ERLEADA- apalutamide tablet, film coated
ERLEADA by
Drug Labeling and Warnings
ERLEADA by is a Prescription medication manufactured, distributed, or labeled by Janssen Products, LP, Janssen Pharmaceuticals, Inc., Janssen Pharmaceutica NV, Janssen Cilag SpA, Johnson & Johnson Private Limited, Janssen Ortho LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ERLEADA safely and effectively. See full prescribing information for ERLEADA.
ERLEADA® (apalutamide) tablets, for oral use
Initial U.S. Approval – 2018INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 60 mg (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Ischemic cardiovascular events occurred in patients receiving ERLEADA. Monitor for signs and symptoms of ischemic heart disease. Optimize management of cardiovascular risk factors. (5.1).
- Fractures occurred in patients receiving ERLEADA. Evaluate patients for fracture risk and treat patients with bone-targeted agents according to established guidelines. (5.2)
- Falls occurred in patients receiving ERLEADA with increased incidence in the elderly. Evaluate patients for fall risk. (5.3)
- Seizure occurred in 0.4% of patients receiving ERLEADA. Permanently discontinue ERLEADA in patients who develop a seizure during treatment. (5.4)
- Embryo-Fetal Toxicity: ERLEADA can cause fetal harm. Advise males with female partners of reproductive potential to use effective contraception. (5.5, 8.1, 8.3)
ADVERSE REACTIONS
The most common adverse reactions (≥10%) are fatigue, arthralgia, rash, decreased appetite, fall, weight decreased, hypertension, hot flush, diarrhea, and fracture. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Products, LP at 1-800-526-7736 (1-800-JANSSEN or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Concomitant use with medications that are sensitive substrates of CYP3A4, CYP2C19, CYP2C9, UGT, P-gp, BCRP, or OATP1B1 may result in loss of activity of these medications. (7.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 9/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dose Modification
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Ischemic Cardiovascular Events
5.2 Fractures
5.3 Falls
5.4 Seizure
5.5 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on ERLEADA
7.2 Effect of ERLEADA on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dose of ERLEADA is 240 mg (four 60 mg tablets) administered orally once daily. Swallow the tablets whole. ERLEADA can be taken with or without food.
Patients should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had a bilateral orchiectomy.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Ischemic Cardiovascular Events
Ischemic cardiovascular events, including events leading to death, occurred in patients receiving ERLEADA. Monitor for signs and symptoms of ischemic heart disease. Optimize management of cardiovascular risk factors, such as hypertension, diabetes, or dyslipidemia. Consider discontinuation of ERLEADA for Grade 3 and 4 events.
In a randomized study (SPARTAN) of patients with nmCRPC, ischemic cardiovascular events occurred in 4% of patients treated with ERLEADA and 3% of patients treated with placebo. In a randomized study (TITAN) in patients with mCSPC, ischemic cardiovascular events occurred in 4% of patients treated with ERLEADA and 2% of patients treated with placebo. Across the SPARTAN and TITAN studies, 6 patients (0.5%) treated with ERLEADA and 2 patients (0.2%) treated with placebo died from an ischemic cardiovascular event. Patients with current evidence of unstable angina, myocardial infarction, or congestive heart failure within six months of randomization were excluded from the SPARTAN and TITAN studies.
5.2 Fractures
Fractures occurred in patients receiving ERLEADA. Evaluate patients for fracture risk. Monitor and manage patients at risk for fractures according to established treatment guidelines and consider use of bone-targeted agents.
In a randomized study (SPARTAN) of patients with non-metastatic castration-resistant prostate cancer, fractures occurred in 12% of patients treated with ERLEADA and in 7% of patients treated with placebo. Grade 3-4 fractures occurred in 3% of patients treated with ERLEADA and in 1% of patients treated with placebo. The median time to onset of fracture was 314 days (range: 20 to 953 days) for patients treated with ERLEADA. Routine bone density assessment and treatment of osteoporosis with bone-targeted agents were not performed in the SPARTAN study.
In a randomized study (TITAN) of patients with metastatic castration-sensitive prostate cancer, fractures occurred in 9% of patients treated with ERLEADA and in 6% of patients treated with placebo. Grade 3-4 fractures were similar in both arms at 2%. The median time to onset of fracture was 56 days (range: 2 to 111 days) for patients treated with ERLEADA. Routine bone density assessment and treatment of osteoporosis with bone-targeted agents were not performed in the TITAN study.
5.3 Falls
Falls occurred in patients receiving ERLEADA with increased frequency in the elderly [See Use in Specific Populations (8.5)]. Evaluate patients for fall risk.
In a randomized study (SPARTAN), falls occurred in 16% of patients treated with ERLEADA compared to 9% of patients treated with placebo. Falls were not associated with loss of consciousness or seizure.
5.4 Seizure
Seizure occurred in patients receiving ERLEADA. Permanently discontinue ERLEADA in patients who develop a seizure during treatment. It is unknown whether anti-epileptic medications will prevent seizures with ERLEADA. Advise patients of the risk of developing a seizure while receiving ERLEADA and of engaging in any activity where sudden loss of consciousness could cause harm to themselves or others.
In two randomized studies (SPARTAN and TITAN), five patients (0.4%) treated with ERLEADA and one patient treated with placebo (0.1%) experienced a seizure. Seizure occurred from 159 to 650 days after initiation of ERLEADA. Patients with a history of seizure, predisposing factors for seizure, or receiving drugs known to decrease the seizure threshold or to induce seizure were excluded. There is no clinical experience in re-administering ERLEADA to patients who experienced a seizure.
5.5 Embryo-Fetal Toxicity
The safety and efficacy of ERLEADA have not been established in females. Based on its mechanism of action, ERLEADA can cause fetal harm and loss of pregnancy when administered to a pregnant female [see Clinical Pharmacology (12.1)]. Advise males with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of ERLEADA [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following are discussed in more detail in other sections of the labeling:
- Ischemic Cardiovascular Events [see Warnings and Precautions (5.1)].
- Fractures [see Warnings and Precautions (5.2)].
- Falls [see Warnings and Precautions (5.3)].
- Seizure [see Warnings and Precautions (5.4)].
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common adverse reactions (≥ 10%) that occurred more frequently in the ERLEADA-treated patients (≥ 2% over placebo) from the randomized placebo-controlled clinical trials (TITAN and SPARTAN) were fatigue, arthralgia, rash, decreased appetite, fall, weight decreased, hypertension, hot flush, diarrhea, and fracture.
