SAPHNELO- anifrolumab-fnia injection, solution
SAPHNELO by
Drug Labeling and Warnings
SAPHNELO by is a Prescription medication manufactured, distributed, or labeled by AstraZeneca Pharmaceuticals LP, AstraZeneca PLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SAPHNELO® safely and effectively. See full prescribing information for SAPHNELO.
SAPHNELO (anifrolumab-fnia) injection, for intravenous, or subcutaneous use
Initial U.S. Approval: 2021RECENT MAJOR CHANGES
Dosage and Administration (2) 04/2026
INDICATIONS AND USAGE
SAPHNELO is a type I interferon (IFN) receptor antagonist indicated for the treatment of adult patients with moderate to severe systemic lupus erythematosus (SLE), who are receiving standard therapy. (1)
Limitations of Use: The efficacy of SAPHNELO has not been evaluated in patients with severe active lupus nephritis or severe active central nervous system lupus. Use of SAPHNELO is not recommended in these situations. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
SAPHNELO is contraindicated in patients with a history of anaphylaxis with anifrolumab-fnia. (4)
WARNINGS AND PRECAUTIONS
- Serious Infections: Serious and sometimes fatal infections have occurred in patients receiving SAPHNELO. SAPHNELO increases the risk of respiratory infections and herpes zoster. Avoid initiating treatment during an active infection. Consider the individual benefit-risk if using in patients with severe or chronic infections. Consider interrupting therapy with SAPHNELO if patients develop a new infection during treatment. (5.1)
- Hypersensitivity Reactions Including Anaphylaxis: Serious hypersensitivity reactions including anaphylaxis and angioedema have been reported. (5.2)
- Malignancy: Consider the individual benefit-risk in patients with known risk factors for malignancy prior to prescribing SAPHNELO. (5.3)
- Immunizations: Avoid use of live or live-attenuated vaccines in patients receiving SAPHNELO. (5.4)
- Not Recommended for Use with Other Biologic Therapies. (5.5)
ADVERSE REACTIONS
Most common adverse drug reactions (incidence ≥5%) are nasopharyngitis, upper respiratory tract infections, bronchitis, infusion related reactions, herpes zoster and cough. (6)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Information
2.2 Recommended Dosage
2.3 Intravenous Preparation and Administration Instructions
2.4 Subcutaneous Preparation and Administration Instructions
2.5 Switching Between Intravenous and Subcutaneous Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
5.2 Hypersensitivity Reactions Including Anaphylaxis
5.3 Malignancy
5.4 Immunizations
5.5 Not Recommended for Concomitant Use with Other Biologic Therapies
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Intravenous Administration in Adults with Moderate to Severe SLE
14.2 Subcutaneous Administration in Adults with Moderate to Severe SLE
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
SAPHNELO is indicated for the treatment of adult patients with moderate to severe systemic lupus erythematosus (SLE), who are receiving standard therapy.
Limitations of Use
The efficacy of SAPHNELO has not been evaluated in patients with severe active lupus nephritis or severe active central nervous system lupus. Use of SAPHNELO is not recommended in these situations.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Information
SAPHNELO is intended for use under the guidance of a healthcare provider and may be administered as an intravenous infusion or as a subcutaneous injection.
SAPHNELO vials are for intravenous use only and must be diluted prior to intravenous administration [see Dosage and Administration (2.3)].
SAPHNELO prefilled syringe and autoinjector (SAPHNELO PEN) are for subcutaneous use only [see Dosage and Administration (2.4)].
2.2 Recommended Dosage
The recommended dosage of SAPHNELO is:
- 300 mg, administered as an intravenous infusion over a 30‑minute period every 4 weeks; or
- 120 mg, administered as a subcutaneous injection once every week.
Missed Dose
If a planned intravenous infusion is missed, administer SAPHNELO as soon as possible. Maintain a minimum interval of 14 days between infusions.
If a planned subcutaneous dose is missed, instruct the patient to administer SAPHNELO as soon as they remember. Thereafter, instruct the patient to start a new weekly schedule from the day the missed dose was administered or resume dosing on their usual day of administration, providing a minimum interval of 3 days between subcutaneous injections.
2.3 Intravenous Preparation and Administration Instructions
SAPHNELO is supplied as a single-dose vial. Prepare the diluted infusion solution using aseptic technique, by the following procedure:
- 1. Visually inspect the vial for particulate matter and discoloration. SAPHNELO is a clear to opalescent, colorless to slightly yellow, solution. Discard the vial if the solution is cloudy, discolored or visible particles are observed. Do not shake the vial.
- 2. Withdraw and discard 2 mL of solution from a 50 mL or 100 mL 0.9% Sodium Chloride Injection, USP infusion bag.
- 3. Withdraw 2 mL of solution from the vial of SAPHNELO and add it to the infusion bag. Mix the solution by gentle inversion. Do not shake.
- 4. Each vial is intended for one time use only. Discard any unused portion remaining in the vial.
- 5. Administer the infusion solution immediately after preparation.
- 6. If the infusion solution is not administered immediately, store the diluted solution of SAPHNELO at room temperature 59°F to 77°F (15°C to 25°C) for up to 4 hours, or refrigerated 36°F to 46°F (2°C to 8°C) for up to 24 hours. Do not freeze. Protect from light. If refrigerated, allow the diluted SAPHNELO solution to reach room temperature prior to administration.
- 7. Administer the infusion solution intravenously over a 30-minute period through an infusion line containing a sterile, low-protein binding 0.2 to 15 micron in-line or add-on filter.
- 8. To ensure the complete dose of SAPHNELO has been administered, flush the entire infusion line with 25 mL of 0.9% Sodium Chloride Injection, USP at the end of the infusion.
- 9. Do not co-administer other medicinal products through the same infusion line.
- 10. Dispose of any unused medicinal product or waste material in accordance with local requirements.
2.4 Subcutaneous Preparation and Administration Instructions
The SAPHNELO prefilled syringe and autoinjector (SAPHNELO PEN) are for subcutaneous use.
- 1. Comprehensive instructions for subcutaneous administration of SAPHNELO using the prefilled syringe or autoinjector are provided in the ‘Instructions for Use’.
- 2. Patients/caregivers may administer SAPHNELO prefilled syringe/SAPHNELO PEN after proper training in the subcutaneous injection technique and after the healthcare provider determines it is appropriate.
- 3. Prior to administration, remove SAPHNELO from the refrigerator and allow it to come to room temperature for 60 minutes.
- 4. Visually inspect SAPHNELO for particulate matter and discoloration prior to administration. SAPHNELO is a clear to opalescent, colorless to slightly yellow, solution. Discard the prefilled syringe or autoinjector if the solution is cloudy, discolored or visible particles are observed.
- 5. Administer the subcutaneous injection into the thigh or abdomen, avoiding the 5 cm (approximately 2 inches) area around the navel. The upper arm can also be used if a healthcare professional or caregiver administers the injection.
- 6. Do not inject into areas where the skin is tender, bruised, erythematous or hardened. When injecting in the same region, advise patients to use an injection site at least 3 cm (1‑inch) away from the last injection site.
- 7. Dispose of any unused medicinal product or waste material in accordance with local requirements.
2.5 Switching Between Intravenous and Subcutaneous Administration
When transitioning patients from intravenous administration to subcutaneous administration of SAPHNELO, administer the first subcutaneous injection approximately 2 weeks after the last intravenous infusion.
When transitioning patients from subcutaneous administration to intravenous administration of SAPHNELO, administer the first intravenous infusion approximately 3 to 4 weeks after the last subcutaneous injection.
-
3 DOSAGE FORMS AND STRENGTHS
Intravenous Infusion
- Injection: 300 mg/2 mL (150 mg/mL) as a clear to opalescent, colorless to slightly yellow solution in a single-dose vial.
Subcutaneous Injection
- Injection: 120 mg/0.8 mL as a clear to opalescent, colorless to slightly yellow solution in a single-dose prefilled syringe or single-dose autoinjector (SAPHNELO PEN).
-
4 CONTRAINDICATIONS
SAPHNELO is contraindicated in patients with a history of anaphylaxis with anifrolumab-fnia [see Warnings and Precautions (5.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
Serious and sometimes fatal infections (including COVID‑19) have occurred in patients receiving immunosuppressive agents, including SAPHNELO. In controlled trials, fatal infections occurred more frequently in patients receiving SAPHNELO [see Adverse Reactions (6.1)].
In controlled trials, SAPHNELO increased the risk of respiratory infections and herpes zoster (disseminated herpes zoster events have been reported) [see Adverse Reactions (6.1)].
Avoid initiating treatment with SAPHNELO in patients with any clinically significant active infection until the infection is resolved or adequately treated. Consider the benefit and risk of administering SAPHNELO in patients with a chronic infection, a history of recurrent infections, or known risk factors for infection. Instruct patients to seek medical advice if signs or symptoms of a clinically significant infection occur. If a patient develops an infection or is not responding to standard anti-infective therapy while on SAPHNELO, monitor the patient closely and consider interrupting SAPHNELO until the infection resolves.
5.2 Hypersensitivity Reactions Including Anaphylaxis
Serious hypersensitivity reactions (including anaphylaxis) have been reported following SAPHNELO administration [see Contraindication (4)]. Events of angioedema have also been reported [see Adverse Reactions (6.1)].
Other hypersensitivity reactions and infusion-related reactions have occurred following administration of SAPHNELO [see Adverse Reactions (6.1)]. Consider pre-medication before infusion of SAPHNELO for patients with a history of these reactions.
SAPHNELO should be administered by healthcare providers prepared to manage hypersensitivity reactions, including anaphylaxis, and infusion-related reactions. If a serious infusion-related or hypersensitivity reaction (e.g., anaphylaxis) occurs, immediately interrupt the administration of SAPHNELO and initiate appropriate therapy.
