IDARUBICIN HYDROCHLORIDE injection, solution
Idarubicin Hydrochloride by
Drug Labeling and Warnings
Idarubicin Hydrochloride by is a Prescription medication manufactured, distributed, or labeled by Teva Parenteral Medicines, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- SPL UNCLASSIFIED SECTION
-
BOXED WARNING
(What is this?)
WARNINGS
- Idarubicin hydrochloride injection should be given slowly into a freely flowing intravenous infusion. It must never be given intramuscularly or subcutaneously. Severe local tissue necrosis can occur if there is extravasation during administration.
- As is the case with other anthracyclines the use of idarubicin hydrochloride injection can cause myocardial toxicity leading to congestive heart failure. Cardiac toxicity is more common in patients who have received prior anthracyclines or who have pre-existing cardiac disease.
- As is usual with antileukemic agents, severe myelosuppression occurs when idarubicin hydrochloride injection is used at effective therapeutic doses.
- It is recommended that idarubicin hydrochloride injection be administered only under the supervision of a physician who is experienced in leukemia chemotherapy and in facilities with laboratory and supportive resources adequate to monitor drug tolerance and protect and maintain a patient compromised by drug toxicity. The physician and institution must be capable of responding rapidly and completely to severe hemorrhagic conditions and/or overwhelming infection.
- Dosage should be reduced in patients with impaired hepatic or renal function. (See DOSAGE AND ADMINISTRATION.)
-
DESCRIPTION
Idarubicin Hydrochloride Injection USP contains idarubicin hydrochloride, USP and is a sterile, semi-synthetic, preservative-free solution (PFS) antineoplastic anthracycline for intravenous use. Chemically, idarubicin hydrochloride, USP is 5, 12-Naphthacenedione, 9-acetyl-7-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-7,8,9,10-tetrahydro-6,9,11-trihydroxyhydrochloride, (7S-cis). The structural formula is as follows:
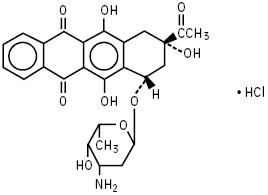
C26H27NO9HCl M.W. 533.95
Idarubicin Hydrochloride Injection USP is a sterile, clear, orange-red, isotonic parenteral preservative-free solution, available in 5 mL (5 mg), 10 mL (10 mg) and 20 mL (20 mg) single-dose-only vials.
Each mL contains idarubicin hydrochloride, USP 1 mg and the following inactive ingredients: glycerin, USP 25 mg and water for injection, USP q.s. Hydrochloric acid, NF and/or sodium hydroxide, NF is used to adjust pH to a target of 3.5.
-
CLINICAL PHARMACOLOGY
Mechanism of Action
Idarubicin hydrochloride is a DNA-intercalating analog of daunorubicin which has an inhibitory effect on nucleic acid synthesis and interacts with the enzyme topoisomerase II. The absence of a methoxy group at position 4 of the anthracycline structure gives the compound a high lipophilicity which results in an increased rate of cellular uptake compared with other anthracyclines.
Pharmacokinetics
General Pharmacokinetics
Pharmacokinetic studies have been performed in adult leukemia patients with normal renal and hepatic function following intravenous administration of 10 to 12 mg/m2 of idarubicin daily for 3 to 4 days as a single agent or combined with cytarabine. The plasma concentrations of idarubicin are best described by a two or three compartment open model. The elimination rate of idarubicin from plasma is slow with an estimated mean terminal half-life of 22 hours (range, 4 to 48 hours) when used as a single agent and 20 hours (range, 7 to 38 hours) when used in combination with cytarabine. The elimination of the primary active metabolite, idarubicinol, is considerably slower than that of the parent drug with an estimated mean terminal half-life that exceeds 45 hours; hence, its plasma levels are sustained for a period greater than 8 days.
