NERLYNX- neratinib tablet
Nerlynx by
Drug Labeling and Warnings
Nerlynx by is a Prescription medication manufactured, distributed, or labeled by Puma Biotechnology, Inc., AndersonBrecon Inc., Excella, Esteve. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NERLYNX safely and effectively. See full prescribing information for NERLYNX.
NERLYNX ® (neratinib) tablets, for oral use
Initial U.S. Approval: 2017RECENT MAJOR CHANGES
Indications and Usage, Advanced or Metastatic Breast Cancer ( 1.2) 2/2020 Dosage and Administration, Antidiarrheal Prophylaxis ( 2.1) 2/2020 Dosage and Administration, Recommended Dose and Schedule ( 2.2) 2/2020 Dosage and Administration, Dose Modifications ( 2.3) 2/2020 Warnings and Precautions, Diarrhea ( 5.1) 2/2020 Warnings and Precautions, Hepatotoxicity ( 5.2) 2/2020 INDICATIONS AND USAGE
NERLYNX is a kinase inhibitor indicated:
- As a single agent, for the extended adjuvant treatment of adult patients with early stage HER2-positive breast cancer, to follow adjuvant trastuzumab-based therapy. ( 1.1)
- In combination with capecitabine, for the treatment of adult patients with advanced or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2 based regimens in the metastatic setting. ( 1.2)
DOSAGE AND ADMINISTRATION
- Antidiarrheal prophylaxis: Initiate loperamide with the first dose of NERLYNX and continue during the first 56 days of treatment. After day 56, use loperamide to maintain 1-2 bowel movements per day. ( 2.1)
- Extended Adjuvant Treatment of Early Stage Breast Cancer: 240 mg (6 tablets) given orally once daily, with food, continuously until disease recurrence for up to one year. ( 2.2)
- Advanced or metastatic breast cancer: 240 mg (6 tablets) given orally once daily with food on Days 1-21 of a 21-day cycle plus capecitabine (750 mg/m 2 given orally twice daily) on Days 1-14 of a 21-day cycle until disease progression or unacceptable toxicities. ( 2.2)
- Dose interruptions and/or dose reductions are recommended based on individual safety and tolerability. ( 2.3)
- Hepatic impairment: Reduce starting dose to 80 mg in patients with severe hepatic impairment. ( 2.3)
DOSAGE FORMS AND STRENGTHS
Tablets: 40 mg. ( 3)
CONTRAINDICATIONS
None. ( 4)
WARNINGS AND PRECAUTIONS
- Diarrhea: Aggressively manage diarrhea. If diarrhea occurs despite recommended prophylaxis, treat with additional antidiarrheals, fluids, and electrolytes as clinically indicated. Withhold NERLYNX in patients experiencing severe and/or persistent diarrhea. Permanently discontinue NERLYNX in patients experiencing Grade 4 diarrhea or Grade ≥2 diarrhea that occurs after maximal dose reduction. ( 2.3, 5.1)
- Hepatotoxicity: Monitor liver function tests monthly for the first 3 months of treatment, then every 3 months while on treatment and as clinically indicated. Withhold NERLYNX in patients experiencing Grade 3 liver abnormalities and permanently discontinue NERLYNX in patients experiencing Grade 4 liver abnormalities. ( 2.3, 5.2)
- Embryo-Fetal Toxicity: NERLYNX can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. ( 5.3, 8.1, 8.3)
ADVERSE REACTIONS
The most common adverse reactions (reported in ≥5% of patients) were:
- NERLYNX as a single agent: diarrhea, nausea, abdominal pain, fatigue, vomiting, rash, stomatitis, decreased appetite, muscle spasms, dyspepsia, AST or ALT increased, nail disorder, dry skin, abdominal distention, epistaxis, weight decreased, and urinary tract infection. ( 6)
- NERLYNX in combination with capecitabine: diarrhea, nausea, vomiting, decreased appetite, constipation, fatigue/asthenia, weight decreased, dizziness, back pain, arthralgia, urinary tract infection, upper respiratory tract infection, abdominal distention, renal impairment, and muscle spasms. ( 6)
To report SUSPECTED ADVERSE REACTIONS, contact Puma Biotechnology, Inc. at 1-844-NERLYNX (1-844-637-5969) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Gastric acid reducing agents: Avoid concomitant use with proton pump inhibitors. When patients require gastric acid reducing agents, use an H 2-receptor antagonist or antacid. Separate NERLYNX by at least 3 hours with antacids. Separate NERLYNX by at least 2 hours before or 10 hours after H 2-receptor antagonists. ( 2.3, 7.1)
- Strong CYP3A4 inhibitors: Avoid concomitant use. ( 7.1)
- Moderate CYP3A4 and P-gp dual inhibitors: Avoid concomitant use ( 7.1)
- Strong or moderate CYP3A4 inducers: Avoid concomitant use. ( 7.1)
- P-glycoprotein (P-gp) substrates: Monitor for adverse reactions of narrow therapeutic agents that are P-gp substrates when used concomitantly with NERLYNX. ( 7.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Extended Adjuvant Treatment of Early-Stage Breast Cancer
1.2 Advanced or Metastatic Breast Cancer
2 DOSAGE AND ADMINISTRATION
2.1 Antidiarrheal Prophylaxis
2.2 Recommended Dose and Schedule
2.3 Dose Modifications
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Diarrhea
5.2 Hepatotoxicity
5.3 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on NERLYNX
7.2 Effect of NERLYNX on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Extended Adjuvant Treatment of Early Stage Breast Cancer
14.2 Advanced or Metastatic Breast Cancer
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Extended Adjuvant Treatment of Early-Stage Breast Cancer
NERLYNX as a single agent is indicated for the extended adjuvant treatment of adult patients with early-stage human epidermal growth factor receptor 2 (HER2)-positive breast cancer, to follow adjuvant trastuzumab based therapy [ see Clinical Studies ( 14.1) ].
1.2 Advanced or Metastatic Breast Cancer
NERLYNX in combination with capecitabine is indicated for the treatment of adult patients with advanced or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2 based regimens in the metastatic setting [ see Clinical Studies ( 14.2) ].
-
2 DOSAGE AND ADMINISTRATION
2.1 Antidiarrheal Prophylaxis
Administer antidiarrheal prophylaxis during the first 2 cycles of treatment and initiate with the first dose of NERLYNX [ see Warnings and Precautions (5.1) and Adverse Reactions ( 6.1) ]. Instruct patients to take loperamide as directed in Table 1. Titrate loperamide to 1-2 bowel movements per day.
Table 1: Loperamide Prophylaxis Time on Nerlynx Loperamide Dose and Frequency Weeks 1-2 (days 1-14) 4 mg three times daily Weeks 3-8 (days 15-56) 4 mg twice daily Weeks 9-52 (days 57-365) 4 mg as needed, not to exceed 16 mg per day; titrate dosing to achieve 1-2 bowel movements per day If diarrhea occurs despite prophylaxis, treat with additional antidiarrheals, fluids and electrolytes as clinically indicated. NERLYNX dose interruptions and dose reductions may also be required to manage diarrhea [ see Dosage and Administration ( 2.3) ].
2.2 Recommended Dose and Schedule
Extended Adjuvant Treatment of Early Stage Breast Cancer
The recommended dose of NERLYNX is 240 mg (six tablets) given orally once daily, with food, continuously until disease recurrence or for up to one year.
Advanced or Metastatic Breast Cancer
The recommended dose of NERLYNX is 240 mg (six tablets) given orally once daily with food on Days 1-21 of a 21-day cycle plus capecitabine (750 mg/m 2 given orally twice daily) on Days 1-14 of a 21-day cycle until disease progression or unacceptable toxicities.
Instruct patients to take NERLYNX at approximately the same time every day. NERLYNX tablets should be swallowed whole (tablets should not be chewed, crushed, or split prior to swallowing).
If a patient misses a dose, do not replace missed dose, and instruct the patient to resume NERLYNX with the next scheduled daily dose.
2.3 Dose Modifications
Dose Modifications for Adverse Reactions
NERLYNX dose modification is recommended based on individual safety and tolerability. Management of some adverse reactions may require dose interruption and/or dose reduction as shown in Table 2 to Table 7. Discontinue NERLYNX for patients with adverse reactions that fail to recover to Grade 0-1 or baseline, with toxicities that result in a treatment delay >3 weeks, or if unable to tolerate 120 mg daily. Additional clinical situations may result in dose adjustments as clinically indicated (e.g. intolerable toxicities, persistent Grade 2 adverse reactions, etc.).
When NERLYNX is used in combination with capecitabine, refer to the capecitabine prescribing information for dose modifications of capecitabine.
Table 2: NERLYNX Monotherapy Dose Modifications for Adverse Reactions Dose Level NERLYNX Dose Recommended starting dose 240 mg daily (six 40 mg tablets) First dose reduction 200 mg daily (five 40 mg tablets) Second dose reduction 160 mg daily (four 40 mg tablets) Third dose reduction 120 mg daily (three 40 mg tablets) Table 3: NERLYNX in Combination with Capecitabine Dose Modifications for Adverse Reactions Dose Level NERLYNX Dose Recommended starting dose 240 mg daily (six 40 mg tablets) First dose reduction 160 mg daily (four 40 mg tablets) Second dose reduction 120 mg daily (three 40 mg tablets) Table 4: NERLYNX Dose Modifications and Management – General Toxicities* * Refer to Table 5, Table 6, and Table 7 below for management of diarrhea and hepatotoxicity
† Per CTCAE v4.0
Severity of Toxicity† Action Grade 3 Hold NERLYNX until recovery to Grade ≤1 or baseline within 3 weeks of stopping treatment. Then resume NERLYNX at the next lower dose level. Grade 4 Discontinue NERLYNX permanently. Dose Modifications for Diarrhea
Guidelines for adjusting doses of NERLYNX in the setting of diarrhea are shown in Table 5 and Table 6. Diarrhea management may require use of antidiarrheal medications, dietary changes, replacement of fluids and electrolytes and appropriate dose modifications of NERLYNX as clinically indicated.