Metastatic Castration-sensitive Prostate Cancer (mCSPC)
TITAN, a randomized (1:1), double-blind, placebo-controlled, multi-center clinical study, enrolled patients who had mCSPC. In this study, patients received either ERLEADA at a dose of 240 mg daily or placebo. All patients in the TITAN study received a concomitant gonadotropin-releasing hormone (GnRH) analog or had prior bilateral orchiectomy. The median duration of exposure was 20 months (range: 0 to 34 months) in patients who received ERLEADA and 18 months (range: 0.1 to 34 months) in patients who received placebo.
Ten patients (2%) who were treated with ERLEADA died from adverse reactions. The reasons for death were ischemic cardiovascular events (n=3), acute kidney injury (n=2), cardio-respiratory arrest (n=1), sudden cardiac death (n=1), respiratory failure (n=1), cerebrovascular accident (n=1), and large intestinal ulcer perforation (n=1). ERLEADA was discontinued due to adverse reactions in 8% of patients, most commonly from rash (2%). Adverse reactions leading to dose interruption or reduction of ERLEADA occurred in 23% of patients; the most frequent (>1%) were rash, fatigue, and hypertension. Serious adverse reactions occurred in 20% of ERLEADA-treated patients and 20% in patients receiving placebo.
Table 1 shows adverse reactions occurring in ≥10% on the ERLEADA arm in TITAN that occurred with a ≥2% absolute increase in frequency compared to placebo. Table 2 shows laboratory abnormalities that occurred in ≥15% of patients, and more frequently (>5%) in the ERLEADA arm compared to placebo.
Table 1: Adverse Reactions in TITAN (mCSPC) ERLEADA
N=524Placebo
N=527System/Organ Class
Adverse reactionAll Grades
%Grade 3–4
%All Grades
%Grade 3–4
%- * Includes fatigue and asthenia
- † Per the Common Terminology Criteria for Adverse Reactions (CTCAE), the highest severity for these events is Grade 3
- ‡ Includes rash, rash maculo-papular, rash generalized, urticaria, rash pruritic, rash macular, conjunctivitis, erythema multiforme, rash papular, skin exfoliation, genital rash, rash erythematous, stomatitis, drug eruption, mouth ulceration, rash pustular, blister, papule, pemphigoid, skin erosion, dermatitis, and rash vesicular
General disorders and administration site conditions Fatigue*,† 26 3 25 2 Musculoskeletal and connective tissue disorders Arthralgia† 17 0.4 15 0.9 Skin and subcutaneous tissue disorders Rash‡ 28 6 9 0.6 Pruritus 11 <1 5 <1 Vascular disorders Hot flush 23 0 16 0 Hypertension 18 8 16 9 Additional adverse reactions of interest occurring in 2%, but less than 10% of patients treated with ERLEADA included diarrhea (9% versus 6% on placebo), muscle spasm (3% versus 2% on placebo), dysgeusia (3% versus 1% on placebo), and hypothyroidism (4% versus 1% on placebo).
Table 2: Laboratory Abnormalities Occurring in ≥ 15% of ERLEADA-Treated Patients and at a Higher Incidence than Placebo (Between Arm Difference > 5% All Grades) in TITAN (mCSPC) ERLEADA
N=524Placebo
N=527Laboratory Abnormality All Grades
%Grade 3–4
%All Grades
%Grade 3–4
%- * Does not reflect fasting values
Hematology White blood cell decreased 27 0.4 19 0.6 Chemistry Hypertriglyceridemia* 17 3 12 2 Non-metastatic Castration-resistant Prostate Cancer (nmCRPC)
SPARTAN, a randomized (2:1), double-blind, placebo-controlled, multi-center clinical study, enrolled patients who had nmCRPC. In this study, patients received either ERLEADA at a dose of 240 mg daily or a placebo. All patients in the SPARTAN study received a concomitant gonadotropin-releasing hormone (GnRH) analog or had a bilateral orchiectomy. The median duration of exposure was 16.9 months (range: 0.1 to 42 months) in patients who received ERLEADA and 11.2 months (range: 0.1 to 37 months) in patients who received placebo.
Eight patients (1%) who were treated with ERLEADA died from adverse reactions. The reasons for death were infection (n=4), myocardial infarction (n=3), and cerebral hemorrhage (n=1). One patient (0.3%) treated with placebo died from an adverse reaction of cardiopulmonary arrest (n=1). ERLEADA was discontinued due to adverse reactions in 11% of patients, most commonly from rash (3%). Adverse reactions leading to dose interruption or reduction of ERLEADA occurred in 33% of patients; the most common (>1%) were rash, diarrhea, fatigue, nausea, vomiting, hypertension, and hematuria. Serious adverse reactions occurred in 25% of ERLEADA-treated patients and 23% in patients receiving placebo. The most frequent serious adverse reactions (>2%) were fracture (3%) in the ERLEADA arm and urinary retention (4%) in the placebo arm.
Table 3 shows adverse reactions occurring in ≥10% on the ERLEADA arm in SPARTAN that occurred with a ≥2% absolute increase in frequency compared to placebo. Table 4 shows laboratory abnormalities that occurred in ≥15% of patients, and more frequently (>5%) in the ERLEADA arm compared to placebo.
