LOARGYS- pegzilarginase-nbln injection
Loargys by
Drug Labeling and Warnings
Loargys by is a Prescription medication manufactured, distributed, or labeled by Immedica Pharma US Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LOARGYS safely and effectively. See full prescribing information for LOARGYS.
LOARGYS (pegzilarginase-nbln) injection, for intravenous or subcutaneous use
Initial U.S. Approval: 2026WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
See full prescribing information for complete boxed warning.
Initiate LOARGYS in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation equipment. (5.1)
If a severe hypersensitivity reaction (e.g. anaphylaxis) occurs, discontinue LOARGYS, and immediately initiate appropriate medical treatment, including use of epinephrine. (5.1)
INDICATIONS AND USAGE
LOARGYS is an arginine specific enzyme indicated for the treatment of hyperargininemia in adult and pediatric patients 2 years of age and older with Arginase 1 Deficiency (ARG1-D), in conjunction with dietary protein restriction. (1)
This indication is approved under accelerated approval based on reduction of plasma arginine. (14) Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. (1)
DOSAGE AND ADMINISTRATION
- Administer LOARGYS under the supervision of a health care provider knowledgeable in the management of hypersensitivity reactions including anaphylaxis. (2.1)
- Initiate LOARGYS in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation equipment. (2.1)
- Consider pre‑medication with antihistamines. (2.1)
- Obtain a baseline plasma arginine concentration prior to initiating treatment. (2.1)
- Recommended starting dosage of LOARGYS is 0.1 mg/kg administered by intravenous infusion once weekly. (2.2)
- Maximum recommended dosage is 0.2 mg/kg once weekly. (2.2)
- See the Full Prescribing Information for recommended titration and maintenance dosage and recommended plasma arginine level testing during treatment. (2.2)
- After eight weeks of once weekly intravenous LOARGYS, patients may be switched to once weekly subcutaneous LOARGYS at the same dosage of intravenous therapy. (2.4)
- See Full Prescribing Information for dosage and administration modifications due to hypersensitivity reactions. (2.5)
- See Full Prescribing Information for instructions on preparation, storage, and administration. (2.7, 2.8, 2.9, 2.10)
DOSAGE FORMS AND STRENGTHS
Injection: 2 mg/0.4 mL and 5 mg/mL in a single-dose vial. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
Hypersensitivity: If a severe hypersensitivity reaction occurs, discontinue LOARGYS and immediately initiate appropriate medical treatment, including epinephrine. (5.1)
ADVERSE REACTIONS
Most common adverse reactions (>10%) are vomiting, pyrexia, infusion associated reactions and constipation. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Immedica at toll-free phone 1-844-627-4687 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommendations Prior to LOARGYS Treatment
2.2 Recommended Dosage and Administration
2.3 Collect Plasma Arginine Using Specific Collection Tubes and Measure Arginine Concentration Using a Specific Assay
2.4 Switching Patients from Intravenous to Subcutaneous LOARGYS Administration
2.5 Dosage and Administration Modifications Due to Hypersensitivity Reactions
2.6 Recommendation Regarding a Missed Dose
2.7 Preparation Instructions for Subcutaneous or Intravenous Administration
2.8 Storage of the Diluted LOARGYS Solution for Intravenous Use and the Undiluted LOARGYS Solution for Subcutaneous Use
2.9 Intravenous or Subcutaneous Administration Instructions
2.10 Subcutaneous Administration of LOARGYS at Home
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions Including Anaphylaxis
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
Patients treated with enzyme replacement therapies have experienced life-threatening hypersensitivity reactions, including anaphylaxis. Anaphylaxis has occurred during the early course of enzyme replacement therapy and after extended duration of therapy.
Initiate LOARGYS in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation equipment. If a severe hypersensitivity reaction (e.g. anaphylaxis) occurs, discontinue LOARGYS and immediately initiate appropriate medical treatment, including use of epinephrine.
Inform patients of the symptoms of life-threatening hypersensitivity reactions, including anaphylaxis and to seek immediate medical care should symptoms occur [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
LOARGYS is indicated for the treatment of hyperargininemia in adult and pediatric patients 2 years of age and older with Arginase 1 Deficiency (ARG1-D), in conjunction with dietary protein restriction.
This indication is approved under accelerated approval based on reduction of plasma arginine [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommendations Prior to LOARGYS Treatment
- Administer LOARGYS under the supervision of a healthcare provider knowledgeable in the management of hypersensitivity reactions including anaphylaxis [see Warnings and Precautions (5.1)].
- Initiate LOARGYS in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation equipment [see Warnings and Precautions (5.1)].
- Obtain a baseline plasma arginine concentration.