Table 5: Dose Modifications of Nerlynx Monotherapy for Diarrhea Severity of Diarrhea* Action * Per CTCAE v4.0
† Complicated features include dehydration, fever, hypotension, renal failure, or Grade 3 or 4 neutropenia
‡ Despite being treated with optimal medical therapy
- Grade 1 diarrhea [increase of <4 stools per day over baseline]
- Grade 2 diarrhea [increase of 4-6 stools per day over baseline] lasting ≤5 days
- Grade 3 diarrhea [increase of ≥7 stools per day over baseline; incontinence; hospitalization indicated; limiting self-care activities of daily living] lasting ≤2 days
- Adjust antidiarrheal treatment
- Diet modifications
- Fluid intake of ~2 L should be maintained to avoid dehydration
- Once event resolves to ≤Grade 1 or baseline, start loperamide 4 mg with each subsequent NERLYNX administration
- Any grade with complicated features †
- Grade 2 diarrhea lasting 5 days or longer ‡
- Grade 3 diarrhea lasting longer than 2 days ‡
- Interrupt NERLYNX treatment
- Diet modifications
- Fluid intake of ~2 L should be maintained to avoid dehydration
- If diarrhea resolves to Grade 0-1 in one week or less, then resume NERLYNX treatment at the same dose.
- If diarrhea resolves to Grade 0-1 in longer than one week, then resume NERLYNX treatment at reduced dose (see Table 2)
- Once event resolves to ≤Grade 1 or baseline, start loperamide 4 mg with each subsequent NERLYNX administration
- Grade 4 diarrhea [life-threatening consequences; urgent intervention indicated]
- Permanently discontinue NERLYNX treatment
- Diarrhea recurs to Grade 2 or higher at 120 mg per day
- Permanently discontinue NERLYNX treatment
Table 6: Dose Modifications of Nerlynx and Capecitabine for Diarrhea Severity of Diarrhea* Actions Abbreviations: L: liter
* NCI CTCAE v.4.0
a Since capecitabine is provided as 150 mg or 500 mg tablets, it is recommended that the capecitabine dose reduction(s) is rounded down to the nearest 500 mg or multiple of 150 mg for the twice daily dose. If the patient's body surface area is >2.0, the standard of care for the study center can be utilized for capecitabine mg/m 2 dosing.
- Grade 1 Diarrhea [Increase of <4 stools per day over baseline]
- Grade 2 Diarrhea [Increase of 4-6 stools per day over baseline] lasting ≤5 days
- Grade 3 Diarrhea: [Increase of ≥7 stools per day over baseline; incontinence; hospitalization indicated; limiting self-care and activities of daily living] lasting ≤2 days
- Adjust antidiarrheal treatment
- Continue NERLYNX and capecitabine at full doses
- Diet modifications
- Fluid intake of ~2 L/day should be maintained to avoid dehydration
- Once the event resolved to Grade ≤1 or baseline, start loperamide 4 mg with each subsequent NERLYNX administration
- Persisting and intolerable Grade 2 Diarrhea: lasting >5 days
- Grade 3 Diarrhea lasting >2 days
- Grade 4 diarrhea [Life-threatening consequences; urgent intervention indicated]
- Adjust antidiarrheal treatment
- Hold NERLYNX and capecitabine until recovery to Grade ≤1 or baseline
- Diet modifications
- Fluid intake of ~2 L/day should be maintained intravenously, if needed
- If recovery occurs:
- ≤1 week after withholding treatment, resume same doses of NERLYNX and capecitabine
- Within 1-3 weeks after withholding treatment, reduce NERLYNX dose to 160 mg and maintain the same dose of capecitabine
- If event occurs a second time and the NERLYNX dose has not already been decreased, reduce NERLYNX dose to 160 mg (maintain the same dose of capecitabine). If NERLYNX dose has already been reduced, then reduce the dose of capecitabine to 550 mg/m 2 given twice daily a (maintain the same dose of NERLYNX).
- If subsequent events occur, reduce the dose of NERLYNX or capecitabine to the next lower dose level in an alternate fashion (i.e., reduce capecitabine to 375 mg/m 2 given twice daily a if NERLYNX was previously reduced, or reduce NERLYNX to 120 mg if capecitabine was previously reduced).
- Once the event resolved to Grade ≤1 or baseline, start loperamide 4 mg with each subsequent NERLYNX administration.
Dose Modifications for Hepatic Impairment
Reduce the NERLYNX starting dose to 80 mg in patients with severe hepatic impairment (Child Pugh C). No dose modifications are recommended for patients with mild to moderate hepatic impairment (Child Pugh A or B) [ see Use in Specific Populations ( 8.6) and Clinical Pharmacology ( 12.3) ].
Dose Modifications for Hepatotoxicity
Guidelines for dose adjustment of NERLYNX in the event of liver toxicity are shown in Table 7. Patients who experience ≥Grade 3 diarrhea requiring IV fluid treatment or any signs or symptoms of hepatotoxicity, such as worsening of fatigue, nausea, vomiting, right upper quadrant pain or tenderness, fever, rash, or eosinophilia, should be evaluated for changes in liver function tests. Fractionated bilirubin and prothrombin time should also be collected during hepatotoxicity evaluation [ see Warnings and Precautions ( 5.2) ].
Table 7: Dose Modifications for Hepatotoxicity Severity of Hepatotoxicity* Action ALT=Alanine Aminotransferase; AST=Aspartate Aminotransferase; ULN=Upper Limit Normal
* Per CTCAE v4.0
- Grade 3 ALT or AST (>5-20x ULN)
OR - Grade 3 bilirubin (>3-10x ULN)
- Hold NERLYNX until recovery to ≤Grade 1
- Evaluate alternative causes
- Resume NERLYNX at the next lower dose level if recovery to ≤Grade 1 occurs within 3 weeks. If Grade 3 ALT or bilirubin occurs again despite one dose reduction, permanently discontinue NERLYNX
- Grade 4 ALT or AST (>20x ULN)
OR - Grade 4 bilirubin (>10x ULN)
- Permanently discontinue NERLYNX
- Evaluate alternative causes
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Diarrhea
Severe diarrhea and sequelae, such as dehydration, hypotension, and renal failure occurred during treatment with NERLYNX. Diarrhea was reported in 95% of NERLYNX-treated patients in ExteNET, a randomized placebo-controlled trial in the extended adjuvant setting who were not required to receive antidiarrheal prophylaxis. In the NERLYNX arm, Grade 3 diarrhea occurred in 40% and Grade 4 diarrhea occurred in 0.1% of patients. The majority of patients (93%) had diarrhea in the first month of treatment, the median time to first onset of Grade ≥3 diarrhea was 8 days (range, 1-350), and the median cumulative duration of Grade ≥3 diarrhea was 5 days (range, 1-139) [ see Adverse Reactions ( 6.1) ].
Diarrhea was reported in 83% of NERLYNX plus capecitabine treated patients in NALA, a randomized placebo-controlled trial in the metastatic breast cancer setting who were required to receive anti-diarrheal prophylaxis in the first cycle. The majority of patients (70%) had diarrhea in the first cycle of treatment, the median time to first onset of Grade ≥3 diarrhea was 11 days (range, 2-728) and the median cumulative duration of Grade ≥3 diarrhea was 3 days (range, 1-21). In the NERLYNX plus capecitabine arm, Grade 3 diarrhea occurred in 24% of patients [ see Adverse Reactions (6.1)].
Antidiarrheal prophylaxis has been shown to lower the incidence and severity of diarrhea. Instruct patients to initiate antidiarrheal prophylaxis with loperamide along with the first dose of NERLYNX and continue during the first 56 days of treatment; after day 56 titrate dose to achieve 1-2 bowel movements per day and not to exceed 16 mg loperamide per day [ see Dosage and Administration (2.1)]. Consider adding other agents to loperamide as clinically indicated [ see Adverse Reactions ( 6.1) ].
Monitor patients for diarrhea and treat with additional antidiarrheals as needed. When severe diarrhea with dehydration occurs, administer fluid and electrolytes as needed, interrupt NERLYNX, and reduce subsequent doses [ see Dosage and Administration (2.3)]. Perform stool cultures as clinically indicated to exclude infectious causes of Grade 3 or 4 diarrhea or diarrhea of any grade with complicating features (dehydration, fever, neutropenia).
5.2 Hepatotoxicity
NERLYNX has been associated with hepatotoxicity characterized by increased liver enzymes. In ExteNET, 10% of patients experienced an alanine aminotransferase (ALT) increase ≥2x ULN, 5% of patients experienced an aspartate aminotransferase (AST) increase ≥2x ULN, and 1.7% of patients experienced an AST or ALT elevation >5 x ULN (≥Grade 3). Hepatotoxicity or increases in liver transaminases led to drug discontinuation in 1.7% of NERLYNX-treated patients.