5.3 Malignancy
There is an increased risk of malignancies with the use of immunosuppressants. The impact of SAPHNELO treatment on the potential development of malignancies is not known.
Consider the individual benefit-risk in patients with known risk factors for the development or reoccurrence of malignancy prior to prescribing SAPHNELO. In patients who develop malignancies, consider the benefit-risk of continued treatment with SAPHNELO.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are also discussed elsewhere in the labeling:
- Serious Infections [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions Including Anaphylaxis [see Warnings and Precautions (5.2)]
- Malignancy [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions with Intravenous Administration
The safety of SAPHNELO was assessed in adult patients with moderate to severe SLE who received SAPHNELO 300 mg by intravenous infusion every 4 weeks (N=459) for 52 weeks, compared to placebo (N=466) in controlled clinical trials (Trials 1, 2 and 3) [see Clinical Studies (14.1)]. The population studied had a mean age of 41 years (range: 18 to 69), of which 93% were female, 60% White, 13% Black/African American, and 10% Asian.
In the controlled-clinical trials, adverse reactions, irrespective of causality, were reported in 87% of patients receiving SAPHNELO and 79% of patients receiving placebo.
Adverse reactions that occurred at greater than or equal to 2% incidence are shown in Table 1.
Table 1 Adverse Reactions Occurring in ≥2% of Adults with Moderate to Severe SLE Treated with Intravenous SAPHNELO for 52 weeks in Trials 1, 2 and 3 - * Upper respiratory tract infections (including Upper respiratory tract infections, Nasopharyngitis, Pharyngitis)
- † Bronchitis (including Bronchitis, Bronchitis viral, Tracheobronchitis)
- ‡ Respiratory tract infection (including Respiratory tract infection, Respiratory tract infection viral, Respiratory tract infection bacterial)
Adverse Reaction
SAPHNELO
(N=459)
%
Placebo
(N=466)
%
Upper respiratory tract infection*
34
23
Bronchitis†
11
5.2
Infusion‑related reactions
9.4
7.1
Herpes Zoster
6.1
1.3
Cough
5.0
3.2
Respiratory tract infection‡
3.3
1.5
Hypersensitivity
2.8
0.6
All patients received standard therapy.
Long-term Safety
Patients who completed Trials 2 and 3 (Phase III feeder trials) were eligible to continue on treatment in a randomized, double-blind, placebo-controlled long-term extension (LTE) trial, for an additional 3 years. The long-term safety of SAPHNELO was assessed in 257 patients who received SAPHNELO 300 mg every 4 weeks and 112 patients who received placebo in both a feeder trial and the LTE. Of these, 177 patients who received SAPHNELO (68.9%) and 52 patients who received placebo (46.4%) completed a total of 4 years on treatment. The overall long-term safety profile of SAPHNELO was consistent with Trials 1, 2 and 3.
Specific Adverse Reactions
Infections: In the 52‑week controlled-clinical trials, infections were reported in a greater proportion of patients while on treatment with SAPHNELO compared to placebo (69.7% versus 55.4%, respectively).
Herpes Zoster: In the 52‑week controlled-clinical trials, the incidence of herpes zoster in patients while on treatment with SAPHNELO was 6.1% and 1.3% in patients on placebo. Cases with multidermatomal involvement and disseminated presentation have been reported. Of the 28 SAPHNELO-treated patients with herpes zoster, 2 experienced disseminated disease requiring hospitalization compared to none among placebo-treated patients.
Hypersensitivity Reactions Including Anaphylaxis: During the SLE intravenous development program, there was one report of an anaphylactic reaction in a patient who received SAPHNELO 150 mg, and 4 reports of angioedema after 300 mg. In general, the hypersensitivity reactions were predominantly mild or moderate in intensity and did not lead to discontinuation of SAPHNELO.
In the 52‑week controlled‑clinical trials, hypersensitivity reactions occurred in 2.8% of patients while on treatment with SAPHNELO and 0.6% of patients on placebo. Serious hypersensitivity reactions were reported in 0.6% of patients receiving SAPHNELO, including angioedema (n=2).
Infusion‑related Reactions: Infusion‑related reactions were mild to moderate in intensity; the most common symptoms were headache, nausea, vomiting, fatigue, and dizziness.
In the 52‑week controlled‑clinical trials, the incidence of infusion‑related reactions while on treatment was 9.4% in patients while on treatment with SAPHNELO and 7.1% in patients on placebo.
Malignancies: In 52‑week controlled-clinical trials, malignancies (excluding non‑melanoma skin cancers) were observed in 0.7% and 0.6% of SAPHNELO-treated and placebo-treated patients. Malignant neoplasm (including non‑melanoma skin cancers) was reported in 1.3% of SAPHNELO-treated patients, compared to 0.6% placebo-treated patients. The malignancies that were reported in more than one patient treated with SAPHNELO included breast cancer and squamous cell carcinoma.
Adverse Reactions with Subcutaneous Administration
The safety profile of SAPHNELO administered subcutaneously in adult patients with moderate to severe SLE is derived from Trial 5 [see Clinical Studies (14.2)]. The data reflect SAPHNELO exposure in 185 patients who received SAPHNELO 120 mg by subcutaneous injection once every week, with a median duration of 52 weeks, as compared with comparable exposure to placebo in 182 patients. All patients received standard therapy. The population studied had a mean age of 43 years (range: 18 to 70), of which 92% were female, 74% White, 9% Asian, 8% American Indian or Alaska Native, and 5% Black/African American.
The safety profile observed for SAPHNELO administered subcutaneously was consistent with the known safety profile of SAPHNELO administered intravenously.
Serious Infections in the Pool of Intravenous and Subcutaneous Trials
In the pool of four placebo-controlled clinical trials in adults with moderate to severe SLE where SAPHNELO was administered either intravenously or subcutaneously (Trials 1, 2, 3, and 5), the incidence of serious infections while on treatment was 5.4% in patients treated with SAPHNELO compared to 4.9% in patients receiving placebo. Fatal infections occurred while on treatment in 0.5% of SAPHNELO-treated patients and 0% in placebo-treated patients. The most frequent serious infection reported in patients treated with SAPHNELO was pneumonia. During the LTE of Trials 2 and 3, the most common serious infections were COVID‑19 and pneumonia.
6.2 Postmarketing Experience
The following adverse reaction has been identified during post-approval use of SAPHNELO. Because the reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Arthralgia
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to SAPHNELO during pregnancy. For more information about the registry or to report a pregnancy while on SAPHNELO, healthcare providers should contact AstraZeneca at 1‑877‑693‑9268 or https://anifrolumabpregnancyandbreastfeedingstudy.us/.
Risk Summary
The limited human data with SAPHNELO use in pregnant women are insufficient to inform on drug‑associated risk for major birth defects, miscarriage, or adverse maternal or fetal outcome. Monoclonal IgG antibodies are known to be actively transported across the placenta as pregnancy progresses; therefore, anifrolumab‑fnia exposure to the fetus may be greater during the third trimester of pregnancy.
In an enhanced pre- and post-natal development study with pregnant cynomolgus monkeys that received intravenous administration of anifrolumab-fnia, there was no evidence of embryotoxicity or fetal malformations with exposures up to approximately 28‑times the exposure at the maximum recommended human dose (MRHD) on an Area Under Curve (AUC) basis (see Data).
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk: Pregnant women with SLE are at increased risk of adverse pregnancy outcomes, including worsening of the underlying disease, premature birth, miscarriage, and intrauterine growth restriction. Maternal lupus nephritis increases the risk of hypertension and preeclampsia/eclampsia. Passage of maternal autoantibodies across the placenta may result in adverse neonatal outcomes, including neonatal lupus and congenital heart block.
Animal Data: In an enhanced pre- and post-natal development study, pregnant cynomolgus monkeys received anifrolumab-fnia at intravenous doses of 30 or 60 mg/kg once every 2 weeks from confirmation of pregnancy at Gestation Day 20, throughout the gestation period, and continuing until 1‑month post-partum (approximately Lactation Day 28). There was no evidence of anifrolumab-fnia related maternal toxicity, embryo-fetal toxicity, or post-natal developmental effects. No anifrolumab-fnia related effect on T-cell-dependent antibody response in the infants was noted up to Day 180 after birth. The no observed adverse effect level (NOAEL) for maternal and developmental toxicity was identified as 60 mg/kg (approximately 28‑times the MRHD on an AUC basis). In the infants, mean serum concentrations of anifrolumab‑fnia on Day 30 after birth increased with dose and were approximately 4.2% to 9.7% of the respective maternal concentrations. The anifrolumab-fnia concentrations in the infant serum were up to approximately 22‑times the concentrations in the maternal milk, suggesting that anifrolumab-fnia had transferred via the placenta.
8.2 Lactation
Risk Summary
No data are available regarding the presence of SAPHNELO in human milk, the effects on the breastfed child, or the effects on milk production. Anifrolumab-fnia was detected in the milk of female cynomolgus monkeys administered anifrolumab-fnia. Due to species-species differences in lactation physiology, animal data may not reliably predict drug levels in humans. Maternal IgG is known to be present in human milk. If anifrolumab-fnia is transferred into human milk, the effects of local gastrointestinal exposure and limited systemic exposure in the breastfed infant to anifrolumab-fnia are unknown.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for SAPHNELO and any potential adverse effects on the breast-fed child from SAPHNELO or from the underlying maternal condition.
-
11 DESCRIPTION
Anifrolumab-fnia is a type I interferon (IFN) receptor antagonist, immunoglobulin G1 kappa (IgG1κ) monoclonal antibody that is produced in mouse myeloma cells (NS0) by recombinant DNA technology. The molecular weight is approximately 148 kDa.
Intravenous Infusion
SAPHNELO (anifrolumab-fnia) injection is a sterile, preservative‑free, clear to opalescent, colorless to slightly yellow, solution in a single-dose vial for intravenous use.