Distribution
The disposition profile shows a rapid distributive phase with a very high volume of distribution presumably reflecting extensive tissue binding. Studies of cellular (nucleated blood and bone marrow cells) drug concentrations in leukemia patients have shown that peak cellular idarubicin concentrations are reached a few minutes after injection. Concentrations of idarubicin and idarubicinol in nucleated blood and bone marrow cells are more than a hundred times the plasma concentrations. Idarubicin disappearance rates in plasma and cells were comparable with a terminal half-life of about 15 hours. The terminal half-life of idarubicinol in cells was about 72 hours. The extent of drug and metabolite accumulation predicted in leukemia patients for Days 2 and 3 of dosing, based on the mean plasma levels and half-life obtained after the first dose, is 1.7- and 2.3-fold, respectively, and suggests no change in kinetics following a daily × 3 regimen. The percentages of idarubicin and idarubicinol bound to human plasma proteins averaged 97% and 94%, respectively, at concentrations similar to maximum plasma levels obtained in the pharmacokinetic studies. The binding is concentration independent. The plasma clearance is twice the expected hepatic plasma flow indicating extensive extrahepatic metabolism.
Pharmacokinetics in Special Populations
Pediatric Patients
Idarubicin studies in pediatric leukemia patients, at doses of 4.2 to 13.3 mg/m2/day × 3, suggest dose independent kinetics. There is no difference between the half-lives of the drug following daily × 3 or weekly × 3 administration. Cerebrospinal fluid (CSF) levels of idarubicin and idarubicinol were measured in pediatric leukemia patients treated intravenously. Idarubicin was detected in 2 of 21 CSF samples (0.14 and 1.57 ng/mL), while idarubicinol was detected in 20 of these 21 CSF samples obtained 18 to 30 hours after dosing (mean = 0.51 ng/mL; range, 0.22 to 1.05 ng/mL). The clinical relevance of these findings is unknown.
Hepatic and Renal Impairment
The pharmacokinetics of idarubicin have not been evaluated in leukemia patients with hepatic impairment. It is expected that in patients with moderate or severe hepatic dysfunction, the metabolism of idarubicin may be impaired and lead to higher systemic drug levels. The disposition of idarubicin may be also affected by renal impairment. Therefore, a dose reduction should be considered in patients with hepatic and/or renal impairment (see DOSAGE AND ADMINISTRATION).
-
CLINICAL STUDIES
Four prospective randomized studies, three U.S. and one Italian, have been conducted to compare the efficacy and safety of idarubicin (IDR) to that of daunorubicin (DNR), each in combination with cytarabine as induction therapy in previously untreated adult patients with acute myeloid leukemia (AML). These data are summarized in the following table and demonstrate significantly greater complete remission rates for the IDR regimen in two of the three U.S. studies and significantly longer overall survival for the IDR regimen in two of the three U.S. studies.
Inductiona Regimen Dose in mg/m2 – Daily x 3 Days
Complete Remission Rate, All Pts Randomized
Median Survival (Days) All Pts Randomized
IDR
DNR
IDR
DNR
IDR
DNR
U.S. (IND Studies)
1. MSKCC
12b
50b
51/65*
38/65
508*
435
(Age ≤ 60 years)
(78%)
(58%)
2. SEG
12c
45c
76/111*
65/119
328
277
(Age ≥ 15 years)
(69%)
(55%)
3. U.S. Multicenter
13c
45c
68/101
66/113
393*
281
(Age ≥ 18 years)
(67%)
(58%)
Foreign (non-IND study)
GIMEMA
12c
45c
49/124
49/125
87
169
(Age ≥ 55 years)
(40%)
(39%)
*Overall p < 0.05, unadjusted for prognostic factors or multiple endpoints
Abbreviations: MSKCC=Memorial Sloan Kettering Cancer Center; SEG= Southeastern Cancer Study Group; GIMEMA= Gruppo Italiano Malattie Ematologiche Maligne dell’ Adulto; IDR=Idarubicin; DNR=Daunorubicin; Pts=Patients; IND=Investigational New Drug
a Patients who had persistent leukemia after the first induction course received a second course
b Cytarabine 25 mg/m2 bolus IV followed by 200 mg/m2 daily x 5 days by continuous infusion
c Cytarabine 100 mg/m2 daily x 7 days by continuous infusion
There is no consensus regarding optional regimens to be used for consolidation; however, the following consolidation regimens were used in U.S. controlled trials. Patients received the same anthracycline for consolidation as was used for induction.
