NELARABINE by Novadoz Pharmaceuticals LLC / MSN LABORATORIES PRIVATE LIMITED NELARABINE injection
NELARABINE by
Drug Labeling and Warnings
NELARABINE by is a Prescription medication manufactured, distributed, or labeled by Novadoz Pharmaceuticals LLC, MSN LABORATORIES PRIVATE LIMITED. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NELARABINE INJECTION safely and effectively. See full prescribing information for NELARABINE INJECTION.
NELARABINE injection, for intravenous use
Initial U.S. Approval: 2005
WARNING: NEUROLOGIC ADVERSE REACTIONS
See full prescribing information for complete boxed warning.
Severe neurologic adverse reactions have been reported with the use of nelarabine injection. These adverse reactions have included altered mental states including severe somnolence, central nervous system effects including convulsions, and peripheral neuropathy ranging from numbness and paresthesias to motor weakness and paralysis. There have also been reports of adverse reactions associated with demyelination, and ascending peripheral neuropathies similar in appearance to Guillain-Barré syndrome. (5.1)
Full recovery from these adverse reactions has not always occurred with cessation of therapy with nelarabine injection. Monitor frequently for signs and symptoms of neurologic toxicity. Discontinue nelarabine for neurologic adverse reactions of NCI Common Toxicity Criteria for Adverse Events (CTCAE) Grade 2 or greater. (5.1)
INDICATIONS AND USAGE
Nelarabine is a nucleoside metabolic inhibitor indicated for the treatment of patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma in adult and pediatric patients age 1 year and older whose disease has not responded to or has relapsed following treatment with at least two chemotherapy regimens. (1)
DOSAGE AND ADMINISTRATION
- Adult Dose: 1,500 mg/m² administered intravenously over 2 hours on Days 1, 3, and 5 repeated every 21 days. (2.1)
- Pediatric Dose: 650 mg/m² administered intravenously over 1 hour daily for 5 consecutive days repeated every 21 days. (2.1)
- Discontinue treatment for neurologic reactions greater than or equal to Grade 2. (2.2)
- Dosage may be delayed for hematologic reactions. (2.2)
- Take measures to prevent hyperuricemia. (2.4)
DOSAGE FORMS AND STRENGTHS
Injection: 250 mg/50 mL (5 mg/mL) single-dose vial (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Neurologic Adverse Reactions: Severe neurologic reactions have been reported. Monitor for signs and symptoms of neurologic toxicity. (5.1)
- Hematologic Reactions: Complete blood counts including platelets should be monitored regularly. (5.2)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception; and advise males to use condoms. (5.3, 8.1, 8.3)
- Effects on Ability to Drive and Use Machines: Somnolence may occur.Advise patients to refrain from these activities until somnolence has resolved. (5.6)
ADVERSE REACTIONS
The most common (≥ 20%) adverse reactions were:
Adult: anemia, thrombocytopenia, neutropenia, nausea, diarrhea, vomiting, constipation, fatigue, pyrexia, cough, and dyspnea (6.1)
Pediatric: anemia, neutropenia, thrombocytopenia, and leukopenia (6.1)
The most common (> 10%) neurological adverse reactions were:
Adult: somnolence, dizziness, peripheral neurologic disorders, hypoesthesia, headache, and paresthesia (6.1)
Pediatric: headache and peripheral neurologic disorders (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novadoz Pharmaceuticals LLC at 1-855-668-2369 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: NEUROLOGIC ADVERSE REACTIONS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Modification
2.3 Adjustment of Dose in Special Populations
2.4 Prevention of Hyperuricemia
2.5 Instructions for Handling, Preparation, and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Neurologic Adverse Reactions
5.2 Hematologic Adverse Reactions
5.3 Embryo-Fetal Toxicity
5.4 Tumor Lysis Syndrome
5.5 Vaccinations
5.6 Effects on Ability to Drive and Use Machines
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Adult Clinical Trial in Relapsed or Refractory T-ALL and T-LBL
14.2 Pediatric Clinical Trial in Relapsed or Refractory T-ALL and T-LBL
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: NEUROLOGIC ADVERSE REACTIONS
Severe neurologic adverse reactions have been reported with the use of nelarabine. These adverse reactions have included altered mental states including severe somnolence, central nervous system effects including convulsions, and peripheral neuropathy ranging from numbness and paresthesias to motor weakness and paralysis. There have also been reports of adverse reactions associated with demyelination, and ascending peripheral neuropathies similar in appearance to Guillain-Barré syndrome [see Warnings and Precautions (5.1)].
Full recovery from these adverse reactions has not always occurred with cessation of therapy with nelarabine. Monitor frequently for signs and symptoms of neurologic toxicity during treatment with nelarabine. Discontinue nelarabine for neurologic adverse reactions of NCI Common Toxicity Criteria for Adverse Events (CTCAE) Grade 2 or greater [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
Nelarabine injection is indicated for the treatment of T-cell acute lymphoblastic leukemia (T-ALL) and T-cell lymphoblastic lymphoma (T-LBL) in adult and pediatric patients age 1 year and older whose disease has not responded to or has relapsed following treatment with at least two chemotherapy regimens.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
This product is for intravenous use only.
Adult Dosage: The recommended adult dose of nelarabine injection is 1,500 mg/m² administered intravenously over 2 hours on Days 1, 3, and 5 repeated every 21 days. Administer nelarabine injection undiluted.
Pediatric Dosage: The recommended pediatric dose of nelarabine injection is 650 mg/m² administered intravenously over 1 hour daily for 5 consecutive days repeated every 21 days. Administer nelarabine injection undiluted.
The recommended duration of treatment for adult and pediatric patients has not been clearly established. In clinical trials, treatment was generally continued until there was evidence of disease progression, the patient experienced unacceptable toxicity, the patient became a candidate for hematopoietic stem cell transplantation (HSCT), or the patient no longer continued to benefit from treatment.
2.2 Dosage Modification
Discontinue nelarabine injection if the patient develops a neurologic adverse reaction of NCI CTCAE Grade 2 or greater. Dosage may be delayed for other toxicity, including hematologic toxicity [see Boxed Warning, Warnings and Precautions (5.1, 5.2)].
