage of etretinate in adipose tissue.
Progestin-only Contraceptives:
It has not been established if there is a pharmacokinetic interaction between acitretin and combined oral contraceptives. However, it has been established that acitretin interferes with the contraceptive effect of microdosed progestin preparations.1 Microdosed “minipill” progestin preparations are not recommended for use with SORIATANE. It is not known whether other progestin-only contraceptives, such as implants and injectables, are adequate methods of contraception during acitretin therapy.
-
CLINICAL STUDIES
In 2 double-blind, placebo-controlled trials, SORIATANE was administered once daily to subjects with severe psoriasis (e.g., covering at least 10% to 20% of the body surface area). At 8 weeks (see Table 1) subjects treated in Trial A with 50 mg of SORIATANE per day showed significant improvements (P ≤0.05) relative to baseline and to placebo in the physician’s global evaluation and in the mean ratings of severity of psoriasis (scaling, thickness, and erythema). In Trial B, differences from baseline and from placebo were statistically significant (P ≤0.05) for all variables at both the 25-mg and 50-mg doses; it should be noted for Trial B that no statistical adjustment for multiplicity was carried out.
Table 1. Summary of the Efficacy Results of the 8-Week Double-Blind Phase of Trials A and B of SORIATANE Efficacy Variables
Trial A
Trial B
Total Daily Dose
Total Daily Dose
Placebo
(N = 29)50 mg
(N = 29)Placebo
(N = 72)25 mg
(N = 74)50 mg
(N = 71)Physician’s
Global EvaluationBaseline
4.62
4.55
4.43
4.37
4.49
Mean Change
After 8 Weeks
−0.29
−2.00a
−0.06
−1.06a
−1.57a
Scaling
Baseline
4.10
3.76
3.97
4.11
4.10
Mean Change
After 8 Weeks
−0.22
−1.62a
−0.21
−1.50a
−1.78a
Thickness
Baseline
4.10
4.10
4.03
4.11
4.20
Mean Change
After 8 Weeks
−0.39
−2.10a
−0.18
−1.43 a
−2.11a
Erythema
Baseline
4.21
4.59
4.42
4.24
4.45
Mean Change
After 8 Weeks
−0.33
−2.10a
−0.37
−1.12a
−1.65a
a Values were statistically significantly different from placebo and from baseline (P ≤0.05). No adjustment for multiplicity was done for Trial B.
The efficacy variables consisted of: the mean severity rating of scale, lesion thickness, erythema, and the physician's global evaluation of the current status of the disease. Ratings of scaling, erythema, and lesion thickness, and the ratings of the global assessments were made using a 7-point scale (0 = none, 1 = trace, 2 = mild, 3 = mild-moderate, 4 = moderate, 5 = moderate-severe, 6 = severe).
A subset of 141 subjects from both pivotal Trials A and B continued to receive SORIATANE in an open fashion for up to 24 weeks. At the end of the treatment period, all efficacy variables, as indicated in Table 2, were significantly improved (P ≤0.01) from baseline, including extent of psoriasis, mean ratings of psoriasis severity, and physician’s global evaluation.
Table 2. Summary of the First Course of Therapy with SORIATANE (24 Weeks) Variables
Trial A
Trial B
Mean Total Daily Dose of SORIATANE (mg)
42.8
43.1
Mean Duration of Therapy (Weeks)
21.1
22.6
Physician’s Global Evaluation
N = 39
N = 98
Baseline
4.51
4.43
Mean Change from Baseline
-2.26a
-2.60a
Scaling
N = 59
N = 132
Baseline
3.97
4.07
Mean Change from Baseline
−2.15a
−2.42a
Thickness
N = 59
N = 132
Baseline
4.00
4.12
Mean Change from Baseline
−2.44a
−2.66a
Erythema
N = 59
N = 132
Baseline
4.35
4.33
Mean Change from Baseline
−2.31a
−2.29a
a Indicates that the difference from baseline was statistically significant (P ≤0.01).
The efficacy variables consisted of: the mean severity rating of scale, lesion thickness, erythema, and the physician's global evaluation of the current status of the disease. Ratings of scaling, erythema, and lesion thickness, and the ratings of the global assessments were made using a 7-point scale (0 = none, 1 = trace, 2 = mild, 3 = mild-moderate, 4 = moderate, 5 = moderate-severe, 6 = severe).All efficacy variables improved significantly in a subset of 55 subjects from Trial A treated for a second, 6-month maintenance course of therapy (for a total of 12 months of treatment); a small subset of subjects (n = 4) from Trial A continued to improve after a third 6-month course of therapy (for a total of 18 months of treatment).
-
INDICATIONS AND USAGE
SORIATANE is indicated for the treatment of severe psoriasis in adults. Because of significant adverse effects associated with its use, SORIATANE should be prescribed only by those knowledgeable in the systemic use of retinoids. In females of reproductive potential, SORIATANE should be reserved for non-pregnant patients who are unresponsive to other therapies or whose clinical condition contraindicates the use of other treatments (see boxed CONTRAINDICATIONS AND WARNINGS — SORIATANE can cause severe birth defects).
Most patients experience relapse of psoriasis after discontinuing therapy. Subsequent courses, when clinically indicated, have produced efficacy results similar to the initial course of therapy.
-
CONTRAINDICATIONS
Pregnancy:
See boxed CONTRAINDICATIONS AND WARNINGS.
