ENBUMYST- bumetanide spray
Enbumyst by
Drug Labeling and Warnings
Enbumyst by is a Prescription medication manufactured, distributed, or labeled by Corstasis USA LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ENBUMYST safely and effectively. See full prescribing information for ENBUMYST.
ENBUMYST (bumetanide nasal spray)
Initial U.S. Approval: 1983INDICATIONS AND USAGE
ENBUMYST is a loop diuretic indicated for the treatment of edema associated with congestive heart failure, hepatic and renal disease, including nephrotic syndrome in adults. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Nasal spray: 0.5 mg bumetanide per 0.1 mL spray (3)
WARNINGS AND PRECAUTIONS
- Fluid, Electrolyte, and Metabolic Abnormalities: Monitor serum electrolytes, CO2, BUN, creatinine, glucose and uric acid. (5.1)
- Worsening Renal Function: Monitor for dehydration and azotemia. (5.2)
- Ototoxicity: Avoid higher than recommended doses. (5.3)
- Potential Altered Absorption in Patients with Nasal Mucosal or Structural Abnormalities: Avoid use in patients with significant nasal mucosal or structural abnormalities, such as acute episodes of rhinitis or congestion due to any cause. (5.4)
ADVERSE REACTIONS
Most common adverse reactions (incidence > 0.5%) are hypovolemia, headache, muscle cramps, dizziness, hypotension, nausea and encephalopathy (in patients with pre-existing liver disease). (6)
To report SUSPECTED ADVERSE REACTIONS, contact Corstasis Therapeutics at 1-877-300-5339 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Lithium may reduce renal clearance of bumetanide and add a high risk of lithium toxicity. (7)
- Probenecid reduces both the natriuresis and hyperreninemia. (7)
- Indomethacin blunts the increase in urine volume and sodium excretion. (7)
- Drugs with ototoxic potential: avoid parenteral bumetanide in patients receiving aminoglycoside antibiotics. (7)
- Drugs with nephrotoxic potential: the simultaneous administration of ENBUMYST with drugs of nephrotoxic potential should be avoided. (7)
- Antihypertensive effects may be potentiated by bumetanide. (7)
USE IN SPECIFIC POPULATIONS
- Lactation: A lactating woman treated with ENBUMYST should monitor her infant for excessive urine output, dehydration, and lethargy. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Fluid, Electrolyte, and Metabolic Abnormalities
5.2 Worsening Renal Function
5.3 Ototoxicity
5.4 Potential Altered Absorption in Patients with Nasal Mucosal or Structural Abnormalities
6 ADVERSE REACTIONS
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on Bumetanide
7.2 Effects of Bumetanide on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Each unit-dose nasal spray contains 0.5 mg of bumetanide. The usual total daily dosage of ENBUMYST is 0.5 mg to 2 mg once daily. The number of nasal spray devices needed for a single dose depends upon the prescribed dose. Individualize dosage based on patient response up to a maximum dose of 2 mg/day.
ENBUMYST is not intended for chronic use and should be replaced with oral diuretics as soon as practical.
ENBUMYST can be substituted at approximately a 1:40 ratio to oral furosemide and a 1:20 ratio to intravenous furosemide.
2.2 Administration Instructions
- ENBUMYST is for nasal use only.
- Each ENBUMYST unit is for single use and delivers 0.5 mg bumetanide upon actuation.
- Do not prime or attempt to reuse ENBUMYST for more than one administration.
- Administer ENBUMYST directly into the nose and not against the wall of the nose.
- If prescribed dose requires more than one nasal spray, alternate between right and left nostrils.
Refer patients and caregivers to the Instructions for Use for detailed administration instructions.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Fluid, Electrolyte, and Metabolic Abnormalities
Bumetanide may cause fluid, electrolyte, and metabolic abnormalities such as hypovolemia, hypokalemia, azotemia, hyponatremia, hypochloremic alkalosis, hypomagnesemia, hypocalcemia, hyperglycemia, or hyperuricemia, particularly in patients receiving higher doses, patients with inadequate oral electrolyte intake, and in elderly patients. Excessive diuresis may cause dehydration and blood volume reduction with circulatory collapse and possibly vascular thrombosis and embolism, particularly in elderly patients. Serum electrolytes, CO2, BUN, creatinine, glucose, and uric acid should be monitored frequently during bumetanide therapy.