- Consider pre-medication with antihistamines [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage and Administration
The recommended starting dosage of LOARGYS is 0.1 mg/kg administered via intravenous infusion once weekly [see Dosage and Administration (2.9)]. For the recommended dosage use actual body weight.
Recommended Adjustment, Maximum Dosage, and Monitoring
To maximize the time within the normal range of 40 to 115 micromolar, dose adjustments should be aimed at achieving a pre-dose level of plasma arginine near the upper limit of normal (ULN). After four weeks of LOARGYS administration, measure pre-dose plasma arginine (168 hours after prior dose) to determine the need for dosage adjustment. If two consecutive weekly pre-dose plasma arginine measurements are not in the desired therapeutic range, increase or decrease the weekly LOARGYS dosage as follows:
- Below 50 micromolar, reduce the weekly LOARGYS dosage by 0.05 mg/kg.
- Above 150 micromolar, increase the weekly LOARGYS dosage by 0.05 mg/kg.
The maximum recommended LOARGYS dosage is 0.2 mg/kg once weekly.
Monitor plasma arginine levels (prior to LOARGYS dosing) weekly for 2 weeks after any LOARGYS dosage adjustment and as clinically indicated.
Patients may be switched from intravenous administration of LOARGYS to subcutaneous administration [see Dosage and Administration (2.4)].
2.3 Collect Plasma Arginine Using Specific Collection Tubes and Measure Arginine Concentration Using a Specific Assay
Collect samples into Immedica Pharma’s Nor-NOHA Blood Collection Tubes, which contain Nω-hydroxy-nor-Arginine (nor-NOHA), an enzyme inhibitor used to inhibit post-sampling degradation of arginine by LOARGYS, and measure arginine concentration using Immedica Pharma’s LOARGYS Arginine Assay.
LOARGYS is only available to a patient if the patient is enrolled in Study IMM-PEG-005 to obtain Immedica Pharma’s Nor-NOHA Blood Collection Tubes and LOARGYS Arginine Assay. Further information is available by contacting Immedica Pharma at 1‑844‑627‑4687.
2.4 Switching Patients from Intravenous to Subcutaneous LOARGYS Administration
After eight weeks of once weekly intravenous LOARGYS, patients may be switched to once weekly subcutaneous LOARGYS at the same dosage of intravenous therapy.
2.5 Dosage and Administration Modifications Due to Hypersensitivity Reactions
In the event of a severe hypersensitivity reaction (e.g., anaphylaxis), discontinue LOARGYS and immediately initiate appropriate medical treatment, including use of epinephrine. For additional recommendations in the event of a severe hypersensitivity reaction, [see Warnings and Precautions (5.1)].
In the event of a mild or moderate hypersensitivity reaction, consider treatment with antihistamines and/or corticosteroids. For intravenous administration, consider temporarily holding the infusion or slowing the infusion rate.
2.6 Recommendation Regarding a Missed Dose
If a dose is missed, administer LOARGYS as soon as possible. Do not administer two LOARGYS doses on the same day or within four days of another dose to make up for a missed dose. Ensure there is a minimum of four days between doses.
2.7 Preparation Instructions for Subcutaneous or Intravenous Administration
Use aseptic technique when preparing and administering LOARGYS for subcutaneous or intravenous administration.
- 1. Determine the number of LOARGYS vials needed based on actual body weight in kg and the recommended dose [see Dosage and Administration (2.2)]. Round the calculated volume of LOARGYS to nearest 0.1 mL.
- 2. Remove the vial(s) from the refrigerator and allow the vial(s) to reach room temperature.
- 3. Visually inspect the solution in the vials for particulate matter and discoloration. The solution should be clear to slightly opalescent, colorless to slightly yellow or slightly pink. Discard the vial(s) if the solution is not consistent with this appearance or if visible particulate matter is present.
- 4. Use a syringe to withdraw the calculated volume from the vial(s).
- 5. Discard unused portion.
Do not dilute the withdrawn volume of LOARGYS solution for subcutaneous administration. However, dilute the withdrawn volume of LOARGYS solution for intravenous administration.
Dilution Instructions for Intravenous Administration
- 1. Dilute the withdrawn volume of LOARGYS solution in 0.9% Sodium Chloride Injection to a maximum concentration of 0.5 mg/mL.
- 2. Gently invert the infusion bag to mix the solution. Avoid vigorous shaking or agitation.
2.8 Storage of the Diluted LOARGYS Solution for Intravenous Use and the Undiluted LOARGYS Solution for Subcutaneous Use
Use LOARGYS immediately after preparation. If not used immediately, store the diluted solution for intravenous use in the infusion container, and the undiluted solution for subcutaneous use in the syringe for up to:
- 2 hours at room temperature at 20°C to 25°C (68°F to 77°F) or
- 4 hours if stored refrigerated at 2°C to 8°C (36°F to 46°F).