SORIATANE is contraindicated in patients with severely impaired liver or kidney function and in patients with chronic abnormally elevated blood lipid values (see boxed WARNINGS:Hepatotoxicity, WARNINGS:Lipids and Possible Cardiovascular Effects, and PRECAUTIONS).
An increased risk of hepatitis has been reported to result from combined use of methotrexate and etretinate. Consequently, the combination of methotrexate with SORIATANE is also contraindicated (see PRECAUTIONS: Drug Interactions).
Since both SORIATANE and tetracyclines can cause increased intracranial pressure, their combined use is contraindicated (see WARNINGS: Pseudotumor Cerebri).
SORIATANE is contraindicated in cases of hypersensitivity (e.g., angioedema, urticaria) to the preparation (acitretin or excipients) or to other retinoids.
-
WARNINGS
(See also boxed CONTRAINDICATIONS AND WARNINGS.)
Hepatotoxicity: Of the 525 subjects treated in US clinical trials, 2 had clinical jaundice with elevated serum bilirubin and transaminases considered related to treatment with SORIATANE. Liver function test results in these subjects returned to normal after SORIATANE was discontinued. Two of the 1,289 subjects treated in European clinical trials developed biopsy-confirmed toxic hepatitis. A second biopsy in one of these subjects revealed nodule formation suggestive of cirrhosis. One subject in a Canadian clinical trial of 63 subjects developed a 3-fold increase of transaminases. A liver biopsy of this subject showed mild lobular disarray, multifocal hepatocyte loss, and mild triaditis of the portal tracts compatible with acute reversible hepatic injury. The subject’s transaminase levels returned to normal 2 months after SORIATANE was discontinued.
The potential of therapy with SORIATANE to induce hepatotoxicity was prospectively evaluated using liver biopsies in an open-label trial of 128 subjects. Pretreatment and posttreatment biopsies were available for 87 subjects. A comparison of liver biopsy findings before and after therapy revealed 49 (58%) subjects showed no change, 21 (25%) improved, and 14 (17%) subjects had a worsening of their liver biopsy status. For 6 subjects, the classification changed from class 0 (no pathology) to class I (normal fatty infiltration; nuclear variability and portal inflammation; both mild); for 7 subjects, the change was from class I to class II (fatty infiltration, nuclear variability, portal inflammation, and focal necrosis; all moderate to severe); and for 1 subject, the change was from class II to class IIIb (fibrosis, moderate to severe). No correlation could be found between liver function test result abnormalities and the change in liver biopsy status, and no cumulative dose relationship was found.
Elevations of AST (SGOT), ALT (SGPT), GGT (GGTP), or LDH have occurred in approximately 1 in 3 subjects treated with SORIATANE. Of the 525 subjects treated in clinical trials in the US, treatment was discontinued in 20 (3.8%) due to elevated liver function test results. If hepatotoxicity is suspected during treatment with SORIATANE, the drug should be discontinued and the etiology further investigated.
Ten of 652 subjects treated in US clinical trials of etretinate, of which acitretin is the active metabolite, had clinical or histologic hepatitis considered to be possibly or probably related to etretinate treatment.
There have been reports of hepatitis-related deaths worldwide; a few of these subjects had received etretinate for a month or less before presenting with hepatic symptoms or signs.
Skeletal Abnormalities:
In adults receiving long-term treatment with SORIATANE, appropriate examinations should be periodically performed in view of possible ossification abnormalities (see ADVERSE REACTIONS). Because the frequency and severity of iatrogenic bony abnormality in adults is low, periodic radiography is only warranted in the presence of symptoms or long-term use of SORIATANE. If such disorders arise, the continuation of therapy should be discussed with the patient on the basis of a careful risk/benefit analysis. In clinical trials with SORIATANE, subjects were prospectively evaluated for evidence of development or change in bony abnormalities of the vertebral column, knees, and ankles.
Of 380 subjects treated with SORIATANE, 15% had preexisting abnormalities of the spine which showed new changes or progression of preexisting findings. Changes included degenerative spurs, anterior bridging of spinal vertebrae, diffuse idiopathic skeletal hyperostosis, ligament calcification, and narrowing and destruction of a cervical disc space. De novo changes (formation of small spurs) were seen in 3 subjects after 1½ to 2½ years.
Six of 128 subjects treated with SORIATANE showed abnormalities in the knees and ankles before treatment that progressed during treatment. In 5, these changes involved the formation of additional spurs or enlargement of existing spurs. The sixth subject had degenerative joint disease which worsened. No subjects developed spurs de novo. Clinical complaints did not predict radiographic changes.
Lipids and Possible Cardiovascular Effects:
Blood lipid determinations should be performed before SORIATANE is administered and again at intervals of 1 to 2 weeks until the lipid response to the drug is established, usually within 4 to 8 weeks. In subjects receiving SORIATANE during clinical trials, 66% and 33% experienced elevation in triglycerides and cholesterol, respectively. Decreased high density lipoproteins (HDL) occurred in 40% of subjects. These effects of SORIATANE were generally reversible upon cessation of therapy.
Subjects with an increased tendency to develop hypertriglyceridemia included those with disturbances of lipid metabolism, diabetes mellitus, obesity, increased alcohol intake, or a familial history of these conditions. Because of the risk of hypertriglyceridemia, serum lipids must be more closely monitored in high-risk patients and during long-term treatment.