Each 2 mL single-dose vial contains 300 mg (150 mg/mL) of anifrolumab-fnia, L-histidine (3 mg), L-histidine hydrochloride monohydrate (6 mg), L-lysine hydrochloride (18 mg), polysorbate 80 (1 mg), trehalose dihydrate (98 mg), and Water for Injection, USP. The pH is 5.9.
Subcutaneous Injection
SAPHNELO (anifrolumab-fnia) injection is a sterile, preservative-free, clear to opalescent, colorless to slightly yellow, solution in a prefilled syringe or autoinjector for subcutaneous injection.
Each 0.8 mL single-dose prefilled syringe or autoinjector delivers 120 mg of anifrolumab-fnia, histidine (1 mg), L-histidine hydrochloride monohydrate (3 mg), lysine hydrochloride (7 mg), polysorbate 80 (0.4 mg), trehalose (36 mg), and Water for Injection, USP. The pH is 5.9.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Anifrolumab-fnia is a human IgG1κ monoclonal antibody that binds to subunit 1 of the type I interferon receptor (IFNAR) with high specificity and affinity. This binding inhibits type I IFN signaling, thereby blocking the biologic activity of type I IFNs. Anifrolumab-fnia also induces the internalization of IFNAR1, thereby reducing the levels of cell surface IFNAR1 available for receptor assembly. Blockade of receptor mediated type I IFN signaling inhibits IFN responsive gene expression as well as downstream inflammatory and immunological processes. Inhibition of type I IFN blocks plasma cell differentiation and normalizes peripheral T-cell subsets.
Type I IFNs play a role in the pathogenesis of SLE. Approximately 60-80% of adult patients with active SLE express elevated levels of type I IFN inducible genes.
12.2 Pharmacodynamics
In patients with moderate to severe SLE, following the administration of SAPHNELO, neutralization (≥80%) of a type I IFN gene signature was observed from Week 4 to Week 52 in blood samples of patients with elevated levels of type I IFN inducible genes and returned to baseline levels within 8 to 12 weeks following withdrawal of SAPHNELO at the end of the 52‑week treatment period. However, the clinical relevance of the type I IFN gene signature neutralization is unclear.
In SLE patients with positive anti-dsDNA antibodies at baseline (Trials 2 and 3), treatment with SAPHNELO led to numerical reductions in anti-dsDNA antibodies over time through Week 52.
In patients with low complement levels (C3 and C4) at baseline (Trials 2 and 3), increases in complement levels were observed in patients receiving SAPHNELO through Week 52.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of anifrolumab-fnia was studied in adult patients with SLE following intravenous doses ranging from one-third (100 mg) to 3.3 times (1000 mg) the approved intravenous dosage, 300 mg once every 4 weeks, and subcutaneous 120 mg weekly doses, as well as in healthy volunteers following a single intravenous dose at 300 mg and a single subcutaneous dose of 120 mg.
Anifrolumab-fnia exhibits non-linear PK in the dose range of 100 mg to 1000 mg with more than dose-proportional increases in the exposure as measured by AUC, following intravenous administration.
The estimated time to reach steady state is approximately 112 days for both intravenous and subcutaneous administration.
Following 300 mg intravenous administrations every 4 weeks, the accumulation ratio for Cmax was 1.11 and for Ctrough was 2.37. Following 120 mg subcutaneous administration weekly, the accumulation ratio for Cmax was 1.85 and Ctrough was 1.85.
Absorption
The bioavailability of anifrolumab-fnia was estimated to be 73% following subcutaneous injection. The estimated maximum serum concentration (Cmax) of anifrolumab-fnia at steady state was 63.7 μg/mL.
Distribution
Based on population PK analysis, the estimated volume of distribution at steady state for a typical patient with SLE (68 kg) is 5.16 L.
Elimination
From population PK analysis, anifrolumab-fnia exhibited non-linear PK due to IFNAR1-mediated drug clearance.
The estimated systemic clearance (CL) for anifrolumab-fnia is 0.146 L/day.
Based on population PK analysis of patients who received SAPHNELO for one year, serum concentrations of anifrolumab-fnia were below detection in 95% of patients approximately 16 weeks after the last dose.
Specific Populations
There was no clinically meaningful difference in systemic clearance based on age, race, ethnicity, region, gender, IFN status or body weight, that requires dose adjustment.
Age: Based on population PK analyses, age (range 18 to 70 years) did not affect anifrolumab-fnia clearance. Limited PK data are available for geriatric patients; 3% (n=33) of the patients included in the PK analysis were 65 years or older [see Use in Specific Populations (8.5)].
Patients with Renal Impairment: No specific clinical trials have been conducted to investigate the effect of renal impairment on anifrolumab-fnia. Based on population PK analyses, anifrolumab-fnia clearance was comparable in SLE patients with mild (60-89 mL/min/1.73 m2) and moderate (30-59 mL/min/1.73 m2) decrease in eGFR values and patients with normal renal function (≥90 mL/min/1.73 m2). There were no SLE patients with a severe decrease in eGFR or end stage renal disease (<30 mL/min/1.73 m2); anifrolumab-fnia is not cleared renally.
Patients with urine protein/creatinine ratio (UPCR) >2 mg/mg were excluded from the clinical trials. Based on population PK analyses, increased UPCR did not significantly affect anifrolumab-fnia clearance.
Patients with Hepatic Impairment: No specific clinical trials have been conducted to investigate the effect of hepatic impairment on anifrolumab-fnia. IgG1 monoclonal antibodies are predominantly eliminated via catabolism and are not expected to undergo hepatic metabolism; changes in hepatic function are not expected to influence anifrolumab-fnia clearance. Based on population PK analyses, baseline hepatic function biomarkers (ALT and AST ≤2.0 × ULN, and total bilirubin) had no clinically relevant effect on anifrolumab-fnia clearance.
Drug Interactions
No formal drug-drug interaction trials have been conducted.
Based on population PK analysis, concomitant use of oral corticosteroids, anti-malarials, immunosuppressants (azathioprine, methotrexate, mycophenolate mofetil, mycophenolic acid, and mizoribine), NSAIDs, ACE inhibitors, and HMG-CoA reductase inhibitors did not significantly affect the PK of anifrolumab-fnia.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies (ADA) in the trials described below with the incidence of anti-drug antibodies in other trials, including those of anifrolumab-fnia.
In Trials 2, 3, and the long-term extension trial, treatment emergent anti-anifrolumab-fnia antibodies were detected in 9 of 350 patients (ADA incidence 2.6%) who received SAPHNELO at the recommended intravenous dosing regimen for up to 4 years.
In Trial 5, treatment emergent anti-anifrolumab-fnia antibodies were detected in 6 of 107 patients (ADA incidence 5.6%) treated with SAPHNELO at the recommended subcutaneous dosing regimen during the 52‑week treatment period. No neutralizing antibodies were detected.
Because of the low occurrence of anti-drug antibodies, the effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and/or effectiveness of anifrolumab-fnia products is unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic and genotoxic potential of anifrolumab-fnia have not been evaluated. In rodent models of IFNAR1 blockade, increased carcinogenic potential has been observed. The clinical relevance of these findings is unknown.
Effects on male and female fertility have not been directly evaluated in animal studies. No anifrolumab-fnia-related adverse effects on indirect measures of male or female fertility, based on semen analysis, spermatogenesis staging, menses cycle, organ weights and histopathological findings in the reproductive organs were observed in 9-month repeat-dose toxicity studies in cynomolgus monkeys at doses up to 50 mg/kg intravenous once weekly (approximately 58‑times the MRHD on an AUC basis) and 60 mg/kg subcutaneous once weekly (approximately 52-times the subcutaneous MRHD on an AUC basis).
-
14 CLINICAL STUDIES
14.1 Intravenous Administration in Adults with Moderate to Severe SLE
Trial Design and Population
The safety and efficacy of SAPHNELO were evaluated in three 52-week treatment period, multicenter, randomized, double-blind, placebo-controlled trials (Trial 1 [NCT01438489], Trial 2 [NCT02446912] and Trial 3 [NCT02446899]). Patients were diagnosed with SLE according to the American College of Rheumatology (1982 revised) classification criteria. All patients were ≥18 years of age and had moderate to severe disease, with a SLE Disease Activity Index 2000 (SLEDAI-2K) score ≥6 points, organ level involvement based on the British Isles Lupus Assessment Group (BILAG) assessment, and a Physician’s Global Assessment [PGA] score ≥1, despite receiving standard SLE therapy consisting of either one or any combination of oral corticosteroids (OCS), antimalarials and/or immunosuppressants at baseline. Patients continued to receive their existing SLE therapy at stable doses during the clinical trials, with the exception of OCS (prednisone or equivalent) where tapering was a component of the protocol. Patients who had severe active lupus nephritis and patients who had severe active central nervous system lupus were excluded. The use of other biologic agents and cyclophosphamide were not permitted during the trials; patients receiving other biologic therapies were required to complete a wash-out period of at least 5 half‑lives prior to enrollment. All three trials were conducted in North America, Europe, South America and Asia. Patients received SAPHNELO or placebo, administered by intravenous infusion, every 4 weeks.
Efficacy of SAPHNELO was established based on assessment of clinical response using the composite endpoints, the British Isles Lupus Assessment Group based Composite Lupus Assessment (BICLA) and the SLE Responder Index (SRI‑4).
BICLA response at Week 52, was defined as improvement in all organ domains with moderate or severe activity at baseline:
- Reduction of all baseline BILAG A to B/C/D and baseline BILAG B to C/D, and no BILAG worsening in other organ systems, as defined by ≥1 new BILAG A or ≥2 new BILAG B;
- No worsening from baseline in SLEDAI-2K, where worsening is defined as an increase from baseline of >0 points in SLEDAI-2K;
- No worsening from baseline in patients’ lupus disease activity, where worsening is defined by an increase ≥0.30 points on a 3-point PGA visual analogue scale (VAS);
- No discontinuation of treatment;
- No use of restricted medication beyond the protocol-allowed threshold.