Discard LOARGYS if not administered within these time frames, including total infusion time if administered intravenously.
2.9 Intravenous or Subcutaneous Administration Instructions
For Intravenous Administration
- Administer the diluted LOARGYS infusion intravenously over at least 30 minutes.
- After LOARGYS infusion, use 0.9% Sodium Chloride Injection to flush the line.
- Do not mix other drugs with LOARGYS or co-administer other drugs through the same intravenous line.
For Subcutaneous Administration
- Administer the undiluted solution subcutaneously into the abdomen, lateral part of the thigh, or the side or back of the upper arms. If injecting into the abdomen, avoid the area directly surrounding the navel.
- If more than 1 injection is needed for a single dose of LOARGYS, the injection sites should be at least 1 inch apart.
- Do not inject into scar tissue or areas that are reddened, inflamed, or swollen.
- Rotate injection sites between doses.
2.10 Subcutaneous Administration of LOARGYS at Home
If patients tolerate maintenance subcutaneous administration of LOARGYS, they may receive subcutaneous administration at home under the supervision of a healthcare provider. When switching patients from subcutaneous administration in a supervised clinical setting to the home, initially use the same dose. Do not change the dose without supervision of a healthcare provider.
In the case of a missed dose or delayed injection, contact a healthcare provider as subsequent injections may need to occur in a supervised clinical setting.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions Including Anaphylaxis
Life-threatening hypersensitivity reactions, including anaphylaxis, have occurred in patients treated with enzyme replacement therapies (ERTs) including LOARGYS. Hypersensitivity reactions that were mild to moderate in severity occurred in 13% (6/48) of LOARGYS-treated patients in clinical trials [see Adverse Reactions (6)]. Hypersensitivity reactions have included facial swelling, rash, flushing and dyspnea. The reactions generally occurred with the first few doses, but may also occur later in treatment.
LOARGYS-treated patients who developed anti-drug antibodies (ADA) generally had a greater incidence of hypersensitivity reactions compared to those who did not develop ADA [see Adverse Reactions (6.1)].
Anaphylaxis has occurred during the early course of ERT and after extended duration of ERT.
Administration of LOARGYS should be supervised by a healthcare provider knowledgeable in the management of hypersensitivity reactions including anaphylaxis. Consider pre‑medication with an antihistamine. Corticosteroids can be considered in patients who have previously developed a hypersensitivity reaction. Initiate LOARGYS in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation equipment.
- If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue LOARGYS and immediately initiate appropriate medical treatment, including use of epinephrine. Consider the risks and benefits of re-administering LOARGYS following a severe hypersensitivity reaction (including anaphylaxis). Caution should be exercised upon rechallenge. Inform patients of the symptoms of life-threatening hypersensitivity reactions, including anaphylaxis and to seek immediate medical care should symptoms occur.
- If a mild or moderate hypersensitivity reaction occurs, consider treatment with antihistamines and/or corticosteroids. For intravenous administration, consider temporarily holding the infusion or slowing the infusion rate.
-
6 ADVERSE REACTIONS
The following adverse reactions are described below and elsewhere in the labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of LOARGYS was evaluated in a randomized, double-blind, placebo-controlled trial in pediatric and adult patients with ARG1-D (Trial 1). Additional safety information was derived from Trial 2, a Phase 1 open-label trial that evaluated 16 patients between the ages of 5 to 31 years to assess safety, PK and PD of LOARGYS, and Trial 3, an open-label extension including 14 patients from Trial 2.
Adverse Reactions from Trial 1 (Double-Blind Period)
A total of 21 patients between the ages of 2 and 28 years of age, at enrollment, received intravenous LOARGYS dosages up to 0.2 mg/kg once weekly and 11 patients received placebo for 24 weeks [see Clinical Studies (14)].
Table 1 summarizes the adverse reactions reported in ≥ 2 LOARGYS-treated patients and at a higher incidence in LOARGYS-treated patients compared to placebo-treated patients in Trial 1.
Table 1: Common Adverse Reactionsa in Pediatric and Adult Patients with ARG1-D in Trial 1 a Common adverse reactions were those that occurred in ≥ 2 LOARGYS-treated patients and at a higher incidence in LOARGYS-treated patients compared to placebo-treated patients.
b Infusion-associated reactions are defined as reactions occurring within 4 hours after the infusion and included pruritus, arm swelling, and abdominal pain in LOARGYS-treated patients.
c Includes reactions that were classified as hypersensitivity during the trial and reactions that were assessed as hypersensitivity based on symptoms and temporal relationship with drug.Adverse Reaction
LOARGYS
N = 21
n (%)Placebo
N = 11
n (%)Vomiting
7 (33)
3 (27)
Pyrexia
4 (19)
1 (9)
Infusion Associated Reactionb
3 (14)
1 (9)
Constipation
3 (14)
1 (9)
Dizziness
2 (10)
0
Fall
2 (10)
0
Hypersensitivityc
2 (10)
0
Nasopharyngitis
2 (10)
0
Rhinorrhoea
2 (10)
0
Alanine aminotransferase increased
2 (10)
0
Aspartate aminotransferase increased
2 (10)
0
Description of Selected Adverse Reactions
Hypersensitivity Reactions: Hypersensitivity reactions with symptoms including facial swelling, rash, flushing and dyspnea were reported in 13% (6/48) of LOARGYS-treated patients during clinical trials [see Warnings and Precautions (5.1)].