Hypertriglyceridemia and lowered HDL may increase a patient’s cardiovascular risk status. Although no causal relationship has been established, there have been postmarketing reports of acute myocardial infarction or thromboembolic events in patients on therapy with SORIATANE. In addition, elevation of serum triglycerides to greater than 800 mg per dL has been associated with fatal fulminant pancreatitis. Therefore, dietary modifications, reduction in dose of SORIATANE, or drug therapy should be employed to control significant elevations of triglycerides. If, despite these measures, hypertriglyceridemia and low HDL levels persist, the discontinuation of SORIATANE should be considered.
Ophthalmologic Effects:
The eyes and vision of 329 subjects treated with SORIATANE were examined by ophthalmologists. The findings included dry eyes (23%), irritation of eyes (9%), and brow and lash loss (5%). The following were reported in less than 5% of subjects: Bell’s palsy, blepharitis and/or crusting of lids, blurred vision, conjunctivitis, corneal epithelial abnormality, cortical cataract, decreased night vision, diplopia, itchy eyes or eyelids, nuclear cataract, pannus, papilledema, photophobia, posterior subcapsular cataract, recurrent sties, and subepithelial corneal lesions.
Any patient treated with SORIATANE who is experiencing visual difficulties should discontinue the drug and undergo ophthalmologic evaluation.
Pancreatitis:
Lipid elevations occur in 25% to 50% of subjects treated with SORIATANE. Triglyceride increases sufficient to be associated with pancreatitis are much less common, although fatal fulminant pancreatitis has been reported. There have been rare reports of pancreatitis during therapy with SORIATANE in the absence of hypertriglyceridemia.
Pseudotumor Cerebri:
SORIATANE and other retinoids administered orally have been associated with cases of pseudotumor cerebri (benign intracranial hypertension). Some of these events involved concomitant use of isotretinoin and tetracyclines. However, the event seen in a single patient receiving SORIATANE was not associated with tetracycline use. Early signs and symptoms include papilledema, headache, nausea and vomiting, and visual disturbances. Patients with these signs and symptoms should be examined for papilledema and, if present, should discontinue SORIATANE immediately and be referred for neurological evaluation and care. Since both SORIATANE and tetracyclines can cause increased intracranial pressure, their combined use is contraindicated (see CONTRAINDICATIONS).
Capillary Leak Syndrome:
Capillary leak syndrome, a potential manifestation of retinoic acid syndrome, has been reported in patients receiving SORIATANE. Features of this syndrome may include localized or generalized edema with secondary weight gain, fever, and hypotension. Rhabdomyolysis and myalgias have been reported in association with capillary leak syndrome, and laboratory tests may reveal neutrophilia, hypoalbuminemia, and an elevated hematocrit. Discontinue SORIATANE if capillary leak syndrome develops during therapy.
-
PRECAUTIONS
A description of the Do Your P.A.R.T. materials is provided below. The main goals of the materials are to explain the program requirements, to reinforce the educational messages, and to assess program effectiveness.
The Do Your P.A.R.T. booklet includes:
- The Do Your P.A.R.T. Patient Brochure: information on the program requirements, risks of acitretin, and the types of contraceptive methods
- The Contraception Counseling Referral Form for female patients who want to receive free contraception counseling reimbursed by the manufacturer
- The Patient Agreement/Informed Consent for Female Patients form
- Medication Guide
The Do Your P.A.R.T. program also includes a voluntary patient survey for women of childbearing potential to assess the effectiveness of the SORIATANE Pregnancy Prevention Program Do Your P.A.R.T. Do Your P.A.R.T. Program materials are available at www.soriatane.com or may be requested by calling 1-888-784-3335 (1-888-STIEFEL).
Information for Patients:
(See Medication Guide for all patients and Patient Agreement/Informed Consent for Female Patients at end of professional labeling).
Patients should be instructed to read the Medication Guide supplied as required by law when SORIATANE is dispensed.
Females of Reproductive Potential:
SORIATANE can cause severe birth defects. Female patients must not be pregnant when therapy with SORIATANE is initiated, they must not become pregnant while taking SORIATANE and for at least 3 years after stopping SORIATANE, so that the drug can be eliminated to below a blood concentration that would be associated with an increased incidence of birth defects. Because this threshold has not been established for acitretin in humans and because elimination rates vary among patients, the duration of posttherapy contraception to achieve adequate elimination cannot be calculated precisely (see boxed CONTRAINDICATIONS AND WARNINGS).
Females of reproductive potential should also be advised that they must not ingest beverages or products containing ethanol while taking SORIATANE and for 2 months after SORIATANE has been discontinued. This allows for elimination of the acitretin which can be converted to etretinate in the presence of alcohol.
Female patients should be advised that any method of birth control can fail, including tubal ligation, and that microdosed progestin “minipill” preparations are not recommended for use with SORIATANE (see CLINICAL PHARMACOLOGY: Pharmacokinetic Drug Interactions). Data from one patient who received a very low-dosed progestin contraceptive (levonorgestrel 0.03 mg) had a significant increase of the progesterone level after 3 menstrual cycles during acitretin treatment.2
Female patients should be advised to contact their physician, women’s health centers, pharmacies, or hospital emergency rooms for information about how to obtain Emergency Contraception if sexual intercourse occurs without using 2 effective forms of contraception simultaneously. A 24-hour, toll-free number (1-800-739-6700) is also available for patients to receive automated birth control and emergency contraception information.
Female patients should sign a consent form prior to beginning therapy with SORIATANE (see boxed CONTRAINDICATIONS AND WARNINGS).