SRI‑4 response, was defined as meeting each of the following criteria at Week 52 compared with baseline:
- Reduction from baseline of ≥4 points in the SLEDAI-2K;
- No new organ system affected as defined by 1 or more BILAG A or 2 or more BILAG B items compared to baseline;
- No worsening from baseline in the patients’ lupus disease activity defined by an increase ≥0.30 points on a 3‑point PGA VAS;
- No discontinuation of treatment;
- No use of restricted medication beyond the protocol-allowed threshold.
Trial 1 randomized 305 patients (1:1:1) who received SAPHNELO, 300 mg or 1000 mg, or placebo once every 4 weeks for up to 52 weeks. The primary endpoint was a combined assessment of the SRI-4 and the sustained reduction in OCS (<10 mg/day and ≤OCS dose at Week 1, sustained for 12 weeks) measured at Week 24.
Trial 2 and 3 were similar in design. Trial 2 randomized 457 patients (1:2:2) who received SAPHNELO 150 mg, 300 mg once every 4 weeks or placebo. Trial 3 randomized 362 patients (1:1) who received SAPHNELO 300 mg once every 4 weeks or placebo. The primary endpoints were improvement in disease activity evaluated at 52 weeks, measured by SRI‑4 in Trial 2 and BICLA in Trial 3 (defined above). The common secondary efficacy endpoints included in both trials were the maintenance of OCS reduction, improvement in cutaneous SLE activity, and flare rate. During Weeks 8-40, patients with a baseline OCS ≥10 mg/day were required to taper their OCS dose to ≤7.5 mg/day, unless there was worsening of disease activity. Both trials evaluated the efficacy of SAPHNELO 300 mg once every 4 weeks versus placebo; a dose of 150 mg was also evaluated for dose-response in Trial 2.
Patient demographics and disease characteristics were generally similar and balanced across treatment arms (Table 2).
Table 2 Demographics and Baseline Characteristics of Adults with Moderate to Severe SLE in Trials 1, 2, and 3 Total Population
Trial 1
(N=305)
Trial 2
(N=457)
Trial 3
(N=362)
Mean Age (years)
40
41
42
Female (%)
93
92
93
White (%)
42
71
60
Black/African American (%)
13
14
12
Asian (%)
7
5
17
Hispanic or Latino (%)
42
19
30
Baseline SLEDAI-2K score
Mean (SD)
10.9 (4.1)
11.3 (3.72)
11.5 (3.76)
≥10 points, n (%)
182 (60)
328 (72)
260 (72)
BILAG organ system scoring (Overall)
At least one A, n (%)
152 (50)
217 (48)
176 (49)
No A and at least 2 Bs, n (%)
134 (44)
211 (46)
169 (47)
Positive Anti-dsDNA levels, n (%)
185 (77)
207 (45)
159 (44)
Abnormal ANA, n (%)
299 (98)
412 (90)
325 (90)
Abnormal Complement C3 level, n (%)
119 (39)
157 (34)
144 (40)
Abnormal Complement C4 level, n (%)
74 (24)
95 (21)
95 (26)
Baseline SLE treatment
OCS, n (%)
258 (85)
381 (83)
292 (81)
Antimalarials, n (%)
219 (72)
334 (73)
252 (70)
Immunosuppressants, n (%)
150 (49)
214 (47)
174 (48)
Randomization was stratified by disease severity (SLEDAI-2K score at baseline, <10 vs ≥10 points), OCS dose on Day 1 (<10 mg/day vs ≥10 mg/day prednisone or equivalent) and interferon gene signature test results (high vs low).
Trials 1, 2, and 3 Results
Although other intravenous SAPHNELO dosages were studied in Trials 1 and 2, only data for the approved intravenous SAPHNELO dosage of 300 mg every 4 weeks are presented below.
The reduction in disease activity seen in the BICLA and SRI-4 was related primarily to improvement in the mucocutaneous and musculoskeletal organ systems. Flare rate was reduced in SAPHNELO patients compared to placebo patients although the difference was not statistically significant.
BICLA responder analysis
BICLA was the primary endpoint in Trial 3, SAPHNELO 300 mg once every 4 weeks demonstrated statistically significant and clinically meaningful efficacy in overall disease activity compared with placebo, with greater improvements in all components of the composite endpoint. In Trial 1 and 2, BICLA was a pre-specified analysis. The BICLA results are presented in Table 3.
Table 3 BICLA Response Rate at Week 52 in Adults with Moderate to Severe SLE in Trials 1, 2, and 3 - * Not formally tested in a pre-specified testing scheme and findings should be interpreted with caution.
- † Based on post hoc analysis.
- ‡ Primary endpoint.
- § In all 3 trials, patients who discontinued investigational product or initiated restricted medications beyond the protocol-specified thresholds are considered non-responders. For consistency, the results presented for Trial 2 represent the post-hoc analysis using the restricted medication thresholds as defined in Trial 3.
Trial 3‡
SAPHNELO 300 mg once every 4 weeks
(N=99)
Placebo
(N=102)
SAPHNELO 300 mg once every 4 weeks
(N=180)
Placebo
(N=184)
SAPHNELO 300 mg once every 4 weeks
(N=180)
Placebo
(N=182)
BICLA Response Rate§
Responder, n (%)
54 (54.6)
27 (25.8)
85 (47.1)
55 (30.2)
86 (47.8)
57 (31.5)
Difference in Response Rates (95% CI)
28.8 (15.7, 41.9)
17.0 (7.2, 26.8)
16.3 (6.3, 26.3)
p-value=0.001
Components of BICLA Response§
BILAG Improvement, n (%)
54 (54.5)
28 (27.5)
85 (47.2)
58 (31.5)
88 (48.9)
59 (32.4)
No Worsening of SLEDAI-2K, n (%)
73 (73.7)
61 (59.8)
121 (67.2)
104 (56.5)
122 (67.8)
94 (51.6)
No Worsening of PGA, n (%)
76 (76.8)
62 (60.8)
117 (65.0)
105 (57.1)
122 (67.8)
95 (52.2)
The response rates and associated difference and 95% CI are calculated using a Cochran-Mantel-Haenszel approach adjusted for stratification factors. The reported percentages for the components are unadjusted.
In Trial 3, examination of subgroups by age, race, gender, ethnicity, disease severity [SLEDAI-2K at baseline], and baseline OCS use did not identify differences in response to SAPHNELO.
Figure 1 shows the proportion of BICLA responders through the 52-week treatment period in Trial 3.
Figure 1 Trial 3: Proportion (%) of BICLA Responders in Adults with Moderate to Severe SLE by Visit*
SRI-4 responder analysis
SRI-4 was the primary endpoint in Trial 2, treatment with SAPHNELO did not result in statistically significant improvements over placebo. In Trials 1 and 3, SRI-4 was a pre-specified analysis. The SRI-4 results are presented in Table 4.
Table 4 SRI-4 Response Rate at Week 52 in Adults with Moderate to Severe SLE in Trials 1, 2, and 3 - * Not formally tested in a pre-specified testing scheme and findings should be interpreted with caution.
- † Primary endpoint.
- ‡ In all 3 trials, patients who discontinued investigational product or initiated restricted medications beyond the protocol-specified thresholds are considered non-responders. For consistency, the results presented for Trial 2 represent the post-hoc analysis using the restricted medication thresholds as defined in Trial 3. The most commonly involved SLEDAI-2K organ domains were mucocutaneous, musculoskeletal and immune.
Trial 1*
Trial 2†
Trial 3*
SAPHNELO 300 mg once every 4 weeks (N=99)
Placebo
(N=102)
SAPHNELO 300 mg once every 4 weeks (N=180)
Placebo
(N=184)
SAPHNELO 300 mg once every 4 weeks (N=180)
Placebo
(N=182)
SRI-4 Response Rate‡
Responder, n (%)
62 (62.8)
41 (38.8)
88 (49.0)
79 (43.0)
100 (55.5)
68 (37.3)
Difference in Response Rates (95% CI)
24.0 (10.9, 37.2)
6.0 (-4.2, 16.2)
18.2 (8.1, 28.3)
Components of SRI-4 Response‡
SLEDAI-2K improvement, n (%)
62 (62.6)
41 (40.2)
89 (49.4)
80 (43.5)
101 (56.1)
71 (39.0)
No worsening of BILAG, n (%)
75 (75.8)
61 (59.8)
119 (66.1)
105 (57.1)
125 (69.4)
94 (51.6)
No worsening of PGA, n (%)
76 (76.8)
62 (60.8)
117 (65.0)
105 (57.1)
122 (67.8)
95 (52.2)
The response rates and associated difference and 95% CI are calculated using a Cochran-Mantel-Haenszel approach adjusted for stratification factors. The reported percentages for the components are unadjusted.
Effect on Concomitant Steroid Treatment
In Trial 3, among the 47% of patients with a baseline OCS use ≥10 mg/day, SAPHNELO demonstrated a statistically significant difference in the proportion of patients able to reduce OCS use by at least 25% to ≤7.5 mg/day at Week 40 and maintain the reduction through Week 52 (p-value = 0.004); 52% (45/87) of patients in the SAPHNELO group versus 30% (25/83) in the placebo group achieved this level of steroid reduction (difference 21% [95% CI 6.8, 35.7]). Consistent trends in favor of SAPHNELO compared to placebo, on effect of reduction of OCS use, were observed in Trial 1 and 2, but the difference was not statistically significant.
