NADOLOL AND BENDROFLUMETHIAZIDE tablet
Nadolol and Bendroflumethiazide by
Drug Labeling and Warnings
Nadolol and Bendroflumethiazide by is a Prescription medication manufactured, distributed, or labeled by Impax Generics, Impax Laboratories, Inc, Packaging Coordinators, LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
Nadolol and bendroflumethiazide tablets for oral administration combine two antihypertensive agents: nadolol, a nonselective beta-adrenergic blocking agent, and bendroflumethiazide, a thiazide diuretic-antihypertensive.
Each tablet contains 40 mg or 80 mg nadolol combined with 5 mg bendroflumethiazide. Inactive ingredients: croscarmellose sodium, hypromellose (type 2910), lactose monohydrate, magnesium stearate, mannitol, microcrystalline cellulose, pregelatinized starch and talc.
Nadolol
Nadolol is a white crystalline powder. It is freely soluble in ethanol, soluble in hydrochloric acid, slightly soluble in water and in chloroform, and very slightly soluble in sodium hydroxide.
Nadolol is designated chemically as 1-(tert-butylamino)-3-{(5,6,7,8-tetrahydro-cis-6,7-dihydroxy-1-naphthyl)oxy}-2-propanol. Structural formula:
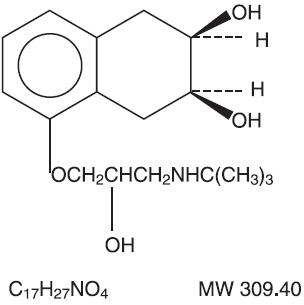
Bendroflumethiazide
Bendroflumethiazide is a white crystalline powder. It is soluble in alcohol and in sodium hydroxide, and insoluble in hydrochloric acid, water, and chloroform.
Bendroflumethiazide is designated chemically as 3-benzyl-3,4-dihydro-6-(trifluoromethyl)-2H-1,2,4-benzothiadiazine-7-sulfonamide 1,1-dioxide. Structural formula:
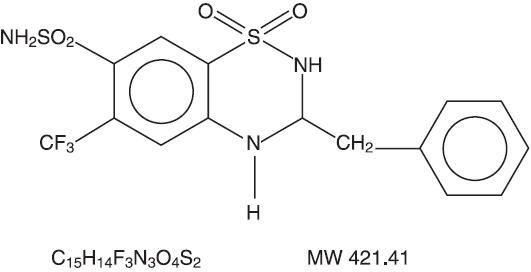
-
CLINICAL PHARMACOLOGY
Nadolol
Nadolol is a nonselective beta-adrenergic receptor blocking agent. Clinical pharmacology studies have demonstrated beta-blocking activity by showing (1) reduction in heart rate and cardiac output at rest and on exercise, (2) reduction of systolic and diastolic blood pressure at rest and on exercise, (3) inhibition of isoproterenol-induced tachycardia, and (4) reduction of reflex orthostatic tachycardia.
Nadolol specifically competes with beta-adrenergic receptor agonists for available beta receptor sites; it inhibits both the beta1 receptors located chiefly in cardiac muscle and the beta2 receptors located chiefly in the bronchial and vascular musculature, inhibiting the chronotropic, inotropic, and vasodilator responses to beta-adrenergic stimulation proportionately. Nadolol has no intrinsic sympathomimetic activity and, unlike some other beta-adrenergic blocking agents, nadolol has little direct myocardial depressant activity and does not have an anesthetic-like membrane-stabilizing action. Animal and human studies show that nadolol slows the sinus rate and depresses AV conduction. In dogs, only minimal amounts of nadolol were detected in the brain relative to amounts in blood and other organs and tissues. Nadolol has low lipophilicity as determined by octanol/water partition coefficient, a characteristic of certain beta-blocking agents that has been correlated with the limited extent to which these agents cross the blood-brain barrier, their low concentration in the brain, and low incidence of CNS-related side effects.