Studies 1 and 3 utilized 2 courses of consolidation therapy consisting of idarubicin 12 or 13 mg/m2 daily for 2 days, respectively (or DNR 50 or 45 mg/m2 daily for 2 days), and cytarabine, either 25 mg/m2 by IV bolus followed by 200 mg/m2 daily by continuous infusion for 4 days (Study 1), or 100 mg/m2 daily for 5 days by continuous infusion (Study 3). A rest period of 4 to 6 weeks is recommended prior to initiation of consolidation and between the courses. Hematologic recovery is mandatory prior to initiation of each consolidation course.
Study 2 utilized 3 consolidation courses, administered at intervals of 21 days or upon hematologic recovery. Each course consisted of idarubicin 15 mg/m2 IV for 1 dose (or DNR 50 mg/m2 IV for 1 dose), cytarabine 100 mg/m2 every 12 hours for 10 doses and 6-thioguanine 100 mg/m2 orally for 10 doses. If severe myelosuppression occurred, subsequent courses were given with 25% reduction in the doses of all drugs. In addition, this study included 4 courses of maintenance therapy (2 days of the same anthracycline as was used in induction and 5 days of cytarabine).
Toxicities and duration of aplasia were similar during induction on the 2 arms in the U.S. studies except for an increase in mucositis on the IDR arm in one study. During consolidation, duration of aplasia on the IDR arm was longer in all three studies and mucositis was more frequent in two studies. During consolidation, transfusion requirements were higher on the IDR arm in the two studies in which they were tabulated, and patients on the IDR arm in Study 3 spent more days on IV antibiotics (Study 3 used a higher dose of idarubicin).
The benefit of consolidation and maintenance therapy in prolonging the duration of remission and survival is not proven.
Intensive maintenance with idarubicin is not recommended in view of the considerable toxicity (including deaths in remission) experienced by patients during the maintenance phase of Study 2.
A higher induction death rate was noted in patients on the IDR arm in the Italian trial. Since this was not noted in patients of similar age in the U.S. trials, one may speculate that it was due to a difference in the level of supportive care.
- INDICATIONS AND USAGE
-
WARNINGS
Idarubicin is intended for administration under the supervision of a physician who is experienced in leukemia chemotherapy.
Idarubicin is a potent bone marrow suppressant. Idarubicin should not be given to patients with pre-existing bone marrow suppression induced by previous drug therapy or radiotherapy unless the benefit warrants the risk.
Severe myelosuppression will occur in all patients given a therapeutic dose of this agent for induction, consolidation or maintenance. Careful hematologic monitoring is required. Deaths due to infection and/or bleeding have been reported during the period of severe myelosuppression. Facilities with laboratory and supportive resources adequate to monitor drug tolerability and protect and maintain a patient compromised by drug toxicity should be available. It must be possible to treat rapidly and completely a severe hemorrhagic condition and/or a severe infection.
Pre-existing heart disease and previous therapy with anthracyclines at high cumulative doses or other potentially cardiotoxic agents are co-factors for increased risk of idarubicin-induced cardiac toxicity and the benefit to risk ratio of idarubicin therapy in such patients should be weighed before starting treatment with idarubicin.
Myocardial toxicity as manifested by potentially fatal congestive heart failure, acute life-threatening arrhythmias or other cardiomyopathies may occur following therapy with idarubicin. Appropriate therapeutic measures for the management of congestive heart failure and/or arrhythmias are indicated.
Cardiac function should be carefully monitored during treatment in order to minimize the risk of cardiac toxicity of the type described for other anthracycline compounds. The risk of such myocardial toxicity may be higher following concomitant or previous radiation to the mediastinal-pericardial area or in patients with anemia, bone marrow depression, infections, leukemic pericarditis and/or myocarditis, active or dormant cardiovascular disease, previous therapy with other anthracyclines or anthracenediones, and concomitant use of drugs with the ability to suppress cardiac contractility or cardiotoxic drugs (e.g., trastuzumab, cyclophosphamide and paclitaxel). Due to the increased risk of cardiotoxicity, avoid concomitant use of idarubicin hydrochloride injection until the cardiotoxic agent has been discontinued for at least 5 half-lives, and specifically avoid idarubicin hydrochloride injection for up to 7 months after stopping trastuzumab.