Injection Site Reactions: Injection site reactions were reported in 14% (6/44) of patients after subcutaneous LOARGYS administration during the open-label extension periods in Trial 1 and Trial 3. Signs and symptoms included pain, erythema, swelling, irritation, and rash at the injection site.
Immunogenicity: Anti-Drug Antibody-Associated Adverse Reactions
Across Trials 1, 2, and 3, in patients with ARG1-D, the incidence of hypersensitivity reactions was 42% (5/12) in LOARGYS-treated patients who developed anti-drug antibodies (ADA) (i.e., anti-pegzilarginase-nbln antibodies and/or anti-PEG antibodies) and 3% (1/36) in those who were ADA-negative [see Clinical Pharmacology (12.6)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on LOARGYS use in pregnant females to evaluate for a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes.
In animal reproduction studies, intravenous administration of pegzilarginase-nbln to pregnant rats and rabbits during organogenesis resulted in maternal toxicity with associated increased incidence of fetal growth deficiencies (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryofetal development study in pregnant rats with normal circulating arginine levels, intravenous pegzilarginase-nbln was administered throughout organogenesis at 0.1, 0.3, and 1 mg/kg/dose on gestation days 6, 11, and 16. Maternal toxicity was observed as reduced body weight, reduced body weight gain, reduced food consumption and reduced mean gravid uterine weights at 1 mg/kg/dose (8-fold the human exposure, based on AUC at the maximum recommended human dose (MRHD), with associated decreases in fetal body weights and increased fetal developmental malformations and variations at this dose.
In an embryofetal development study in pregnant rabbits with normal circulating arginine levels, intravenous pegzilarginase-nbln was administered throughout organogenesis at 0.06, 0.1, and 0.3 mg/kg/dose on gestation days 6, 13, and 20. Maternal toxicity was observed as decreased maternal body weights, food consumption and mean gravid uterine weights at 0.3 mg/kg/dose (3-fold the human exposure, based on AUC at the MRHD), with associated decreases in fetal body weights and increased developmental malformations and variations at this dose.
In a pre- and postnatal development study in pregnant rats with normal circulating arginine levels, intravenous pegzilarginase-nbln was administered at 0.1, 0.3 and 1 mg/kg/dose once weekly from gestation day 6 to lactation day 20. Maternal toxicity included reduced body weight and food consumption during gestation and lactation with associated reduced pup body weight at 1 mg/kg/dose (8-fold the human exposure based on AUC at the MRHD). Male pups exposed to 1 mg/kg/dose through gestation and lactation also had impaired learning and memory in the passive avoidance test.
8.2 Lactation
Risk Summary
There are no data on the presence of pegzilarginase-nbln in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for LOARGYS and any potential adverse effects on the breast-fed infant from LOARGYS or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of LOARGYS have been established under accelerated approval for the treatment of hyperargininemia in pediatric patients 2 years of age and older with ARG1-D, in conjunction with dietary protein restriction [see Indications and Usage (1)]. Use of LOARGYS for this indication is supported by evidence from an adequate and well-controlled trial in adult and pediatric patients that included 29 pediatric patients 2 to 17 years of age with ARG1-D (Trial 1) and additional safety information from an open-label extension trial in adult and pediatric patients that included 9 pediatric patients (Trial 3) [see Adverse Reactions (6.1)].
The safety and effectiveness of LOARGYS have not been established for the treatment of hyperargininemia in pediatric patients less than 2 years of age with ARG1-D.
Juvenile Animal Toxicity Data
Juvenile rats with normal circulating arginine levels were administered once weekly intravenous pegzilarginase-nbln doses of 0.1, 0.3 and 1 mg/kg/dose from postnatal day 21 (the equivalent of a 2-year-old human) up to 13 weeks of age. At 1 mg/kg (15-fold the human exposure based on AUC at the MRHD), male and female rats showed increased incidence of tremors, salivation, and piloerection, along with reduced body weight and body weight gain. Reproductive organ effects were observed in male rats administered 1 mg/kg, including decreased sperm motility and total sperm count, increased percentage of abnormal sperm, and decreased epididymis, testes and prostate gland weights.