Table 3: Adverse Reactions in SPARTAN (nmCRPC) ERLEADA
N=803Placebo
N=398System/Organ Class
Adverse reactionAll Grades
%Grade 3–4
%All Grades
%Grade 3–4
%- * Includes fatigue and asthenia
- † Per the Common Terminology Criteria for Adverse Reactions (CTCAE), the highest severity for these events is Grade 3
- ‡ Includes rash, rash maculo-papular, rash generalized, urticaria, rash pruritic, rash macular, conjunctivitis, erythema multiforme, rash papular, skin exfoliation, genital rash, rash erythematous, stomatitis, drug eruption, mouth ulceration, rash pustular, blister, papule, pemphigoid, skin erosion, dermatitis, and rash vesicular
- § Includes appetite disorder, decreased appetite, early satiety, and hypophagia
- ¶ Includes peripheral edema, generalized edema, edema, edema genital, penile edema, peripheral swelling, scrotal edema, lymphedema, swelling, and localized edema
- # Includes rib fracture, lumbar vertebral fracture, spinal compression fracture, spinal fracture, foot fracture, hip fracture, humerus fracture, thoracic vertebral fracture, upper limb fracture, fractured sacrum, hand fracture, pubis fracture, acetabulum fracture, ankle fracture, compression fracture, costal cartilage fracture, facial bones fracture, lower limb fracture, osteoporotic fracture, wrist fracture, avulsion fracture, fibula fracture, fractured coccyx, pelvic fracture, radius fracture, sternal fracture, stress fracture, traumatic fracture, cervical vertebral fracture, femoral neck fracture, and tibia fracture
General disorders and administration site conditions Fatigue*,† 39 1 28 0.3 Musculoskeletal and connective tissue disorders Arthralgia† 16 0 8 0 Skin and subcutaneous tissue disorders Rash‡ 25 5 6 0.3 Metabolism and nutrition disorders Decreased appetite§ 12 0.1 9 0 Peripheral edema¶ 11 0 9 0 Injury, poisoning and procedural complications Fall† 16 2 9 0.8 Fracture# 12 3 7 0.8 Investigations Weight decreased† 16 1 6 0.3 Vascular disorders Hypertension 25 14 20 12 Hot flush 14 0 9 0 Gastrointestinal disorders Diarrhea 20 1 15 0.5 Nausea 18 0 16 0 Additional clinically significant adverse reactions occurring in 2% or more of patients treated with ERLEADA included hypothyroidism (8.1% versus 2% on placebo), pruritus (6.2% versus 2% on placebo), and heart failure (2.2% versus 1% on placebo).
Table 4: Laboratory Abnormalities Occurring in ≥ 15% of ERLEADA-Treated Patients and at a Higher Incidence than Placebo (Between Arm Difference > 5% All Grades) in SPARTAN (nmCRPC) ERLEADA
N=803Placebo
N=398Laboratory Abnormality All Grades
%Grade 3–4
%All Grades
%Grade 3–4
%- * Does not reflect fasting values
Hematology Anemia 70 0.4 64 0.5 Leukopenia 47 0.3 29 0 Lymphopenia 41 2 21 2 Chemistry Hypercholesterolemia* 76 0.1 46 0 Hyperglycemia* 70 2 59 1 Hypertriglyceridemia* 67 2 49 0.8 Hyperkalemia 32 2 22 0.5 Rash
In the combined data of two randomized, placebo-controlled clinical studies, rash associated with ERLEADA was most commonly described as macular or maculo-papular. Adverse reactions of rash were reported for 26% of patients treated with ERLEADA versus 8% of patients treated with placebo. Grade 3 rashes (defined as covering > 30% body surface area [BSA]) were reported with ERLEADA treatment (6%) versus placebo (0.5%).
The onset of rash occurred at a median of 83 days of ERLEADA treatment. Rash resolved in 78% of patients within a median of 78 days from onset of rash. Rash was commonly managed with oral antihistamines, topical corticosteroids, and 19% of patients received systemic corticosteroids. Dose reduction or dose interruption occurred in 14% and 28% of patients, respectively. Of the patients who had dose interruption, 59% experienced recurrence of rash upon reintroduction of ERLEADA.
Hypothyroidism
In the combined data of two randomized, placebo-controlled clinical studies, hypothyroidism was reported for 8% of patients treated with ERLEADA and 2% of patients treated with placebo based on assessments of thyroid-stimulating hormone (TSH) every 4 months. Elevated TSH occurred in 25% of patients treated with ERLEADA and 7% of patients treated with placebo. The median onset was at the first scheduled assessment. There were no Grade 3 or 4 adverse reactions. Thyroid replacement therapy was initiated in 5% of patients treated with ERLEADA. Thyroid replacement therapy, when clinically indicated, should be initiated or dose-adjusted [see Drug Interactions (7.2)].
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on ERLEADA
Strong CYP2C8 or CYP3A4 Inhibitors
Co-administration of a strong CYP2C8 or CYP3A4 inhibitor is predicted to increase the steady-state exposure of the active moieties (sum of unbound apalutamide plus the potency-adjusted unbound N-desmethyl-apalutamide). No initial dose adjustment is necessary however, reduce the ERLEADA dose based on tolerability [see Dosage and Administration (2.2)]. Mild or moderate inhibitors of CYP2C8 or CYP3A4 are not expected to affect the exposure of apalutamide.
7.2 Effect of ERLEADA on Other Drugs
CYP3A4, CYP2C9, CYP2C19 and UGT Substrates
ERLEADA is a strong inducer of CYP3A4 and CYP2C19, and a weak inducer of CYP2C9 in humans. Concomitant use of ERLEADA with medications that are primarily metabolized by CYP3A4, CYP2C19, or CYP2C9 can result in lower exposure to these medications. Substitution for these medications is recommended when possible or evaluate for loss of activity if medication is continued. Concomitant administration of ERLEADA with medications that are substrates of UDP-glucuronosyl transferase (UGT) can result in decreased exposure. Use caution if substrates of UGT must be co-administered with ERLEADA and evaluate for loss of activity [see Clinical Pharmacology (12.3)].
P-gp, BCRP or OATP1B1 Substrates
Apalutamide was shown to be a weak inducer of P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and organic anion transporting polypeptide 1B1 (OATP1B1) clinically. At steady-state, apalutamide reduced the plasma exposure to fexofenadine (a P-gp substrate) and rosuvastatin (a BCRP/OATP1B1 substrate). Concomitant use of ERLEADA with medications that are substrates of P-gp, BCRP, or OATP1B1 can result in lower exposure of these medications. Use caution if substrates of P-gp, BCRP or OATP1B1 must be co-administered with ERLEADA and evaluate for loss of activity if medication is continued [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The safety and efficacy of ERLEADA have not been established in females. Based on its mechanism of action, ERLEADA can cause fetal harm and loss of pregnancy [see Clinical Pharmacology (12.1)]. There are no human data on the use of ERLEADA in pregnant women. ERLEADA is not indicated for use in females, so animal embryo-fetal developmental toxicology studies were not conducted with apalutamide.
8.3 Females and Males of Reproductive Potential
Contraception
Males
Based on the mechanism of action and findings in an animal reproduction study, advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of ERLEADA. [see Use in Specific Populations (8.1)].
Infertility
Males
Based on animal studies, ERLEADA may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of ERLEADA in pediatric patients have not been established.
8.5 Geriatric Use
Of the 1327 patients who received ERLEADA in clinical studies, 19% of patients were less than 65 years, 41% of patients were 65 years to 74 years, and 40% were 75 years and over.