5.2 Worsening Renal Function
Bumetanide can cause dehydration and azotemia. If increasing azotemia and oliguria occur during treatment of severe progressive renal disease, discontinue bumetanide [see Clinical Pharmacology (12.3)].
5.3 Ototoxicity
Tinnitus and hearing loss (usually reversible) have been reported with loop diuretics, including bumetanide. Reports indicate that ototoxicity is associated with rapid injection, severe renal impairment, the use of higher than recommended doses, hypoproteinemia or concomitant therapy with aminoglycoside antibiotics, ethacrynic acid, or other ototoxic drugs.
5.4 Potential Altered Absorption in Patients with Nasal Mucosal or Structural Abnormalities
ENBUMYST has not been assessed in individuals with nasal mucosal or structural abnormalities. Avoid use in patients with significant nasal mucosal or structural abnormalities, such as acute episodes of rhinitis or congestion due to any cause. Consider alternative products or therapies in such patients.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Fluid, electrolyte, and metabolic abnormalities [see Warnings and Precautions (5.1)]
- Worsening Renal Function [see Warnings and Precautions (5.2)]
- Ototoxicity [see Warnings and Precautions (5.3)]
- Potential Altered Absorption in Patients with Nasal Mucosal or Structural Abnormalities [see Warnings and Precautions (5.4)]
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of ENBUMYST is supported by clinical trials and postmarketing reports of oral bumetanide, as well as open-label, single- and repeat-dose studies of ENBUMYST in healthy subjects.
Adverse Reactions in Two Clinical Pharmacology Studies with ENBUMYST in Adult Subjects
In open-label studies of ENBUMYST in healthy subjects (n = 84), the most common adverse reaction that occurred with ENBUMYST was hypovolemia (4.8%) [see Warnings and Precautions (5.1)]. Headache occurred in 3% of subjects. There were no adverse reactions specifically associated with the nasal route of administration such as nasal irritation or pain. There was a single case of nasal dryness.
Adverse Reactions in Studies with Oral Bumetanide
The following adverse reactions were identified in clinical studies or postmarketing reports with the use of oral bumetanide. Because some of these reactions were reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The most frequent clinical adverse reactions considered probably or possibly related to oral bumetanide are muscle cramps (seen in 1.1% of treated patients), dizziness (1.1%), hypotension (0.8%), headache (0.6%), nausea (0.6%) and encephalopathy (in patients with pre-existing liver disease) (0.6%). One or more of these adverse reactions have been reported in approximately 4.1% of patients treated with bumetanide.
The following additional adverse reactions have been reported with bumetanide.
Blood and Lymphatic System Disorders: Deviations in hemoglobin, prothrombin time, hematocrit, WBC and differential counts, thrombocytopenia
Cardiac Disorders: Chest pain, electrocardiogram changes
Ear and Labyrinth Disorders: Ear discomfort, impaired hearing, vertigo
Gastrointestinal Disorders: Abdominal pain, diarrhea, dry mouth, GI upset, vomiting
General Disorders and Administration Site Conditions: Fatigue, weakness
Investigations: Changes in LDH, total serum bilirubin, serum proteins, SGOT, SGPT, alkaline phosphatase, cholesterol, creatinine clearance, urinary glucose, and urinary protein
Metabolism and Nutrition Disorders: Dehydration
Musculoskeletal and Connective Tissue Disorders: Arthritic pain, musculoskeletal pain
Nervous System Disorders: Asterixis
Renal and Urinary Disorders: Renal failure
Reproductive System and Breast Disorders: Erectile dysfunction, nipple tenderness, premature ejaculation
Respiratory, Thoracic and Mediastinal Disorders: Hyperventilation
Skin and Subcutaneous Tissue Disorders: Pruritus, rash, Stevens-Johnson syndrome, sweating, toxic epidermal necrolysis
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on Bumetanide
Lithium
Lithium should generally not be given with diuretics (such as bumetanide) because they reduce its renal clearance and add a high risk of lithium toxicity.