In controlled clinical studies, nadolol at doses of 40 to 320 mg/day has been shown to decrease both standing and supine blood pressure, the effect persisting for approximately 24 hours after dosing.
The mechanism of the antihypertensive effects of beta-adrenergic receptor blocking agents has not been established; however, factors that may be involved include (1) competitive antagonism of catecholamines at peripheral (non-CNS) adrenergic neuron sites (especially cardiac) leading to decreased cardiac output, (2) a central effect leading to reduced tonic-sympathetic nerve outflow to the periphery, and (3) suppression of renin secretion by blockade of the beta-adrenergic receptors responsible for renin release from the kidneys.
While cardiac output and arterial pressure are reduced by nadolol therapy, renal hemodynamics are stable, with preservation of renal blood flow and glomerular filtration rate.
By blocking catecholamine-induced increases in heart rate, velocity and extent of myocardial contraction, and blood pressure, nadolol generally reduces the oxygen requirements of the heart at any given level of effort, making it useful for many patients in the long-term management of angina pectoris. On the other hand, nadolol can increase oxygen requirements by increasing left ventricular fiber length and end diastolic pressure, particularly in patients with heart failure.
Although beta-adrenergic receptor blockade is useful in treatment of angina and hypertension, there are also situations in which sympathetic stimulation is vital. For example, in patients with severely damaged hearts, adequate ventricular function may depend on sympathetic drive. Beta2-adrenergic blockade may worsen AV block by preventing the necessary facilitating effects of sympathetic activity on conduction. Beta2-adrenergic blockade results in passive bronchial constriction by interfering with endogenous adrenergic bronchodilator activity in patients subject to bronchospasm and may also interfere with exogenous bronchodilators in such patients.
Absorption of nadolol after oral dosing is variable, averaging about 30 percent. Peak serum concentrations of nadolol usually occur in three to four hours after oral administration and the presence of food in the gastrointestinal tract does not affect the rate or extent of nadolol absorption. Approximately 30 percent of the nadolol present in serum is reversibly bound to plasma protein.
Unlike many other beta-adrenergic blocking agents, nadolol is not metabolized by the liver and is excreted unchanged, principally by the kidneys.
The half-life of therapeutic doses of nadolol is about 20 to 24 hours, permitting once-daily dosage. Because nadolol is excreted predominantly in the urine, its half-life increases in renal failure (see PRECAUTIONS, General, and DOSAGE AND ADMINISTRATION). Steady state serum concentrations of nadolol are attained in six to nine days with once-daily dosage in persons with normal renal function. Because of variable absorption and different individual responsiveness, the proper dosage must be determined by titration.
Exacerbation of angina and, in some cases, myocardial infarction and ventricular dysrhythmias have been reported after abrupt discontinuation of therapy with beta-adrenergic blocking agents in patients with coronary artery disease. Abrupt withdrawal of these agents in patients without coronary artery disease has resulted in transient symptoms, including tremulousness, sweating, palpitation, headache, and malaise. Several mechanisms have been proposed to explain these phenomena, among them increased sensitivity to catecholamines because of increased numbers of beta receptors.
Bendroflumethiazide
The mechanism of action of bendroflumethiazide results in an interference with the renal tubular mechanism of electrolyte reabsorption. At maximal therapeutic dosage all thiazides are approximately equal in their diuretic potency.
Thiazides increase excretion of sodium and chloride in approximately equivalent amounts. Natriuresis causes a secondary loss of potassium and bicarbonate.
The mechanism of the antihypertensive effect of thiazides is unknown. Thiazides do not affect normal blood pressure.
Onset of action of thiazides occurs in two hours and the peak effect at about four hours. Duration of action persists for approximately six to 12 hours. Thiazides are eliminated rapidly by the kidney.