2.3 Adjustment of Dose in Special Populations
Nelarabine injection has not been studied in patients with renal or hepatic dysfunction [see Use in Specific Populations (8.6,8.7)]. No dose adjustment is recommended for patients with a creatinine clearance (CLCr) greater than or equal to 50 mL/min [see Clinical Pharmacology (12.3)]. There are insufficient data to support a dose recommendation for patients with a CLCr less than 50 mL/min.
2.4 Prevention of Hyperuricemia
Take precautions against hyperuricemia (e.g., hydration, urine alkalinization, and prophylaxis with allopurinol) [see Warnings and Precautions (5.4)].
2.5 Instructions for Handling, Preparation, and Administration
Handling: Nelarabine injection is a cytotoxic agent. Caution should be used during handling and preparation. Use of gloves and other protective clothing to prevent skin contact is recommended. Proper aseptic technique should be used. Guidelines for proper handling and disposal of anticancer drugs have been published.1
Preparation and Administration: Administer nelarabine injection undiluted. Transfer the appropriate dose of nelarabine injection into polyvinylchloride (PVC) infusion bags or glass containers and administer as a 2-hour infusion in adult patients and as a 1-hour infusion in pediatric patients.
Prior to administration, inspect the drug product visually for particulate matter and discoloration.
Stability:Nelarabine Injection is stable in polyvinylchloride (PVC) infusion bags and glass containers for up to 8 hours at up to 30°C.
Discard unused portion.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Neurologic Adverse Reactions
Nervous system adverse reactions of any grade were reported for 223 (76%) adult patients across the Phase I and Phase II trials, and Grade 3 or higher (severe, life-threatening, or fatal) adverse reactions were reported for 55 (19%) patients following initiation of nelarabine therapy [see Adverse Reactions (6.1)]. Based on patients with complete data, the median time to onset of first event is 5 days from start of first infusion (range: 1-166), and the median duration is 6 days (range: 1-393 days).
Nervous system adverse reactions of any grade were reported for 69 (42%) pediatric patients across the Phase I and Phase II trials, and Grade 3 or higher (severe, life-threatening, or fatal) adverse reactions were reported for 25 (15%) patients following initiation of nelarabine therapy [see Adverse Reactions (6.1)]. Based on patients with complete data, the median time to onset of first event is 8 days from start of first infusion (range: 1-269), and the median duration is 2 days (range: 1-82 days).
Common signs and symptoms of nelarabine-related neurotoxicity include somnolence, headache, paresthesia and dysesthesia, dizziness, neuropathy (sensory and motor), cerebellar disturbances and tremor. Severe neurologic toxicity can manifest as coma, status epilepticus, craniospinal demyelination, or ascending neuropathy similar in presentation to Guillain-Barré syndrome.
Full recovery from these adverse reactions has not always occurred with cessation of therapy with nelarabine. Patients treated previously or concurrently with intrathecal chemotherapy or previously with craniospinal irradiation may be at increased risk for neurologic adverse events.
Monitor patients frequently for signs and symptoms of neurologic toxicity during and for at least 24 hours after completion of treatment with nelarabine. Discontinue nelarabine for neurologic adverse reactions of NCI CTCAE Grade 2 or greater and provide supportive care [see Dosage and Administration (2.2), Adverse Reactions (6.1)].
5.2 Hematologic Adverse Reactions
Leukopenia, thrombocytopenia, anemia, and neutropenia, including febrile neutropenia, have been associated with nelarabine therapy. Complete blood counts including platelets should be monitored regularly [see Dosage and Administration (2.2), Adverse Reactions (6.1)].
5.3 Embryo-Fetal Toxicity
Based on its mechanism of action and findings in animal studies, nelarabine can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. In animal reproduction studies, intravenous administration of nelarabine to pregnant rabbits during the period of organogenesis resulted in teratogenicity at maternal doses below the recommended human adult dose of 1,500 mg/m2/day (see Data).
Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with nelarabine. Advise males with female partners of reproductive potential to use condoms during treatment with nelarabine and for 3 months after the last dose [see Use in Specific Populations (8.1,8.3), Nonclinical Toxicology (13.1)].
5.4 Tumor Lysis Syndrome
Patients receiving nelarabine should receive intravenous hydration according to standard medical practice for the management of hyperuricemia in patients at risk for tumor lysis syndrome. Consideration should be given to the use of allopurinol in patients at risk of hyperuricemia [see Dosage and Administration (2.4)].
5.6 Effects on Ability to Drive and Use Machines
Patients treated with nelarabine may experience somnolence during and for several days after treatment [see Adverse Reactions (6.1)]. Advise patients to refrain from driving or engaging in hazardous occupations or activities until somnolence has resolved.
-
6 ADVERSE REACTIONS
The following clinically-significant adverse reactions are discussed in greater detail in other sections of the label:
- Neurologic [see Boxed Warning, Warnings and Precautions (5.1)]
- Hematologic [see Warnings and Precautions (5.2)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.4)]
- Effects on Ability to Drive and Use Machines [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Relapsed or Refractory T-ALL and T-LBL
Nelarabine was studied in 459 patients in Phase I and Phase II clinical trials.
Adult Patient: The safety profile of nelarabine is based on data from 103 adult patients treated with the recommended dose and schedule in 2 studies: an adult T-cell acute lymphoblastic leukemia (T-ALL)/T-cell lymphoblastic lymphoma (T-LBL) trial and an adult chronic lymphocytic leukemia trial.
The most common adverse reactions in adults were fatigue; gastrointestinal (GI) disorders (nausea, diarrhea, vomiting, and constipation); hematologic disorders (anemia, neutropenia, and thrombocytopenia); respiratory disorders (cough and dyspnea); nervous system disorders (somnolence and dizziness); and pyrexia.
The most common adverse reactions in adults by Body System, including severe or life-threatening adverse reactions (NCI CTCAE Grade 3 or Grade 4) and fatal adverse reactions (Grade 5) are shown in Table 1.