No overall differences in effectiveness were observed between older and younger patients.
Of patients treated with ERLEADA (n=1073), Grade 3–4 adverse reactions occurred in 39% of patients younger than 65 years, 41% of patients 65–74 years, and 49% of patients 75 years or older. Falls in patients receiving ERLEADA with androgen deprivation therapy was elevated in the elderly, occurring in 8% of patients younger than 65 years, 10% of patients 65-74 years, and 19% of patients 75 years or older.
- 10 OVERDOSAGE
-
11 DESCRIPTION
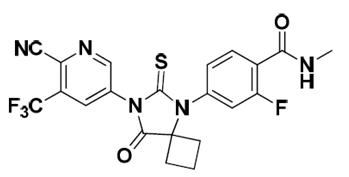
Apalutamide, the active ingredient of ERLEADA, is an androgen receptor inhibitor. The chemical name is (4-[7-(6-Cyano-5-trifluoromethylpyridin-3-yl)-8-oxo-6-thioxo-5,7-diazaspiro[3.4]oct-5-yl]-2-fluoro-N-methylbenzamide). Apalutamide is a white to slightly yellow powder. Apalutamide is practically insoluble in aqueous media over a wide range of pH values.
The molecular weight is 477.44 and molecular formula is C21H15F4N5O2S. The structural formula is:
ERLEADA (apalutamide) is supplied as film-coated tablets for oral administration containing 60 mg of apalutamide. Inactive ingredients of the core tablet are: colloidal anhydrous silica, croscarmellose sodium, hydroxypropyl methylcellulose-acetate succinate, magnesium stearate, microcrystalline cellulose, and silicified microcrystalline cellulose.
The tablets are finished with a commercially available film-coating comprising the following excipients: iron oxide black, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Apalutamide is an Androgen Receptor (AR) inhibitor that binds directly to the ligand-binding domain of the AR. Apalutamide inhibits AR nuclear translocation, inhibits DNA binding, and impedes AR-mediated transcription. A major metabolite, N-desmethyl apalutamide, is a less potent inhibitor of AR, and exhibited one-third the activity of apalutamide in an in vitro transcriptional reporter assay. Apalutamide administration caused decreased tumor cell proliferation and increased apoptosis leading to decreased tumor volume in mouse xenograft models of prostate cancer.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of apalutamide 240 mg once daily on the QTc interval was assessed in an open-label, uncontrolled, multi-center, single-arm dedicated QT study in 45 patients with CRPC. The maximum mean QTcF change from baseline was 12.4 ms (2-sided 90% upper CI: 16.0 ms). An exposure-QT analysis suggested a concentration-dependent increase in QTcF for apalutamide and its active metabolite.
12.3 Pharmacokinetics
Apalutamide pharmacokinetic parameters are presented as the mean [standard deviation (SD)] unless otherwise specified. Apalutamide Cmax and area under the concentration curve (AUC) increased proportionally following repeated once-daily dosing of 30 to 480 mg (0.125 to 2 times the recommended dosage). Following administration of the recommended dosage, apalutamide steady-state was achieved after 4 weeks and the mean accumulation ratio was approximately 5-fold. Apalutamide Cmax was 6.0 mcg/mL (1.7) and AUC was 100 mcg∙h/mL (32) at steady-state. Daily fluctuations in apalutamide plasma concentrations were low, with mean peak-to-trough ratio of 1.63. An increase in apparent clearance (CL/F) was observed with repeat dosing, likely due to induction of apalutamide's own metabolism. The auto-induction effect likely reached its maximum at the recommended dosage because exposure of apalutamide across the dose range of 30 to 480 mg is dose-proportional.
The major active metabolite N-desmethyl apalutamide Cmax was 5.9 mcg/mL (1.0) and AUC was 124 mcg∙h/mL (23) at steady-state after the recommended dosage. N-desmethyl apalutamide was characterized by a flat concentration-time profile at steady-state with a mean peak-to-trough ratio of 1.27. Mean AUC metabolite/parent drug ratio for N-desmethyl apalutamide following repeat-dose administration was 1.3. Based on systemic exposure, relative potency, and pharmacokinetic properties, N-desmethyl apalutamide likely contributed to the clinical activity of apalutamide.
Absorption
Mean absolute oral bioavailability was approximately 100%. Median time to achieve peak plasma concentration (tmax) was 2 hours (range: 1 to 5 hours).
Effect of Food
Administration of apalutamide to healthy subjects under fasting conditions and with a high-fat meal (approximately 500 to 600 fat calories, 250 carbohydrate calories, and 150 protein calories) resulted in no clinically relevant changes in Cmax and AUC. Median time to reach tmax was delayed approximately 2 hours with food.
Distribution
The mean apparent volume of distribution at steady-state of apalutamide was approximately 276 L.
Apalutamide was 96% and N-desmethyl apalutamide was 95% bound to plasma proteins with no concentration dependency.
Elimination
The CL/F of apalutamide was 1.3 L/h after single dosing and increased to 2.0 L/h at steady-state after once-daily dosing likely due to CYP3A4 auto-induction. The mean effective half-life for apalutamide in patients was approximately 3 days at steady-state.
Metabolism
Metabolism is the main route of elimination of apalutamide. Apalutamide is primarily metabolized by CYP2C8 and CYP3A4 to form active metabolite, N-desmethyl apalutamide. The contribution of CYP2C8 and CYP3A4 in the metabolism of apalutamide is estimated to be 58% and 13% following single dose but changes to 40% and 37%, respectively at steady-state.
Apalutamide represented 45% and N-desmethyl apalutamide represented 44% of the total AUC following a single oral administration of radiolabeled apalutamide 240 mg.
Excretion
Up to 70 days following a single oral administration of radiolabeled apalutamide, 65% of the dose was recovered in urine (1.2% of dose as unchanged apalutamide and 2.7% as N-desmethyl apalutamide) and 24% was recovered in feces (1.5% of dose as unchanged apalutamide and 2% as N-desmethyl apalutamide).
Specific Populations
No clinically significant differences in the pharmacokinetics of apalutamide or N-desmethyl apalutamide were observed based on age (18–94 years), race (Black, non-Japanese Asian, Japanese), mild to moderate (eGFR 30–89 mL/min/1.73m2, estimated by the modification of diet in renal disease [MDRD] equation) renal impairment, or mild (Child-Pugh A) to moderate (Child-Pugh B) hepatic impairment.