-
INDICATIONS
Nadolol and bendroflumethiazide tablets are indicated for the treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes including the classes to which this drug principally belongs. There are no controlled trials demonstrating risk reduction with nadolol and bendroflumethiazide tablets.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program's Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
Nadolol and bendroflumethiazide tablets are not indicated for initial therapy of hypertension. If the fixed combination represents the dose titrated to the individual patient's needs, it may be more convenient than the separate components.
-
CONTRAINDICATIONS
Nadolol
Nadolol is contraindicated in bronchial asthma, sinus bradycardia and greater than first degree conduction block, cardiogenic shock, and overt cardiac failure (see WARNINGS).
-
WARNINGS
Nadolol
Cardiac Failure
Sympathetic stimulation may be a vital component supporting circulatory function in patients with congestive heart failure, and its inhibition by beta-blockade may precipitate more severe failure. Although beta-blockers should be avoided in overt congestive heart failure, if necessary, they can be used with caution in patients with a history of failure who are well compensated, usually with digitalis and diuretics. Beta-adrenergic blocking agents do not abolish the inotropic action of digitalis on heart muscle.
IN PATIENTS WITHOUT A HISTORY OF HEART FAILURE, continued use of beta-blockers can, in some cases, lead to cardiac failure. Therefore, at the first sign or symptom of heart failure, the patient should be digitalized and/ or treated with diuretics, and the response observed closely, or nadolol should be discontinued (gradually, if possible).
Exacerbation of Ischemic Heart Disease Following Abrupt Withdrawal
Hypersensitivity to catecholamines has been observed in patients withdrawn from beta-blocker therapy; exacerbation of angina and, in some cases, myocardial infarction have occurred after abrupt discontinuation of such therapy. When discontinuing chronically administered nadolol, particularly in patients with ischemic heart disease, the dosage should be gradually reduced over a period of one to two weeks and the patient should be carefully monitored. If angina markedly worsens or acute coronary insufficiency develops, nadolol administration should be reinstituted promptly, at least temporarily, and other measures appropriate for the management of unstable angina should be taken. Patients should be warned against interruption or discontinuation of therapy without the physician's advice. Because coronary artery disease is common and may be unrecognized, it may be prudent not to discontinue nadolol therapy abruptly even in patients treated only for hypertension.
Nonallergic Bronchospasm (e.g., chronic bronchitis, emphysema)
PATIENTS WITH BRONCHOSPASTIC DISEASES SHOULD IN GENERAL NOT RECEIVE BETA-BLOCKERS. Nadolol should be administered with caution since it may block bronchodilation produced by endogenous or exogenous catecholamine stimulation of beta2 receptors.
Major Surgery
Chronically administered beta-blocking therapy should not be routinely withdrawn prior to major surgery; however, the impaired ability of the heart to respond to reflex adrenergic stimuli may augment the risks of general anesthesia and surgical procedures.
Diabetes and Hypoglycemia
Beta-adrenergic blockade may prevent the appearance of premonitory signs and symptoms (e.g., tachycardia and blood pressure changes) of acute hypoglycemia. This is especially important with labile diabetics. Beta-blockade also reduces the release of insulin in response to hyperglycemia; therefore, it may be necessary to adjust the dose of antidiabetic drugs.
Bendroflumethiazide
Thiazides should be used with caution in severe renal disease. In patients with renal disease, thiazides may precipitate azotemia. Cumulative effects of the drug may develop in patients with impaired renal function.
Thiazides should be used with caution in patients with impaired hepatic function or progressive liver disease, since minor alterations of fluid and electrolyte balance may precipitate hepatic coma.
Sensitivity reactions may occur in patients with or without a history of allergy or bronchial asthma.
The possibility of exacerbation or activation of systemic lupus erythematosus has been reported.
Lithium generally should not be given with diuretics; diuretic agents reduce the renal clearance of lithium and add a high risk of lithium toxicity. Refer to the package insert for lithium preparations before use of such concomitant therapy.