Nursing Mothers:
Studies on lactating rats have shown that etretinate is excreted in the milk. There is one prospective case report where acitretin is reported to be excreted in human milk. Therefore, nursing mothers should not receive SORIATANE prior to or during nursing because of the potential for serious adverse reactions in nursing infants.
All Patients:
Depression and/or other psychiatric symptoms such as aggressive feelings or thoughts of self-harm have been reported. These events, including self‑injurious behavior, have been reported in patients taking other systemically administered retinoids, as well as in patients taking SORIATANE. Since other factors may have contributed to these events, it is not known if they are related to SORIATANE. Patients should be counseled to stop taking SORIATANE and notify their prescriber immediately if they experience psychiatric symptoms.
Patients should be advised that a transient worsening of psoriasis is sometimes seen during the initial treatment period. Patients should be advised that they may have to wait 2 to 3 months before they get the full benefit of SORIATANE, although some patients may achieve significant improvements within the first 8 weeks of treatment as demonstrated in clinical trials.
Decreased night vision has been reported during therapy with SORIATANE. Patients should be advised of this potential problem and warned to be cautious when driving or operating any vehicle at night. Visual problems should be carefully monitored (see WARNINGS and ADVERSE REACTIONS). Patients should be advised that they may experience decreased tolerance to contact lenses during the treatment period and sometimes after treatment has stopped.
Patients should not donate blood during and for at least 3 years following therapy because SORIATANE can cause birth defects and women of childbearing potential must not receive blood from patients being treated with SORIATANE.
Because of the relationship of SORIATANE to vitamin A, patients should be advised against taking vitamin A supplements in excess of minimum recommended daily allowances to avoid possible additive toxic effects.
Patients should avoid the use of sun lamps and excessive exposure to sunlight (non-medical UV exposure) because the effects of UV light are enhanced by retinoids.
Patients should be advised that they must not give their SORIATANE to any other person.
Phototherapy:
Significantly lower doses of phototherapy are required when SORIATANE is used because effects on the stratum corneum induced by SORIATANE can increase the risk of erythema (burning) (see DOSAGE AND ADMINISTRATION).
Drug Interactions:
Ethanol:
Clinical evidence has shown that etretinate can be formed with concurrent ingestion of acitretin and ethanol (see boxed CONTRAINDICATIONS AND WARNINGS and CLINICAL PHARMACOLOGY: Pharmacokinetics).
Glyburide:
In a trial of 7 healthy male volunteers, acitretin treatment potentiated the blood glucose-lowering effect of glyburide (a sulfonylurea similar to chlorpropamide) in 3 of the 7 subjects. Repeating the trial with 6 healthy male volunteers in the absence of glyburide did not detect an effect of acitretin on glucose tolerance. Careful supervision of diabetic patients under treatment with SORIATANE is recommended (see CLINICAL PHARMACOLOGY: Pharmacokinetics and DOSAGE AND ADMINISTRATION).
Hormonal Contraceptives:
It has not been established if there is a pharmacokinetic interaction between acitretin and combined oral contraceptives. However, it has been established that acitretin interferes with the contraceptive effect of microdosed progestin “minipill” preparations. Microdosed “minipill” progestin preparations are not recommended for use with SORIATANE (see CLINICAL PHARMACOLOGY: Pharmacokinetic Drug Interactions). It is not known whether other progestin-only contraceptives, such as implants and injectables, are adequate methods of contraception during acitretin therapy.
Methotrexate:
An increased risk of hepatitis has been reported to result from combined use of methotrexate and etretinate. Consequently, the combination of methotrexate with acitretin is also contraindicated (see CONTRAINDICATIONS).
Phenytoin:
If acitretin is given concurrently with phenytoin, the protein binding of phenytoin may be reduced.
Tetracyclines:
Since both acitretin and tetracyclines can cause increased intracranial pressure, their combined use is contraindicated (see CONTRAINDICATIONS and WARNINGS: Pseudotumor Cerebri).
Laboratory Tests:
If significant abnormal laboratory results are obtained, either dosage reduction with careful monitoring or treatment discontinuation is recommended, depending on clinical judgment.
Blood Sugar:
Some patients receiving retinoids have experienced problems with blood sugar control. In addition, new cases of diabetes have been diagnosed during retinoid therapy, including diabetic ketoacidosis. In diabetics, blood-sugar levels should be monitored very carefully.
Lipids:
In clinical trials, the incidence of hypertriglyceridemia was 66%, hypercholesterolemia was 33%, and that of decreased HDL was 40%. Pretreatment and follow-up measurements should be obtained under fasting conditions. It is recommended that these tests be performed weekly or every other week until the lipid response to SORIATANE has stabilized (see WARNINGS).
Liver Function Tests:
Elevations of AST (SGOT), ALT (SGPT), or LDH were experienced by approximately 1 in 3 patients treated with SORIATANE. It is recommended that these tests be performed prior to initiation of therapy with SORIATANE, at 1- to 2-week intervals until stable, and thereafter at intervals as clinically indicated (see CONTRAINDICATIONS and boxed WARNINGS).