The effect of severe renal impairment or end stage renal disease (eGFR ≤29 mL/min/1.73m2, MDRD) or severe hepatic impairment (Child-Pugh C) on apalutamide pharmacokinetics is unknown.
Drug Interactions
Effect of Other Drugs on ERLEADA
Strong CYP2C8 inhibitors
Apalutamide Cmax decreased by 21% while AUC increased by 68% following co-administration of ERLEADA as a 240 mg single dose with gemfibrozil (a strong CYP2C8 inhibitor). Gemfibrozil is predicted to increase the steady-state apalutamide Cmax by 32% and AUC by 44%. For the active moieties (sum of unbound apalutamide plus the potency-adjusted unbound N-desmethyl apalutamide), the predicted steady-state Cmax increased by 19% and AUC by 23%.
Strong CYP3A4 inhibitors
Apalutamide Cmax decreased by 22% while AUC was similar following co-administration of ERLEADA as a 240 mg single dose with itraconazole (a strong CYP3A4 inhibitor). Ketoconazole (a strong CYP3A4 inhibitor) is predicted to increase the single-dose apalutamide AUC by 24% but have no impact on Cmax. Ketoconazole is predicted to increase the steady-state apalutamide Cmax by 38% and AUC by 51%. For the active moieties, the predicted steady-state Cmax increased by 23% and AUC by 28%.
CYP3A4/CYP2C8 inducers
Rifampin (a strong CYP3A4 and moderate CYP2C8 inducer) is predicted to decrease the steady-state apalutamide Cmax by 25% and AUC by 34%. For the active moieties, the predicted steady-state Cmax decreased by 15% and AUC by 19%.
Acid lowering agents
Apalutamide is not ionizable under relevant physiological pH condition, therefore acid lowering agents (e.g. proton pump inhibitor, H2-receptor antagonist, antacid) are not expected to affect the solubility and bioavailability of apalutamide.
Drugs affecting transporters
In vitro, apalutamide and N-desmethyl apalutamide are substrates for P-gp but not BCRP, OATP1B1, and OATP1B3. Because apalutamide is completely absorbed after oral administration, P-gp does not limit the absorption of apalutamide and therefore, inhibition or induction of P-gp is not expected to affect the bioavailability of apalutamide.
Effect of ERLEADA on Other Drugs
CYP substrates
In vitro studies showed that apalutamide and N-desmethyl apalutamide are moderate to strong CYP3A4 and CYP2B6 inducers, are moderate inhibitors of CYP2B6 and CYP2C8, and weak inhibitors of CYP2C9, CYP2C19, and CYP3A4. Apalutamide and N-desmethyl apalutamide do not affect CYP1A2 and CYP2D6 at therapeutically relevant concentrations.
Co-administration of ERLEADA with single oral doses of sensitive CYP substrates resulted in a 92% decrease in the AUC of midazolam (a CYP3A4 substrate), 85% decrease in the AUC of omeprazole (a CYP2C19 substrate), and 46% decrease in the AUC of S-warfarin (a CYP2C9 substrate). ERLEADA did not cause clinically significant changes in exposure to a CYP2C8 substrate.
P-gp, BCRP and OATP1B1 substrates
Co-administration of ERLEADA with single oral doses of transporter substrates resulted in a 30% decrease in the AUC of fexofenadine (a P-gp substrate) and 41% decrease in the AUC of rosuvastatin (a BCRP/OATP1B1 substrate) but had no impact on Cmax.
UGT substrates
Apalutamide may induce UGT. Concomitant administration of ERLEADA with medications that are substrates of UGT may result in lower exposure to these medications.
OCT2, OAT1, OAT3 and MATEs substrates
In vitro, apalutamide and N-desmethyl apalutamide inhibit organic cation transporter 2 (OCT2), organic anion transporter 3 (OAT3) and multidrug and toxin extrusions (MATEs), and do not inhibit organic anion transporter 1. Apalutamide is not predicted to cause clinically significant changes in exposure to an OAT3 substrate.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been conducted to evaluate the carcinogenic potential of apalutamide. Apalutamide did not induce mutations in the bacterial reverse mutation (Ames) assay and was not genotoxic in either in vitro chromosome aberration assay or the in vivo rat bone marrow micronucleus assay or the in vivo rat Comet assay.
In repeat-dose toxicity studies in male rats (up to 26 weeks) and dogs (up to 39 weeks), atrophy of the prostate gland and seminal vesicles, aspermia/hypospermia, tubular degeneration and/or hyperplasia or hypertrophy of the interstitial cells in the reproductive system were observed at ≥ 25 mg/kg/day in rats (1.4 times the human exposure based on AUC) and ≥ 2.5 mg/kg/day in dogs (0.9 times the human exposure based on AUC).
In a fertility study in male rats, a decrease in sperm concentration and motility, increased abnormal sperm morphology, lower copulation and fertility rates (upon pairing with untreated females) along with reduced weights of the secondary sex glands and epididymis were observed following 4 weeks of dosing at ≥ 25 mg/kg/day (0.8 times the human exposure based on AUC). A reduced number of live fetuses due to increased pre- and/or post-implantation loss was observed following 4 weeks of 150 mg/kg/day administration (5.7 times the human exposure based on AUC). Effects on male rats were reversible after 8 weeks from the last apalutamide administration.
-
14 CLINICAL STUDIES
The efficacy and safety of ERLEADA was established in two randomized placebo-controlled clinical trials.
TITAN (NCT02489318): Metastatic Castration-sensitive Prostate Cancer (mCSPC)
TITAN was a randomized, double-blind, placebo-controlled, multinational, clinical trial in which 1052 patients with mCSPC were randomized (1:1) to receive either ERLEADA orally at a dose of 240 mg once daily (N = 525) or placebo once daily (N = 527). All patients in the TITAN trial received concomitant GnRH analog or had prior bilateral orchiectomy. Patients were stratified by Gleason score at diagnosis, prior docetaxel use, and region of the world. Patients with both high- and low-volume mCSPC were eligible for the study. High volume of disease was defined as metastases involving the viscera with 1 bone lesion or the presence of 4 or more bone lesions, at least 1 of which must be in a bony structure beyond the vertebral column and pelvic bones.