-
11 DESCRIPTION
Pegzilarginase-nbln is a trimeric cobalt substituted recombinant human arginase 1 enzyme conjugated with six to twelve moles of 5 kDa monomethoxy polyethylene glycol (mPEG) per monomer. The molecular weight of pegzilarginase-nbln is approximately 224 to 344 kDa.
LOARGYS (pegzilarginase-nbln) injection is a sterile, preservative-free, clear to slightly opalescent, and colorless to slightly yellow or slightly pink solution in a single-dose vial for intravenous infusion or subcutaneous injection. Available as 2 mg of pegzilarginase-nbln in 0.4 mL or 5 mg of pegzilarginase-nbln in 1 mL.
Each 1 mL contains 5 mg pegzilarginase-nbln, dibasic potassium phosphate (0.7 mg), glycerin (15 mg), monobasic potassium phosphate (0.14 mg), sodium chloride (2.92 mg), Water for Injection, USP, and hydrochloric acid and/ or sodium hydroxide to adjust pH to 7 to 7.6 The resultant concentration is 5 mg/mL.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
ARG1-D is an inherited metabolic disease characterized by deficiency of the arginase 1 enzyme and associated with the persistent elevation of plasma arginine.
LOARGYS provides an exogenous source of the deficient human arginase 1 enzyme activity in patients with ARG1-D and reduces plasma arginine by converting it to urea and ornithine.
12.2 Pharmacodynamics
Treatment with intravenous LOARGYS in patients with ARG1-D led to dose-dependent reduction in plasma arginine levels with median time to nadir (lowest arginine level) of 2‑5 hours. After once weekly intravenous administration at the recommended dosage, plasma arginine levels reached steady state on or before Week 8 (see Figure 1).
Predose plasma arginine levels were similar after switching from intravenous to subcutaneous LOARGYS administration at the same dose. For a given dose level, the nadir following subcutaneous dosing was higher and led to shorter durations of LOARGYS-induced hypoargininemia.
Treatment with intravenous or subcutaneous LOARGYS in patients with ARG1-D led to increases in plasma ornithine levels and decreases in plasma levels of the guanidino compounds argininic acid, guanidinoacetic acid, α-k-δ-guanidinovaleric acid, and Nα-acetyl-L-arginine.
12.3 Pharmacokinetics
The maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) of pegzilarginase-nbln at steady state following intravenous and subcutaneous administration of LOARGYS at the 0.1 mg/kg weekly dosage in adults and pediatric patients with ARG1‑D are summarized in Table 2. Pegzilarginase-nbln exposures increased in an approximately dose-proportional manner at the recommended dosages.
Table 2: Pharmacokinetic Parameters of Pegzilarginase-nbln at Steady State in Adult and Pediatric Patients with ARG1-D Abbreviations: AUC0-168=area under the concentration-time curve from time 0 to 168 hours; Cmax=maximum concentration
* Data displayed are geometric mean and geometric coefficient of variation (%)- IntravenousLOARGYS
- SubcutaneousLOARGYS
[Cmax (µg/mL)]*
- 2.48 (20%)
- 0.58 (20%)
[AUC0‑168 (h*µg/mL)]*
- 108 (18%)
- 61.3 (18%)
Absorption
Following subcutaneous LOARGYS administration, the mean absolute bioavailability was 57% and the median time to reach maximum concentration (Tmax) was approximately 34 hours.
Distribution
The volume of distribution of pegzilarginase-nbln was approximately 47 mL/kg.
Elimination
Pegzilarginase-nbln clearance (CL) was 0.029 L/h. The mean half-life was approximately 50 hours.
Metabolism
Pegzilarginase-nbln is expected to be metabolized into small peptides and amino acids by catabolic pathways.
Specific Populations
Age and sex did not have clinically meaningful effect on the pharmacokinetics of pegzilarginase-nbln once the effect of body weight was taken into account.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADAs) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADAs in the trials described below with the incidence of ADA in other studies, including those of LOARGYS or of other pegzilarginase products.
The incidence of ADAs (i.e., anti-pegzilarginase-nbln antibodies and/or anti-PEG antibodies) in LOARGYS-treated pediatric and adult patients with ARG1-D over 6 to 274 weeks in Trials 1, 2, and 3 is summarized in Table 3.
- In Trial 2, anti-PEG antibodies were observed after the second LOARGYS dose and peak anti-PEG antibody titers occurred at Week 3. In Trial 1, peak anti-PEG antibody titers were observed at Week 7.
- Anti-pegzilarginase-nbln antibodies showed similar titer profiles as anti-PEG antibodies in Trial 2, while no obvious peak titer was observed in Trial 1.
- The anti-pegzilarginase-nbln antibody and anti-PEG antibody titers decreased during continued LOARGYS treatment with the median duration of ADA positivity lasting approximately two weeks.