-
PRECAUTIONS
General
Nadolol
Nadolol should be used with caution in patients with impaired renal function (see DOSAGE AND ADMINISTRATION).
Bendroflumethiazide
Periodic determination of serum electrolytes to detect possible electrolyte imbalance should be performed at appropriate intervals.
All patients receiving thiazide therapy should be observed for clinical signs of fluid or electrolyte imbalance, namely: hyponatremia, hypochloremic alkalosis, and hypokalemia. Serum and urine electrolyte determinations are particularly important when the patient is vomiting excessively or receiving parenteral fluids. Warning signs or symptoms of fluid and electrolyte imbalance may include: dryness of the mouth, thirst, weakness, lethargy, drowsiness, restlessness, muscle pains or cramps, muscular fatigue, hypotension, oliguria, tachycardia, and gastrointestinal disturbances, such as nausea and vomiting.
Hypokalemia may develop, especially with brisk diuresis or when severe cirrhosis is present.
Interference with adequate oral electrolyte intake will also contribute to hypokalemia. Hypokalemia can sensitize or exaggerate the response of the heart to the toxic effects of digitalis (e.g., increased ventricular irritability). Concurrent administration of a potassium-sparing diuretic or potassium supplements may be indicated in these patients.
Any chloride deficit is generally mild and usually does not require specific treatment except under extraordinary circumstances (as in liver disease or renal disease). Dilutional hyponatremia may occur in edematous patients in hot weather; appropriate therapy is water restriction, rather than administration of salt, except in rare instances when the hyponatremia is life-threatening. In actual salt depletion, appropriate replacement is the therapy of choice.
Hyperuricemia may occur or frank gout may be precipitated in certain patients receiving thiazide therapy.
Latent diabetes mellitus may become manifest during thiazide administration.
The antihypertensive effect of thiazide diuretics may be enhanced in the postsympathectomy patient.
If progressive renal impairment becomes evident, as indicated by a rising nonprotein nitrogen or blood urea nitrogen (BUN), a careful reappraisal of therapy is necessary with consideration given to withholding or discontinuing diuretic therapy.
Thiazides may decrease serum PBI levels without signs of thyroid disturbance.
Calcium excretion is decreased by thiazides. Pathological changes in the parathyroid gland with hypercalcemia and hypophosphatemia have been observed in a few patients on prolonged thiazide therapy. The common complications of hyperparathyroidism such as renal lithiasis, bone resorption, and peptic ulceration have not been seen. Thiazides should be discontinued before carrying out tests for parathyroid function.
Thiazides have been shown to increase the urinary excretion of magnesium; this may result in hypomagnesemia.
Information for Patients
Patients, especially those with evidence of coronary artery insufficiency, should be warned against interruption or discontinuation of therapy without the physician's advice. Although cardiac failure rarely occurs in properly selected patients, patients being treated with beta-adrenergic blocking agents should be advised to consult the physician at the first sign or symptom of impending failure.
The patient should also be advised of a proper course in the event of an inadvertently missed dose.
The patient should be informed of symptoms that would suggest potential adverse effects and told to report them promptly.
Laboratory Tests
Serum electrolyte levels should be regularly monitored (see WARNINGS, Bendroflumethiazide, also PRECAUTIONS, General, Bendroflumethiazide).
Drug Interactions
Nadolol
When administered concurrently the following drugs may interact with beta-adrenergic receptor blocking agents:
Anesthetics, general-exaggeration of the hypotension induced by general anesthetics (see WARNINGS, Nadolol, Major Surgery).
Antidiabetic drugs (oral agents and insulin)-hypoglycemia or hyperglycemia; adjust dosage of antidiabetic drug accordingly (see WARNINGS, Nadolol, Diabetes and Hypoglycemia).
Catecholamine-depleting drugs (e.g., reserpine)- additive effect; monitor closely for evidence of hypotension and/or excessive bradycardia (e.g., vertigo, syncope, postural hypotension).