The following patient demographics and baseline disease characteristics were balanced between the treatment arms. The median age was 68 years (range 43–94) and 23% of patients were 75 years of age or older. The racial distribution was 68% Caucasian, 22% Asian, and 2% Black. Sixty-three percent (63%) of patients had high-volume disease and 37% had low-volume disease. Sixteen percent (16%) of patients had prior surgery, radiotherapy of the prostate or both. A majority of patients had a Gleason score of 8 or higher (67%). Sixty-eight percent (68%) of patients received prior treatment with an anti-androgen (bicalutamide, flutamide, or nilutamide). All patients except one in the placebo group, had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 0 or 1 at study entry.
The major efficacy outcome measures of the study were overall survival (OS) and radiographic progression-free survival (rPFS). Radiographic progression-free survival was based on investigator assessment and was defined as time from randomization to radiographic disease progression or death. Radiographic disease progression was defined by identification of 2 or more new bone lesions on a bone scan with confirmation (Prostate Cancer Working Group 2 criteria) and/or progression in soft tissue disease.
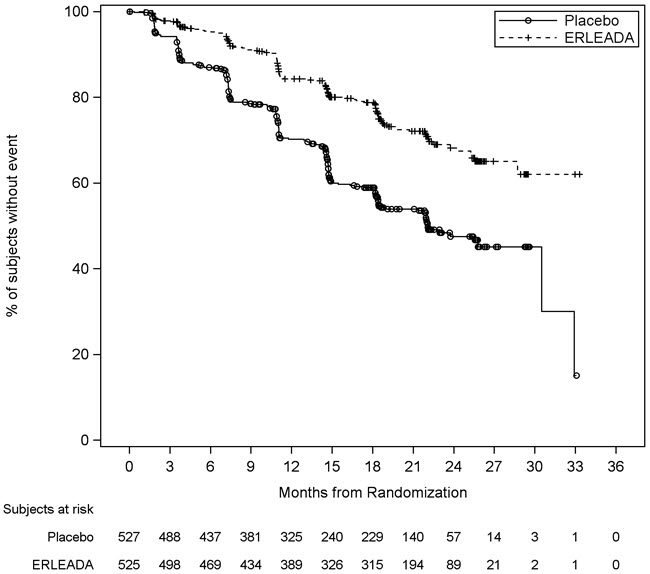
A statistically significant improvement in OS and rPFS was demonstrated in patients randomized to receive ERLEADA compared with patients randomized to receive placebo. The results for OS are based upon a prespecified interim efficacy analysis. Efficacy results of TITAN are summarized in Table 5 and Figures 1 and 2.
Table 5: Summary of Efficacy Results – Intent-to-treat mCSPC Population (TITAN) Endpoint ERLEADA
N=525Placebo
N=527- * Interim analysis is based on 50% of the number of events planned for the final analysis. Allocated alpha = 0.01.
- † NE=Not Estimable
- ‡ Hazard ratio is from stratified proportional hazards model. Hazard ratio <1 favors ERLEADA.
- § p-value is from the log-rank test stratified by Gleason score at diagnosis (≤7 vs. >7), Region (NA/EU vs. Other Countries) and Prior docetaxel use (Yes vs. No).
Overall Survival* Deaths (%) 83 (16%) 117 (22%) Median, months (95% CI)† NE (NE, NE) NE (NE, NE) Hazard ratio (95% CI)‡ 0.67 (0.51, 0.89) p-value§ 0.0053 Radiographic Progression-free Survival Disease progression or death (%) 134 (26%) 231 (44%) Median, months (95% CI)† NE (NE, NE) 22.1 (18, 33) Hazard ratio (95% CI)‡ 0.48 (0.39, 0.60) p-value§ <0.0001 Consistent improvement in rPFS was observed across the following patient subgroups: disease volume (high vs low), prior docetaxel use (yes or no), and Gleason score at diagnosis (≤7 vs. >7).
Consistent improvement in OS was observed across the following patient subgroups: disease volume (high vs low) and Gleason score at diagnosis (≤7 vs. >7).
Treatment with ERLEADA statistically significantly delayed the initiation of cytotoxic chemotherapy (HR = 0.39, 95% CI = 0.27, 0.56; p < 0.0001).
Figure 1: Kaplan-Meier Plot of Overall Survival (OS); Intent-to-treat mCSPC Population (TITAN)
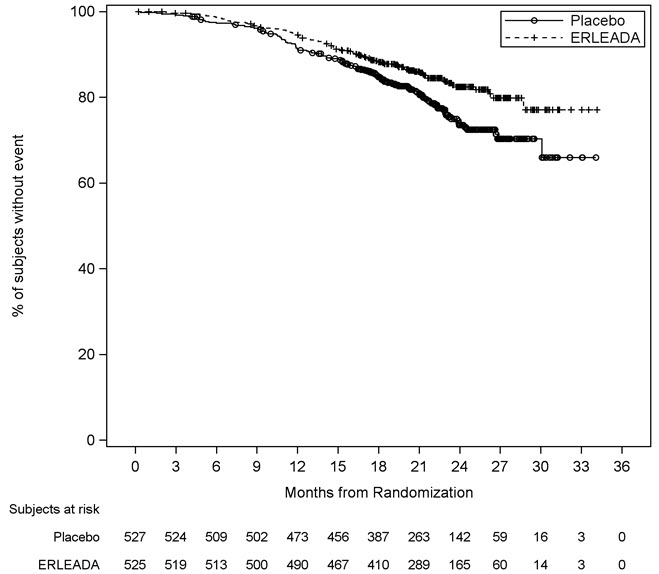
Figure 2: Kaplan-Meier Plot of Radiographic Progression-Free Survival (rPFS); Intent-to-treat mCSPC Population (TITAN)
SPARTAN (NCT01946204): Non-metastatic, Castration-resistant Prostate Cancer (nmCRPC)
SPARTAN was a multicenter, double-blind, randomized (2:1), placebo-controlled clinical trial in which 1207 patients with nmCRPC were randomized (2:1) to receive either ERLEADA orally at a dose of 240 mg once daily (N = 806) or placebo once daily (N = 401). All patients in the SPARTAN trial received a concomitant GnRH analog or had a bilateral orchiectomy. Patients were stratified by Prostate Specific Antigen (PSA) Doubling Time (PSADT), the use of bone-sparing agents, and locoregional disease. Patients were required to have a PSADT ≤ 10 months and confirmation of non-metastatic disease by blinded independent central review (BICR). PSA results were blinded and were not used for treatment discontinuation. Patients randomized to either arm discontinued treatment for radiographic disease progression confirmed by BICR, locoregional-only progression, initiation of new treatment, unacceptable toxicity, or withdrawal.