In the NALA study, in NERLYNX and capecitabine-treated patients, 7% experienced an ALT or AST >3x ULN, 2% experienced ALT or AST >5x ULN, 7% experienced a bilirubin >1.5x ULN, and 1.3% experienced a bilirubin >3x ULN. Hepatotoxicity or increases in liver transaminases led to drug discontinuation in 0.3% of NERLYNX and capecitabine-treated patients.
Total bilirubin, AST, ALT, and alkaline phosphatase should be measured prior to starting treatment with NERLYNX monthly for the first 3 months of treatment, then every 3 months while on treatment and as clinically indicated. These tests should also be performed in patients experiencing Grade 3 diarrhea or any signs or symptoms of hepatotoxicity, such as worsening of fatigue, nausea, vomiting, right upper quadrant tenderness, fever, rash, or eosinophilia [ see Dosage and Administration ( 2.3) and Adverse Reactions ( 6.1) ].
5.3 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, NERLYNX can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of neratinib to pregnant rabbits during organogenesis caused abortions, embryo-fetal death, and fetal abnormalities in rabbits at maternal AUCs approximately 0.2 times the AUC in patients receiving the recommended dose. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for at least 1 month after the last dose. [ see Use in Specific Populations ( 8.1, 8.3) and Clinical Pharmacology ( 12.1) ].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Diarrhea [ see Warnings and Precautions ( 5.1) ]
- Hepatotoxicity [ see Warnings and Precautions ( 5.2) ]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
ExteNET
The data described below reflect the safety data of NERLYNX as a single agent in ExteNET a multicenter, randomized, double-blind, placebo-controlled study of NERLYNX within 2 years after completion of adjuvant treatment with trastuzumab-based therapy in women with HER2-positive early-stage breast cancer. Patients who received NERLYNX in this trial were not required to receive any prophylaxis with antidiarrheal agents to prevent the NERLYNX-related diarrhea. Patients were treated with 240 mg of NERLYNX given orally once daily with food, continuously until disease recurrence or for up to one year. The median duration of treatment was 11.6 months in the NERLYNX arm and 11.8 months in the placebo arm. The median age was 52 years (60% were ≥50 years old, 12% were ≥65 years old); 81% were Caucasian, 3% Black or African American, 14% Asian, and 3% other. A total of 1408 patients were treated with NERLYNX.
NERLYNX dose reduction due to an adverse reaction of any grade occurred in 31% of patients receiving NERLYNX compared to 2.6% of patients receiving placebo. Permanent discontinuation due to any adverse reaction was reported in 28% of NERLYNX-treated patients. The most common adverse reaction leading to discontinuation was diarrhea, accounting for 17% of NERLYNX-treated patients.
The most common adverse reactions (≥5%) were diarrhea, nausea, abdominal pain, fatigue, vomiting, rash, stomatitis, decreased appetite, muscle spasms, dyspepsia, AST or ALT increased, nail disorder, dry skin, abdominal distention, epistaxis, weight decreased and urinary tract infection. The most frequently reported Grade 3 or 4 adverse reactions were diarrhea, vomiting, nausea, and abdominal pain.
Serious adverse reactions in the NERLYNX arm included diarrhea (1.6%), vomiting (0.9%), dehydration (0.6%), cellulitis (0.4%), renal failure (0.4%), erysipelas (0.4%), ALT (0.3%), AST increased (0.3%), nausea (0.3%), fatigue (0.2%), and abdominal pain (0.2%).
Table 8 summarizes the adverse reactions in ExteNET.
Table 8: Adverse Reactions Reported in ≥2% of NERLYNX-Treated Patients in ExteNET System Organ Class
(Preferred Term)NERLYNXn=1408 Placebo
n=1408All Grades
(%)Grade 3 (%) Grade 4 (%) All Grades (%) Grade 3 (%) Grade 4 (%) * Includes abdominal pain, abdominal pain upper, and abdominal pain lower
† Includes stomatitis, aphthous stomatitis, mouth ulceration, oral mucosal blistering, mucosal inflammation, oropharyngeal pain, oral pain, glossodynia, glossitis, and cheilitis
‡ Includes rash, rash erythematous, rash follicular, rash generalized, rash pruritic, rash pustular, rash maculo-papular, rash papular, dermatitis, dermatitis acneiform, and toxic skin eruption
§ Includes nail disorder, paronychia, onychoclasis, nail discoloration, nail toxicity, nail growth abnormal, and nail dystrophy
Gastrointestinal Disorders Diarrhea 95 40 0.1 35 2 0 Nausea 43 2 0 22 0.1 0 Abdominal pain * 36 2 0 15 0.4 0 Vomiting 26 3 0 8 0.4 0 Stomatitis † 14 0.6 0 6 0.1 0 Dyspepsia 10 0.4 0 4 0 0 Abdominal distension 5 0.3 0 3 0 0 Dry mouth 3 0.1 0 2 0 0 General Disorders and Administration Site Conditions Fatigue 27 2 0 20 0.4 0 Hepatobiliary Disorders Alanine aminotransferase increased 9 1 0.2 3 0.2 0 Aspartate aminotransferase increased 7 0.5 0.2 3 0.3 0 Infections and Infestations Urinary tract infection 5 0.1 0 2 0 0 Investigations Weight decreased 5 0.1 0 0.5 0 0 Metabolism and Nutrition Disorders Decreased appetite 12 0.2 0 3 0 0 Dehydration 4 0.9 0.1 0.4 0.1 0 Musculoskeletal and Connective Tissue Disorders Muscle spasms 11 0.1 0 3 0.1 0 Respiratory, Thoracic and Mediastinal Disorders Epistaxis 5 0 0 1 0.1 0 Skin and Subcutaneous Tissue Disorders Rash ‡ 18 0.6 0 9 0 0 Dry skin 6 0 0 2 0 0 Nail Disorder § 8 0.3 0 2 0 0 Skin fissures 2 0.1 0 0.1 0 0 NALA
The data described below reflect the safety data of NERLYNX plus capecitabine in NALA, a randomized, multicenter, multinational, open-label, active-controlled study of HER2+ metastatic breast cancer in patients, with or without brain metastases, who have received two or more prior anti HER2-based regimens in the metastatic setting.
Patients were treated with NERLNX 240 mg orally once daily Days 1-21 of a 21-day cycle in combination with capecitabine (750 mg/m 2 given orally twice daily) Days 1-14 of a 21-day cycle or lapatinib 1250 mg orally once daily Days 1-21 of a 21-day cycle in combination with capecitabine (1000 mg/m 2 given orally twice daily) Days 1-14 of a 21-day cycle until disease progression. The median duration of treatment was 5.7 months in the NERLYNX plus capecitabine arm and 4.4 months in the lapatinib plus capecitabine arm.
NERLYNX dose reduction due to an adverse reaction of any grade occurred in 10% of patients receiving NERLYNX plus capecitabine. Permanent discontinuation due to any adverse reaction was reported in 14% of NERLYNX plus capecitabine treated patients. The most common adverse reactions leading to discontinuation were vomiting (3.6%), diarrhea (2.6%), nausea (2.6%), and palmar-plantar erythrodysaesthesia syndrome (2.3%) of NERLYNX plus capecitabine -treated patients.
The most common adverse reactions of any grade (≥5%) in the NERLYNX plus capecitabine arm were diarrhea, nausea, vomiting, decreased appetite, constipation, fatigue/asthenia, weight decreased, dizziness, back pain, arthralgia, urinary tract infection, upper respiratory tract infection, abdominal distention, renal impairment, and muscle spasms. The most frequently reported Grade 3 or 4 adverse reactions were diarrhea, nausea, vomiting, fatigue, and decreased appetite.
Serious adverse reactions ≥2% in the NERLYNX plus capecitabine arm included diarrhea (7%), vomiting (3%), nausea (2.3%), and acute kidney injury (2.3%).
Table 9 summarizes the adverse reactions in NALA.
Table 9: Adverse Reactions Reported in ≥2% of NERLYNX-Treated Patients in Combination with Capecitabine in NALA System Organ Class
(Preferred Term)NERLYNX plus capecitabinen=303 Lapatinib plus capecitabine
n=311All Grades
(%)Grade 3 (%) Grade 4 (%) All Grades (%) Grade 3 (%) Grade 4 (%) * Renal impairment includes acute kidney injury, blood creatinine increased, renal failure, and renal impairment
Gastrointestinal Disorders Diarrhea 83 25 0 66 13 0 Nausea 53 4.3 0 42 2.9 0 Vomiting 46 4 0 31 1.9 0 Constipation 31 1 0 13 0 0 Abdominal distension 8 0.3 0 3.2 0.6 0 General Disorders and Administration Site Conditions Fatigue/asthenia 45 6 0 40 4.5 0 Malaise 4.3 0 0 2.3 0.3 0 Influenza like illness 4 0 0 1.3 0 0 Infections and Infestations Urinary tract infection 9 0.7 0 4.2 0.6 0 Upper respiratory tract infection 8 0.3 0 4.5 0.3 0 Investigations Weight decreased 20 0.3 0 13 0.6 0 Metabolism and Nutrition Disorders Decreased appetite 35 2.6 0 22 2.3 0 Musculoskeletal and Connective Tissue Disorders Back Pain 10 0.3 0 8 0.3 0 Arthralgia 10 0 0 6 1 0 Muscle spasms 5 0 0 1.9 0 0 Nervous System Disorder Dizziness 14 0.3 0 10 0.6 0 Renal and urinary disorders Renal impairment* 7 2 0.3 1 0 0.3 Dysuria 4.6 0 0 1.9 0 0 Investigations Weight decreased 20 0.3 0 13 0.6 0 CONTROL
The CONTROL (NCT02400476) study was a multicenter, open-label, multi-cohort trial evaluating patients with early stage HER2-positive breast cancer treated with neratinib 240 mg daily for up to one year receiving loperamide prophylaxis with and without an additional anti-diarrheal treatment. All patients received loperamide 4 mg loading dose, followed by 4 mg three times a day from days 1-14, followed by 4 mg twice a day on days 15-56, followed by loperamide as needed through 1 year of treatment with neratinib [ see Dosage and Administration ( 2.1) ]. One cohort of patients received budesonide 9 mg once daily on cycle 1 days 1-28, in addition to loperamide. At the interim analysis, the incidence of all grade diarrhea for patients receiving loperamide alone (n=109) was 78% compared to 86% of patients who received budesonide and loperamide (n=64). The incidence of Grade 2 diarrhea was 25% compared to 33%, respectively. The incidence of Grade 3 diarrhea was 32% compared to 28%, respectively. Diarrhea leading to treatment discontinuation occurred in 18% of patients treated with loperamide alone compared to 11% of the patients who received loperamide and budesonide.