While there are no reliable means for predicting congestive heart failure, cardiomyopathy induced by anthracyclines is usually associated with a decrease of the left ventricular ejection fraction (LVEF) from pretreatment baseline values.
Since hepatic and/or renal function impairment can affect the disposition of idarubicin, liver and kidney function should be evaluated with conventional clinical laboratory tests (using serum bilirubin and serum creatinine as indicators) prior to and during treatment. In a number of Phase III clinical trials, treatment was not given if bilirubin and/or creatinine serum levels exceeded 2 mg%. However, in one Phase III trial, patients with bilirubin levels between 2.6 and 5 mg% received the anthracycline with a 50% reduction in dose. Dose reduction of idarubicin should be considered if the bilirubin and/or creatinine levels are above the normal range. (See DOSAGE AND ADMINISTRATION.)
Pregnancy
Idarubicin was embryotoxic and teratogenic in the rat at a dose of 1.2 mg/m2/day or one tenth the human dose, which was nontoxic to dams. Idarubicin was embryotoxic but not teratogenic in the rabbit even at a dose of 2.4 mg/m2/day or two tenths the human dose, which was toxic to dams. There is no conclusive information about idarubicin adversely affecting human fertility or causing teratogenesis. There has been one report of a fetal fatality after maternal exposure to idarubicin during the second trimester.
There are no adequate and well-controlled studies in pregnant women. If idarubicin is to be used during pregnancy, or if the patient becomes pregnant during therapy, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid pregnancy.
-
PRECAUTIONS
General
Therapy with idarubicin requires close observation of the patient and careful laboratory monitoring. Hyperuricemia secondary to rapid lysis of leukemic cells may be induced. Appropriate measures must be taken to prevent hyperuricemia and to control any systemic infection before beginning therapy.
Extravasation of idarubicin can cause severe local tissue necrosis. Extravasation may occur with or without an accompanying stinging or burning sensation even if blood returns well on aspiration of the infusion needle. If signs or symptoms of extravasation occur the injection or infusion should be terminated immediately and restarted in another vein. (See DOSAGE AND ADMINISTRATION.)
Laboratory Tests
Frequent complete blood counts and monitoring of hepatic and renal function tests are recommended.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Formal long-term carcinogenicity studies have not been conducted with idarubicin. Idarubicin and related compounds have been shown to have mutagenic and carcinogenic properties when tested in experimental models (including bacterial systems, mammalian cells in culture and female Sprague-Dawley rats).
In male dogs given 1.8 mg/m2/day 3 times/week (about one seventh the weekly human dose on a mg/m2 basis) for 13 weeks, or 3 times the human dose, testicular atrophy was observed with inhibition of spermatogenesis and sperm maturation with few or no mature sperm. These effects were not readily reversed after a recovery of 8 weeks.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from idarubicin, mothers should discontinue nursing prior to taking this drug.
Geriatric Use
Patients over 60 years of age who were undergoing induction therapy experienced congestive heart failure, serious arrhythmias, chest pain, myocardial infarction, and asymptomatic declines in LVEF more frequently than younger patients (see ADVERSE REACTIONS).
-
ADVERSE REACTIONS
Approximately 550 patients with AML have received idarubicin in combination with cytarabine in controlled clinical trials worldwide. In addition, over 550 patients with acute leukemia have been treated in uncontrolled trials utilizing idarubicin as a single agent or in combination. The table below lists the adverse experiences reported in U.S. Study 2 (see CLINICAL STUDIES) and is representative of the experiences in other studies. These adverse experiences constitute all reported or observed experiences, including those not considered to be drug related. Patients undergoing induction therapy for AML are seriously ill due to their disease, are receiving multiple transfusions, and concomitant medications including potentially toxic antibiotics and antifungal agents. The contribution of the study drug to the adverse experience profile is difficult to establish.