Table 1. Most Commonly Reported (≥ 5% Overall) Adverse Reactions in Adult Patients Treated with 1,500 mg/m2of Nelarabine Administered Intravenously over 2 Hours on Days 1, 3, and 5 Repeated Every 21 Days
Body System Adverse Reaction Percentage of Patients (N = 103)
Toxicity GradeGrade 3
%
Grades 4 and 5a
%
All Grades
%
Blood and Lymphatic System Disorders Anemia
20
14
99Thrombocytopenia
37
22
86Neutropenia
14
49
81Febrile neutropenia
9
1
12Cardiac Disorders Sinus tachycardia
1
0
8Gastrointestinal Disorders Nausea
0
0
41Diarrhea
1
0
22Vomiting
1
0
22Constipation
1
0
21Abdominal pain
1
0
9Stomatitis
1
0
8Abdominal distension
1
0
6General Disorders and Administration Site Conditions Fatigue
10
2
50Pyrexia
5
0
23Asthenia
0
1
17Edema, peripheral
0
0
15Edema
0
0
11Pain
3
0
11Rigors
0
0
8Gait, abnormal
0
0
6Chest pain
0
0
5Noncardiac chest pain
0
1
5Infections Infection
2
1
9Pneumonia
4
1
8Sinusitis
1
0
7Hepatobiliary Disorders AST increased
1
1
6Metabolism and Nutrition Disorders Anorexia
0
0
9Dehydration
3
1
7Hyperglycemia
1
0
6Musculoskeletal and Connective Tissue Disorders Myalgia
1
0
13Arthralgia
1
0
9Back pain
0
0
8Muscular weakness
5
0
8Pain in extremity
1
0
7Nervous System Disorders (see Table 2) Psychiatric Disorders Confusional state
2
0
8Insomnia
0
0
7Depression
1
0
6Respiratory, Thoracic, and Mediastinal Disorders Cough
0
0
25Dyspnea
4
2
20Pleural effusion
5
1
10Epistaxis
0
0
8Dyspnea, exertional
0
0
7Wheezing
0
0
5Vascular Disorders Petechiae
2
0
12Hypotension
1
1
8Abbreviation: AST, aspartate transaminase.
a Five patients had a fatal adverse reaction. Fatal adverse reactions included hypotension (n = 1), respiratory arrest (n = 1), pleural effusion/pneumothorax (n = 1), pneumonia (n = 1), and cerebral hemorrhage/coma/leukoencephalopathy (n = 1).
Other Adverse Reactions: Blurred vision was also reported in 4% of adult patients.
There was a single report of biopsy-confirmed progressive multifocal leukoencephalopathy in the adult patient population.
Neurologic Adverse Reactions: Nervous system adverse reactions, were reported for 76% of adult patients across the Phase I and Phase II trials. The most common neurologic adverse reactions (≥ 2%) in adult patients including all grades (NCI CTCAE) are shown in Table 2.
Table 2. Neurologic Adverse Reactions ( ≥ 2%) in Adult Patients Treated With 1,500 mg/m2of Nelarabine Administered Intravenously Over 2 Hours on Days 1, 3, and 5 Repeated Every 21 Days
Nervous System Disorders Adverse ReactionPercentage of Patients (N =103)
Grade 1
%
Grade 2
%
Grade 3
%
Grade 4
%
All Grades
%
Somnolence
20
3
0
0
23
Dizziness
14
8
0
0
21
Peripheral neurologic disorders, any adverse reaction
8
12
2
0
21
Neuropathy
0
4
0
0
4
Peripheral neuropathy
2
2
1
0
5
Peripheral motor neuropathy
3
3
1
0
7
Peripheral sensory neuropathy
7
6
0
0
13
Hypoesthesia
5
10
2
0
17
Headache
11
3
1
0
15
Paresthesia
11
4
0
0
15
Ataxia
1
6
2
0
9
Depressed level of consciousness
4
1
0
1
6
Tremor
2
3
0
0
5
Amnesia
2
1
0
0
3
Dysgeusia
2
1
0
0
3
Balance disorder
1
1
0
0
2
Sensory loss
0
2
0
0
2
One patient had a fatal neurologic adverse reaction, cerebral hemorrhage/coma/leukoencephalopathy.
Most nervous system adverse reactions in the adult patients were evaluated as Grade 1 or 2. The additional Grade 3 adverse reactions in adult patients, were aphasia, convulsion, hemiparesis, and loss of consciousness, each reported in 1 patient (1%). The additional Grade 4 adverse reactions were cerebral hemorrhage, coma, intracranial hemorrhage, leukoencephalopathy, and metabolic encephalopathy, each reported in one patient (1%).
The other neurologic adverse reactions reported as Grade 1, 2, or unknown in adult patients were abnormal coordination, burning sensation, disturbance in attention, dysarthria, hyporeflexia, neuropathic pain, nystagmus, peroneal nerve palsy, sciatica, sensory disturbance, sinus headache, and speech disorder, each reported in one patient (1%).
Pediatric Patient: The safety profile for children is based on data from 84 pediatric patients treated with the recommended dose and schedule in a T-ALL/T-LBL treatment trial.
The most common adverse reactions in pediatric patients were hematologic disorders (anemia, leukopenia, neutropenia, and thrombocytopenia). Of the non-hematologic adverse reactions in pediatric patients, the most frequent adverse reactions reported were headache, increased transaminase levels, decreased blood potassium, decreased blood albumin, increased blood bilirubin, and vomiting.
The most common adverse reactions in pediatric patients by System Organ Class including severe or life threatening adverse reactions (NCI CTCAE Grade 3 or Grade 4) and fatal adverse reactions (Grade 5) are shown in Table 3.
Table 3. Most Commonly Reported (≥ 5% Overall) Adverse Reactions in Pediatric Patients Treated With 650 mg/m2of Nelarabine Administered Intravenously Over 1 Hour Daily for 5 Consecutive Days Repeated Every 21 Days
Body System Adverse ReactionPercentage of Patients (N = 84)
Toxicity Grade
Grade 3
%
Grade 4 and 5a
%
All Grades
%
Blood and Lymphatic System Disorders
Anemia
45
10
95
Neutropenia
17
62
94
Thrombocytopenia
27
32
88
Leukopenia
14
7
38
Hepatobiliary Disorders
Transaminases increased
4
0
12
Blood albumin decreased
5
1
10
Blood bilirubin increased
7
2
10
Metabolic/Laboratory
Blood potassium decreased
4
2
11
Blood calcium decreased
1
1
8
Blood creatinine increased
0
0
6
Blood glucose decreased
4
0
6
Blood magnesium decreased
2
0
6
Nervous System Disorders (see Table 4)
Gastrointestinal Disorders
Vomiting
0
0
10
General Disorders & Administration Site Conditions
Asthenia
1
0
6
Infections & Infestations
Infection
2
1
5
aThree patients had a fatal adverse reaction. Fatal adverse reactions included neutropenia and pyrexia (n = 1), status epilepticus/seizure (n = 1), and fungal pneumonia (n = 1).