Response to Treatment for Anaphylactic Reaction-While taking beta-blockers, patients with a history of severe anaphylactic reaction to a variety of allergens may be more reactive to repeated challenge, either accidental, diagnostic, or therapeutic. Such patients may be unresponsive to the usual doses of epinephrine used to treat allergic reaction.
Bendroflumethiazide
When administered concurrently the following drugs may interact with thiazide diuretics:
Alcohol, barbiturates, or narcotics-potentiation of orthostatic hypotension may occur.
Amphotericin B, corticosteroids, or corticotropin (ACTH)-may intensify electrolyte imbalance, particularly hypokalemia. Monitor potassium levels; use potassium replacements if necessary.
Anticoagulants (oral)-dosage adjustments of anticoagulant medication may be necessary since bendroflumethiazide may decrease their effects.
Antigout medications-dosage adjustments of antigout medication may be necessary since bendroflumethiazide may raise the level of blood uric acid.
Other antihypertensive medications (e.g., ganglionic or peripheral adrenergic blocking agents)-dosage adjustments may be necessary since bendroflumethiazide may potentiate their effects.
Antidiabetic drugs (oral agents and insulin)-since thiazides may elevate blood glucose levels, dosage adjustments of antidiabetic agents may be necessary.
Calcium salts-increased serum calcium levels due to decreased excretion may occur. If calcium must be prescribed monitor serum calcium levels and adjust calcium dosage accordingly.
Cardiac glycosides-enhanced possibility of digitalis toxicity associated with hypokalemia. Monitor potassium levels; use potassium replacement if necessary.
Cholestyramine resin and colestipol HCl-may delay or decrease absorption of bendroflumethiazide. Sulfonamide diuretics should be taken at least one hour before or four to six hours after these medications.
Diazoxide-enhanced hyperglycemic, hyperuricemic, and antihypertensive effects. Be cognizant of possible interaction; monitor blood glucose and serum uric acid levels.
Lithium salts-may enhance lithium toxicity due to reduced renal clearance. Avoid concurrent use; if lithium must be prescribed monitor serum lithium levels and adjust lithium dosage accordingly. (See WARNINGS)
MAO inhibitors-dosage adjustments of one or both agents may be necessary since hypotensive effects are enhanced.
Nondepolarizing muscle relaxants, preanesthetics and anesthetics used in surgery (e.g., tubocurarine chloride and gallamine triethiodide)-effects of these agents may be potentiated; dosage adjustments may be required. Monitor and correct any fluid and electrolyte imbalances prior to surgery if feasible.
Nonsteroidal anti-inflammatory agents-in some patients, the administration of a nonsteroidal anti-inflammatory agent can reduce the diuretic, natriuretic, and antihypertensive effect of loop, potassium-sparing or thiazide diuretics. Therefore, when bendroflumethiazide and nonsteroidal anti-inflammatory agents are used concomitantly, the patient should be observed closely to determine if the desired effect of the diuretic is obtained.
Methenamine-possible decreased effectiveness due to alkalinization of the urine.
Pressor amines (e.g., norepinephrine)-decreased arterial responsiveness, but not sufficient to preclude effectiveness of the pressor agent for therapeutic use. Use caution in patients taking both medications who undergo surgery. Administer preanesthetic and anesthetic agents in reduced dosage, and if possible, discontinue bendroflumethiazide one week prior to surgery.
Probenecid or sulfinpyrazone-increased dosage of these agents may be necessary since bendroflumethiazide may have hyperuricemic effects.
Drug/Laboratory Test Interactions
Bendroflumethiazide may produce false-negative results with the phentolamine and tyramine tests; may interfere with the phenosulfonphthalein test due to decreased excretion; and it may cause diagnostic interference of serum electrolyte levels, blood and urine glucose levels, and a decrease in serum PBI levels without signs of thyroid disturbance.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Nadolol
In chronic oral toxicologic studies (one to two years) in mice, rats, and dogs, nadolol did not produce any significant toxic effects. In two-year oral carcinogenicity studies in rats and mice, nadolol did not produce any neoplastic, preneoplastic, or nonneoplastic pathologic lesions. In fertility and general reproductive performance studies in rats, nadolol caused no adverse effect.