Carcinogenesis, Mutagenesis, Impairment of Fertility:
Carcinogenesis:
A carcinogenesis study of acitretin in Wistar rats, at doses up to 2 mg per kg per day administered 7 days per week for 104 weeks, has been completed. There were no neoplastic lesions observed that were considered to have been related to treatment with acitretin. An 80-week carcinogenesis study in mice has been completed with etretinate, the ethyl ester of acitretin. Blood level data obtained during this study demonstrated that etretinate was metabolized to acitretin and that blood levels of acitretin exceeded those of etretinate at all times studied. In the etretinate study, an increased incidence of blood vessel tumors (hemangiomas and hemangiosarcomas at several different sites) was noted in male, but not female, mice at doses approximately one-half the maximum recommended human therapeutic dose based on a mg-per-m2 comparison.
Mutagenesis:
Acitretin was evaluated for mutagenic potential in the Ames test, in the Chinese hamster (V79/HGPRT) assay, in unscheduled DNA synthesis assays using rat hepatocytes and human fibroblasts, and in an in vivo mouse micronucleus assay. No evidence of mutagenicity of acitretin was demonstrated in any of these assays.
Impairment of Fertility:
In a fertility study in rats, the fertility of treated animals was not impaired at the highest dosage of acitretin tested, 3 mg per kg per day (approximately one-half the maximum recommended therapeutic dose based on a mg-per-m2 comparison). Chronic toxicity studies in dogs revealed testicular changes (reversible mild to moderate spermatogenic arrest and appearance of multinucleated giant cells) in the highest dosage group (50 then 30 mg per kg per day).
No decreases in sperm count or concentration and no changes in sperm motility or morphology were noted in 31 men (17 psoriatic subjects, 8 subjects with disorders of keratinization, and 6 healthy volunteers) given 30 to 50 mg per day of acitretin for at least 12 weeks. In these trials, no deleterious effects were seen on either testosterone production, LH, or FSH in any of the 31 men.4-6 No deleterious effects were seen on the hypothalamic-pituitary axis in any of the 18 men where it was measured.4,5
Pregnancy:
Teratogenic Effects:
(see boxed CONTRAINDICATIONS AND WARNINGS).
In a study in which acitretin was administered to male rats only at a dosage of 5 mg per kg per day for 10 weeks (approximate duration of one spermatogenic cycle) prior to and during mating with untreated female rats, no teratogenic effects were observed in the progeny (see boxed CONTRAINDICATIONS AND WARNINGS for information about male use of SORIATANE).
Nonteratogenic Effects:
In rats dosed at 3 mg per kg per day (approximately one-half the maximum recommended therapeutic dose based on a mg-per-m2 comparison), slightly decreased pup survival and delayed incisor eruption were noted. At the next lowest dose tested, 1 mg per kg per day, no treatment-related adverse effects were observed.
Pediatric Use:
Safety and effectiveness in pediatric patients have not been established. No clinical trials have been conducted in pediatric subjects. Ossification of interosseous ligaments and tendons of the extremities, skeletal hyperostoses, decreases in bone mineral density, and premature epiphyseal closure have been reported in children taking other systemic retinoids, including etretinate, a metabolite of SORIATANE. A causal relationship between these effects and SORIATANE has not been established. While it is not known that these occurrences are more severe or more frequent in children, there is special concern in pediatric patients because of the implications for growth potential (see WARNINGS: Hyperostosis).
Geriatric Use:
Clinical trials of SORIATANE did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently than younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger subjects. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. A 2-fold increase in acitretin plasma concentrations was seen in healthy elderly subjects compared with young subjects, although the elimination half-life did not change (see CLINICAL PHARMACOLOGY: Special Populations).
-
ADVERSE REACTIONS
Hypervitaminosis A produces a wide spectrum of signs and symptoms primarily of the mucocutaneous, musculoskeletal, hepatic, neuropsychiatric, and central nervous systems. Many of the clinical adverse reactions reported to date with administration of SORIATANE resemble those of the hypervitaminosis A syndrome.
Adverse Events/Postmarketing Reports:
In addition to the events listed in the tables for the clinical trials, the following adverse events have been identified during postapproval use of SORIATANE. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders:
Hypersensitivity, including angioedema and urticaria (see CONTRAINDICATIONS).
Nervous System:
Myopathy with peripheral neuropathy has been reported during therapy with SORIATANE. Both conditions improved with discontinuation of the drug.
Psychiatric:
Aggressive feelings and/or suicidal thoughts have been reported. These events, including self-injurious behavior, have been reported in patients taking other systemically administered retinoids, as well as in patients taking SORIATANE. Since other factors may have contributed to these events, it is not known if they are related to SORIATANE (see PRECAUTIONS).
Clinical Trials:
During clinical trials with SORIATANE, 513 of 525 (98%) subjects reported a total of 3,545 adverse events. One-hundred sixteen subjects (22%) left trials prematurely, primarily because of adverse experiences involving the mucous membranes and skin. Three subjects died. Two of the deaths were not drug-related (pancreatic adenocarcinoma and lung cancer); the other subject died of an acute myocardial infarction, considered remotely related to drug therapy. In clinical trials, SORIATANE was associated with elevations in liver function test results or triglyceride levels and hepatitis.
The tables below list by body system and frequency the adverse events reported during clinical trials of 525 subjects with psoriasis.