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on NERLYNX
Table 10 includes drug interactions that affect the pharmacokinetics of neratinib.
Table 10: Drug Interactions That Affect Neratinib AUC=Area Under Curve; C max=Maximum Concentration
* These examples are a guide and not considered a comprehensive list of all possible drugs that may fit this category. The healthcare provider should consult appropriate references for comprehensive information.
Gastric Acid Reducing Agents Clinical Impact Concomitant use of NERLYNX with a proton pump inhibitor, H 2-receptor antagonist, or antacid may decrease neratinib plasma concentration. Decreased neratinib AUC may reduce NERLYNX activity. Lansoprazole (PPI) resulted in a decrease of neratinib C max by 71% and AUC by 65% [ see Clinical Pharmacology ( 12.3) ]. Prevention or Management - PPIs
Avoid concomitant use [ see Dosage and Administration ( 2.3) ]. - H 2-receptor antagonists
Take NERLYNX at least 2 hours before the next dose of the H 2-receptor antagonist or 10 hours after the H 2-receptor antagonist [ see Dosage and Administration ( 2.3) ]. - Antacids
Separate NERLYNX dosing by 3 hours after antacids [ see Dosage and Administration ( 2.3) ]. Strong CYP3A4 Inhibitors Clinical Impact - Concomitant use of NERLYNX with a strong CYP3A4 inhibitor (ketoconazole) increased neratinib C max by 221% and AUC by 381% [ see Clinical Pharmacology ( 12.3) ].
- Concomitant use of NERLYNX with other strong CYP3A4 inhibitors may increase neratinib concentrations.
- Increased neratinib concentrations may increase the risk of toxicity.
Prevention or Management Avoid concomitant use of NERLYNX with strong CYP3A4 inhibitors. Moderate CYP3A4 and P-gp Dual Inhibitors Clinical Impact - Simulated concomitant use of NERLYNX with a moderate CYP3A4 and P-gp dual inhibitor (verapamil) suggest the neratinib C max and AUC may increase by 203% and 299%, respectively [ see Clinical Pharmacology ( 12.3) ].
- Concomitant use of NERLYNX with other moderate CYP3A4 and P-gp dual inhibitors may increase neratinib concentrations.
- Increased neratinib concentrations may increase the risk of toxicity.
Prevention or Management Avoid concomitant use of NERLYNX with other moderate CYP3A4 and P-gp dual inhibitors. Strong or Moderate CYP3A4 Inducers Clinical Impact - Concomitant use of NERLYNX with a strong CYP3A4 inducer (rifampin) reduced neratinib C max by 76% and AUC by 87% [ see Clinical Pharmacology ( 12.3) ].
- Concomitant use of NERLYNX with other strong or moderate CYP3A4 inducers may decrease NERLYNX concentrations.
- Decreased neratinib AUC may reduce NERLYNX activity.
Prevention or Management Avoid concomitant use of NERLYNX with strong or moderate CYP3A4 inducers. 7.2 Effect of NERLYNX on Other Drugs
P-glycoprotein (P-gp) Substrates
Concomitant use of NERLYNX with digoxin, a P-gp substrate, increased digoxin concentrations [ see Clinical Pharmacology ( 12.3) ]. Increased concentrations of digoxin may lead to increased risk of adverse reactions including cardiac toxicity. Refer to the digoxin prescribing information for dosage adjustment recommendations due to drug interactions. NERLYNX may inhibit the transport of other P-gp substrates (e.g., dabigatran, fexofenadine).
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and the mechanism of action, NERLYNX can cause fetal harm when administered to a pregnant woman [ see Clinical Pharmacology ( 12.1) ].
There are no available data in pregnant women to inform the drug-associated risk. In animal reproduction studies, administration of neratinib to pregnant rabbits during organogenesis resulted in abortions, embryo-fetal death and fetal abnormalities in rabbits at maternal exposures (AUC) approximately 0.2 times exposures in patients at the recommended dose ( see Data). Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. However, the background risk of major birth defects is 2%-4% and of miscarriage is 15%-20% of clinically recognized pregnancies in the U.S. general population.
Data
Animal Data
In a fertility and early embryonic development study in female rats, neratinib was administered orally for 15 days before mating to Day 7 of pregnancy, which did not cause embryonic toxicity at doses up to 12 mg/kg/day in the presence of maternal toxicity. A dose of 12 mg/kg/day in rats is approximately 0.5 times the maximum recommended dose of 240 mg/day in patients on a mg/m 2 basis.
In an embryo-fetal development study in rats, pregnant animals received oral doses of neratinib up to 15 mg/kg/day during the period of organogenesis. No effects on embryo-fetal development or survival were observed. Maternal toxicity was evident at 15 mg/kg/day (approximately 0.6 times the AUC in patients receiving the maximum recommended dose of 240 mg/day).
In an embryo-fetal development study in rabbits, pregnant animals received oral doses of neratinib up to 9 mg/kg/day during the period of organogenesis. Administration of neratinib at doses ≥6 mg/kg/day resulted in maternal toxicity, abortions, and embryo-fetal death (increased resorptions). Neratinib administration resulted in increased incidence of fetal gross external (domed head), soft tissue (dilation of the brain ventricles and ventricular septal defect), and skeletal (misshapen anterior fontanelles and enlarged anterior and/or posterior fontanelles) abnormalities at ≥3 mg/kg/day. The AUC (0-t) at 6 mg/kg/day and 9 mg/kg/day in rabbits were approximately 0.5 and 0.8 times, respectively, the AUCs in patients receiving the maximum recommended dose of 240 mg/day.
In a peri- and postnatal development study in rats, oral administration of neratinib from gestation day 7 until lactation day 20 resulted in maternal toxicity at ≥10 mg/kg/day (approximately 0.4 times the maximum recommended dose of 240 mg/day in patients on a mg/m 2 basis) including decreased body weights, body weight gains, and food consumption. Effects on long-term memory were observed in male offspring at maternal doses ≥5 mg/kg/day (approximately 0.2 times the maximum recommended dose of 240 mg/day in patients on a mg/m 2 basis).
8.2 Lactation
Risk Summary
No data are available regarding the presence of neratinib or its metabolites in human milk or its effects on the breastfed infant or on milk production. Because of the potential for serious adverse reactions in breastfed infants from NERLYNX, advise lactating women not to breastfeed while taking NERLYNX and for at least 1 month after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy
Based on animal studies, NERLYNX can cause fetal harm when administered to a pregnant woman [ see Use in Specific Populations ( 8.1) ]. Females of reproductive potential should have a pregnancy test prior to starting treatment with NERLYNX.
Contraception
Females
Based on animal studies, NERLYNX can cause fetal harm when administered to a pregnant woman [ see Use in Specific Populations ( 8.1) ]. Advise females of reproductive potential to use effective contraception during treatment with NERLYNX and for at least 1 month after the last dose.
Males
Based on findings in animal reproduction studies, advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of NERLYNX [ see Use in Specific Populations ( 8.1) ].
8.4 Pediatric Use
The safety and efficacy of NERLYNX in pediatric patients has not been established.
8.5 Geriatric Use
In the ExteNET trial, in the NERLYNX arm; 1236 patients were <65 years, 172 patients were ≥65 years, of whom 25 patients were 75 years or older. There was a higher frequency of treatment discontinuations due to adverse reactions in the ≥65 years age group than in the <65 years age group; in the NERLYNX arm, the percentages were 45% compared with 25%, respectively, and in the placebo arm 6% and 5%, respectively. The incidence of serious adverse reactions in the NERLYNX arm vs. placebo arm was 7% vs. 6% (<65 years old) and 10% vs. 8% (≥65 years old). The serious adverse reactions most frequently reported in the ≥65 years old group were vomiting (2.3%), diarrhea (1.7%), renal failure (1.7%), and dehydration (1.2%).
In the NALA trial, in the NERLYNX plus capecitabine arm; 242 patients were <65 years, 61 patients were ≥65 years, of whom 12 patients were 75 years or older. The incidence of serious adverse reactions in the NERLYNX plus capecitabine arm in the ≥65 years age group was 36% and in the <65 years age group was 34%. The serious adverse reactions most frequently reported in the ≥65 years-old group were diarrhea (16%), acute kidney injury (8%), and dehydration (7%). No overall differences in effectiveness were observed between patients ≥65 years old and patients <65 years old.