The incidence of neutralizing ADA has not been characterized; however, it cannot be excluded that the ADAs that showed an effect on pharmacokinetics or pharmacodynamics of pegzilarginase-nbln had neutralizing activity.
Table 3: Incidence of ADAs in LOARGYS-Treated Pediatric and Adult Patients with ARG1-D (Trials 1, 2, and 3)1 1 ADA incidences are rounded up to the nearest integer. Trial 1
(up to 152 weeks)
(N= 32)Trials 2 and 3
(up to 274 weeks)
(N= 16)ADA incidence
13%
50%
Both anti-pegzilarginase-nbln antibodies and anti-PEG antibodies
3%
31%
Only anti-pegzilarginase-nbln antibodies
9%
6%
Only anti-PEG antibodies
0%
13%
Anti-Drug Antibody Effects on Pharmacokinetics
In Trial 1, pharmacokinetic (PK) samples evaluable for the impact of ADA were available in 2 LOARGYS-treated patients. These 2 patients had pegzilarginase-nbln AUC that were comparable to pegzilarginase-nbln AUC in patients who did not develop anti-pegzilarginase-nbln antibodies.
Across Trials 1, 2, and 3, LOARGYS-treated patients who had higher anti-PEG antibody titers had lower pegzilarginase-nbln AUC compared to those with lower anti-PEG antibody titers or those who did not develop ADA.
Anti-Drug Antibody Effects on Pharmacodynamics
In Trial 1, pharmacodynamic (PD) samples evaluable for the impact of anti-pegzilarginase-nbln antibodies were available in 2 LOARGYS-treated patients. These 2 patients had similar plasma arginine levels to patients who did not develop anti-pegzilarginase-nbln antibodies at the same dosage.
Across Trials 1, 2, and 3, LOARGYS-treated patients who had higher anti-PEG antibody titers had lower plasma arginine reduction compared to those who did not develop anti-PEG antibodies and those with lower anti-PEG antibody titers.
In the LOARGYS-treated patients who exhibited the highest anti-pegzilarginase-nbln antibody titers and the highest anti-PEG antibody titers, the arginine concentrations were higher after the ADAs developed.
Anti-Drug Antibody Effects on Safety and Efficacy
Hypersensitivity reactions occurred at a higher incidence in LOARGYS-treated patients who were ADA-positive compared to those who were ADA-negative [see Adverse Reactions (6.1)]. Across Trials 1, 2, and 3, of the 4 LOARGYS-treated patients who experienced hypersensitivity reactions and had an evaluation of anti-pegzilarginase-nbln IgE antibodies, 25% (1/4) of patients had positive results for anti-pegzilarginase-nbln IgE antibodies.
No clinically significant effect of ADAs on the incidence of injection site reactions was identified over the LOARGYS treatment duration of 274 weeks.
The effect of ADAs on the effectiveness of LOARGYS in the treatment of hyperargininemia in adult and pediatric patients 2 years of age and older with ARG1-D is unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term studies in animals to evaluate carcinogenic potential have not been performed with LOARGYS.
Mutagenesis
Studies to evaluate the mutagenic potential of LOARGYS have not been conducted.
Impairment of Fertility
In a fertility and early embryonic development study in rats with normal circulating arginine levels, pegzilarginase-nbln was administered intravenously at 0.1, 0.3 and 1 mg/kg/dose once weekly to males 56 days prior to mating and continuing through mating, and to females 14 days prior to mating and continuing through gestation day 7. Male rats administered 1 mg/kg/dose (0.8-fold the MRHD, based on body surface area) showed increased salivation, thin body condition and unkempt appearance, reduced body weights, reduced food consumption, decreased sperm motility, decreased sperm concentration and increased abnormal sperm morphology, which was associated with decreased uterine implantation sites and increased pre-implantation loss in untreated paired females. Female rats administered 1 mg/kg/dose (0.8-fold the MRHD, based on body surface area) showed reduced body weight, and decreased fecundity and fertility indices when paired with untreated males.
13.2 Animal Toxicology and/or Pharmacology
In a repeated dose toxicity study in rats with normal circulating arginine levels, pegzilarginase-nbln was administered intravenously at 0.1, 0.3, and 1 mg/kg once weekly for 6 months beginning on postnatal day 21. Decreased body weight gain, and increased convulsions, tremors, salivation, and piloerection were observed in males and females at 1 mg/kg/dose (15-fold the human exposure based on AUC at the MRHD). In male rats administered doses ≥0.3 mg/kg (4.5-fold the human exposure based on AUC at the MRHD), findings included decreased reproductive organ weights (testis, seminal vesicles, epididymis and prostate), testicular tubular degeneration, epididymal germ cell debris, and decreased sperm motility, decreased total sperm count and increased percentage of abnormal sperm.