Induction Phase
Percentage of Patients
Adverse Experiences
IDR
(N=110)
DNR
(N=118)
Infection
95%
97%
Nausea & Vomiting
82%
80%
Hair Loss
77%
72%
Abdominal Cramps/Diarrhea
73%
68%
Hemorrhage
63%
65%
Mucositis
50%
55%
Dermatologic
46%
40%
Mental Status
41%
34%
Pulmonary-Clinical
39%
39%
Fever (not elsewhere classified)
26%
28%
Headache
20%
24%
Cardiac-Clinical
16%
24%
Neurologic-Peripheral Nerves
7%
9%
Pulmonary Allergy
2%
4%
Seizure
4%
5%
Cerebellar
4%
4%
Abbreviations: IDR=Idarubicin; DNR=Daunorubicin
The duration of aplasia and incidence of mucositis were greater on the IDR arm than the DNR arm, especially during consolidation in some U.S. controlled trials (see CLINICAL STUDIES).
The following information reflects experience based on U.S. controlled clinical trials.
Myelosuppression
Severe myelosuppression is the major toxicity associated with idarubicin therapy, but this effect of the drug is required in order to eradicate the leukemic clone. During the period of myelosuppression, patients are at risk of developing infection and bleeding which may be life-threatening or fatal.
Gastrointestinal
Nausea and/or vomiting, mucositis, abdominal pain and diarrhea were reported frequently, but were severe (equivalent to WHO Grade 4) in less than 5% of patients. Severe enterocolitis with perforation has been reported rarely. The risk of perforation may be increased by instrumental intervention. The possibility of perforation should be considered in patients who develop severe abdominal pain and appropriate steps for diagnosis and management should be taken.
Dermatologic
Alopecia was reported frequently and dermatologic reactions including generalized rash, urticaria and a bullous erythrodermatous rash of the palms and soles have occurred. The dermatologic reactions were usually attributed to concomitant antibiotic therapy. Local reactions including hives at the injection site have been reported. Recall of skin reaction due to prior radiotherapy has occurred with idarubicin administration.
Hepatic and Renal
Changes in hepatic and renal function tests have been observed. These changes were usually transient and occurred in the setting of sepsis and while patients were receiving potentially hepatotoxic and nephrotoxic antibiotics and antifungal agents. Severe changes in renal function (equivalent to WHO Grade 4) occurred in no more than 1% of patients, while severe changes in hepatic function (equivalent to WHO Grade 4) occurred in less than 5% of patients.
Cardiac
Congestive heart failure (frequently attributed to fluid overload), serious arrhythmias including atrial fibrillation, chest pain, myocardial infarction and asymptomatic declines in LVEF have been reported in patients undergoing induction therapy for AML. Myocardial insufficiency and arrhythmias were usually reversible and occurred in the setting of sepsis, anemia and aggressive intravenous fluid administration. The events were reported more frequently in patients over age 60 years and in those with pre-existing cardiac disease.
To report SUSPECTED ADVERSE REACTIONS, contact Teva Pharmaceuticals USA, Inc. at 1-888-838-2872 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
-
OVERDOSAGE
There is no known antidote to idarubicin. Two cases of fatal overdosage in patients receiving therapy for AML have been reported. The doses were 135 mg/m2 over 3 days and 45 mg/m2 of idarubicin and 90 mg/m2 of daunorubicin over a three day period.
It is anticipated that overdosage with idarubicin will result in severe and prolonged myelosuppression and possibly in increased severity of gastrointestinal toxicity. Adequate supportive care including platelet transfusions, antibiotics and symptomatic treatment of mucositis is required. The effect of acute overdose on cardiac function is not fully known, but severe arrhythmia occurred in 1 of the 2 patients exposed. It is anticipated that very high doses of idarubicin may cause acute cardiac toxicity and may be associated with a higher incidence of delayed cardiac failure.