14.2 Subcutaneous Administration in Adults with Moderate to Severe SLE
The safety and efficacy of SAPHNELO administered subcutaneously were evaluated in a 52-week treatment period, multicenter, randomized, double-blind, placebo-controlled trial (Trial 5 [NCT04877691]). All patients were ≥18 years of age, diagnosed with SLE according to the American College of Rheumatology (1997 revised) classification criteria, and had moderate to severe disease, with a SLEDAI-2K score ≥6 points, organ level involvement based on BILAG assessment, and a PGA score ≥1, despite receiving standard SLE therapy consisting of either one or any combination of OCS, antimalarials and/or immunosuppressants at baseline. Patients continued to receive their existing SLE therapy at stable doses during the trial, with the exception of OCS (prednisone or equivalent) where tapering was a component of the protocol. Patients who had severe active lupus nephritis or severe active central nervous system lupus were excluded. Patients were randomized (1:1) to receive SAPHNELO 120 mg plus standard therapy or placebo plus standard therapy by subcutaneous injection once every week. Randomization was stratified by SLEDAI‑2K score at baseline (<10 vs ≥10 points), OCS dose on Day 1 (<10 mg/day vs ≥10 mg/day prednisone or equivalent) and interferon gene signature test results (high vs low).
A pre-specified interim analysis was conducted when the first 220 randomized patients completed Week 52 or had withdrawn from the trial. Of these, 89% were female, 45% Hispanic or Latino, 78% White, 7% American Indian or Alaska Native, 7% Asian, and 4% Black/African American. The mean age was 43 years. At baseline, the mean SLEDAI‑2K score was 10.9 (SD: 3.4) and 67% had high disease activity (SLEDAI‑2K score ≥10), 45% had severe disease (BILAG A) in at least 1 organ system and 50% had moderate disease (BILAG B) in at least 2 organ systems. The most commonly affected organ systems (BILAG A or B at baseline) were the musculoskeletal (95%) and mucocutaneous (92%) systems; 2% cardiorespiratory and 2% renal organ domain involvement. At baseline, 95% had abnormal ANA, 40% were positive for anti-dsDNA antibodies; 33% of patients had abnormal C3, and 24% abnormal C4. Background SLE standard therapy included OCS (82%; mean daily dose, prednisone or equivalent, 9.8 mg), immunosuppressants (56%), and anti-malarials (80%). During Weeks 8-40, patients with a baseline OCS ≥10 mg/day were required to taper their OCS dose to ≤7.5 mg/day, unless there was worsening of disease activity.
The primary endpoint was BICLA response rate measured at Week 52. At the interim analysis, SAPHNELO by subcutaneous administration demonstrated a statistically significant and clinically meaningful reduction of overall disease activity compared with placebo (Table 5).
Table 5 BICLA Response Rate at Week 52 in Adults with Moderate to Severe SLE in Trial 5 - * Per protocol, patients who used restricted medications beyond the protocol-specified thresholds, discontinued investigational product or died were considered non-responders.
- † Based on an interim analysis using Pocock alpha spending function with an information fraction of 0.6. The primary endpoint was tested at the alpha level of 0.0354 at the interim analysis.
SAPHNELO 120 mg once weekly
(N=109)
Placebo
(N=111)
BICLA Response
- Responder, n (%)*
64 (58.5)
48 (43.2)
- Difference in Response Rates, % (95% CI)
15.3 (2.1, 28.5)
p-value=0.0231†
Components of BICLA Response
- BILAG Improvement, n (%)
64 (58.5)
48 (43.3)
- No Worsening of SLEDAI-2K, n (%)
80 (73.4)
77 (68.9)
- No Worsening of PGA, n (%)
80 (73.5)
78 (70.0)
The response rates, associated difference, and 95% CI are calculated using a Cochran-Mantel-Haenszel approach adjusted for stratification factors.
Subgroup analysis by disease severity (based on baseline SLEDAI-2K, <10 points, ≥10 points) and baseline OCS use (<10 mg/day, ≥10 mg/day) did not identify differences in response to SAPHNELO.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
SAPHNELO (anifrolumab-fnia) injection is a sterile, preservative-free, clear to opalescent, colorless to slightly yellow solution in a single-dose vial for intravenous infusion or a prefilled syringe or autoinjector for subcutaneous injection.
SAPHNELO is available as follows:
Packaging Unit and Strength
Pack Size and NDC
300 mg/2 mL (150 mg/mL) single-dose vial
(NDC: 0310-3040-00)
One vial in a carton: NDC: 0310-3040-00
120 mg/0.8 mL single-dose prefilled syringe
(NDC: 0310-3080-75)
One syringe in a carton: NDC: 0310-3080-02
120 mg/0.8 mL single-dose autoinjector
(SAPHNELO PEN) (NDC: 0310-3080-25)
One autoinjector in a carton: NDC: 0310-3080-01
The prefilled syringe (including needle cover) and autoinjector (including cap) are not made with natural rubber latex.
Storage and Handling
Store SAPHNELO in a refrigerator at 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
- Do not freeze. Do not shake. Do not expose to heat. Do not use SAPHNELO past the expiration date.
If needed, SAPHNELO prefilled syringe or autoinjector can be stored at room temperature 68°F to 77°F (20°C to 25°C) for up to 7 days in the original carton to protect from light. After the SAPHNELO prefilled syringe or autoinjector has reached room temperature, do not return to the refrigerator. Discard SAPHNELO prefilled syringe or autoinjector if not used within 7 days at room temperature storage.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Serious Infections
Inform patients that SAPHNELO may decrease their ability to fight infections and that serious infections, including fatal ones, occurred in patients receiving SAPHNELO in clinical trials. Also inform patients that they are at increased risk of respiratory infections and herpes zoster during treatment with SAPHNELO [see Warnings and Precautions (5.1)]. Advise patients to contact their healthcare provider if they develop any symptoms of an infection.
Hypersensitivity Reactions/Anaphylaxis
Inform patients that serious hypersensitivity reactions, including anaphylaxis, have been reported in patients who received SAPHNELO. Instruct patients to immediately tell their healthcare provider or go to the emergency department of their nearest hospital, if they experience symptoms of an allergic reaction (e.g., anaphylaxis) during or after the administration of SAPHNELO [see Warnings and Precautions (5.2)].
Immunizations
Inform patients that they should not receive live or live-attenuated vaccines while receiving SAPHNELO. Advise patients to discuss with their healthcare provider before seeking immunizations on their own [see Warnings and Precautions (5.4)].
Pregnancy
Advise female patients to inform their healthcare provider if they intend to become pregnant during therapy, suspect they are pregnant or become pregnant while receiving SAPHNELO [see Use in Specific Populations (8.1)].
Inform women that there is a pregnancy registry that monitors pregnancy outcomes in women exposed to SAPHNELO and they can contact AstraZeneca at 1-877-693-9268 for more information or to report the pregnancy.
Manufactured by: AstraZeneca AB Södertälje, Sweden SE-15185
US License No. 2059
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
SAPHNELO is a registered trademark of AstraZeneca.
©AstraZeneca 2026
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
SAPHNELO® (saf-NEH-low)
(anifrolumab-fnia)
injection, for intravenous or subcutaneous use
What is SAPHNELO?
- SAPHNELO is a prescription medicine used to treat adults with moderate to severe systemic lupus erythematosus (SLE or lupus) who are receiving other lupus medicines.
- SAPHNELO contains anifrolumab-fnia which is in a group of medicines called monoclonal antibodies. Lupus is a disease of the immune system (the body system that fights infection). When given together with other medicines for lupus, SAPHNELO may help to reduce your lupus disease activity more than other lupus medicines alone.
- It is not known if SAPHNELO is effective in people with severe active lupus nephritis or central nervous system lupus.
- It is not known if SAPHNELO is safe and effective in children.
Do not use SAPHNELO if you:
- are allergic to anifrolumab-fnia or any of the ingredients in SAPHNELO. See the end of this Patient Information leaflet for a complete list of ingredients in SAPHNELO.
Before you receive SAPHNELO, tell your healthcare provider about all of your medical conditions, including if you:
- think you have an infection or have infections that keep coming back. You should not receive SAPHNELO if you have an infection unless your healthcare provider tells you to. See “What are the possible side effects of SAPHNELO?”
- are scheduled to receive a vaccination or if you think you may need a vaccination. You should not receive live vaccines during treatment with SAPHNELO.
- have or have had any type of cancer.
- are receiving other biologic medicines or monoclonal antibodies.
- are pregnant or plan to become pregnant. It is not known if SAPHNELO will harm your unborn baby. Tell your healthcare provider if you are pregnant, think you might be pregnant, or plan to become pregnant during your treatment with SAPHNELO.
- Pregnancy Exposure Registry. If you become pregnant while receiving SAPHNELO, talk to your healthcare provider. A pregnancy exposure registry monitors pregnancy outcomes in women exposed to SAPHNELO. You can find out more information about the registry by calling AstraZeneca at 1-877-693-9268.
- are breastfeeding or plan to breastfeed. It is not known if SAPHNELO passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby while receiving SAPHNELO.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. SAPHNELO may affect the way other medicines work, and other medicines may affect how SAPHNELO works.
How will I receive SAPHNELO?
SAPHNELO is intended for use under your healthcare provider’s care and may be administered as an intravenous infusion or as a subcutaneous injection.
When given into a vein (intravenously)
- Your healthcare provider will give you SAPHNELO through a needle placed in a vein (IV or intravenous infusion). It takes about 30 minutes to give you the full dose of SAPHNELO.
- SAPHNELO is usually given 1 time every 4 weeks.
- If you miss an appointment, call your healthcare provider as soon as possible to reschedule your appointment.
When given under the skin (subcutaneously)
- SAPHNELO may be prescribed as a single-dose prefilled syringe or as a single-dose autoinjector (SAPHNELO PEN).
- Use SAPHNELO exactly as your healthcare provider tells you to use it.
- SAPHNELO is injected under your skin (subcutaneously) in your stomach (abdomen), thigh or upper arm (for caregiver administration only) 1 time every week.
- Before you use SAPHNELO, your healthcare provider will train you or your caregiver how to give the injections.