Probenecid
Probenecid should not be administered concurrently with bumetanide. Pretreatment with probenecid reduces both the natriuresis and hyperreninemia produced by bumetanide. This antagonistic effect of probenecid on bumetanide natriuresis is not due to a direct action on sodium excretion but is probably secondary to its inhibitory effect on renal tubular secretion of bumetanide.
Indomethacin
Concurrent therapy with bumetanide is not recommended. Indomethacin blunts the increases in urine volume and sodium excretion seen during bumetanide treatment and inhibits the bumetanide-induced increase in plasma renin activity.
7.2 Effects of Bumetanide on Other Drugs
Drugs with Ototoxic Potential [see Warnings and Precautions (5.3)]
Especially in the presence of impaired renal function, the use of parenterally administered bumetanide in patients to whom aminoglycoside antibiotics are also being given should be avoided, except in life-threatening conditions.
Drugs with Nephrotoxic Potential
Monitor renal function. Concomitant use may worsen renal function and increase the risk of nephrotoxicity.
Antihypertensives
Bumetanide may potentiate the effect of various antihypertensive drugs, necessitating a reduction in the dosage of these drugs.
Digoxin
Interaction studies in humans have shown no effect on digoxin blood levels.
Anticoagulants
Interaction studies in humans have shown bumetanide to have no effect on warfarin metabolism or on plasma prothrombin activity.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The available data on bumetanide use in pregnant women from scientific review publications have not identified a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Untreated congestive heart failure, hepatic disease such as cirrhosis and chronic kidney disease can lead to adverse outcomes for the mother and fetus (see Clinical Considerations).
In animal reproduction studies, no malformations were observed with oral administration of bumetanide to pregnant rats and rabbits during organogenesis at doses approximately 485 times and equivalent to a human dose of 2 mg once daily, respectively (see Data).
The background risk for major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the background risk of major birth defects and miscarriage in the clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Disease-Associated Maternal and/or Embryo/Fetal Risk
Pregnant women with congestive heart failure are at increased risk for pre-term birth. Stroke volume and heart rate increase during pregnancy, increasing cardiac output, especially during the first trimester. Clinical classification of heart disease may worsen with pregnancy and lead to maternal death and/or stillbirth. Closely monitor pregnant patients for destabilization of their heart failure.
Pregnant women with symptomatic cirrhosis generally have poor outcomes including hepatic failure, variceal hemorrhage, pre-term delivery, fetal growth restriction and maternal death. Outcomes are worse with coexisting esophageal varices. Pregnant women with cirrhosis of the liver should be carefully monitored and managed accordingly.
Chronic kidney disease in pregnancy increases the risk for maternal hypertension and preeclampsia, miscarriage, preterm delivery, polyhydramnios, still birth, and low birth weight infants.
Animal Data
Bumetanide has been shown to be nonteratogenic, but it has a slight embryocidal effect in rats when given at a dose approximately 485 times and in rabbits at a dose equivalent to a 2 mg/day human dose, based on body surface area (BSA). In one study, moderate growth retardation and increased incidence of delayed ossification of sternebrae were observed in rats at oral doses of 100 mg/kg/day, approximately 485 times a 2 mg/day human dose, based on BSA. These effects were associated with maternal weight reductions noted during dosing. No such adverse effects were observed at 30 mg/kg/day (approximately 146 times a 2 mg/day human dose, based on BSA). No fetotoxicity was observed at dose up to approximately 292 times a 2 mg/day human dose, based on BSA.
In rabbits, a dose-related decrease in litter size and an increase in resorption rate were noted at oral doses of 0.1 mg/kg/day and 0.3 mg/kg/day (approximately equivalent to 3 times a 2 mg/day human dose, based on BSA). A slightly increased incidence of delayed ossification of sternebrae occurred at 0.3 mg/kg/day; however, no such adverse effects were observed at the dose of 0.03 mg/kg/day. The sensitivity of the rabbit to bumetanide parallels the marked pharmacologic and toxicologic effects of the drug in this species.
Bumetanide was not teratogenic in the hamster at an oral dose of 0.5 mg/kg/day (approximately 3 times a 2 mg/day human dose, based on BSA).