Pregnancy-Teratogenic Effects
Nadolol
Category C
In animal reproduction studies with nadolol, evidence of embryo- and fetotoxicity was found in rabbits, but not in rats or hamsters, at doses 5 to 10 times greater (on a mg/kg basis) than the maximum indicated human dose. No teratogenic potential was observed in any of these species.
There are no adequate and well-controlled studies in pregnant women. Nadolol should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Neonates whose mothers are receiving nadolol at parturition have exhibited bradycardia, hypoglycemia, and associated symptoms.
Bendroflumethiazide
Category C
Animal reproduction studies have not been conducted with bendroflumethiazide. It is also not known whether this drug can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Bendroflumethiazide should be given to a pregnant woman only if clearly needed.
Pregnancy-Nonteratogenic Effects
Thiazides cross the placental barrier and appear in cord blood. The use of thiazides in pregnant women requires that the anticipated benefit be weighed against possible hazards to the fetus. These hazards include fetal or neonatal jaundice, thrombocytopenia, and possibly other adverse reactions which have occurred in the adult.
Nursing Mothers
Both nadolol and bendroflumethiazide are excreted in human milk. Because of the potential for serious adverse reactions in nursing infants from both drugs, a decision should be made whether to discontinue nursing or to discontinue therapy taking into account the importance of nadolol and bendroflumethiazide tablets to the mother.
Geriatric Use
Clinical studies of nadolol and bendroflumethiazide tablets did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. This drug is known to be substantially excreted by the kidney, and the risk of toxic reaction to this drug may be greater in patients with impaired function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
-
ADVERSE REACTIONS
Nadolol
Most adverse effects have been mild and transient and have rarely required withdrawal of therapy.
Cardiovascular-Bradycardia with heart rates of less than 60 beats per minute occurs commonly, and heart rates below 40 beats per minute and/or symptomatic bradycardia were seen in about 2 of 100 patients. Symptoms of peripheral vascular insufficiency, usually of the Raynaud type, have occurred in approximately 2 of 100 patients. Cardiac failure, hypotension, and rhythm/conduction disturbances have each occurred in about 1 of 100 patients. Single instances of first degree and third degree heart block have been reported; intensification of AV block is a known effect of beta-blockers (see also CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS).
Central Nervous System-Dizziness or fatigue has been reported in approximately 2 of 100 patients; paresthesias, sedation, and change in behavior have each been reported in approximately 6 of 1000 patients.
Respiratory-Bronchospasm has been reported in approximately 1 of 1000 patients (see CONTRAINDICATIONS and WARNINGS).
Gastrointestinal-Nausea, diarrhea, abdominal discomfort, constipation, vomiting, indigestion, anorexia, bloating, and flatulence have been reported in 1 to 5 of 1000 patients.
Miscellaneous-Each of the following has been reported in 1 to 5 of 1000 patients: rash; pruritus; headache; dry mouth, eyes, or skin; impotence or decreased libido; facial swelling; weight gain; slurred speech; cough; nasal stuffiness; sweating; tinnitus; blurred vision. Reversible alopecia has been reported infrequently.
The following adverse reactions have been reported in patients taking nadolol and/or other beta-adrenergic blocking agents, but no causal relationship to nadolol has been established.
Central Nervous System-Reversible mental depression progressing to catatonia; visual disturbances; hallucinations; an acute reversible syndrome characterized by disorientation for time and place, short-term memory loss, emotional lability with slightly clouded sensorium, and decreased performance on neuropsychometrics.
Gastrointestinal-Mesenteric arterial thrombosis; ischemic colitis; elevated liver enzymes.