Table 3. Adverse Events Frequently Reported during Clinical Trials
Percent of Subjects Reporting (N = 525) Body System
>75%
50% to 75%
25% to 50%
10% to 25%
CNS
Rigors
Eye Disorders
Xerophthalmia
Mucous Membranes
Cheilitis
Rhinitis
Dry mouth
Epistaxis
Musculoskeletal
Arthralgia
Spinal hyperostosis
(progression of existing lesions)
Skin and Appendages
Alopecia
Skin peeling
Dry skin
Nail disorder
Pruritus
Erythematous rash
Hyperesthesia
Paresthesia
Paronychia
Skin atrophy
Sticky skin
Table 4. Adverse Events Less Frequently Reported during Clinical Trials (Some of Which May Bear No Relationship to Therapy)
Percent of Subjects Reporting (N = 525) Body System
1% to 10%
<1%
Body as a Whole
Anorexia
Edema
Fatigue
Hot flashes
Increased
appetite
Alcohol
intolerance
Dizziness
Fever
Influenza-like
symptoms
Malaise
Moniliasis
Muscle weakness
Weight increase
Cardiovascular
Flushing
Chest pain
Cyanosis
Increased
bleeding time
Intermittent
claudication
Peripheral
ischemia
CNS (also see Psychiatric)
Headache
Pain
Abnormal gait
Migraine
Neuritis
Pseudotumor
cerebri
(intracranial
hypertension)
Eye Disorders
Abnormal/
blurred vision
Blepharitis
Conjunctivitis/
irritation
Corneal epithelial
abnormality
Decreased night
vision/night
blindness
Eye abnormality
Eye pain
Photophobia
Abnormal
lacrimation
Chalazion
Conjunctival
hemorrhage
Corneal ulceration
Diplopia
Ectropion
Itchy eyes and lids
Papilledema
Recurrent sties
Subepithelial
corneal lesions
Gastrointestinal
Abdominal pain
Diarrhea
Nausea
Tongue disorder
Constipation
Dyspepsia
Esophagitis
Gastritis
GastroenteritisGlossitis
Hemorrhoids
Melena
Tenesmus
Tongue ulcerationLiver and Biliary
Hepatic function
abnormal
Hepatitis
JaundiceMucous Membranes
Gingival bleeding
Gingivitis
Increased saliva
Stomatitis
Thirst
Ulcerativestomatitis
Altered saliva
Anal disorder
Gum hyperplasiaHemorrhage
PharyngitisMusculoskeletal
Arthritis
Arthrosis
Back pain
Hypertonia
Myalgia
Osteodynia
Peripheral jointhyperostosis
(progression of
existing lesions)
Bone disorder
Olecranon bursitis
Spinal hyperostosis(new lesions)
TendonitisPsychiatric
Depression Insomnia
Somnolence
Anxiety
DysphoniaLibido decreased Nervousness
Reproductive
Atrophic vaginitis Leukorrhea
Respiratory
Sinusitis
Coughing
Increased sputum
Laryngitis
Skin and Appendages
Abnormal skin
odor
Abnormal hair
texture
Bullous eruption
Cold/clammy
skin
Dermatitis
Increased
sweating
Infection
Psoriasiform rash
Purpura
Pyogenicgranuloma
Rash
Seborrhea
Skin fissures
Skin ulceration
SunburnAcne
Breast pain
Cyst
Eczema
Fungal infection
Furunculosis
Hair discoloration
Herpes simplex
Hyperkeratosis
Hypertrichosis
Hypoesthesia
Impaired healing
Otitis mediaOtitis externa
Photosensitivityreaction
Psoriasis aggravatedScleroderma
Skin nodule
Skin hypertrophy
Skin disorder
Skin irritation
Sweat glanddisorder
Urticaria
VerrucaeSpecial Senses/ Other
Earache
Taste perversion
Tinnitus
Ceruminosis
Deafness
Taste lossUrinary
Abnormal urine
Dysuria
Penis disorderLaboratory:
Therapy with SORIATANE induces changes in liver function tests in a significant number of patients. Elevations of AST (SGOT), ALT (SGPT) or LDH were experienced by approximately 1 in 3 subjects treated with SORIATANE. In most subjects, elevations were slight to moderate and returned to normal either during continuation of therapy or after cessation of treatment. In subjects receiving SORIATANE during clinical trials, 66% and 33% experienced elevation in triglycerides and cholesterol, respectively. Decreased high density lipoproteins (HDL) occurred in 40% (see WARNINGS). Transient, usually reversible elevations of alkaline phosphatase have been observed.
Table 5 lists the laboratory abnormalities reported during clinical trials.
Table 5. Abnormal Laboratory Test Results Reported during Clinical Trials
Percent of Subjects Reporting Body System
50% to 75%
25% to 50%
10% to 25%
1% to 10%
Electrolytes
Increased:
–Phosphorus
–Potassium
–SodiumDecreased:
–Phosphorus
–Potassium
–SodiumIncreased and
decreased:
–MagnesiumIncreased and
decreased:
–Calcium
–ChlorideHematologic
Increased:
–ReticulocytesDecreased:
–Hematocrit
–Hemoglobin
–WBC
Increased:
–Haptoglobin
–Neutrophils
–WBCIncreased:
–Bands
–Basophils
–Eosinophils
–Hematocrit
–Hemoglobin
–Lymphocytes
–MonocytesDecreased:
–Haptoglobin
–Lymphocytes
–Neutrophils
–Reticulocytes
Increased ordecreased:
–Platelets
–RBCHepatic
Increased:
–Cholesterol
–LDH
–SGOT
–SGPT
Decreased:
–HDLcholesterol
Increased:
–Alkalinephosphatase
–Direct bilirubin
–GGTPIncreased:
–Globulin
–Total bilirubin
–Total protein
Increased anddecreased:
–Serum albuminMiscellaneous
Increased:
–TriglyceridesIncreased:
–CPK
–Fasting bloodsugar
Decreased:
–Fasting bloodsugar
–High occultblood
Increased and
decreased:
–IronRenal
Increased:
–Uric acidIncreased:
–BUN
–CreatinineUrinary
WBC in urine
Acetonuria
Hematuria
RBC in urineGlycosuria
Proteinuria -
OVERDOSAGE
In the event of acute overdosage, SORIATANE must be withdrawn at once. Symptoms of overdose are identical to acute hypervitaminosis A (e.g., headache and vertigo). The acute oral toxicity (LD50) of acitretin in both mice and rats was greater than 4,000 mg per kg.