Disposition studies with idarubicin in patients undergoing dialysis have not been carried out. The profound multicompartment behavior, extensive extravascular distribution and tissue binding, coupled with the low unbound fraction available in the plasma pool make it unlikely that therapeutic efficacy or toxicity would be altered by conventional peritoneal or hemodialysis.
-
DOSAGE AND ADMINISTRATION
(See WARNINGS)
For induction therapy in adult patients with AML the following dose schedule is recommended:
Idarubicin hydrochloride injection 12 mg/m2 daily for 3 days by slow (10 to 15 min) intravenous injection in combination with cytarabine. The cytarabine may be given as 100 mg/m2 daily by continuous infusion for 7 days or as cytarabine 25 mg/m2 intravenous bolus followed by cytarabine 200 mg/m2 daily for 5 days continuous infusion. In patients with unequivocal evidence of leukemia after the first induction course, a second course may be administered. Administration of the second course should be delayed in patients who experience severe mucositis, until recovery from this toxicity has occurred, and a dose reduction of 25% is recommended. In patients with hepatic and/or renal impairment, a dose reduction of idarubicin hydrochloride injection should be considered. Idarubicin hydrochloride injection should not be administered if the bilirubin level exceeds 5 mg%. (See WARNINGS.)
The benefit of consolidation in prolonging the duration of remissions and survival is not proven. There is no consensus regarding optional regimens to be used for consolidation. (See CLINICAL STUDIES for doses used in U.S. Clinical studies.)
Preparation and Administration Precautions
Caution in handling the solution must be exercised as skin reactions associated with idarubicin hydrochloride injection may occur. Skin accidentally exposed to idarubicin hydrochloride injection should be washed thoroughly with soap and water and if the eyes are involved, standard irrigation techniques should be used immediately. The use of goggles, gloves, and protective gowns is recommended during preparation and administration of the drug.
Care in the administration of idarubicin hydrochloride injection will reduce the chance of perivenous infiltration. It may also decrease the chance of local reactions such as urticaria and erythematous streaking. During intravenous administration of idarubicin hydrochloride injection extravasation may occur with or without an accompanying stinging or burning sensation even if blood returns well on aspiration of the infusion needle. If any signs or symptoms of extravasation have occurred, the injection or infusion should be immediately terminated and restarted in another vein. If it is known or suspected that subcutaneous extravasation has occurred, it is recommended that intermittent ice packs (1/2 hour immediately, then 1/2 hour 4 times per day for 3 days) be placed over the area of extravasation and that the affected extremity be elevated. Because of the progressive nature of extravasation reactions, the area of injection should be frequently examined and plastic surgery consultation obtained early if there is any sign of a local reaction such as pain, erythema, edema or vesication. If ulceration begins or there is severe persistent pain at the site of extravasation, early wide excision of the involved area should be considered.
Idarubicin hydrochloride injection should be administered slowly (over 10 to 15 minutes) into the tubing of a freely running intravenous infusion of Sodium Chloride Injection, USP (0.9%) or 5% Dextrose Injection, USP. The tubing should be attached to a Butterfly needle or other suitable device and inserted preferably into a large vein.
Incompatibility
Unless specific compatibility data are available, idarubicin hydrochloride injection should not be mixed with other drugs. Precipitation occurs with heparin. Prolonged contact with any solution of an alkaline pH will result in degradation of the drug.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and containers permit.
-
HOW SUPPLIED
Each mL contains Idarubicin Hydrochloride, USP 1 mg.
NDC Number Idarubicin Hydrochloride Injection USP 0703-4154-11
5 mg single dose vial (1 mg/mL) packaged individually
0703-4155-11
10 mg single dose vial (1 mg/mL) packaged individually
0703-4156-11
20 mg single dose vial (1 mg/mL) packaged individually
Sterile single dose only, contains no preservative.
-
REFERENCES
- ONS Clinical Practice Committee. Cancer Chemotherapy Guidelines and Recommendations for Practice. Pittsburgh, PA: Oncology Nursing Society. 1999: 32-41.