8.6 Hepatic Impairment
No dose modifications are recommended for patients with mild to moderate hepatic impairment (Child Pugh A or B). Patients with severe, pre-existing hepatic impairment (Child Pugh Class C) experienced a reduction in neratinib clearance and an increase in C max and AUC. Reduce the NERLYNX dosage for patients with severe hepatic impairment. [ see Dosage and Administration ( 2.3) and Clinical Pharmacology ( 12.3) ].
-
10 OVERDOSAGE
There is no specific antidote, and the benefit of hemodialysis in the treatment of NERLYNX overdose is unknown. In the event of an overdose, administration should be withheld and general supportive measures undertaken.
In the clinical trial setting, a limited number of patients reported overdose. The adverse reactions experienced by these patients were diarrhea, nausea, vomiting, and dehydration. The frequency and severity of gastrointestinal disorders (diarrhea, abdominal pain, nausea and vomiting) appear to be dose related.
-
11 DESCRIPTION
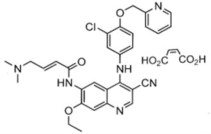
NERLYNX (neratinib) immediate release, film-coated tablets for oral administration contain 40 mg of neratinib, equivalent to 48.31 mg neratinib maleate. Neratinib is a member of the 4-anilino quinolidine class of protein kinase inhibitors. The molecular formula for neratinib maleate is C 30H 29ClN 6O 3C 4H 4O 4 and the molecular weight is 673.11 Daltons. The chemical name is (E)-N-{4-[3-chloro-4-(pyridin-2-yl methoxy)anilino]-3-cyano-7-ethoxyquinolin-6-yl}-4-(dimethylamino)but-2-enamide maleate, and its structural formula is:
Neratinib maleate is an off-white to yellow powder with pK as of 7.65 and 4.66. The solubility of neratinib maleate increases dramatically as neratinib becomes protonated at acidic pH. Neratinib maleate is sparingly soluble at pH 1.2 (32.90 mg/mL) and insoluble at approximate pH 5.0 and above (0.08 mg/mL or less).
Inactive ingredients: Tablet Core: colloidal silicon dioxide, mannitol, microcrystalline cellulose, crospovidone, povidone, magnesium stearate, and purified water. Coating: red film coat: polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide red.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Neratinib is an intracellular kinase inhibitor that irreversibly binds to epidermal growth factor receptor (EGFR), HER2, and HER4. In vitro, neratinib reduces EGFR and HER2 autophosphorylation, downstream MAPK and AKT signaling pathways, and showed antitumor activity in EGFR and/or HER2 expressing carcinoma cell lines. Neratinib human metabolites M3, M6, M7 and M11 inhibited the activity of EGFR, HER2, and HER4 in vitro. In vivo, oral administration of neratinib inhibited tumor growth in mouse xenograft models with tumor cell lines expressing HER2 and EGFR.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of NERLYNX on the QTc interval was evaluated in a randomized, placebo, and positive-controlled, double-blind, single-dose, crossover study in 60 healthy subjects. At 140% the therapeutic exposures of NERLYNX, there was no clinically relevant effect on the QTc interval.
12.3 Pharmacokinetics
Neratinib exhibits a non-linear PK profile with less than dose proportional increase of AUC with the increasing daily dose over the range of 40 to 400 mg.
Absorption
The neratinib and major active metabolites M3, M6 and M7 peak concentrations are reached in the range of 2 to 8 hours after oral administration.
Effect of Food
The food-effect assessment was conducted in healthy volunteers who received NERLYNX 240 mg under fasting conditions and with high-fat food (approximately 55% fat, 31% carbohydrate, and 14% protein) or standard breakfast (approximately 50% carbohydrate, 35% fat, and 15% protein). A high-fat meal increased neratinib C max and AUC inf by 70% (90% CI: 1.1–2.7) and 120% (90% CI: 1.4–3.5), respectively. A standard breakfast increased the C max and AUC inf by 20% (90% CI: 0.97–1.42) and 10% (90% CI: 1.02–1.24), respectively [ See Dosage and Administration ( 2.2) ].
Distribution
In patients, following multiple doses of NERLYNX, the mean (%CV) apparent volume of distribution at steady-state (V ss/F) was 6433 (19%) L. In vitro protein binding of neratinib in human plasma was greater than 99% and independent of concentration. Neratinib bound predominantly to human serum albumin and human alpha-1 acid glycoprotein.
Elimination
Following 7 days of daily 240 mg oral doses of NERLYNX in healthy subjects, the mean (%CV) plasma half-life of neratinib, M3, M6, and M7 was 14.6 (38%), 21.6 (77%), 13.8 (50%) and 10.4 (33%) hours, respectively. The mean elimination half-life of neratinib ranged from 7 to 17 hours following a single oral dose in patients. Following multiple doses of NERLYNX at once-daily 240 mg in cancer patients, the mean (%CV) CL/F after first dose and at steady state (day 21) were 216 (34%) and 281 (40%) L/hour, respectively.
Metabolism
Neratinib is metabolized primarily in the liver by CYP3A4 and to a lesser extent by flavin-containing monooxygenase (FMO).
After oral administration of NERLYNX, neratinib represents the most prominent component in plasma. At steady state after 240 mg daily oral doses of NERLYNX in a healthy subject study (n=25), the systemic exposures (AUC) of the active metabolites M3, M6, M7 and M11were 15%, 33%, 22% and 4% of the systemic neratinib exposure (AUC) respectively.
Excretion
After oral administration of 200 mg (0.83 times of approved recommended dosage) radiolabeled neratinib oral formulation, fecal excretion accounted for approximately 97% and urinary excretion accounted for 1.1% of the total dose. Sixty-one percent of the excreted radioactivity was recovered within 96 hours and 98% was recovered after 10 days.
Specific Populations
Age, gender, race, and renal function do not have a clinically significant effect on neratinib pharmacokinetics.
Patients With Hepatic Impairment
Neratinib is mainly metabolized in the liver. Single doses of 120 mg NERLYNX were evaluated in non-cancer patients with chronic hepatic impairment (n=6 each in Child Pugh Class A, B, and C) and in healthy subjects (n=9) with normal hepatic function. Neratinib exposures in the patients with Child Pugh Class A (mild impairment) and Child Pugh Class B (moderate impairment) were similar to that in normal healthy volunteers. Patients with severe hepatic impairment (Child Pugh Class C) had neratinib C max and AUC increased by 173% and 181%, respectively, as compared to the normal hepatic function controls [ see Dosage and Administration ( 2.3) and Use in Specific Populations ( 8.6) ].
Gastric Acid Reducing Agents: NERLYNX solubility decreases with increasing GI tract pH values. Drugs that alter the pH values of the GI tract may alter the solubility of neratinib and hence its absorption and systemic exposure. When multiple doses of lansoprazole (30 mg daily), a proton pump inhibitor, were co-administered with a single 240 mg oral dose of NERLYNX, the neratinib C max and AUC decreased by 71% and 65%, respectively. When a single oral dose of 240 mg NERLYNX was administered 2 hours following a daily dose of 300 mg ranitidine, an H 2 receptor antagonist, the neratinib C max and AUC were reduced by 57% and 48%, respectively. When a single oral dose of 240 mg NERLYNX was administered 2 hours prior to 150 mg ranitidine twice daily (administered in the morning and evening, approximately 12 hours apart), the neratinib C max and AUC were reduced by 44% and 32%, respectively [ See Dosage and Administration ( 2.3) and Drug Interactions ( 7.1) ].
Strong CYP3A4 inhibitors: Concomitant use of ketoconazole (400 mg once-daily for 5 days), a strong inhibitor of CYP3A4 and an inhibitor of P-gp, with a single oral 240 mg NERLYNX dose in healthy subjects (n=24) increased neratinib C max by 221% and AUC by 381% [ see Drug Interactions ( 7.1) ].
Moderate CYP3A4 and P-gp dual Inhibitors: Simulations using physiologically based pharmacokinetic (PBPK) models suggested that a moderate CYP3A4 and P-gp dual inhibitor (verapamil) may increase the C max and AUC of neratinib by 203% and 299%, respectively [ see Drug Interactions ( 7.1) ].
Moderate CYP3A4 inhibitors: Simulations using PBPK models suggested that a moderate CYP3A4 inhibitor (fluconazole) may increase the C max and AUC of neratinib by 30% and 68%, respectively.
Strong and Moderate CYP3A4 Inducers: Concomitant use of rifampin, a strong inducer of CYP3A4, with a single oral 240 mg NERLYNX dose in healthy subjects (n=24) reduced neratinib C max by 76% and AUC by 87%. The AUC of active metabolites M6 and M7 were also reduced by 37-49% when compared to NERLYNX administered alone. Simulations using PBPK models suggested that a moderate CYP3A4 inducer (efavirenz) may decrease the C max and AUC of neratinib by 36% and 52%, respectively [ see Drug Interactions ( 7.1) ].
Effect of NERLYNX on P-gp Transporters: Concomitant use of digoxin (a single 0.5 mg oral dose), a P-gp substrate, with multiple oral doses of NERLYNX 240 mg in healthy subjects (n=18) increased the mean digoxin C max by 54% and AUC by 32% [ see Drug Interactions ( 7.2) ].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A two-year carcinogenicity study was conducted in rats at oral neratinib doses of 1, 3, and 10 mg/kg/day. Neratinib was not carcinogenic in male and female rats at exposure levels >25 times the AUC in patients receiving the maximum recommended dose of 240 mg/day. Neratinib was not carcinogenic in a 26-week study in Tg.rasH2 transgenic mice when administered daily by oral gavage at doses up to 50 mg/kg/day in males and 125 mg/kg/day in females.