Neurologic Adverse Reactions: Nervous system adverse reactions were reported for 42% of pediatric patients across the Phase I and Phase II trials. The most common neurologic adverse reactions (≥ 2%) in pediatric patients including all grades (NCI CTCAE) are shown in Table 4.
Table 4. Neurologic Adverse Reactions (≥ 2%) in Pediatric Patients Treated With 650 mg/m2of Nelarabine Administered Intravenously Over 1 Hour Daily for 5 Consecutive Days Repeated Every 21 Days
Nervous System Disorders Adverse Reaction
Percentage of Patients (N = 84)
Grade 1
%
Grade 2
%
Grade 3
%
Grade 4 and 5a
%
All Grades
%
Headache
8
2
4
2
17
Peripheral neurologic disorders, any adverse reaction
1
4
7
0
12
Peripheral neuropathy
0
4
2
0
6
Peripheral motor neuropathy
1
0
2
0
4
Peripheral sensory neuropathy
0
0
6
0
6
Somnolence
1
4
1
1
7
Hypoesthesia
1
1
4
0
6
Seizures
0
0
0
6
6
Convulsions
0
0
0
3
4
Grand mal convulsions
0
0
0
1
1
Status epilepticus
0
0
0
1
1
Motor dysfunction
1
1
1
0
4
Nervous system disorder
1
2
0
0
4
Paresthesia
0
2
1
0
4
Tremor
1
2
0
0
4
Ataxia
1
0
1
0
2
a One (1) patient had a fatal neurologic adverse reaction, status epilepticus.
The other Grade 3 neurologic adverse reaction in pediatric patients was hypertonia reported in 1 patient (1%). The additional Grade 4 neurologic adverse reactions, were 3rdnerve paralysis, and 6thnerve paralysis, each reported in 1 patient (1%).
The other neurologic adverse reactions reported as Grade 1, 2, or unknown in pediatric patients were dysarthria, encephalopathy, hydrocephalus, hyporeflexia, lethargy, mental impairment, paralysis, and sensory loss, each reported in 1 patient (1%).
Nelarabine in Combination with Multi-Agent Chemotherapy in T-ALL and T-LBL
Nelarabine was studied in combination with multi-agent chemotherapy in a randomized clinical trial [NCT00408005]. The safety population in this trial included 804 patients with newly-diagnosed T-ALL (85%) or T-LBL (15%) treated with (n = 411) or without (n =393) nelarabine in combination with the augmented Berlin-Frankfurt-Münster chemotherapy regimen (aBFM) after initial induction therapy. Patients assigned to nelarabine received 650 mg/m2intravenously over 1 hour daily for 5 consecutive days, during consolidation days 1 to 5 and 43 to 47, delayed intensification days 29 to 33, and during the initial 3 courses of maintenance days 29 to 33. The median age on enrollment was 9.5 years (range, 1-29), the majority of patients were male (73%) and white (69%). Sixty-five percent of patients assigned to the nelarabine arms received at least 85% of the planned dose through the third course of maintenance therapy compared to 79% of patients on the control arms who received 3 courses of maintenance therapy.
There was one fatal neurological adverse reaction in the nelarabine arm. The incidence of the following grades 3 and 4 adverse reactions were higher in the nelarabine treated arms compared to the control arms: abnormal transaminases, motor and sensory neuropathy, nausea and vomiting, and dehydration. The incidence of seizures of any grade was 3% (14 of 411). Rhabdomyolysis was diagnosed in 2% (7 of 411) of nelarabine treated patients and occurred after the first course of nelarabine during the consolidation phase of therapy.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of nelarabine. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Infections and Infestations: Fatal opportunistic infections.
Metabolism and Nutrition Disorders: Tumor lysis syndrome.
Nervous System Disorders: Demyelination and ascending peripheral neuropathies similar in appearance to Guillain-Barré syndrome.
Musculoskeletal and Connective Disorders: Rhabdomyolysis, blood creatine phosphokinase increased.
-
7 DRUG INTERACTIONS
Administration of nelarabine in combination with adenosine deaminase inhibitors, such as pentostatin, is not recommended [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and findings in animal studies, nelarabine can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. Limited available data with nelarabine use in pregnant women are insufficient to determine a drug-associated risk for major birth defects, miscarriage or adverse maternal or fetal outcomes. There are risks to the pregnant woman associated with untreated leukemia or lymphoma (see Clinical Considerations). In animal reproduction studies, intravenous administration of nelarabine to pregnant rabbits during the period of organogenesis resulted in teratogenicity at maternal doses below the recommended human adult dose of 1500 mg/m2/day (see Data). Advise pregnant women of the potential risk to the fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies are 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo-fetal Risk
There are risks to the mother from untreated leukemia or lymphoma, including anemia, thrombocytopenia, and death.
Data
Animal Data
In an embryo-fetal development study in which pregnant rabbits were administered daily doses of nelarabine during organogenesis, increased incidences of fetal malformations, anomalies, and variations were observed at doses greater than or equal to 360 mg/m2/day (8-hour IV infusion; approximately 25% of the recommended human adult dose compared on a mg/m2basis), which was the lowest dose tested. Cleft palate was seen in rabbits given 3,600 mg/m2/day (approximately 2-fold the adult dose), absent pollices (digits) in rabbits given greater than or equal to 1,200 mg/m2/day (approximately 75% of the recommended adult dose), while absent gall bladder, absent accessory lung lobes, fused or extra sternebrae, and delayed ossification was seen at all doses. Maternal body weight gain and fetal body weights were reduced in rabbits given 3,600 mg/m2/day (approximately 2-fold the adult dose), but could not account for the increased incidence of malformations seen at this or lower administered doses.
8.2 Lactation
Risk Summary
There are no data on the presence of nelarabine or ara-G in human or animal milk, the effect on the breastfed child, or the effect on milk production. Because of the potential for serious adverse reactions in the breastfed child from nelarabine, such as severe neurological reactions, advise women not to breastfeed during treatment with nelarabine.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Nelarabine can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Verify the pregnancy status of females of reproductive potential prior to starting treatment with nelarabine.