In one reported case of overdose, a 32-year-old male with Darier’s disease took 21 x 25-mg capsules (525-mg single dose). He vomited several hours later but experienced no other ill effects.
All female patients of childbearing potential who have taken an overdose of SORIATANE must:
1) Have a pregnancy test at the time of overdose; 2) Be counseled as per the boxed CONTRAINDICATIONS AND WARNINGS and PRECAUTIONS sections regarding birth defects and contraceptive use for at least 3 years’ duration after the overdose.
-
DOSAGE AND ADMINISTRATION
There is intersubject variation in the pharmacokinetics, clinical efficacy, and incidence of side effects with SORIATANE. A number of the more common side effects are dose-related. Individualization of dosage is required to achieve sufficient therapeutic response while minimizing side effects. Therapy with SORIATANE should be initiated at 25 to 50 mg per day, given as a single dose with the main meal. Maintenance doses of 25 to 50 mg per day may be given dependent upon an individual patient’s response to initial treatment. Relapses may be treated as outlined for initial therapy.
When SORIATANE is used with phototherapy, the prescriber should decrease the phototherapy dose, dependent on the patient’s individual response (see PRECAUTIONS: General).
Females who have taken TEGISON (etretinate) must continue to follow the contraceptive recommendations for TEGISON. TEGISON is no longer marketed in the US; for information, call Stiefel at 1-888-784-3335 (1-888-STIEFEL).
-
HOW SUPPLIED:
Brown and white capsules, 10 mg, imprinted “A-10 mg”; bottles of 30 (NDC: 0145-0090-25).
Brown and yellow capsules, 25 mg, imprinted “A-25 mg”; bottles of 30 (NDC: 0145-0091-25).
Store between 15° and 25°C (59° and 77°F). Protect from light. Avoid exposure to high temperatures and humidity after the bottle is opened.
-
REFERENCES:
1. Berbis Ph, et al.: Arch Dermatol Res (1988) 280:388-389.
2. Maier H, Honigsmann H: Concentration of etretinate in plasma and subcutaneous fat after long-term acitretin. Lancet 348:1107, 1996.
3. Geiger JM, Walker M: Is there a reproductive safety risk in male patients treated with acitretin (Neotigason/ Soriatane)? Dermatology 205:105-107, 2002.
4. Sigg C, et al.: Andrological investigations in patients treated with etretin. Dermatologica 175:48-49, 1987.
5. Parsch EM, et al.: Andrological investigation in men treated with acitretin (Ro 10-1670). Andrologia 22:479-482, 1990.
6. Kadar L, et al.: Spermatological investigations in psoriatic patients treated with acitretin. In: Pharmacology of Retinoids in the Skin; Reichert U. et al., ed, KARGER, Basel, vol. 3, pp 253-254, 1988.
-
PATIENT AGREEMENT/INFORMED CONSENT FOR FEMALE PATIENTS
To be completed by the patient* and signed by her prescriber
*Must also be initialed by the parent or guardian of a minor patient (under age 18)
Read each item below and initial in the space provided to show that you understand each item. Do not sign this consent and do not take SORIATANE (acitretin) if there is anything that you do not understand.
_____________________________________________________________
(Patient’s name)
1. I understand that there is a very high risk that my unborn baby could have severe birth defects if I am pregnant or become pregnant while taking SORIATANE in any amount even for short periods of time. Birth defects have also happened in babies of women who became pregnant after stopping treatment with SORIATANE.
INITIAL: ___________
2. I understand that I must not become pregnant while taking SORIATANE and for at least 3 years after the end of my treatment with SORIATANE.
INITIAL: ___________
3. I know that I must avoid all alcohol, including drinks, food, medicines, and over-the-counter products that contain alcohol. I understand that the risk of birth defects may last longer than 3 years if I swallow any form of alcohol during therapy with SORIATANE, and for 2 months after I stop taking SORIATANE.
INITIAL: ___________
4. I understand that I must not have sexual intercourse, or I must use 2 separate, effective forms of birth control at the same time. The only exceptions are if I have had surgery to remove the womb (a hysterectomy) or my prescriber has told me I have gone completely through menopause.
INITIAL: ___________
5. I understand that I have to use 2 effective forms of birth control (contraception) at the same time for at least 1 month before starting SORIATANE, for the entire time of therapy with SORIATANE, and for at least 3 years after stopping SORIATANE.
INITIAL: ___________
6. I understand that any form of birth control can fail. Therefore, I must use 2 different methods at the same time, every time I have sexual intercourse.