- Recommendations for the Safe Handling of Parenteral Antineoplastic Drugs. Washington, DC; Division of Safety, Clinical Center Pharmacy Department and Cancer Nursing Services, National Institutes of Health; 1992. US Department of Health and Human Services, Public Health Service Publication NIH 92-2621.
- AMA Council on Scientific Affairs. Guidelines for Handling Parenteral Antineoplastics. JAMA. 1985; 253: 1590-1591.
- National Study Commission on Cytotoxic Exposure - Recommendations for Handling Cytotoxic Agents. 1987. Available from Louis P. Jeffrey, Sc.D., Chairman, National Study Commission on Cytotoxic Exposure, Massachusetts College of Pharmacy and Allied Health Sciences, 179 Longwood Avenue, Boston, MA 02115.
- Clinical Oncological Society of Australia: Guidelines and Recommendations for Safe Handling of Antineoplastic Agents. Med J Australia. 1983; 1:426-428.
- Jones RB, Frank R, Mass T. Safe Handling of Chemotherapeutic Agents: A Report from the Mount Sinai Medical Center. CA Cancer J Clin. 1983; 33: 258-263.
- American Society of Hospital Pharmacists. ASHP Technical Assistance Bulletin on Handling Cytotoxic and Hazardous Drugs. Am J Hosp Pharm. 1990; 47:1033-1049.
- Controlling Occupational Exposure to Hazardous Drugs (OSHA Work-Practice Guidelines). Am J Health-Syst Pharm. 1996; 53: 1669-1685.
Teva Pharmaceuticals USA, Inc.
North Wales, PA 19454
Rev. C 8/2019
-
Package/Label Display Panel
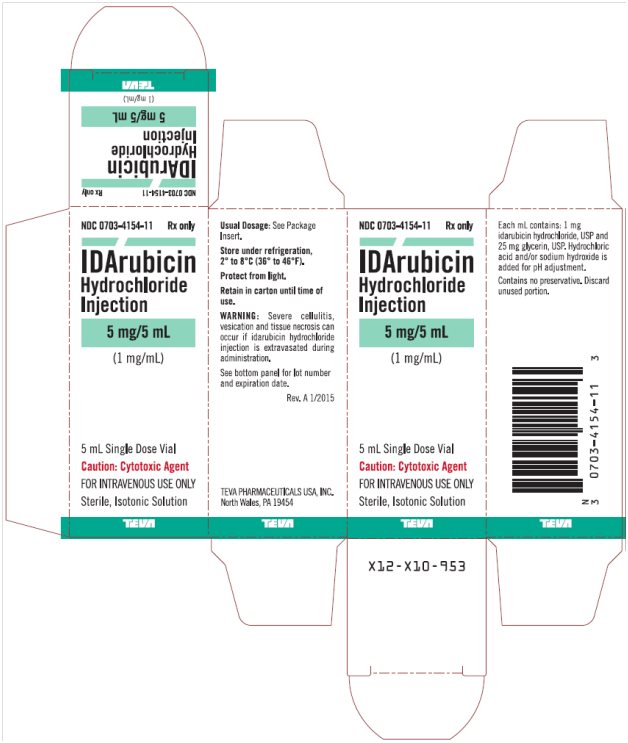
Idarubicin Hydrochloride Injection 1 mg/mL, 5 mL Single Dose Vial, Carton Text
NDC: 0703-4154-11 Rx only
IDArubicin
Hydrochloride
Injection
5 mg/5 mL
(1 mg/mL)
5 mL Single Dose Vial
Caution: Cytotoxic Agent
FOR INTRAVENOUS USE ONLY
Sterile, Isotonic Solution
TEVA
-
Package/Label Display Panel
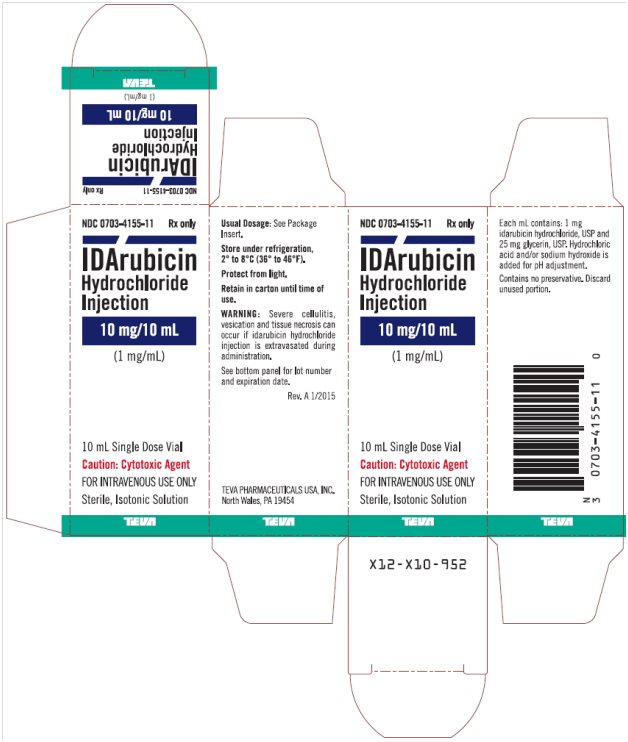
Idarubicin Hydrochloride Injection 1 mg/mL, 10 mL Single Dose Vial, Carton Text
NDC: 0703-4155-11 Rx only
IDArubicin
Hydrochloride
Injection
10 mg/10 mL
(1 mg/mL)
10 mL Single Dose Vial
Caution: Cytotoxic Agent
FOR INTRAVENOUS USE ONLY
Sterile, Isotonic Solution
TEVA
-
Package/Label Display Panel

Idarubicin Hydrochloride Injection 1 mg/mL, 20 mL Single Dose Vial, Carton Text
NDC: 0703-4156-11 Rx only
IDArubicin
Hydrochloride