Neratinib was not mutagenic in an in vitro bacterial reverse mutation (AMES) assay or clastogenic in an in vitro human lymphocyte chromosomal aberration assay or an in vivo rat bone marrow micronucleus assay.
In a fertility study in rats, neratinib administration up to 12 mg/kg/day (approximately 0.5 times the maximum recommended dose of 240 mg/day in patients on a mg/m 2 basis) caused no effects on mating or the ability of animals to become pregnant. In repeat-dose toxicity studies in dogs with oral administration of neratinib daily for up to 39 weeks, tubular hypoplasia of the testes was observed at ≥0.5 mg/kg/day. This finding was observed at AUCs that were approximately 0.4 times the AUC in patients at the maximum recommended dose of 240 mg.
-
14 CLINICAL STUDIES
14.1 Extended Adjuvant Treatment of Early Stage Breast Cancer
The safety and efficacy of NERLYNX were investigated in the ExteNET trial (NCT00878709), a multicenter, randomized, double-blind, placebo-controlled study of NERLYNX after adjuvant treatment with a trastuzumab based therapy in women with HER2-positive breast cancer.
A total of 2840 patients with early-stage (Stage 1 to 3c) HER2-positive breast cancer within two years of completing treatment with adjuvant trastuzumab was randomized to receive either NERLYNX (n=1420) or placebo (n=1420). Randomization was stratified by the following factors: hormone receptor status, nodal status (0, 1-3 vs 4 or more positive nodes) and whether trastuzumab was given sequentially versus concurrently with chemotherapy. NERLYNX 240 mg or placebo was given orally once daily for one year. The major efficacy outcome measure was invasive disease-free survival (iDFS) defined as the time between the date of randomization to the first occurrence of invasive recurrence (local/regional, ipsilateral, or contralateral breast cancer), distant recurrence, or death from any cause, with 2 years and 28 days of follow-up.
Patient demographics and tumor characteristics were generally balanced between treatment arms. Patients had a median age of 52 years (range 23 to 83) and 12% of patients were 65 or older. The majority of patients were White (81%), and most patients (99.7%) had an ECOG performance status of 0 or 1. Fifty-seven percent (57%) of patients had hormone receptor positive disease (defined as ER-positive and/or PR-positive), 24% were node negative, 47% had one to three positive nodes and 30% had four or more positive nodes. Ten percent (10%) of patients had Stage I disease, 41% had Stage II disease and 31% had Stage III disease. The majority of patients (81%) were enrolled within one year of completion of trastuzumab treatment. Median time from the last adjuvant trastuzumab treatment to randomization was 4.4 months in the NERLYNX arm versus 4.6 months in the placebo arm. Median duration of treatment was 11.6 months in the NERLYNX arm vs. 11.8 months in the placebo arm.
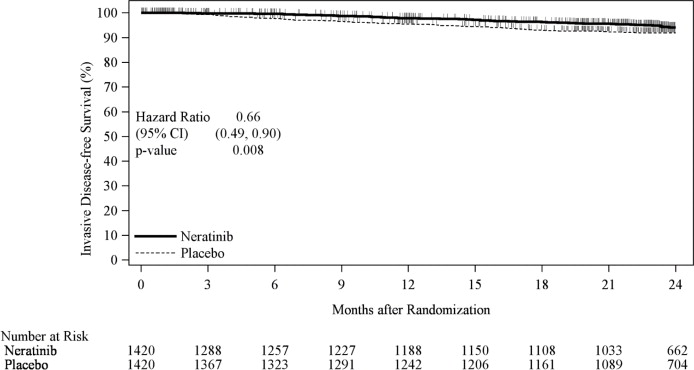
The efficacy results from the ExteNET trial are summarized in Table 11 and Figure 1.
Table 11: Efficacy iDFS Results for the ITT Population Number of Events/ Total N
(%)iDFS at 24 months*
(%, 95% CI)Stratified† HR (95% CI) P-value‡ CI= Confidence Interval; HR=Hazard Ratio; iDFS=Invasive Disease Free-Survival; ITT=Intent to Treat
* Kaplan-Meier estimate
† Stratified by prior trastuzumab (concurrent vs. sequential), nodal status (0-3 positive nodes vs. ≥4 positive nodes), and ER/PR status (positive vs. negative)
‡ Stratified log-rank test
NERLYNX Placebo NERLYNX Placebo 67/1420
(4.7)106/1420
(7.5)94.2
(92.6, 95.4)91.9
(90.2, 93.2)0.66
(0.49, 0.90)0.008 Figure 1: iDFS in the ExteNET Trial - ITT Population
CI=Confidence Interval; HR=Hazard Ratio; iDFS=Invasive Disease Free-Survival; ITT=Intent to Treat
Table 12: Subgroup Analyses * Population Number of Events/Total N (%) iDFS at 24 Months†(%, 95% CI) Unstratified HR (95% CI) CI=Confidence Interval; HR=Hazard Ratio
* Exploratory analyses without adjusting multiple comparisons
† Kaplan-Meier estimate
NERLYNX Placebo NERLYNX Placebo Hormone Receptor Status Positive 29/816
(3.6)63/815
(7.7)95.6
(93.8, 96.9)91.5
(89.2, 93.3)0.49
(0.31, 0.75)Negative 38/604
(6.3)43/605
(7.1)92.2
(89.4, 94.3)92.4
(89.8, 94.3)0.93
(0.60, 1.43)Nodal Status Negative 7/335
(2.1)11/336
(3.3)97.2
(94.1, 98.7)96.5
(93.7, 98.0)0.72
(0.26, 1.83)1-3 Positive Nodes 31/664
(4.7)47/664
(7.1)94.4
(92.2, 96.1)92.4
(90.0, 94.2)0.68
(0.43, 1.07)≥4 Positive Nodes 29/421
(6.9)48/420
(11.4)91.4
(87.9, 94.0)87.3
(83.4, 90.2)0.62
(0.39, 0.97)Prior Trastuzumab Concurrent 49/884
(5.5)66/886
(7.4)93.2
(91.0, 94.8)92.0
(89.9, 93.7)0.80
(0.55, 1.16)Sequential 18/536
(3.4)40/534
(7.5)95.8
(93.4, 97.3)91.6
(88.7, 93.8)0.46
(0.26, 0.78)Completion of Prior Trastuzumab ≤1 year 58/1152
(5.0)95/1145
(8.3)93.8
(92.0, 95.2)90.9
(89.0, 92.5)0.63
(0.45, 0.88)1-2 years 9/262
(3.4)11/270
(4.1)95.8
(92.0, 97.8)95.7
(92.3, 97.6)0.92
(0.37, 2.22)Approximately 75% of patients were re-consented for extended follow-up beyond 24 months. Observations with missing data were censored at the last date of assessment. This exploratory analysis suggests that the iDFS results at 5 years are consistent with the 2-year iDFS results observed in ExteNET. At the time of the iDFS analysis, 2% of patients had died, and overall survival data were immature.
14.2 Advanced or Metastatic Breast Cancer
The safety and efficacy of NERLYNX in combination with capecitabine was studied in NALA (NCT01808573), a randomized, multicenter, open-label clinical trial in patients (N=621) with metastatic HER2 positive breast cancer who had received 2 or more prior anti-HER2 based regimens in the metastatic setting. HER2 expression was based on archival tissue tested at a central laboratory prior to enrollment. HER2 positivity was defined as a HER2 immunohistochemistry (IHC) score of 3+ or IHC 2+ with confirmatory in situ hybridization (ISH) positive. Fifty-nine percent of these patients were hormone receptor positive (HR+) and 41% were hormone receptor negative (HR-); 69% had received two prior anti-HER2 based regimens, 31% had received three or more prior anti-HER2 based regimens, 81% had visceral disease, and 19% had non-visceral-only disease. Patients with asymptomatic or stable brain metastases were included in NALA trial (16%).
Patients were randomized (1:1) to receive NERLYNX 240 mg orally once daily on Days 1-21 in combination with capecitabine 750 mg/m 2 given orally twice daily on Days 1-14 for each 21-day cycle (n=307) or lapatinib 1250 mg orally once daily Days 1-21 in combination with capecitabine 1000 mg/m 2 given orally twice daily on Days 1-14 for each 21-day cycle (n=314). Patients were treated until disease progression or unacceptable toxicity.