INITIAL: ___________
7. I understand that the following are considered effective forms of birth control: Primary: Tubal ligation (having my tubes tied), partner’s vasectomy, birth control pills (not progestin-only “minipills”), injectable/implantable/insertable/topical (patch) hormonal birth control products, and IUDs (intrauterine devices). Secondary: Condoms (with or without spermicide, which is a special cream or jelly that kills sperm), diaphragms and cervical caps (which must be used with a spermicide), and vaginal sponges (contains spermicide). I understand that at least 1 of my 2 methods of birth control must be a primary method.
INITIAL: ___________
8. I will talk with my prescriber about any medicines or dietary supplements I plan to take while taking SORIATANE because certain birth control methods may not work if I am taking certain medicines or herbal products (for example, St. John’s wort).
INITIAL: ___________
9. Unless I have had a hysterectomy or my prescriber says I have gone completely through menopause, I understand that I must have 2 negative pregnancy test results before I can get a prescription to start SORIATANE. I understand that if the second pregnancy test is negative, I must start taking my SORIATANE within 7 days of the specimen collection. I will then have pregnancy tests on a monthly basis during therapy with SORIATANE as instructed by my prescriber. In addition, for at least 3 years after I stop taking SORIATANE, I will have a pregnancy test every 3 months.
INITIAL: ___________
10. I understand that I should not start taking SORIATANE until I am sure that I am not pregnant and have negative results from 2 pregnancy tests.
INITIAL: ___________
11. I have received information on emergency contraception (birth control), including information on its availability over-the-counter.
INITIAL: ___________
12. I understand that my prescriber can give me a referral for a free contraception (birth control) counseling session and pregnancy testing.
INITIAL: ___________
13. I understand that on a monthly basis during therapy with SORIATANE and every 3 months for at least 3 years after stopping SORIATANE that I should receive counseling from my prescriber about contraception (birth control) and behaviors associated with an increased risk of pregnancy.
INITIAL: ___________
14. I understand that I must stop taking SORIATANE right away and call my prescriber if I get pregnant, miss my menstrual period, stop using birth control, or have sexual intercourse without using my 2 birth control methods during and at least 3 years after stopping SORIATANE.
INITIAL: ___________
15. If I do become pregnant while on SORIATANE or at any time within 3 years of stopping SORIATANE, I understand that I should report my pregnancy to Stiefel at 1-888-784-3335 (1-888-STIEFEL) or to the Food and Drug Administration (FDA) MedWatch program at 1-800-FDA-1088. The information I share will be kept confidential (private) unless disclosure is legally required. This will help the company and the FDA evaluate the pregnancy prevention program to prevent birth defects.
INITIAL: ___________
I have received a copy of the Do Your P.A.R.T. brochure. My prescriber has answered all my questions about SORIATANE. I understand that it is my responsibility to follow my doctor’s instructions, and not to get pregnant during treatment with SORIATANE or for at least 3 years after I stop taking SORIATANE.
I now authorize my prescriber, ______________________________________________________, to begin my treatment with SORIATANE.
Patient signature: ________________________________________
Date: ___________________
Parent/guardian signature (if under age 18): ____________________
Date: ___________________
Please print: Patient name and address: _______________________________________________________________
_______________________________________________________________
Telephone: _____________________________________________________________
I have fully explained to the patient, _________________________________________________, the nature and purpose of the treatment described above and the risks to females of childbearing potential. I have asked the patient if she has any questions regarding her treatment with SORIATANE and have answered those questions to the best of my ability.
Prescriber signature: _______________________________________
Date: __________________
Revised 10/2018SRN:8PI
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: 09/2017
-
Principal Display Panel
NDC: 0145-0090-25
SORIATANE®
(acitretin) Capsules
10 mg
30 Capsules
Rx only
CAUSES BIRTH DEFECTS
DO NOT GET PREGNANT
Made in Austria
©2016 Stiefel Laboratories, Inc.
- 10000000140426 Rev. 3/16
-
Principal Display Panel
NDC: 0145-0091-25
SORIATANE®
(acitretin) Capsules
25 mg
30 Capsules
Rx only
CAUSES BIRTH DEFECTS
DO NOT GET PREGNANT
Made in Austria
©2016 Stiefel Laboratories, Inc.
- 10000000140428 Rev. 3/16
-
INGREDIENTS AND APPEARANCE
SORIATANE
acitretin capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0145-0090 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ACITRETIN (UNII: LCH760E9T7) (ACITRETIN - UNII:LCH760E9T7) ACITRETIN 10 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM ASCORBATE (UNII: S033EH8359) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) MALTODEXTRIN (UNII: 7CVR7L4A2D) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE RED (UNII: 1K09F3G675) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color BROWN, WHITE Score no score Shape CAPSULE Size 14mm Flavor Imprint Code A;10;mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0145-0090-25 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/01/1996 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA019821 11/01/1996 SORIATANE
acitretin capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0145-0091 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ACITRETIN (UNII: LCH760E9T7) (ACITRETIN - UNII:LCH760E9T7) ACITRETIN 25 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM ASCORBATE (UNII: S033EH8359) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) MALTODEXTRIN (UNII: 7CVR7L4A2D) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE RED (UNII: 1K09F3G675) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color BROWN, YELLOW Score no score Shape CAPSULE Size 19mm Flavor Imprint Code A;25;mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0145-0091-25 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/01/1996 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA019821 11/01/1996 Labeler - Stiefel Laboratories Inc (808842343)





Trademark Results [SORIATANE]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 SORIATANE 73686706 1497384 Live/Registered |
HOFFMANN-LA ROCHE INC. 1987-09-28 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.