Contraception
Females
Nelarabine can cause fetal harm when administered to a pregnant woman [see Warnings and Precautions (5.3), Use in Specific Populations (8.1)]. Because of the potential for genotoxicity, advise females of reproductive potential to use effective contraception during treatment with nelarabine.
Males
Because of the potential for genotoxicity, advise males (including those who have had vasectomies) with female partners of reproductive potential to use condoms during treatment with nelarabine and for 3 months after the last dose [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of nelarabine for relapsed or refractory T-ALL and T-LBL has been established in pediatric patients age 1 year and older. The effectiveness of nelarabine in pediatric patients is supported by one single-arm clinical trial, and safety has been asssessed in 165 pediatric patients age 1 year and older across multiple Phase I and Phase II trials. The trial establishing efficacy included 84 patients age 21 years and younger, who had relapsed or refractory T-ALL or T-LBL. The most frequent adverse reactions of any grade occurring on treatment in this study were hematologic laboratory abnormalities. Hematologic toxicity observed in the pediatric population was higher than that seen in the adult population. [see Dosage and Administration (2.1), Adverse Reactions (6.1), Clinical Studies (14.2)].
Nervous system adverse reactions have been reported for 42% of pediatric patients across the Phase I and Phase II trials. The incidence of nervous system adverse reactions was less in the pediatric population than that seen in adult patients with relapsed/refractory T-ALL/T-LBL [see Adverse Reactions (6.1)].
In a phase III study of nelarabine in combination with multi-agent chemotherapy as first-line therapy, there were 411 patients with T-ALL or T-LBL treated with nelarabine. The safety profile in the 357 patients age 1 to 16 years was consistent with that seen in older patients in the study. [see Adverse Reactions (6.1)].
Due to lack of long-term follow up data, a determination of the impact of nelarabine on the growth and pubertal development of pediatric patients cannot be made.
8.5 Geriatric Use
Clinical studies of nelarabine did not include sufficient numbers of patients age 65 and over to determine whether they respond differently from younger patients. In an exploratory analysis, increasing age, especially age 65 years and older, appeared to be associated with increased rates of neurologic adverse reactions. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, [see Use in Specific Populations (8.6)].
8.6 Renal Impairment
Ara-G clearance decreased as renal function decreased [see Clinical Pharmacology (12.3)]. Because the risk of adverse reactions to this drug may be greater in patients with moderate (CLCr 30 to 50 mL/min) or severe (CLCr less than 30 mL/min) renal impairment, these patients should be closely monitored for toxicities when treated with nelarabine [see Dosage and Administration (2.3)].
8.7 Hepatic Impairment
The influence of hepatic impairment on the pharmacokinetics of nelarabine has not been evaluated. Because the risk of adverse reactions to this drug may be greater in patients with severe hepatic impairment (total bilirubin greater than 3 times upper limit of normal), these patients should be closely monitored for toxicities when treated with nelarabine.
-
10 OVERDOSAGE
There is no known antidote for overdoses of nelarabine. It is anticipated that overdosage would result in severe neurotoxicity (possibly including paralysis, coma), myelosuppression, and potentially death. In the event of overdose, supportive care consistent with good clinical practice should be provided.
At a dose of 2,200 mg/m2given on Days 1, 3, and 5 every 21 days, 2 patients developed a significant Grade 3 ascending sensory neuropathy. MRI evaluations of the 2 patients demonstrated findings consistent with a demyelinating process in the cervical spine.
-
11 DESCRIPTION
Nelarabine is a prodrug of the cytotoxic deoxyguanosine analogue, 9-β-D arabinofuranosylguanine (ara-G).
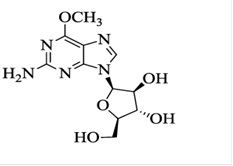
The chemical name for nelarabine is 2-amino-9-β-D-arabinofuranosyl-6-methoxy-9H-purine. It has the molecular formula C11H15N5O5 and a molecular weight of 297.27. Nelarabine has the following structural formula:
Nelarabine is soluble in N, N dimethylformamide, slightly soluble in water and insoluble in cyclohexane.
Nelarabine Injection is supplied as a clear, colorless, sterile, solution in glass single-dose vials. Each vial contains 250 mg of nelarabine (5 mg nelarabine per mL) and the inactive ingredient sodium chloride (4.5 mg per mL) in 50 mL Water for Injection, USP. Nelarabine is intended for intravenous infusion.
Hydrochloric acid and sodium hydroxide may have been used to adjust the pH. The solution pH ranges from 5.0 to 7.0.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Nelarabine is a prodrug of the deoxyguanosine analogue 9-β-D-arabinofuranosylguanine (ara-G), a nucleoside metabolic inhibitor. Nelarabine is demethylated by adenosine deaminase (ADA) to ara-G, mono-phosphorylated by deoxyguanosine kinase and deoxycytidine kinase, and subsequently converted to the active 5’-triphosphate, ara-GTP. Accumulation of ara-GTP in leukemic blasts allows for incorporation into deoxyribonucleic acid (DNA), leading to inhibition of DNA synthesis and cell death. Other mechanisms may contribute to the cytotoxic and systemic toxicity of nelarabine.
12.3 Pharmacokinetics
Absorption: Following intravenous administration of nelarabine to adult patients with refractory leukemia or lymphoma, plasma ara-G Cmax values generally occurred at the end of the nelarabine infusion and were generally higher than nelarabine Cmaxvalues, suggesting rapid and extensive conversion of nelarabine to ara-G. Mean plasma nelarabine and ara-G Cmax values were 5.0 ± 3.0 mcg/mL and 31.4 ± 5.6 mcg/mL, respectively, after a 1,500 mg/m2nelarabine dose infused over 2 hours in adult patients. The area under the concentration-time curve (AUC) of ara-G is 37 times higher than that for nelarabine on Day 1 after nelarabine IV infusion of 1,500 mg/m2 dose (162 ± 49 mcg.h/mL versus 4.4 ± 2.2 mcg.h/mL, respectively). Comparable Cmax and AUC values were obtained for nelarabine between Days 1 and 5 at the nelarabine adult dosage of 1,500 mg/m2, indicating that nelarabine does not accumulate after multiple-dosing. There are not enough ara-G data to make a comparison between Day 1 and Day 5. After a nelarabine adult dose of 1,500 mg/m2, intracellular Cmax for ara- GTP appeared within 3 to 25 hours on Day 1. Exposure (AUC) to intracellular ara-GTP was 532 times higher than that for nelarabine and 14 times higher than that for ara-G (2,339 ± 2,628 mcg.h/mL versus 4.4 ± 2.2 mcg.h/mL and 162 ± 49 mcg.h/mL, respectively). Because the intracellular levels of ara-GTP were so prolonged, its elimination half-life could not be accurately estimated.