The following patient demographics and baseline disease characteristics were balanced between the treatment arms. The median age was 74 years (range 48–97) and 26% of patients were 80 years of age or older. The racial distribution was 66% Caucasian, 12% Asian, and 6% Black. Seventy-seven percent (77%) of patients in both treatment arms had prior surgery or radiotherapy of the prostate. A majority of patients had a Gleason score of 7 or higher (78%). Fifteen percent (15%) of patients had <2 cm pelvic lymph nodes at study entry. Seventy-three percent (73%) of patients received prior treatment with an anti-androgen; 69% of patients received bicalutamide and 10% of patients received flutamide. All patients had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 0 or 1 at study entry. Among the patients who discontinued study treatment (N = 279 for placebo and N = 314 for ERLEADA), a greater proportion (80%) of patients treated with placebo received subsequent therapy compared to patients treated with ERLEADA (56%). Locoregional-only progression occurred in 2% of patients overall.
The major efficacy outcome measure of the study was metastasis-free survival (MFS), defined as the time from randomization to the time of first evidence of BICR-confirmed distant metastasis, defined as new bone or soft tissue lesions or enlarged lymph nodes above the iliac bifurcation, or death due to any cause, whichever occurred first. Additional efficacy endpoints were time to metastasis (TTM), progression-free survival (PFS) which also includes locoregional progression, time to symptomatic progression, and overall survival (OS).
A statistically significant improvement in MFS was demonstrated in patients randomized to receive ERLEADA compared with patients randomized to receive placebo. Consistent results were observed across patient subgroups including PSADT (≤ 6 months or > 6 months), use of a prior bone-sparing agent (yes or no), and locoregional disease (N0 or N1). The major efficacy outcome was supported by statistically significant improvements in TTM, PFS, and time to symptomatic progression. Overall survival (OS) data were not mature at the time of final MFS analysis (24% of the required number of events). The efficacy results of MFS, TTM, and PFS from SPARTAN are summarized in Figure 3 and Table 6.
Figure 3: Kaplan-Meier Metastasis-Free Survival (MFS) Curve in SPARTAN (nmCRPC)
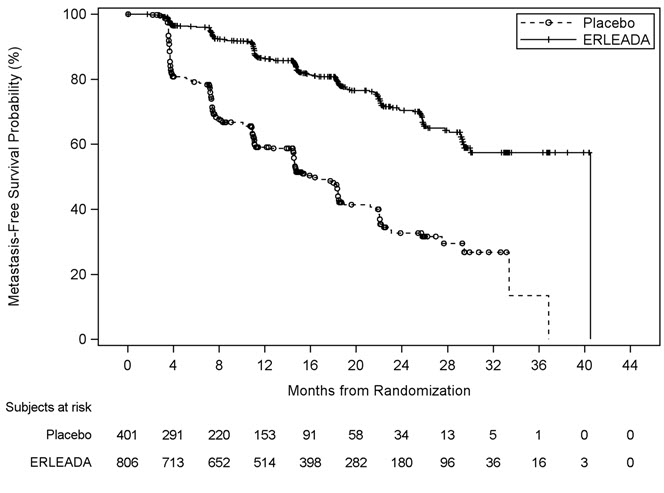
Table 6: BICR-assessed Efficacy Results (SPARTAN) Endpoint Number of Events (%) Median [Months (95% CI)] HR (95% CI)
p-value (log-rank test)*ERLEADA
(N=806)Placebo
(N=401)ERLEADA Placebo NE=Not Estimable - * All analyses stratified by PSA doubling time, bone-sparing agent use, and locoregional disease status.
Metastasis Free Survival 184 (23%) 194 (48%) 40.5
(NE, NE)16.2
(15, 18)0.28 (0.23, 0.35)
<0.0001Time to Metastasis 175 (22%) 191 (48%) 40.5
(NE, NE)16.6
(15, 18)0.27 (0.22, 0.34)
<0.0001Progression-Free Survival 200 (25%) 204 (51%) 40.5
(NE, NE)14.7
(14, 18)0.29 (0.24, 0.36)
<0.0001 - 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Ischemic Cardiovascular Events
- Inform patients that ERLEADA has been associated with ischemic cardiovascular events. Advise patients to seek immediate medical attention if any symptoms suggestive of a cardiovascular event occur [see Warnings and Precautions (5.1)].
Falls and Fractures
- Inform patients that ERLEADA is associated with an increased incidence of falls and fractures [see Warnings and Precautions (5.2, 5.3)].
Seizures
- Inform patients that ERLEADA has been associated with an increased risk of seizure. Discuss conditions that may predispose to seizures and medications that may lower the seizure threshold. Advise patients of the risk of engaging in any activity where sudden loss of consciousness could cause serious harm to themselves or others. Inform patients to contact their healthcare provider right away if they experience a seizure [see Warnings and Precautions (5.4)].
Rash
- Inform patients that ERLEADA is associated with rashes and to inform their healthcare provider if they develop a rash [see Adverse Reactions (6.1)].
Dosage and Administration
- Inform patients receiving concomitant gonadotropin-releasing hormone (GnRH) analog therapy that they need to maintain this treatment during the course of treatment with ERLEADA.
- Instruct patients to take their dose at the same time each day (once daily). ERLEADA can be taken with or without food. Each tablet should be swallowed whole.
- Inform patients that in the event of a missed daily dose of ERLEADA, they should take their normal dose as soon as possible on the same day with a return to the normal schedule on the following day. The patient should not take extra tablets to make up the missed dose [see Dosage and Administration (2.1)].
Embryo-Fetal Toxicity
- Inform patients that ERLEADA can be harmful to a developing fetus. Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of ERLEADA. Advise male patients to use a condom if having sex with a pregnant woman [see Warnings and Precautions (5.5)].
Infertility
- Advise male patients that ERLEADA may impair fertility and not to donate sperm during therapy and for 3 months following the last dose of ERLEADA [see Use in Specific Populations (8.3)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 09/2019 PATIENT INFORMATION
ERLEADA® (er lee'dah)
(apalutamide)
TabletsWhat is ERLEADA?
ERLEADA is a prescription medicine used for the treatment of prostate cancer:- that has spread to other parts of the body and still responds to a medical or surgical treatment that lowers testosterone, OR
- that has not spread to other parts of the body and no longer responds to a medical or surgical treatment that lowers testosterone.