- Read the Instructions for Use that comes with SAPHNELO for information on how to prepare and inject SAPHNELO.
- If you miss your dose of SAPHNELO inject a dose as soon as you remember. Then, continue to inject 1 time each week based on the new day SAPHNELO was injected or at your regularly scheduled time as long as there are at least 3‑days between the doses. In case you are not sure when to inject SAPHNELO, call your healthcare provider.
What are the possible side effects of SAPHNELO?
SAPHNELO may cause serious side effects, including:
- Serious Infections. SAPHNELO can lower the ability of your immune system to fight infections. You may be at a higher risk of developing respiratory infections, and shingles (herpes zoster) during treatment with SAPHNELO. Infections (including COVID-19) could be serious, leading to hospitalization or death. Tell your healthcare provider right away if you have any of the following symptoms of an infection:
- fever, sweating, or chills
- muscle aches
- cough
- shortness of breath
- burning when urinating
- urinating more often
- diarrhea or stomach pain
- warm, red, or painful skin or sores on your body.
- Allergic (hypersensitivity) reactions, including anaphylaxis. Serious allergic reactions can happen during or after you get your SAPHNELO infusion or take your SAPHNELO injection. Tell your healthcare provider or get emergency help right away if you have any of the following symptoms of a serious allergic reaction:
- swelling of your face, mouth, and tongue
- breathing problems
- fainting or dizziness
- feeling lightheaded (low blood pressure)
- Cancer. SAPHNELO may reduce the activity of your immune system. Medicines that affect the immune system may increase your risk of certain cancers.
The most common side effects of SAPHNELO include:
-
- upper respiratory infections
- infusion reactions (when given intravenously)
- cough
- bronchitis
- shingles (herpes zoster)
How should I store SAPHNELO?
- Store SAPHNELO prefilled syringe and autoinjector in a refrigerator between 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
- If needed, the SAPHNELO prefilled syringe or autoinjector (SAPHNELO PEN) can be stored at room temperature between 68°F to 77°F (20°C to 25°C) for up to 7 days in the original carton to protect from light. After the SAPHNELO prefilled syringe or autoinjector has reached room temperature, do not return to the refrigerator. Throw away SAPHNELO prefilled syringe or autoinjector if not used within 7 days at room temperature storage.
- Do not shake SAPHNELO.
- Do not freeze SAPHNELO.
- Do not expose SAPHNELO to heat.
- Do not use SAPHNELO past the expiration date.
- Keep SAPHNELO and all medicines out of the reach of children.
These are not all of the possible side effects of SAPHNELO.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1‑800‑FDA‑1088.
General information about the safe and effective use of SAPHNELO
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use SAPHNELO for a condition for which it was not prescribed. Do not give SAPHNELO to other people, even if they have the same symptoms that you have. It may harm them. If you would like more information about SAPHNELO, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about SAPHNELO that is written for health professionals.
What are the ingredients in SAPHNELO?
Active ingredient: anifrolumab-fnia
Inactive ingredients:
- Intravenous infusion: L-histidine, L-histidine hydrochloride monohydrate, L-lysine hydrochloride, trehalose dihydrate, polysorbate 80 and Water for Injection.
- Subcutaneous injection: histidine, L-histidine hydrochloride monohydrate, lysine hydrochloride, polysorbate 80, trehalose, and Water for Injection.
The SAPHNELO prefilled syringe and autoinjector are not made with natural rubber latex.
Manufactured by: AstraZeneca AB Södertälje, Sweden SE-15185
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
©AstraZeneca 2026
SAPHNELO is a registered trademark of AstraZeneca.
For more information, go to https://www.SAPHNELO.com or call 1-800-236-9933.
This Patient Information has been approved by the U.S. Food and Drug Administration. Issued: April 2026
-
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
SAPHNELO® (saf-NEL-low)
(anifrolumab-fnia)
injection, for subcutaneous use
Single-dose prefilled syringe
120 mg/0.8 mL
This Instructions for Use contains information on how to inject using SAPHNELO prefilled syringe.
Read this Instructions for Use before you start using SAPHNELO prefilled syringe and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
Your healthcare provider should show you or your caregiver how to use SAPHNELO prefilled syringe the right way. If you or your caregiver have any questions, talk to your healthcare provider. SAPHNELO prefilled syringe is for use under the skin (subcutaneous) only.
Important storage information and warnings
- Store SAPHNELO prefilled syringe in a refrigerator between 36°F to 46°F (2°C to 8°C) in the original carton until ready to use.
- If needed, SAPHNELO prefilled syringe can be stored at room temperature between 68°F to 77°F (20°C to 25°C) for up to 7 days in the original carton to protect from light. After SAPHNELO prefilled syringe has reached room temperature, do not return to the refrigerator. Throw away SAPHNELO prefilled syringe if not used within 7 days at room temperature.
- Each SAPHNELO prefilled syringe contains 1 dose for one time use only. Do not share SAPHNELO prefilled syringe with other people.
Do not use SAPHNELO prefilled syringe if it has:
- been frozen or exposed to heat.
- been dropped, damaged, or appears to be tampered with.
Do not shake SAPHNELO prefilled syringe.
If any of the above happens, throw away the prefilled syringe in an FDA-cleared sharps disposal container and use a new prefilled syringe.
Keep SAPHNELO prefilled syringe and all medicines out of the sight and reach of children.
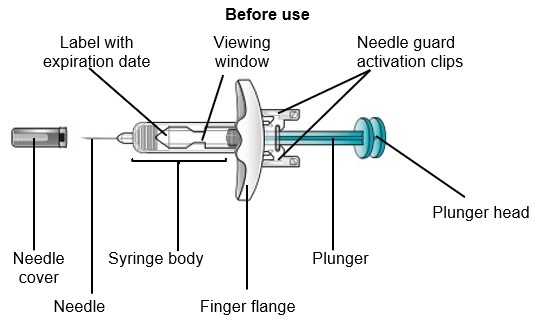
SAPHNELO prefilled syringe parts
Do not remove the needle cover until right before injecting SAPHNELO.
Do not touch the needle guard activation clips. This will keep you from activating the needle guard too soon.
Preparing to inject using SAPHNELO prefilled syringe
Step 1 – Gather supplies for your injection
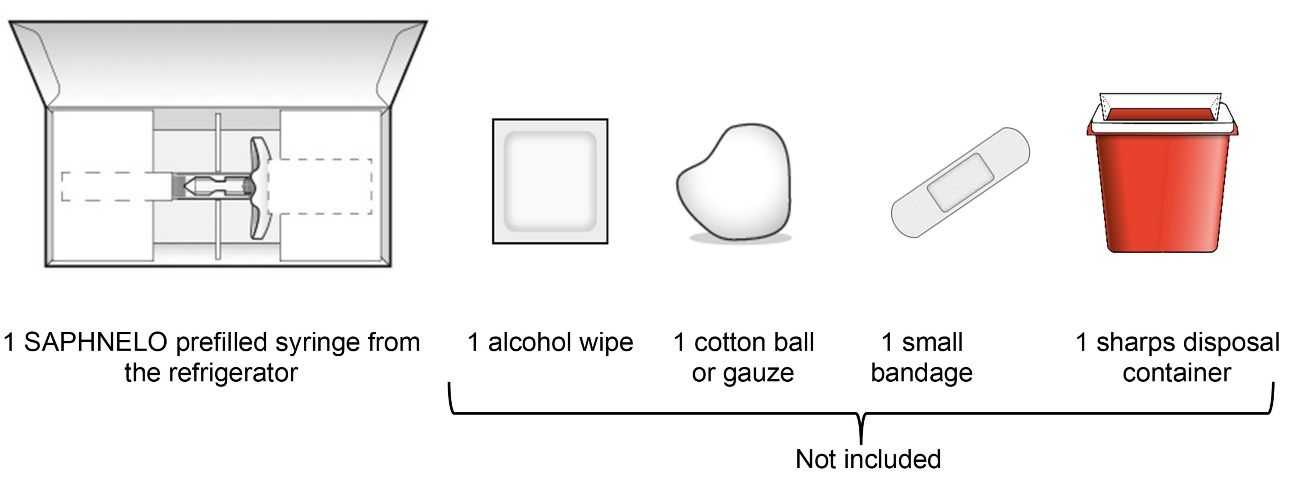
See Step 9 for instructions on how to throw away (dispose of) the used SAPHNELO prefilled syringe.
Step 2 – Inspect the carton and wait 60 minutes
Select a clean, well-lit, flat work surface, such as a table.
Check the expiration date (EXP) on the carton.
- Do not use if the expiration date has passed.
Check the carton for damage.
- Do not use if the carton looks damaged.
Let SAPHNELO prefilled syringe come to room temperature for 60 minutes before injecting.
- Keep SAPHNELO prefilled syringe in the original carton to protect from light.
- Do not warm SAPHNELO prefilled syringe in any other way. For example, donot warm it in a microwave, hot water, direct sunlight or near other heat sources.

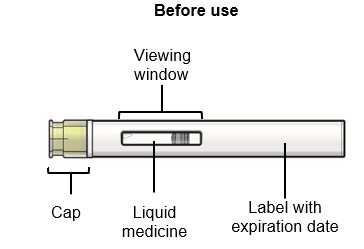
Step 3 – Remove the prefilled syringe from the carton and inspect
Open the carton and remove SAPHNELO prefilled syringe by holding the middle of the syringe body.
- Do not hold or remove by the plunger.
Check the expiration date on the prefilled syringe.
- Do not use if the expiration date has passed.
Check the prefilled syringe for damage.
- Do not use if damaged.
Check the liquid through the viewing window.
- The liquid should be clear and colorless to slightly yellow.
- Do not use if the liquid is cloudy, discolored, or contains visible particles.
- It is normal to see small air bubbles in the liquid. Do not try to remove the air bubbles.