Distribution: Nelarabine and ara-G are extensively distributed throughout the body. For nelarabine, Vss values were 197 ± 216 L/m2in adult patients. For ara-G, Vss/F values were 50 ± 24 L/m2in adult patients.
Nelarabine and ara-G are not substantially bound to human plasma proteins (< 25%) in vitro, and binding is independent of nelarabine or ara-G concentrations up to 600 μM.
Metabolism: The principal route of metabolism for nelarabine is O-demethylation by adenosine deaminase to form ara-G, which undergoes hydrolysis to form guanine. In addition, some nelarabine is hydrolyzed to form methylguanine, which is O-demethylated to form guanine. Guanine is N-deaminated to form xanthine, which is further oxidized to yield uric acid.
Excretion: Nelarabine and ara-G are partially eliminated by the kidneys. Mean urinary excretion of nelarabine and ara-G was 6.6 ± 4.7% and 27 ± 15% of the administered dose, respectively, in 28 adult patients over the 24 hours after nelarabine infusion on Day 1. Renal clearance averaged 24 ± 23 L/h for nelarabine and 6.2 ± 5.0 L/h for ara-G in 21 adult patients. Combined Phase I pharmacokinetic data at nelarabine doses of 199 to 2,900 mg/m2(n = 66 adult patients) indicate that the mean clearance (CL) of nelarabine is 197 ± 189 L/h/m2on Day 1. The apparent clearance of ara-G (CL/F) is 10.5 ± 4.5 L/h/m2on Day 1. Nelarabine and ara-G are rapidly eliminated from plasma with a mean half-life of 18 minutes and 3.2 hours, respectively, in adult patients.
Pediatrics: No pharmacokinetic data are available in pediatric patients at the once-daily 650 mg/m2nelarabine dosage. Combined Phase I pharmacokinetic data at nelarabine doses of 104 to 2,900 mg/m2indicate that the mean clearance (CL) of nelarabine is about 30% higher in pediatric patients than in adult patients (259 ± 409 L/h/m2versus 197 ± 189 L/h/m2, respectively) (n = 66 adults, n = 22 pediatric patients) on Day 1. The apparent clearance of ara-G (CL/F) is comparable between the two groups (10.5 ± 4.5 L/h/m2in adult patients and 11.3 ± 4.2 L/h/m2in pediatric patients) on Day 1. Nelarabine and ara-G are extensively distributed throughout the body. For nelarabine, Vss values were 213 ± 358 L/m2in pediatric patients. For ara-G, Vss/F values were 33 ± 9.3 L/m2in pediatric patients. Nelarabine and ara-G are rapidly eliminated from plasma in pediatric patients, with a half-life of 13 minutes and 2 hours, respectively.
Effect of Age: Age has no effect on the pharmacokinetics of nelarabine or ara-G in adults. Decreased renal function, which is more common in the elderly, may reduce ara-G clearance [see Use in Specific Populations (8.5)].
Effect of Gender: Gender has no effect on nelarabine or ara-G pharmacokinetics.
Effect of Race: In general, nelarabine mean clearance and volume of distribution values tend to be higher in whites (n = 63) than in blacks (by about 10%) (n = 15). The opposite is true for ara-G; mean apparent clearance and volume of distribution values tend to be lower in whites than in blacks (by about 15% to 20%). No differences in safety or effectiveness were observed between these groups.
Effect of Renal Impairment: The pharmacokinetics of nelarabine and ara-G have not been specifically studied in renally impaired or hemodialyzed patients. Nelarabine is excreted by the kidney to a small extent (5% to 10% of the administered dose). Ara-G is excreted by the kidney to a greater extent (20% to 30% of the administered nelarabine dose). In the combined Phase I trials, patients were categorized into 3 groups: normal with CLCr greater than 80 mL/min (n = 67), mild with CLCr = 50 to 80 mL/min (n = 15), and moderate with CLCr less than 50 mL/min (n = 3). The mean apparent clearance (CL/F) of ara-G was about 15% and 40% lower in patients with mild and moderate renal impairment, respectively, than in patients with normal renal function [see Use in Specific Populations (8.6), Dosage and Administration (2.3)]. No differences in safety or effectiveness were observed.
Effect of Hepatic Impairment: The influence of hepatic impairment on the pharmacokinetics of nelarabine has not been evaluated [see Use in Specific Populations (8.7)].
Drug Interactions: Cytochrome P450: Nelarabine and ara-G did not significantly inhibit the activities of the human hepatic cytochrome P450 isoenzymes 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A4 in vitro at concentrations of nelarabine and ara-G up to 100 μM.
Fludarabine: Administration of fludarabine 30 mg/m2as a 30-minute infusion 4 hours before a 1,200-mg/m2infusion of nelarabine did not affect the pharmacokinetics of nelarabine, ara-G, or ara-GTP in 12 patients with refractory leukemia.
Pentostatin: There is in vitro evidence that pentostatin is a strong inhibitor of adenosine deaminase. Inhibition of adenosine deaminase may result in a reduction in the conversion of the prodrug nelarabine to its active moiety and consequently in a reduction in efficacy of nelarabine and/or change in adverse reaction profile of either drug [see Drug Interactions (7)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity testing of nelarabine has not been done. However, nelarabine was mutagenic when tested in vitro in L5178Y/TK mouse lymphoma cells with and without metabolic activation. No studies have been conducted in animals to assess genotoxic potential or effects on fertility. The effect on human fertility is unknown.