Hematologic-Agranulocytosis; thrombocytopenic or nonthrombocytopenic purpura.
Allergic-Fever combined with aching and sore throat; laryngospasm; respiratory distress.
Miscellaneous-Pemphigoid rash; hypertensive reaction in patients with pheochromocytoma; sleep disturbances; Peyronie's disease.
The oculomucocutaneous syndrome associated with the beta-blocker practolol has not been reported with nadolol.
Bendroflumethiazide
Gastrointestinal-Nausea, vomiting, cramping and anorexia are not uncommon; diarrhea, constipation, gastric irritation, abdominal bloating, jaundice (intrahepatic cholestatic jaundice), hepatitis, and sialadenitis occasionally occur; and pancreatitis has been reported.
Central Nervous System-Dizziness, vertigo, paresthesia, headache, and xanthopsia occasionally occur.
Hematologic-Leukopenia, agranulocytosis, thrombocytopenia, hemolytic anemia, and aplastic anemia have been reported.
Dermatologic-Hypersensitivity-Purpura, exfoliative dermatitis, pruritus, ecchymosis, urticaria, necrotizing angiitis (vasculitis, cutaneous vasculitis), respiratory distress including pneumonitis, fever, and anaphylactic reactions occasionally occur; photosensitivity and rash have been reported.
Cardiovascular-Orthostatic hypotension may occur and may be potentiated by coadministration with certain other drugs (e.g., alcohol, barbiturates, narcotics, other antihypertensive medications, etc.; see PRECAUTIONS, Drug Interactions).
Other-Muscle spasm, weakness, or restlessness is not uncommon; hyperglycemia, glycosuria, metabolic acidosis in diabetic patients, hyperuricemia, allergic glomerulonephritis, and transient blurred vision occasionally occur.
Whenever adverse reactions are moderate or severe, thiazide dosage should be reduced or therapy withdrawn.
To report SUSPECTED ADVERSE REACTIONS, contact IMPAX Laboratories, Inc. at 1-800-934-6729 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
-
OVERDOSAGE
In the event of overdosage, nadolol may cause excessive bradycardia, cardiac failure, hypotension, or bronchospasm.
In addition to the expected diuresis, overdosage of bendroflumethiazide may produce varying degrees of lethargy which may progress to coma with minimal depression of respiration and cardiovascular function and without significant serum electrolyte changes or dehydration. The mechanism of thiazide-induced CNS depression is unknown. Gastrointestinal irritation may occur. Transitory increase in BUN has been reported, and serum electrolyte changes may occur, especially in patients with impaired renal function.
Treatment
Nadolol can be removed from the general circulation by hemodialysis. In determining the duration of corrective therapy, note must be taken of the long duration of the effect of nadolol. In addition to gastric lavage, the following measures should be employed, as appropriate.
Excessive Bradycardia
Administer atropine (0.25 to 1.0 mg). If there is no response to vagal blockade, administer isoproterenol cautiously.
Cardiac Failure
Administer a digitalis glycoside and diuretic. It has been reported that glucagon may also be useful in this situation.
-
DOSAGE AND ADMINISTRATION
DOSAGE MUST BE INDIVIDUALIZED (SEE INDICATIONS). NADOLOL AND BENDROFLUMETHIAZIDE TABLETS MAY BE ADMINISTERED WITHOUT REGARD TO MEALS.
Bendroflumethiazide is usually given at a dose of 5 mg daily. The usual initial dose of nadolol is 40 mg once daily whether used alone or in combination with a diuretic. Bendroflumethiazide in nadolol and bendroflumethiazide tablets is 30 percent more bioavailable than that of 5 mg bendroflumethiazide tablets. Conversion from 5 mg bendroflumethiazide tablets to nadolol and bendroflumethiazide tablets represents a 30 percent increase in dose of bendroflumethiazide.