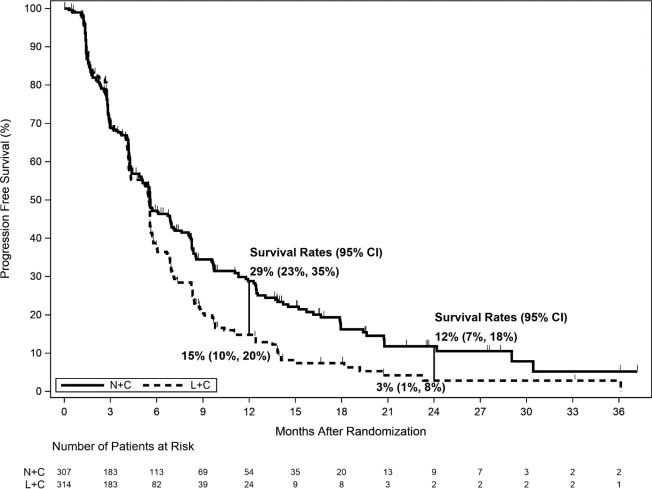
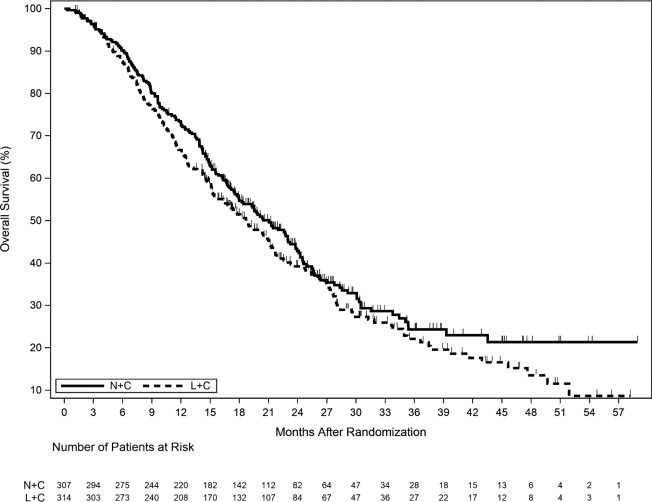
The efficacy results from the NALA trial are summarized in Table 13, Figure 2, and Figure 3.
Table 13. Efficacy Results – NALA Trial (Central Assessment) HR=Hazard Ratio
* Hazard ratio is presented as NERLYNX plus Capecitabine (N+C) vs. Lapatinib plus Capecitabine (L+C).
† Stratified log-rank test
‡ The total number of patients remaining on study at 24 months is 11; with 9 patients on N+C and 2 patients on L+C.
α Exploratory analysis
§ Confirmed ORR
NERLYNX + Capecitabine
(N=307)Lapatinib + Capecitabine
(N=314)Progression Free Survival (PFS) Number of Events (%) 210 (68.4) 223 (71.0) Median PFS, months (95% Cl) 5.6 (4.9, 6.9) 5.5 (4.3, 5.6) HR (95% CI) * 0.76 (0.63,0.93) p-value † 0.0059 PFS rates at 12 months, % (95% CI) α 29 (23, 35) 15 (10, 20) PFS rates at 24 months, % (95% CI) ‡, α 12 (7, 18) 3 (1, 8) Overall Survival (OS) Number of Events (%) 192 (62.5) 218 (69.4) Median OS, months (95% Cl) 21.0 (17.7, 23.8) 18.7 (15.5, 21.2) HR (95% CI) * 0.88 (0.72, 1.07) p-value † 0.2086 Objective Response Rate (ORR)§ ORR, % (95% CI) 32.8 (27.1, 38.9) 26.7 (21.5, 32.4) Duration of Response Median Duration of Response, months (95% CI) 8.5 (5.6, 11.2) 5.6 (4.2, 6.4) Figure 2. Progression Free Survival (Central Assessment – ITT Population)
Figure 3. Overall Survival (ITT Population)
Table 14. Progression-Free Survival Rates – Subgroup Analyses α Population Number of Events/Total N (%) PFS Rates (%) at 12 Months (95% CI) α Exploratory Analysis
NERLYNX + Capecitabine Lapatinib + Capecitabine NERLYNX + Capecitabine Lapatinib + Capecitabine Disease Location Visceral 181/247 (73.3) 185/253 (73.1) 23 (17, 30) 14 (10, 20) Non Visceral 29/60 (48.3) 38/61 (62.3) 53 (38, 66) 18 (7, 32) Hormone Receptor Status Positive 128/181 (70.7) 115/186 (61.8) 27 (19, 34) 23 (15, 31) Negative 82/126 (65.1) 108/128 (84.4) 32 (23, 41) 5 (2, 11) Previous HER2 regimens 2 regimens 148/215 (68.8) 151/215 (70.2) 26 (20, 33) 13 (8, 19) ≥3 regimens 62/92 (67.4) 72/99 (72.7) 34 (24, 45) 19 (11, 29) -
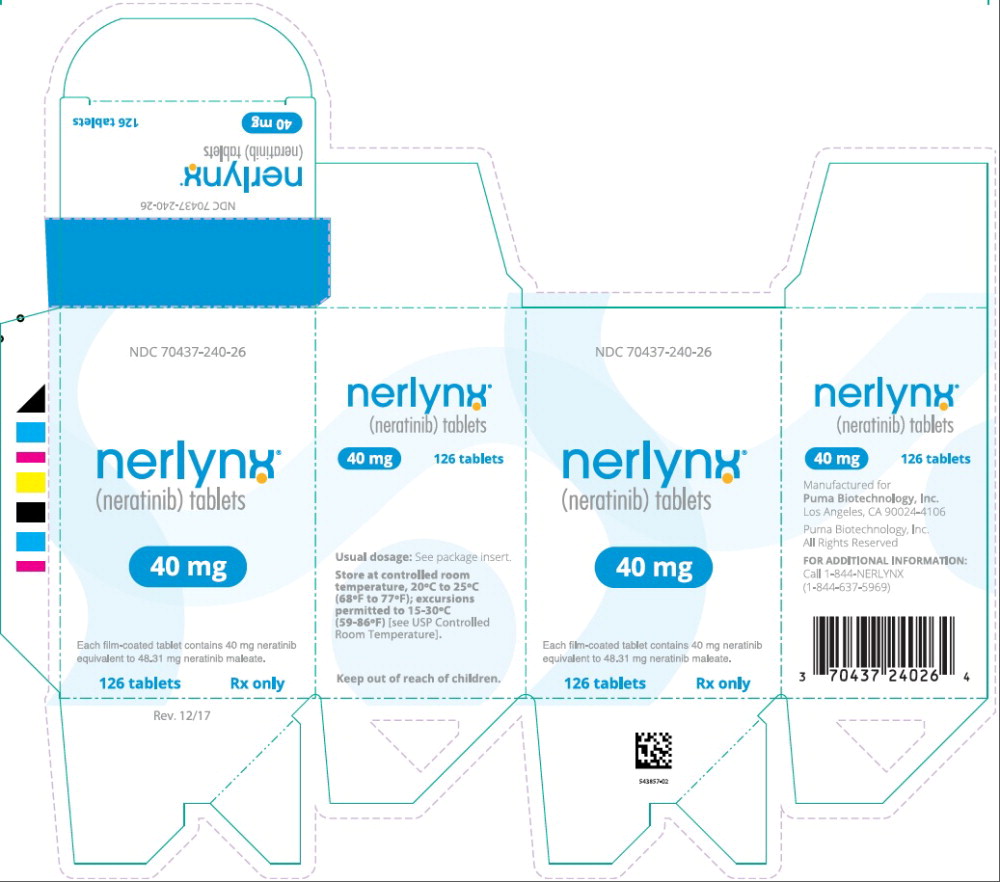
16 HOW SUPPLIED/STORAGE AND HANDLING
NERLYNX 40 mg film-coated tablets are red, oval shaped and debossed with ‘W104’ on one side and plain on the other side.
NERLYNX is available in:
- Bottles of 180 tablets: NDC: 70437-240-18
- Bottles of 126 tablets: NDC: 70437-240-26
Store at controlled room temperature, 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling ( Patient Information).
Diarrhea
- Inform patients that NERLYNX has been associated with diarrhea, which may be severe in some cases.
- Advise patients to initiate antidiarrheal prophylaxis with the first dose of NERLYNX.
- Instruct patients to maintain 1-2 bowel movements per day and on how to use antidiarrheal treatment regimens.
- Advise patients to inform their healthcare provider immediately if severe (≥Grade 3) diarrhea or diarrhea associated with weakness, dizziness, or fever occurs during treatment with NERLYNX [ see Dosage and Administration ( 2.1) and Warnings and Precautions ( 5.1) ].
Hepatotoxicity
- Inform patients that NERLYNX has been associated with hepatotoxicity which may be severe in some cases.
- Inform patients that they should report signs and symptoms of liver dysfunction to their healthcare provider immediately [ see Warnings and Precautions ( 5.2) ].
Embryo-Fetal Toxicity
- Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [ see Use in Specific Populations ( 8.1) ].
- Advise females of reproductive potential to use effective contraception during treatment and for 1 month after receiving the last dose of NERLYNX [ see Warnings and Precautions ( 5.3) and Use in Specific Populations ( 8.1, 8.3) ].
- Advise lactating women not to breastfeed during treatment with NERLYNX and for at least 1 month after the last dose [ see Use in Specific Populations ( 8.2) ].
Drug Interactions
- NERLYNX may interact with many drugs; therefore, advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products [ see Dosage and Administration ( 2.3) and Clinical Pharmacology ( 12.3) ].
- NERLYNX may interact with gastric acid reducing agents. Advise patients to avoid concomitant use of proton pump inhibitors. When patients require gastric acid reducing agents, use an H 2-receptor antagonist or antacid. Advise patients to separate the dosing of NERLYNX by 3 hours after antacid medicine, and to take NERLYNX at least 2 hours before or 10 hours after a H 2-receptor antagonist [ see Dosage and Administration ( 2.3) and Drug Interactions ( 7.1) ].
- NERLYNX may interact with grapefruit. Advise patients to avoid taking NERLYNX with grapefruit products [ see Drug Interactions ( 7.1) ].
Dosing and Administration
- Instruct patients to take NERLYNX with food at approximately the same time each day consecutively for one year.
- If a patient misses a dose, instruct the patient not to replace the missed dose, and to resume NERLYNX with the next scheduled daily dose [ see Dosage and Administration ( 2.2) ].
Manufactured for Puma Biotechnology, Inc.