-
14 CLINICAL STUDIES
14.1 Adult Clinical Trial in Relapsed or Refractory T-ALL and T-LBL
The safety and efficacy of nelarabine in adult patients were studied in a clinical trial which included 39 treated patients, 28 who had T-ALL or T-LBL that had relapsed following or was refractory to at least two prior induction regimens. A 1,500-mg/m2dose of nelarabine was administered intravenously over 2 hours on Days 1, 3, and 5 repeated every 21 days. Patients who experienced signs or symptoms of Grade 2 or greater neurologic toxicity on therapy were to be discontinued from further therapy with nelarabine. Seventeen patients had a diagnosis of T-ALL and 11 had a diagnosis of T-LBL. For patients with ≥ 2 prior inductions, the age range was 16 to 65 years (mean: 34 years) and most patients were male (82%) and Caucasian (61%). Patients with central nervous system (CNS) disease were not eligible.
Complete response (CR) in this trial was defined as bone marrow blast counts ≤ 5%, no other evidence of disease, and full recovery of peripheral blood counts. Complete response without complete hematologic recovery (CR*) was also assessed. The results of the trial for patients who had received ≥ 2 prior inductions are shown in Table 5.
Table 5. Efficacy Results in Adult Patients With ≥ 2 Prior Inductions Treated With 1,500 mg/m2of Nelarabine Administered Intravenously Over 2 Hours on Days 1, 3, and 5 Repeated Every 21 Days
N = 28
CR plus CR* % (n) [95% CI]
21% (6) [8%, 41%]
CR % (n) [95% CI]
18% (5) [6%, 37%]
CR* % (n) [95% CI]
4% (1) [0%, 18%]
Duration of CR plus CR* (range in weeks)a
4 to 195+
Median overall survival (weeks) [95% CI]
20.6 weeks [10.4, 36.4]
Abbreviations: CR, complete response; CI, confidence interval; CR*, complete response without hematologic recovery.
a Does not include 1 patient who was transplanted (duration of response was 156+ weeks).
The mean number of days on therapy was 56 days (range of 10 to 136 days). Time to CR plus CR* ranged from 2.9 to 11.7 weeks.
14.2 Pediatric Clinical Trial in Relapsed or Refractory T-ALL and T-LBL
The safety and efficacy of nelarabine in pediatric patients were studied in a clinical trial which included patients age 21 years and younger, who had relapsed or refractory T-ALL or T-LBL. Eighty-four (84) patients, 39 of whom had received two or more prior induction regimens, were treated with 650 mg/m2/day of nelarabine administered intravenously over 1 hour daily for 5 consecutive days repeated every 21 days (see Table 6). Patients who experienced signs or symptoms of Grade 2 or greater neurologic toxicity on therapy were to be discontinued from further therapy with nelarabine.
Table 6. Pediatric Clinical Trial - Patient Allocation
Patient Population
N
Patients treated at 650 mg/m2/day x 5 days, every 21 days.
84
Patients with T-ALL or T-LBL with two or more prior induction treated at 650 mg/m2/day x 5 days, every 21 days.
39
Patients with T-ALL or T-LBL with one prior induction treated at 650 mg/m2/day x 5 days, every 21 days.
31
Abbreviations: T-ALL, T-cell acute lymphoblastic leukemia; T-LBL, T-cell lymphoblastic lymphoma.
The 84 patients ranged in age from 2.5 to 21.7 years (overall mean: 11.9 years), 52% were 3 to 12 years of age and most were male (74%) and Caucasian (62%). The majority (77%) of patients had a diagnosis of T-ALL.
Complete response (CR) in this trial was defined as bone marrow blast counts ≤ 5%, no other evidence of disease, and full recovery of peripheral blood counts. Complete response without full hematologic recovery (CR*) was also assessed as a meaningful outcome in this trial. Duration of response is reported from date of response to date of relapse, and may include subsequent stem cell transplant. Efficacy results are presented in Table 7.
Table 7. Efficacy Results in Patients Age 21 Years and Younger at Diagnosis With ≥ 2 Prior Inductions Treated With 650 mg/m2of Nelarabine Administered Intravenously Over 1 Hour Daily for 5 Consecutive Days Repeated Every 21 Days
N = 39
CR plus CR* % (n) [95% CI]
23% (9) [11%, 39%]
CR % (n) [95% CI]
13% (5) [4%, 27%]
CR* % (n) [95% CI]
10% (4) [3%, 24%]
Duration of CR plus CR* (range in weeks)a
3.3 to 9.3
Median overall survival (weeks) [95% CI]
13.1 [8.7, 17.4]
Abbreviations: CR, complete response; CI, confidence interval; CR*, complete response without hematologic recovery.
aDoes not include 5 patients who were transplanted or had subsequent systemic chemotherapy (duration of response in these 5 patients was 4.7 to 42.1 weeks).
The mean number of days on therapy was 46 days (range: 7-129 days). Median time to CR plus CR* was 3.4 weeks (95% CI: 3.0, 3.7).
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Nelarabine Injection is supplied as a clear, colorless, sterile, solution free from visible particles in a 50 mL Type I, clear colorless moulded glass vial with 20mm rubber stopper and sealed with 20mm aluminium seal having polypropylene disc. Each vial contains 250 mg of nelarabine (5 mg nelarabine per mL) and the inactive ingredient sodium chloride (4.5 mg per mL) in 50 mL Water for Injection, USP.
Strength
Package Size
Carton NDC Number
250 mg/50 mL
(5 mg/mL)
50 mL Single-Dose Vial
72205-154-01
6 x 50 mL Single-Dose Vials
72205-154-04
Store Nelarabine Injection at 25°C (77°F); excursions permitted to 15°C to 30°C (59°F-86°F). See USP Controlled Room Temperature.
Discard Unused Portion.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Hematologic Adverse Reactions
Advise patients that leukopenia, thrombocytopenia, anemia, and neutropenia, including febrile neutropenia, have been associated with nelarabine.
Advise patients that complete blood counts, including platelets, will be monitored regularly during treatment [see Warnings and Precautions (5.2), Adverse Reactions (6.1)].
Embryo-Fetal Toxicity
Advise pregnant females of reproductive potential and males with female partners of reproductive potential of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with nelarabine. Instruct females to inform their physician of a known or suspected pregnancy.
Advise male patients with partners of reproductive potential to use condoms during treatment with nelarabine and for 3 months after the last dose [see Warnings and Precautions (5.3), Use in Specific Populations (8.1, 8.3)].