Injecting using your SAPHNELO prefilled syringe
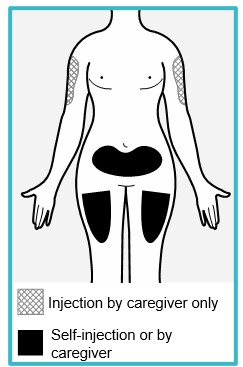
Step 4 – Choose an injection site
You or your caregiver can inject in the front of your thigh or the lower part of your stomach (abdomen).
A caregiver may also inject your upper arm.
Do not try to inject yourself in the upper arm.
Choose an injection site that is at least 1-inch (3 cm) away from where you last injected.
Do not inject:
- into the 2-inch (5 cm) area around your belly button.
- where the skin is red, warm, tender, bruised, scaly or hard.
- into scarred, damaged, discolored or tattooed skin.
- through clothing.
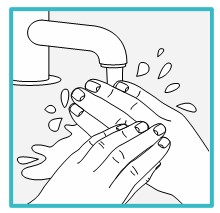
Step 5 – Wash your hands and clean the injection site
Wash your hands well with soap and water.
Clean the injection site with an alcohol wipe or with soap and water. Let the site air dry.
- Do not touch the cleaned injection site again or blow on it before injecting.
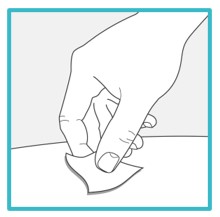
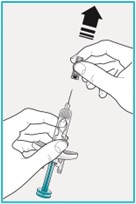
Step 6 – Pull off the needle cover
Hold the SAPHNELO prefilled syringe body with 1 hand and carefully pull the needle cover straight off with your other hand.
- Do not twist or wiggle the needle cover to remove it.
- Do not touch or pull the plunger or plunger head while removing the needle cover.
- You may see a drop of liquid at the end of the needle. This is normal.
- Do not recap the needle. Put the needle cover to the side to throw it in the trash later.
- Do not touch the needle or let it touch any surface.
- Do not use if the needle is damaged or dirty.
Go to Step 7 right away after removing the needle cover.
Step 7 – Injecting SAPHNELO
Hold SAPHNELO prefilled syringe in 1 hand as shown. Use your other hand to gently pinch and hold the cleaned injection site.
- Do not press down on the plunger head until the needle is inserted into the skin.
- Do not pull back on the plunger head at any time.
Inject using the SAPHNELO prefilled syringe by following the steps in figures a, b, and c.
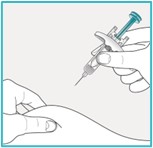
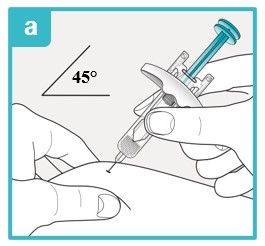
Using a 45 degree angle, fully insert the needle into the pinched skin.
Do not reposition the SAPHNELO prefilled syringe after you insert the needle into the skin.
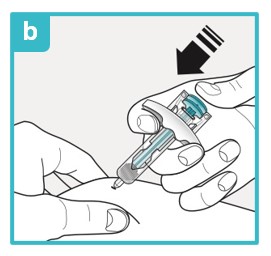
Use your thumb to push down the plunger head.
To make sure you inject all the medicine and activate the needle guard, keep pushing firmly on the plunger head until it is fully down as far as it will go.
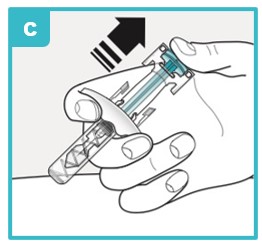
Slowly let go of the plunger head until the needle guard covers the needle.
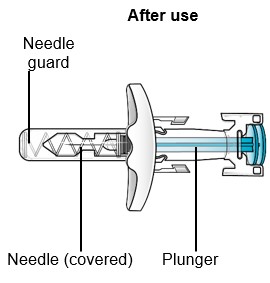
If the needle is not covered, carefully throw away (dispose of) the SAPHNELO prefilled syringe right away (see Step 9).
Step 8 – Check the injection site
There may be a small amount of blood or liquid at the injection site. This is normal.
If needed, press a cotton ball or gauze on the area and apply a small bandage.
- Do not rub the injection site.
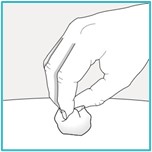
Step 9 – Throw away (dispose of) the used SAPHNELO prefilled syringe
Each prefilled syringe contains 1 single-dose of SAPHNELO and cannot be used again. Do not recap the needle.
Put your used SAPHNELO prefilled syringe in an FDA-cleared sharps disposal container right away after use.
Do not throw away (dispose of) SAPHNELO prefilled syringe in your household trash.
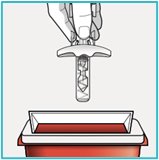
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting puncture resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak resistant, and
- properly labelled to warn of hazardous waste inside the container.
When your sharp disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container.
There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal.
Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
Do not recycle your used sharps disposal container.
Manufactured by:
AstraZeneca AB, Södertälje, Sweden SE-15185
US License No. 2059
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
©AstraZeneca 2026
SAPHNELO is a registered trademark of AstraZeneca.
For more information, go to https://www.SAPHNELO.com or call 1-800-236-9933.
This Instructions for Use has been approved by the U.S. Food and Drug Administration Approved: April 2026
-
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
SAPHNELO® PEN (saf-NEL-low)
(anifrolumab-fnia)
injection, for subcutaneous use
Single-dose Autoinjector
120 mg/0.8 mL
This Instructions for Use contains information on how to inject using SAPHNELO PEN.
Read this Instructions for Use before you start using SAPHNELO PEN and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
Your healthcare provider should show you or your caregiver how to use SAPHNELO PEN the right way. If you or your caregiver have any questions, talk to your healthcare provider. SAPHNELO PEN is for use under the skin (subcutaneous) only.
Important storage information and warnings
- Store SAPHNELO PEN in a refrigerator between 36°F to 46°F (2°C to 8°C) in the original carton until ready to use.
- If needed, SAPHNELO PEN can be stored at room temperature between 68°F to 77°F (20°C to 25°C) for up to 7 days in the original carton to protect from light. After SAPHNELO PEN has reached room temperature, do not return to the refrigerator. Throw away SAPHNELO PEN if not used within 7 days at room temperature.
- Each SAPHNELO PEN contains 1 dose for one time use only. Do not share SAPHNELO PEN with other people.
Do not use SAPHNELO PEN if it has:
- been frozen or exposed to heat.
- been dropped, damaged, or appears to be tampered with.
Do not shake SAPHNELO PEN.
If any of the above happens, throw away SAPHNELO PEN in an FDA-cleared sharps disposal container and use a new SAPHNELO PEN.
Keep SAPHNELO PEN and all medicines out of the sight and reach of children.
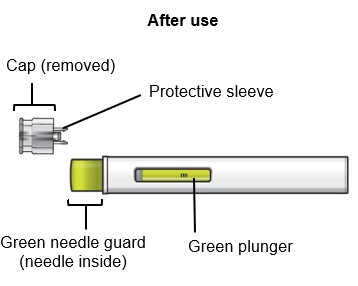
SAPHNELO PEN parts
Do not remove the cap until right before injecting SAPHNELO.
Do not touch the green needle guard.
Preparing to inject using SAPHNELO PEN
Step 1 – Gather supplies for your injection
See Step 10 for instructions on how to throw away (dispose of) the used SAPHNELO PEN.
Step 2 – Inspect the carton and wait 60 minutes
Select a clean, well-lit, flat work surface, such as a table.
Check the expiration date (EXP) on the carton.
- Do not use if the expiration date has passed.
Check the carton for damage.
- Do not use if the carton looks damaged.
Let SAPHNELO PEN come to room temperature for 60 minutes before injecting.
- Keep SAPHNELO PEN in the original carton to protect from light.
- Do not warm SAPHNELO PEN in any other way. For example, do not warm it in a microwave, hot water, direct sunlight or near other heat sources.

Step 3 – Remove SAPHNELO PEN from the carton and inspect
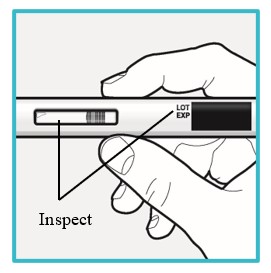
Open the carton and remove SAPHNELO PEN by gently grasping the middle of the device.
Check the expiration date on SAPHNELO PEN.
- Do not use if the expiration date has passed.
Check SAPHNELO PEN for damage.
- Do not use if damaged.
Check the liquid through the viewing window.
- The liquid should be clear and colorless to slightly yellow.
- Do not use if the liquid is cloudy, discolored, or contains visible particles.
- It is normal to see small air bubbles in the liquid.
Do not try to remove the air bubbles.
Injecting using your SAPHNELO PEN
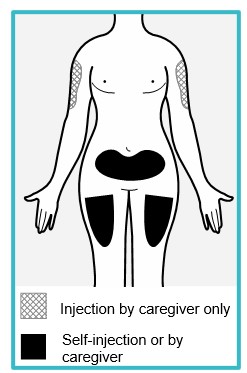
Step 4 – Choose an injection site
You or your caregiver can inject in the front of your thigh or the lower part of your stomach (abdomen).
A caregiver may also inject you in your upper arm.
Do not try to inject yourself in the upper arm.
Choose an injection site that is at least 1-inch (3 cm) away from where you last injected.
Do not inject:
- into the 2-inch (5 cm) area around your belly button.
- where the skin is red, warm, tender, bruised, scaly or hard.
- into scarred, damaged, discolored or tattooed skin.
- through clothing.
Step 5 – Wash your hands and clean the injection site
Wash your hands well with soap and water.
Clean the injection site with an alcohol wipe or with soap and water. Let the site air dry.