In a repeated dose toxicity study in monkeys with normal circulating arginine levels, pegzilarginase-nbln was administered intravenously at 0.1, 0.3 and 1 mg/kg/dose once weekly for 13 weeks. Decreased body weight, inappetence, hunched posture and watery feces led to early death of two males administered 1 mg/kg (11-fold the human exposure based on AUC at the MRHD). Tremor and decreased activity were also observed in these animals. Surviving male and female monkeys administered doses of 1 mg/kg showed reduced body weight, increased incidences of sparse hair, tremors, inappetence, and decreased activity. Animals were considered immature during dose administration therefore effects on reproductive organs could not be assessed.
-
14 CLINICAL STUDIES
The effectiveness of LOARGYS for the treatment of hyperargininemia in adult and pediatric patients with ARG1-D was evaluated in Trial 1 (NCT03921541) which was a multicenter, double-blind, 24-week placebo-controlled trial with a long-term open-label extension period of up to 150 weeks. A total of 32 patients were randomized 2:1 to receive intravenous LOARGYS (n=21) or placebo (n=11) once weekly for 24 weeks.
Of the 32 patients randomized in Trial 1, 19 were males (59%) and 13 were females (41%). Patients were between 2 to 29 years of age with 90% of patients between 2 to 17 years of age (median pediatric age was 9 years) at enrollment. The patient population consisted of 14 Whites (44%), 6 Asians (19%), 2 Black or African Americans (6%), and 2 in mixed racial groups (6%). Ethnicity consisted of 9 patients who were Hispanic or Latino (28%) and 23 who were Not Hispanic or Latino (72%).
LOARGYS-treated patients received an initial dosage of 0.1 mg/kg and were titrated within a range of 0.05 mg to 0.2 mg/kg, as clinically indicated. Patients who previously had an ARG1-D-related dietary regimen or ammonia scavengers continued to receive these treatment(s) throughout the initial trial period. A total of 31 patients completed the double-blind period; one patient in the LOARGYS arm discontinued for personal reasons.
The primary endpoint in Trial 1 was the change from baseline in plasma arginine at Week 24.
LOARGYS-treated patients had a significant mean reduction in plasma arginine levels from baseline to Week 24 (Table 4). Additionally, 90% of LOARGYS-treated patients achieved target plasma arginine levels (below 200 μM) and normalized levels, compared to 0% of placebo-treated patients. Figure 1 displays the mean plasma arginine levels over time by the randomized treatment group.
Table 4: Change in Plasma Arginine (micromolar) from Baseline at Week 24 in Patients with ARG1-D, Trial 1 Study Visit LOARGYS Placebo 1 Calculated as the average of at most four measurements from Week 21 through Week 24.
2 The treatment difference in least-square means was estimated by a mixed model for repeated measures including treatment, visit, treatment-by-visit interaction, and baseline arginine value. The p-value for testing the treatment difference is <0.0001.Baseline
N = 21
N = 11
Mean (SD)
365 (94)
472 (80)
Week 241
N = 20
N = 11
Mean (SD)
92 (51)
449 (86)
Mean Absolute Change at Week 24 (SD)
-272 (104)
-23 (99)
Treatment Difference (95% CI) 2
-312 (-384, -239)
Mean Percent Change at Week 24 (%) (SD)
-74 (14)
-3 (20)
Treatment Difference (95% CI) 2
-72 (-89, -55)
All 31 patients (n=20 LOARGYS and n=11 placebo) who completed the double-blind period entered the extension period and received open label treatment of LOARGYS:
- First 8 weeks: Patients who were previously treated with intravenous LOARGYS continued their weekly intravenous LOARGYS dosage and patients who were previously treated with placebo initially received 0.1 mg/kg of weekly intravenous LOARGYS. The LOARGYS dosage in both groups could be modified based on plasma arginine levels.
- After the first 8 weeks: Patients had the option to receive weekly subcutaneous LOARGYS (same as the intravenous dosage) that could be modified based on plasma arginine levels up to a maximum of 0.2 mg/kg weekly.
The median duration of LOARGYS exposure in the extension period, excluding the double-blind period of 24 weeks, was 94 weeks (range: 62 to 152 weeks). During the extension period, patients who switched from placebo to LOARGYS achieved similar reductions in mean plasma arginine levels as those who had received LOARGYS from the start of the study (Figure 1).