Injection
20 mg/20 mL
(1 mg/mL)
20 mL Single Dose Vial
Caution: Cytotoxic Agent
FOR INTRAVENOUS USE ONLY
Sterile, Isotonic Solution
TEVA
-
INGREDIENTS AND APPEARANCE
IDARUBICIN HYDROCHLORIDE
idarubicin hydrochloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0703-4154 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IDARUBICIN HYDROCHLORIDE (UNII: 5VV3MDU5IE) (IDARUBICIN - UNII:ZRP63D75JW) IDARUBICIN HYDROCHLORIDE 1 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCERIN (UNII: PDC6A3C0OX) WATER (UNII: 059QF0KO0R) Other Ingredients Ingredient Kind Ingredient Name Quantity May contain HYDROCHLORIC ACID (UNII: QTT17582CB) May contain SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0703-4154-11 1 in 1 CARTON 10/01/2002 1 5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA065036 10/01/2002 IDARUBICIN HYDROCHLORIDE
idarubicin hydrochloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0703-4155 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IDARUBICIN HYDROCHLORIDE (UNII: 5VV3MDU5IE) (IDARUBICIN - UNII:ZRP63D75JW) IDARUBICIN HYDROCHLORIDE 1 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCERIN (UNII: PDC6A3C0OX) WATER (UNII: 059QF0KO0R) Other Ingredients Ingredient Kind Ingredient Name Quantity May contain HYDROCHLORIC ACID (UNII: QTT17582CB) May contain SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0703-4155-11 1 in 1 CARTON 10/01/2002 1 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA065036 10/01/2002 IDARUBICIN HYDROCHLORIDE
idarubicin hydrochloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0703-4156 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IDARUBICIN HYDROCHLORIDE (UNII: 5VV3MDU5IE) (IDARUBICIN - UNII:ZRP63D75JW) IDARUBICIN HYDROCHLORIDE 1 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCERIN (UNII: PDC6A3C0OX) WATER (UNII: 059QF0KO0R) Other Ingredients Ingredient Kind Ingredient Name Quantity May contain HYDROCHLORIC ACID (UNII: QTT17582CB) May contain SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0703-4156-11 1 in 1 CARTON 10/01/2002 1 20 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA065036 10/01/2002 Labeler - Teva Parenteral Medicines, Inc. (794362533)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.