- Do not touch the cleaned injection site again or blow on it before injecting.
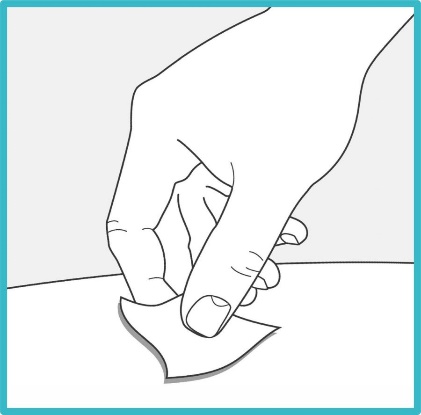
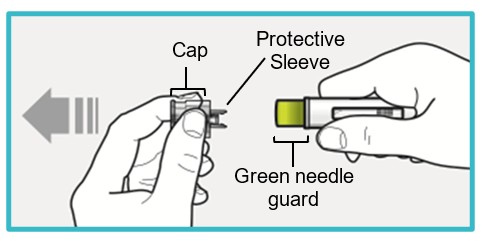
Step 6 – Pull off the cap
Do not remove the cap until you are ready to inject.
Pull off the cap (protective sleeve inside).
- SAPHNELO PEN is now unlocked and ready to inject.
- Do not touch the green needle guard or the needle inside.
- Do not recap SAPHNELO PEN. This could cause the medicine to come out too soon or damage SAPHNELO PEN.
Go to Step 7 right away after removing the cap.
Step 7 – Injecting SAPHNELO
Inject the medicine using SAPHNELO PEN by following the steps in figures a, b, c, and d.
To deliver a full dose, press and hold SAPHNELO PEN for about 15 seconds until the green plunger fills the viewing window.
You may hear a first ‘click’ at the start of the injection and a second ‘click’ at the end of the injection.
Do not move or change the position of SAPHNELO PEN after the injection has started.
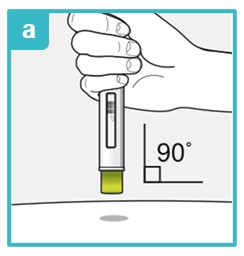
Position SAPHNELO PEN.
- Place the green needle guard flat against the skin (90-degree angle).
- Make sure you can see the viewing window.
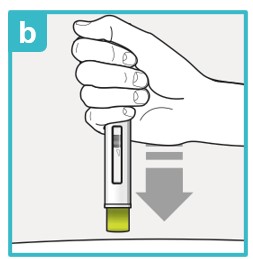
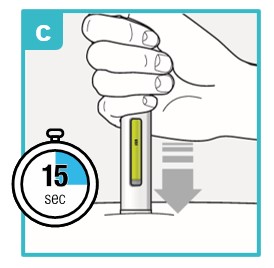
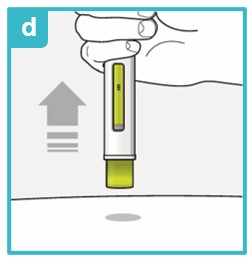
Press down firmly and hold against skin.
- You may hear the first click right away. This tells you the injection has started.
- The green plunger will move down in the viewing window.
Hold down firmly for about 15 seconds.
- The green plunger will fill the viewing window.
- You may hear the second click at the end of injection.
After you have completed your injection, lift SAPHNELO PEN straight up.
- The green needle guard will slide down and lock into place over the needle.
Step 8 – Check the viewing window
Check the viewing window to make sure all the medicine has been injected.
If the green plunger does not fill the viewing window, you may not have received the full dose.
- If this happens or if you have any other concerns, contact your healthcare provider.
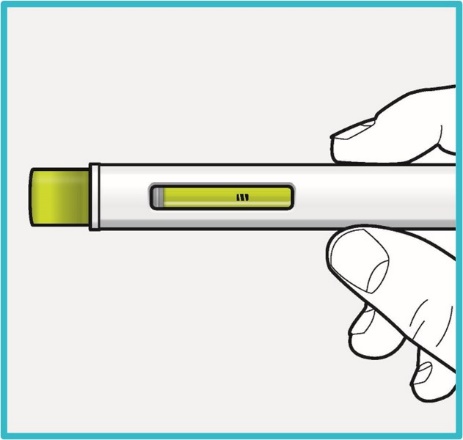

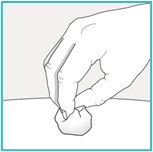
Step 9 – Check the injection site
There may be a small amount of blood or liquid at the injection site. This is normal.
If needed, press a cotton ball or gauze on the area and apply a small bandage.
- Do not rub the injection site.
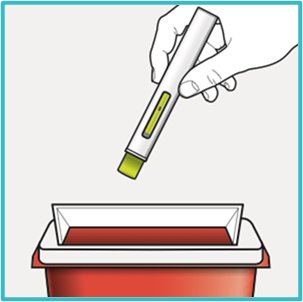
Step 10 – Throw away (dispose of) the used SAPHNELO PEN
Put your used SAPHNELO PEN in an FDA-cleared sharps disposal container right away after use.
Do not throw away (dispose of) SAPHNELO PEN in your household trash.
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting puncture resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak resistant, and
- properly labelled to warn of hazardous waste inside the container.
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container.
There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal.
Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
Do not recycle your used sharps disposal container.
Manufactured by:
AstraZeneca AB, Södertälje, Sweden SE-15185
US License No. 2059
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
©AstraZeneca 2026
SAPHNELO is a registered trademark of AstraZeneca.
For more information, go to https://www.SAPHNELO.com or call 1-800-236-9933.
This Instructions for Use has been approved by the U.S. Food and Drug Administration Approved: April 2026
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 0310-3080-02 Rx only
SAPHNELO®
(anifrolumab-fnia)
Injection
120 mg/0.8 mL
For Subcutaneous Injection only
Do not shake, freeze or expose to heat.
1 single-dose pre-filled syringe
AstraZeneca
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 0310-3080-01 Rx Only
Saphnelo® Pen 120 mg/0.8 mL
(anifrolumab-fnia)
Injection
For Subcutaneous Injection Only
Do not shake, freeze, or expose to heat.
1 single-dose autoinjector
AstraZeneca
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 0310-3040-00 Rx Only
SAPHNELO®
(anifrolumab-fnia)
Injection
300 mg/ 2 mL
(150 mg/mL)
For Intravenous Infusion After Dilution
Must dilute before use. See prescribing information.
Single-dose vial. Discard unused portion.
Store refrigerated at 2oC to 8oC (36oF to 46oF)
Keep vial in original carton to protect from light.
Do not shake or freeze.
1 single-dose vial
AstraZeneca
-
INGREDIENTS AND APPEARANCE
SAPHNELO
anifrolumab-fnia injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-3040 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ANIFROLUMAB (UNII: 38RL9AE51Q) (ANIFROLUMAB - UNII:38RL9AE51Q) ANIFROLUMAB 300 mg in 2.0 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 3 mg in 2.0 mL HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) 6 mg in 2.0 mL LYSINE HYDROCHLORIDE (UNII: JNJ23Q2COM) 18 mg in 2.0 mL TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) 98 mg in 2.0 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 1 mg in 2.0 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-3040-00 1 in 1 CARTON 07/30/2021 1 2.0 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761123 07/30/2021 SAPHNELO
anifrolumab-fnia injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-3080 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ANIFROLUMAB (UNII: 38RL9AE51Q) (ANIFROLUMAB - UNII:38RL9AE51Q) ANIFROLUMAB 120 mg in 0.8 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) LYSINE HYDROCHLORIDE (UNII: JNJ23Q2COM) TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) POLYSORBATE 80 (UNII: 6OZP39ZG8H) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-3080-01 1 in 1 CARTON 04/24/2026 1 NDC: 0310-3080-25 0.8 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC: 0310-3080-95 1 in 1 CARTON 04/24/2026 2 NDC: 0310-3080-25 0.8 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 3 NDC: 0310-3080-02 1 in 1 CARTON 04/24/2026 3 NDC: 0310-3080-75 0.8 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761123 04/24/2026 Labeler - AstraZeneca Pharmaceuticals LP (054743190) Establishment Name Address ID/FEI Business Operations Samsung Biologics Co., Ltd. 557810567 API MANUFACTURE(0310-3080) , ANALYSIS(0310-3080)
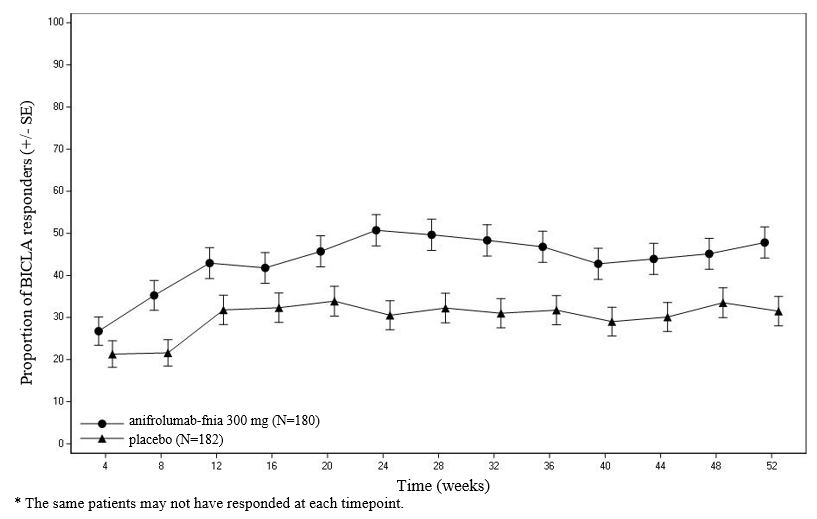

Trademark Results [SAPHNELO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 SAPHNELO 88833575 not registered Live/Pending |
AstraZeneca AB 2020-03-13 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.