It is not known if ERLEADA is safe and effective in children.Before taking ERLEADA, tell your healthcare provider about all your medical conditions, including if you: - have a history of heart disease
- have high blood pressure
- have diabetes
- have abnormal amounts of fat or cholesterol in your blood (dyslipidemia)
- have a history of seizures, brain injury, stroke, or brain tumors
- are pregnant or plan to become pregnant. ERLEADA can cause harm to your unborn baby and loss of pregnancy (miscarriage).
- have a partner who is pregnant or may become pregnant.
- Males who have female partners who are able to become pregnant should use effective birth control (contraception) during treatment and for 3 months after the last dose of ERLEADA.
- Males should use a condom during sex with a pregnant female.
- are breastfeeding or plan to breastfeed. It is not known if ERLEADA passes into breast milk.
You should not start or stop any medicine before you talk with the healthcare provider that prescribed ERLEADA.
Know the medicines you take. Keep a list of them with you to show to your healthcare provider and pharmacist when you get a new medicine.How should I take ERLEADA? - Take ERLEADA exactly as your healthcare provider tells you.
- Your healthcare provider may change your dose if needed.
- Do not stop taking your prescribed dose of ERLEADA without talking with your healthcare provider first.
- Take your prescribed dose of ERLEADA 1 time a day, at the same time each day.
- Take ERLEADA with or without food.
- Swallow ERLEADA tablets whole.
- If you miss a dose of ERLEADA, take your normal dose as soon as possible on the same day. Return to your normal schedule on the following day. You should not take extra tablets to make up the missed dose.
- You should start or continue a gonadotropin-releasing hormone (GnRH) analog therapy during your treatment with ERLEADA unless you have had a surgery to lower the amount of testosterone in your body (surgical castration).
- If you take too much ERLEADA, call your healthcare provider or go to the nearest hospital emergency room.
What are the possible side effects of ERLEADA?
ERLEADA may cause serious side effects including:- Heart Disease. Blockage of the arteries in the heart that can lead to death has happened in some people during treatment with ERLEADA. Your healthcare provider will monitor you for signs and symptoms of heart problems during your treatment with ERLEADA. Call your healthcare provider or go to the nearest emergency room right away if you get chest pain or discomfort at rest or with activity, or shortness of breath during your treatment with ERLEADA.
- Fractures and falls. ERLEADA treatment can cause bones and muscles to weaken and may increase your risk for falls and fractures. Falls and fractures have happened in people during treatment with ERLEADA. Your healthcare provider will monitor your risks for falls and fractures during treatment with ERLEADA.
- Seizure. Treatment with ERLEADA may increase your risk of having a seizure. You should avoid activities where a sudden loss of consciousness could cause serious harm to yourself or others. Tell your healthcare provider right away if you have a loss of consciousness or seizure. Your healthcare provider will stop ERLEADA if you have a seizure during treatment.
- feeling very tired
- joint pain
- rash. Tell your healthcare provider if you get a rash.
- decreased appetite
- fall
- weight loss
- hypertension
- hot flash
- diarrhea
- fracture
ERLEADA may cause fertility problems in males, which may affect the ability to father children. Talk to your healthcare provider if you have concerns about fertility. Do not donate sperm during treatment with ERLEADA and for 3 months after the last dose of ERLEADA.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of ERLEADA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store ERLEADA? - Store ERLEADA at room temperature between 68°F to 77°F (20°C to 25°C).
- Store ERLEADA in the original package.
- The bottle of ERLEADA contains a desiccant packet to help keep your medicine dry (protect it from moisture). Do not throw away (discard) the desiccant.
- Protect ERLEADA from light and moisture.
General information about the safe and effective use of ERLEADA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ERLEADA for a condition for which it was not prescribed. Do not give ERLEADA to other people, even if they have the same symptoms that you have. It may harm them. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about ERLEADA that is written for health professionals.What are the ingredients in ERLEADA?
Active ingredient: apalutamide
Inactive ingredients: colloidal anhydrous silica, croscarmellose sodium, hydroxypropyl methylcellulose-acetate succinate, magnesium stearate, microcrystalline cellulose, and silicified microcrystalline cellulose. The film-coating contains iron oxide black, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
Manufactured by: Janssen Ortho LLC, Gurabo, PR 00778
Manufactured for: Janssen Products, LP, Horsham, PA 19044
© 2019 Janssen Pharmaceutical Companies
For more information, call Janssen Products, LP at 1-800-526-7736 (1-800-JANSSEN) or go to www.erleada.com. - PRINCIPAL DISPLAY PANEL - 60 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
ERLEADA
apalutamide tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59676-600 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Apalutamide (UNII: 4T36H88UA7) (Apalutamide - UNII:4T36H88UA7) Apalutamide 60 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) Croscarmellose sodium (UNII: M28OL1HH48) HYPROMELLOSE ACETATE SUCCINATE 16070722 (3 MM2/S) (UNII: 24P2YXD2PW) Magnesium stearate (UNII: 70097M6I30) Microcrystalline cellulose (UNII: OP1R32D61U) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) Talc (UNII: 7SEV7J4R1U) Titanium dioxide (UNII: 15FIX9V2JP) Product Characteristics Color GREEN (slightly yellowish to greyish green) Score no score Shape OVAL (Oblong) Size 17mm Flavor Imprint Code AR;60 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59676-600-12 120 in 1 BOTTLE; Type 0: Not a Combination Product 02/14/2018 2 NDC: 59676-600-99 120 in 1 BOTTLE; Type 0: Not a Combination Product 02/14/2018 3 NDC: 59676-600-56 2 in 1 CARTON 04/01/2019 3 28 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210951 02/14/2018 Labeler - Janssen Products, LP (804684207) Establishment Name Address ID/FEI Business Operations Janssen Pharmaceuticals, Inc. 080236951 API MANUFACTURE(59676-600) Establishment Name Address ID/FEI Business Operations Johnson & Johnson Private Limited 677603030 ANALYSIS(59676-600) Establishment Name Address ID/FEI Business Operations Janssen Pharmaceutica NV 374747970 API MANUFACTURE(59676-600) Establishment Name Address ID/FEI Business Operations Janssen Ortho LLC 805887986 MANUFACTURE(59676-600) Establishment Name Address ID/FEI Business Operations Janssen Cilag SpA 542797928 PACK(59676-600)

Trademark Results [ERLEADA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ERLEADA 86967766 5492719 Live/Registered |
JOHNSON & JOHNSON 2016-04-07 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.