The initial dose of nadolol and bendroflumethiazide tablets may therefore be the 40 mg/5 mg tablet once daily. When the antihypertensive response is not satisfactory, the dose may be increased by administering the 80 mg/5 mg tablet once daily.
When necessary, another antihypertensive agent may be added gradually beginning with 50 percent of the usual recommended starting dose to avoid an excessive fall in blood pressure.
Dosage Adjustment in Renal Failure
Absorbed nadolol is excreted principally by the kidneys and, although nonrenal elimination does occur, dosage adjustments are necessary in patients with renal impairment. The following dose intervals are recommended:
Creatinine Clearance
(mL/min/1.73 m2)Dosage Interval
(hours)>50 24 31 to 50 24 to 36 10 to 30 24 to 48 <10 40 to 60 -
HOW SUPPLIED
Nadolol and bendroflumethiazide tablets, 40 mg/5 mg, contain 40 mg nadolol combined with 5 mg bendroflumethiazide, and are white to off-white, round, bi-convex tablets debossed with "G" bisect "531" on one side and plain on the other.
Bottles of 100 NDC: 0115-5311-01 Bottles of 500 NDC: 0115-5311-02 Nadolol and bendroflumethiazide tablets, 80 mg/5 mg, contain 80 mg nadolol combined with 5 mg bendroflumethiazide, and are white to off-white, round, bi-convex tablets debossed with "G" bisect "532" on one side and plain on the other.
Bottles of 100 NDC: 0115-5322-01 Bottles of 500 NDC: 0115-5322-02 - SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 40 mg/5 mg Bottle Label
- PRINCIPAL DISPLAY PANEL - 80 mg/5 mg Bottle Label
-
INGREDIENTS AND APPEARANCE
NADOLOL AND BENDROFLUMETHIAZIDE
nadolol and bendroflumethiazide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0115-5311 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NADOLOL (UNII: FEN504330V) (NADOLOL - UNII:FEN504330V) NADOLOL 40 mg BENDROFLUMETHIAZIDE (UNII: 5Q52X6ICJI) (BENDROFLUMETHIAZIDE - UNII:5Q52X6ICJI) BENDROFLUMETHIAZIDE 5 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) STARCH, CORN (UNII: O8232NY3SJ) TALC (UNII: 7SEV7J4R1U) Product Characteristics Color WHITE (white to off-white) Score 2 pieces Shape ROUND (round, bi-convex tablets) Size 9mm Flavor Imprint Code G;531 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0115-5311-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 03/30/2007 2 NDC: 0115-5311-02 500 in 1 BOTTLE; Type 0: Not a Combination Product 03/30/2007 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077833 03/30/2007 NADOLOL AND BENDROFLUMETHIAZIDE
nadolol and bendroflumethiazide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0115-5322 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NADOLOL (UNII: FEN504330V) (NADOLOL - UNII:FEN504330V) NADOLOL 80 mg BENDROFLUMETHIAZIDE (UNII: 5Q52X6ICJI) (BENDROFLUMETHIAZIDE - UNII:5Q52X6ICJI) BENDROFLUMETHIAZIDE 5 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) STARCH, CORN (UNII: O8232NY3SJ) TALC (UNII: 7SEV7J4R1U) Product Characteristics Color WHITE (white to off-white) Score 2 pieces Shape ROUND (round, bi-convex tablets) Size 10mm Flavor Imprint Code G;532 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0115-5322-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 03/30/2007 2 NDC: 0115-5322-02 500 in 1 BOTTLE; Type 0: Not a Combination Product 03/30/2007 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077833 03/30/2007 Labeler - Impax Generics (079832487) Establishment Name Address ID/FEI Business Operations Impax Laboratories, Inc 790947167 MANUFACTURE(0115-5311, 0115-5322) Establishment Name Address ID/FEI Business Operations Packaging Coordinators, LLC 078525133 REPACK(0115-5311, 0115-5322)


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.