10880 Wilshire Blvd., Suite 2150
Los Angeles, CA 90024-4106©2020, Puma Biotechnology, Inc. All Rights Reserved.
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: 2/2020
PATIENT INFORMATION
NERLYNX® (ner links)
(neratinib)
tabletsWhat is the most important information I should know about NERLYNX?
NERLYNX may cause serious side effects, including:
-
Diarrhea. Diarrhea is a common side effect of NERLYNX, but it can also be severe. You may lose too much body salts and fluid, and get dehydrated. When you start NERLYNX, your healthcare provider should prescribe the medicine loperamide for you during your first 2 months (56 days) of NERLYNX and then as needed. Be sure that your healthcare provider prescribes anti-diarrheals with NERLYNX. Anti-diarrheals must be started with the first dose of NERLYNX. To help prevent or reduce diarrhea:
- You should start taking loperamide with your first dose of NERLYNX.
- Keep taking loperamide during the first 2 months (56 days) of NERLYNX treatment and then as needed. Your healthcare provider will tell you exactly how much and how often to take this medicine.
- Your healthcare provider may also need to give you other medicines to manage diarrhea when you start treatment with NERLYNX. Follow your healthcare provider's instructions on how to use these anti-diarrheal medicines.
- Always take anti-diarrheals exactly as your healthcare provider tells you.
- While taking anti-diarrheals, you and your healthcare provider should try to keep the number of bowel movements that you have at 1 or 2 bowel movements each day.
- Tell your healthcare provider if you have more than 2 bowel movements in 1 day, or you have diarrhea that does not go away.
- Call your healthcare provider right away, as instructed, if you have severe diarrhea or if you have diarrhea along with weakness, dizziness, or fever.
- After 2 months (56 days) of treatment with NERLYNX, follow your healthcare provider's instructions about taking loperamide as needed to control diarrhea.
Your healthcare provider may change your dose of NERLYNX, temporarily stop or completely stop NERLYNX if needed to manage your diarrhea.
See “ What are the possible side effects of NERLYNX?” for more information about side effects.What is NERLYNX?
- NERLYNX is a prescription medicine used alone to treat adults with early-stage human epidermal growth factor receptor 2 (HER2)-positive breast cancer and who have previously been treated with trastuzumab-based therapy.
- NERLYNX is also used with a medicine called capecitabine to treat adults with HER2-positive breast cancer that has spread to other parts of the body (metastatic)
and who have received 2 or more anti-HER2 therapy medicines for metastatic breast cancer.
It is not known if NERLYNX is safe and effective in children. Before taking NERLYNX, tell your healthcare provider about all of your medical conditions, including if you:
- have liver problems. You may need a lower dose of NERLYNX.
- are pregnant or plan to become pregnant. NERLYNX can harm your unborn baby. If you are a female who can become pregnant:
- Your healthcare provider should do a pregnancy test before you start taking NERLYNX.
- You should use effective birth control (contraception) during treatment and for at least 1 month after your last dose of NERLYNX.
- Talk with your healthcare provider about forms of birth control that you can use during this time.
- Tell your healthcare provider right away if you become pregnant during treatment with NERLYNX.
- Males with female partners who can become pregnant should use effective birth control during treatment and for 3 months after the last dose of NERLYNX.
- are breastfeeding or plan to breastfeed. It is not known if NERLYNX passes into your breast milk. Do not breastfeed during treatment and for at least 1 month after your last dose of NERLYNX.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Especially tell your healthcare provider if you take medicines used to decrease stomach acid, called proton pump inhibitors or PPIs. You should avoid taking these medicines during treatment with NERLYNX. How should I take NERLYNX?
- Take NERLYNX exactly as your healthcare provider tells you to take it.
- Your healthcare provider may change your dose of NERLYNX if needed.
- Take NERLYNX with food.
- Take NERLYNX at about the same time each day.
- Swallow NERLYNX tablets whole. Do not chew, crush, or split NERLYNX tablets.
- If you take an antacid medicine, take NERLYNX 3 hours after the antacid medicine.
- If you take an acid reducer (H 2 receptor blocker), NERLYNX should be taken at least 2 hours before or 10 hours after you take these medicines.
- NERLYNX is usually taken for 1 year.
- If you miss a dose of NERLYNX, skip that dose and take your next dose at your regular scheduled time.
- If you take too much NERLYNX, call your healthcare provider right away or go to the nearest hospital emergency room.
What should I avoid while taking NERLYNX?
You should avoid eating products that contain grapefruit during treatment with NERLYNX.What are the possible side effects of NERLYNX?
NERLYNX may cause serious side effects, including:
See “ What is the most important information I should know about NERLYNX?”
- Liver problems. Changes in liver function tests are common with NERLYNX. Your healthcare provider should do blood tests before you begin treatment, monthly during the first 3 months, and then every 3 months as needed during treatment with NERLYNX. Your healthcare provider will stop your treatment with NERLYNX if your liver tests show severe problems. Call your healthcare provider right away if you get any of the following signs or symptoms of liver problems:
- tiredness
- nausea
- vomiting
- pain in the right upper stomach-area (abdomen)
- fever
- rash
- itching
- yellowing of your skin or whites of your eyes
The most common side effects of NERLYNX when used alone include: - diarrhea
- nausea
- stomach-area (abdomen) pain
- tiredness
- vomiting
- rash
- dry or inflamed mouth, or mouth sores
- decreased appetite
- muscle spasms
- upset stomach
- nail problems including color change
- dry skin
- swelling of your stomach-area
- nosebleed
- weight loss
- urinary tract infection
The most common side effects of NERLYNX when used with capecitabine include: - diarrhea
- nausea
- vomiting
- decreased appetite
- constipation
- tiredness/weakness
- weight loss
- dizziness
- back pain
- joint pain
- urinary tract infection
- upper respiratory tract infection
- swelling of your stomach-area
- kidney problems
- muscle spasms
These are not all of the possible side effects of NERLYNX. For more information, ask your Healthcare Provider.
Tell your doctor if you have any side effects that bother you or that does not go away. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store NERLYNX?
- Store NERLYNX at room temperature between 68°F to 77°F (20°C to 25°C).
Keep NERLYNX and all medicines out of the reach of children. General information about the safe and effective use of NERLYNX.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use NERLYNX for a condition for which it was not prescribed. Do not give NERLYNX to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about NERLYNX that is written for health professionals.What are the ingredients in NERLYNX?
Active ingredient: neratinib
Inactive ingredients: Tablet Core: colloidal silicon dioxide, mannitol, microcrystalline cellulose, crospovidone, povidone, magnesium stearate, and purified water. Coating: red film coat: polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide red.
Manufactured for: Puma Biotechnology, Inc. 10880 Wilshire Blvd., Suite 2150 Los Angeles, CA 90024-4106
©2020, Puma Biotechnology, Inc. All rights reserved.
For more information, go to www.NERLYNX.com or call 1-844-637-5969. -
Diarrhea. Diarrhea is a common side effect of NERLYNX, but it can also be severe. You may lose too much body salts and fluid, and get dehydrated. When you start NERLYNX, your healthcare provider should prescribe the medicine loperamide for you during your first 2 months (56 days) of NERLYNX and then as needed. Be sure that your healthcare provider prescribes anti-diarrheals with NERLYNX. Anti-diarrheals must be started with the first dose of NERLYNX. To help prevent or reduce diarrhea:
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel - Nerlynx 180 Tablets Carton Label
NDC: 70437-240-18
nerlynx™
(neratinib) tablets40 mg
Each film-coated tablet contains 40 mg neratinib
equivalent to 48.31 mg neratinib maleate.180 tablets Rx only

-
PRINCIPAL DISPLAY PANEL
Principal Display Panel - Nerlynx 180 Tablets Bottle Label
NDC: 70437-240-18
nerlynx™
(neratinib) tablets40 mg
Rx only 180 tablets
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel - Nerlynx 126 Tablets Carton Label
NDC: 70437-240-26
nerlynx™
(neratinib) tablets40 mg
Each film-coated tablet contains 40 mg neratinib
equivalent to 48.31 mg neratinib maleate.126 tablets Rx only
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel - Nerlynx 126 Tablets Bottle Label
NDC: 70437-240-26
nerlynx™
(neratinib) tablets40 mg
Rx only 126 tablets
-
INGREDIENTS AND APPEARANCE
NERLYNX
neratinib tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 70437-240 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NERATINIB MALEATE (UNII: 9RM7XY23ZS) (NERATINIB - UNII:JJH94R3PWB) NERATINIB 40 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) MAGNESIUM STEARATE (UNII: 70097M6I30) WATER (UNII: 059QF0KO0R) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color red (red) Score no score Shape OVAL (OVAL) Size 11mm Flavor Imprint Code W104 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 70437-240-18 1 in 1 BOX 07/17/2017 1 180 in 1 BOTTLE; Type 0: Not a Combination Product 2 NDC: 70437-240-26 1 in 1 BOX 07/17/2017 2 126 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208051 07/17/2017 Labeler - Puma Biotechnology, Inc. (034074860) Establishment Name Address ID/FEI Business Operations AndersonBrecon Inc. 053217022 pack(70437-240) Establishment Name Address ID/FEI Business Operations Excella 329809800 manufacture(70437-240) Establishment Name Address ID/FEI Business Operations Esteve 633485529 api manufacture(70437-240)


Trademark Results [Nerlynx]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 NERLYNX 86678985 5311871 Live/Registered |
Puma Biotechnology Inc. 2015-06-30 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.