Tumor Lysis Syndrome
Advise patients of the risk of tumor lysis syndrome [see Warnings and Precautions (5.4), Adverse Reactions (6.1)].
Vaccinations
Instruct patients not to receive live vaccines during treatment with nelarabine [see Warnings and Precautions (5.5), Adverse Reactions (6.1)].
Effects on Ability to Drive and Use Machines
Patients receiving nelarabine may experience somnolence during and for several days after treatment. Instruct patients to not drive or engage in hazardous occupations or activities until somnolence has resolved [see Warnings and Precautions (5.6), Adverse Reactions (6.1)].
Neurologic Adverse Reactions
Instruct patients to contact their physician if they experience new or worsening symptoms of peripheral neuropathy [see Boxed Warning, Dosage and Administration (2.3), Warnings and Precautions (5.1), Adverse Reactions (6.1)]. These signs and symptoms include: tingling or numbness in fingers, hands, toes, or feet; difficulty with the fine motor coordination tasks such as buttoning clothing; unsteadiness while walking; weakness arising from a low chair; weakness in climbing stairs; increased tripping while walking over uneven surfaces.
Advise patients of the risk of seizures [see Adverse Reactions (6.1)]. If a seizure occurs, instruct patients to promptly notify the physician administering nelarabine.
Infection
Instruct patients to promptly notify their physician if they develop fever or signs of infection while on therapy [see Adverse Reactions (6.1, 6.2)].
Lactation
Advise women not to breastfeed during treatment with nelarabine [see Use in Specific Populations (8.2)].
Manufactured by:
MSN laboratories Private Limited
Telangana – 509 228,
INDIA
Distributed by:
Novadoz Pharmaceuticals LLC
Piscataway, NJ 08854-3714
Issued on: 05/2023
-
PATIENT INFORMATION
Nelarabine (nel ar' a been )
Injection
Read the Patient Information that comes with nelarabine injection before you or your child starts treatment with nelarabine. Read the information you get each time before each treatment with nelarabine. There may be new information. This information does not take the place of talking with the doctor about your or your child’s medical condition or treatment. Talk to your or your child’s doctor, if you have any questions. What is the most important information I should know about nelarabine injection?
Nelarabine injection may cause blood related adverse reactions. Your doctor will do blood tests regularly during treatment to monitor blood counts, including platelets.
Nelarabine injection may cause serious nervous system problems including:
- extreme sleepiness
- seizures
- coma
- numbness and tingling in the hands, fingers, feet, or toes (peripheral neuropathy)
- weakness and paralysis
Call the doctor right away if you or your child has the following symptoms:
- seizures
- numbness and tingling in the hands, fingers, feet, or toes
- problems with fine motor skills such as buttoning clothes
- unsteadiness while walking
- increased tripping while walking
- weakness when getting out of a chair or walking upstairs
What is nelarabine injection?
Nelarabine injection is an anti-cancer medicine used to treat adults and children who have:
- T-cell acute lymphoblastic leukemia
- T-cell lymphoblastic lymphoma
What should you tell the doctor before you or your child starts nelarabine injection?
Tell the doctor about all health conditions you or your child have, including if you or your child:
- have any nervous system problems.
- have kidney problems.
- are breastfeeding or plan to breastfeed. It is not known whether nelarabine passes through breast milk. You should not breastfeed during treatment with nelarabine.
- are pregnant or plan to become pregnant. Nelarabine can harm your unborn baby. You should not become pregnant while you are taking nelarabine. You should use effective birth control to avoid getting pregnant. Talk with your doctor about your choices.
- are male (including if you have had a vasectomy) with a sexual partner who is pregnant, think that they may be pregnant, or who could become pregnant. You should use condoms during sexual intercourse during treatment with nelarabine and for 3 months after last dose.
Tell the doctor about all the medicines you or your child take, including prescription and nonprescription medicines, vitamins, and herbal supplements. How is nelarabine injection given?
Nelarabine injection is an intravenous medicine. This means it is given through a tube in your vein.What should you or your child avoid during treatment with nelarabine injection?
- You or your child should not drive or operate dangerous machines. Nelarabine may cause sleepiness during and for several days after treatment.
- You or your child should not receive vaccines made with live germs during treatment with nelarabine.
What are the possible side effects of nelarabine injection?
Nelarabine injection may cause serious nervous system problems. See “What is the most important information I should know about nelarabine injection?”
Nelarabine injection may also cause:
- decreased blood counts such as low red blood cells, low white blood cells, and low platelets. Blood tests should be done regularly to check blood counts. Call the doctor right away if you or your child:
- is more tired than usual, pale, or has trouble breathing
- has a fever or other signs of an infection
- bruises easy or has any unusual bleeding
- stomach area problems such as nausea, vomiting, diarrhea, and constipation
- headache
- sleepiness
- blurry eyesight
Call your doctor right away if you experience unexplained muscle pain, tenderness, or weakness while taking nelarabine. This is because on rare occasions, muscle problems can be serious.
These are not all the side effects associated with nelarabine. Ask your doctor or pharmacist for more information.
General Advice about nelarabine injection
This leaflet summarizes important information about nelarabine. If you have questions or problems, talk with your or your child’s doctor. You can ask your doctor or pharmacist for information about nelarabine that is written for healthcare providers or it is available at www.novadozpharma.com or call 1-855-668-2369.
Manufactured by:
MSN laboratories Private Limited
Telangana – 509 228,
INDIA
Distributed by:
Novadoz Pharmaceuticals LLC
Piscataway, NJ 08854-3714
Issued on: 05/2023
- extreme sleepiness
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
NELARABINE
nelarabine injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72205-154 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NELARABINE (UNII: 60158CV180) (NELARABINE - UNII:60158CV180) NELARABINE 5 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72205-154-01 1 in 1 CARTON 10/22/2024 1 50 mL in 1 VIAL; Type 0: Not a Combination Product 2 NDC: 72205-154-04 6 in 1 CARTON 10/22/2024 2 50 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA216948 09/13/2024 Labeler - Novadoz Pharmaceuticals LLC (081109687) Establishment Name Address ID/FEI Business Operations MSN LABORATORIES PRIVATE LIMITED 650786952 ANALYSIS(72205-154) , MANUFACTURE(72205-154)



© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.