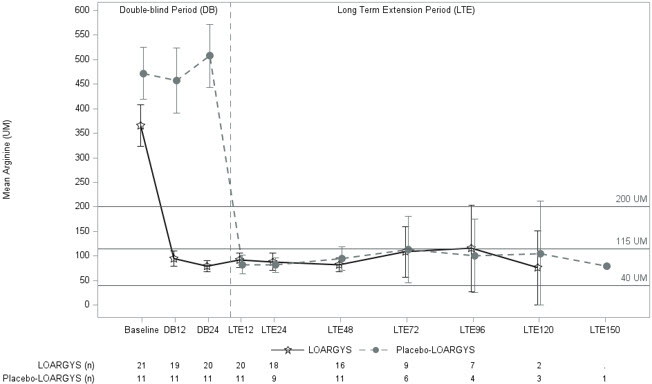
Figure 1: Mean (95% CI) Plasma Arginine Level (micromolar) in Patients with ARG1-D By Visit (Weeks), Trial 1
Notes: 95% confidence interval of mean (pre-dose) is displayed; Recommended plasma arginine levels: below 200 micromolar; Normal range defined as 40–115 micromolar in Trial 1. The x-axis unit of time is visit week.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
LOARGYS (pegzilarginase-nbln) is supplied as a sterile, preservative-free, clear to slightly opalescent and colorless to slightly yellow or slightly pink solution. LOARGYS is available as:
- One single-dose vial 2 mg/0.4 mL in a carton (NDC: 81583-102-01)
- One single-dose vial 5 mg/mL in a carton (NDC: 81583-105-01).
Storage and Handling
Store refrigerated at 2 °C to 8 °C (36 °F to 46 °F) in the original carton to protect from light. Do not freeze. Do not shake.
-
17 PATIENT COUNSELING INFORMATION
Hypersensitivity Reactions Including Anaphylaxis:
Advise the patient and caregiver that life-threatening hypersensitivity reactions, including anaphylaxis may occur with LOARGYS treatment.
Advise the patient or caregiver that anaphylaxis has occurred during the early course of enzyme replacement therapy and after extended duration of therapy.
Inform the patient and caregiver of the symptoms of life-threatening hypersensitivity reactions, including anaphylaxis, and to seek immediate medical care should symptoms occur [see Warnings and Precautions (5.1)].
-
Manufactured by:
Immedica Pharma AB, Stockholm, Sweden
U.S. License No. 2342Manufactured at:
Lyophilization Services of New England, Inc. (LSNE), D/B/A: PCI Pharma Services, Bedford, NH 03110Marketed by:
Immedica Pharma US Inc., Chicago, IL 60642LOARGYS is a registered trademark of Immedica Pharma AB.
-
PRINCIPAL DISPLAY PANEL - 2 mg/0.4 mL Carton
NDC: 81583-102-01
Rx only
Loargys
(pegzilarginase-nbln)
Injection2 mg/0.4 mL
For Intravenous Infusion After
Dilution or Subcutaneous InjectionOne 0.4 mL single-dose vial
discard unused portionImmedica
pharma -
PRINCIPAL DISPLAY PANEL - 5 mg/1 mL Carton
NDC: 81583-105-01
Rx only
Loargys
(pegzilarginase-nbln)
Injection5 mg/mL
For Intravenous Infusion After
Dilution or Subcutaneous InjectionOne 1 mL single-dose vial
Discard unused portionImmedica
pharma -
INGREDIENTS AND APPEARANCE
LOARGYS
pegzilarginase-nbln injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 81583-102 Route of Administration INTRAVENOUS, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PEGZILARGINASE (UNII: 4YV4KW88GD) (PEGZILARGINASE - UNII:4YV4KW88GD) PEGZILARGINASE 2 mg in 0.4 mL Inactive Ingredients Ingredient Name Strength GLYCERIN (UNII: PDC6A3C0OX) SODIUM CHLORIDE (UNII: 451W47IQ8X) MONOBASIC POTASSIUM PHOSPHATE (UNII: 4J9FJ0HL51) DIBASIC POTASSIUM PHOSPHATE (UNII: CI71S98N1Z) WATER (UNII: 059QF0KO0R) HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 81583-102-01 1 in 1 CARTON 03/09/2026 1 0.4 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761211 03/09/2026 LOARGYS
pegzilarginase-nbln injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 81583-105 Route of Administration INTRAVENOUS, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PEGZILARGINASE (UNII: 4YV4KW88GD) (PEGZILARGINASE - UNII:4YV4KW88GD) PEGZILARGINASE 5 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCERIN (UNII: PDC6A3C0OX) SODIUM CHLORIDE (UNII: 451W47IQ8X) MONOBASIC POTASSIUM PHOSPHATE (UNII: 4J9FJ0HL51) DIBASIC POTASSIUM PHOSPHATE (UNII: CI71S98N1Z) WATER (UNII: 059QF0KO0R) HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 81583-105-01 1 in 1 CARTON 12/31/2027 1 1 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761211 03/09/2026 Labeler - Immedica Pharma US Inc. (141007232)


Trademark Results [Loargys]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 LOARGYS 88627216 not registered Live/Pending |
Aeglea BioTherapeutics, Inc. 2019-09-23 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.