8.2 Lactation
Risk Summary
There are no data on the presence of bumetanide in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. Because there have been no reports of adverse events in breastfed infants over decades of bumetanide use, the developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for bumetanide and any potential adverse effects on the breastfed infant from bumetanide or from the underlying maternal condition.
Clinical Considerations
Monitor infants potentially exposed to ENBUMYST through breast milk for excessive urine output, dehydration and lethargy.
8.4 Pediatric Use
The safety and effectiveness of ENBUMYST (bumetanide nasal spray) have not been established in pediatric patients.
In vitro studies using pooled sera from critically ill neonates have shown bumetanide to be a potent displacer of bilirubin. The administration of bumetanide could present a particular concern if given to critically ill or jaundiced neonates at risk for kernicterus. ENBUMYST (bumetanide nasal spray) is not approved for use in pediatric patients, including neonates.
8.5 Geriatric Use
Clinical studies of bumetanide did not include sufficient numbers of subjects aged 65 and over to determine whether they responded differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal or cardiac function and of concomitant disease or other drug therapy.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
-
10 OVERDOSAGE
Overdosage can lead to acute profound water loss, volume and electrolyte depletion, dehydration, reduction of blood volume and circulatory collapse with a possibility of vascular thrombosis and embolism. Electrolyte depletion may be manifested by weakness, dizziness, mental confusion, anorexia, lethargy, vomiting and cramps. Treatment consists of replacement of fluid and electrolyte losses by careful monitoring of the urine and electrolyte output and serum electrolyte levels.
-
11 DESCRIPTION
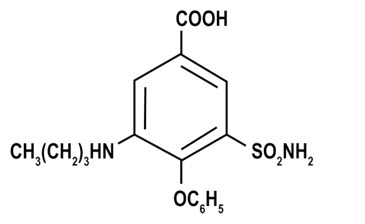
ENBUMYST contains bumetanide, a loop diuretic. Chemically, bumetanide is 3-(butylamino)-4-phenoxy-5-sulfamoylbenzoic acid. It is a practically white powder with a calculated molecular weight of 364.42 g/mol, and the following structural formula:
ENBUMYST is supplied as a unit-dose nasal spray containing 0.5 mg of bumetanide (equivalent to about 0.554 mg of potassium salt of bumetanide) per 0.1 mL. ENBUMYST contains the following inactive ingredients: benzyl alcohol (0.5 mg per 0.1 mL spray), carboxymethylcellulose sodium (viscosity control agent), mannitol, potassium hydroxide (pH modifier), hydrochloric acid (to adjust pH) and water for injection.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bumetanide primarily inhibits the reabsorption of sodium and chloride in the proximal and distal tubules and in the loop of Henle. The high degree of diuresis is largely due to the unique site of action. The action on the distal tubule is independent of any inhibitory effect on carbonic anhydrase and aldosterone.
12.2 Pharmacodynamics
Following administration of ENBUMYST, the onset of diuresis occurs in less than 60 minutes. Peak activity is reached between 0.75 and 1.5 hours. At a maximum dose of 2 mg, diuresis is largely complete within 4 hours. Bumetanide decreases uric acid excretion and increases serum uric acid.
In a study in 68 healthy adults, the effect of 2 mg ENBUMYST on diuresis, natriuresis and potassium urine excretion over 0 to 8 hours and 0 to 24 hours was similar to that of 2 mg bumetanide oral tablet and 2 mg bumetanide IV injection.
12.3 Pharmacokinetics
In a study in healthy adults, the exposure (Cmax and AUC) of 2 mg ENBUMYST was similar to that of 2 mg bumetanide oral tablet. The exposure of 2 mg ENBUMYST was approximately 11% (Cmax) and 65-67% (AUC) relative to that of 2 mg bumetanide IV injection.
Absorption
Bumetanide median time to maximum concentration (Tmax) is 1.0 hour following nasal administration of ENBUMYST.
Distribution
Bumetanide exhibits high plasma protein binding in the range of 94% to 96%.
Elimination
Bumetanide is eliminated rapidly in humans following both oral and parenteral (intravenous or intramuscular) administration, with a half-life of 1 to 1.5 hours. ENBUMYST demonstrated a half-life of approximately 3 hours in healthy adult subjects.
Metabolism
Mass balance studies using carbon-14 labeled bumetanide in healthy adults identified metabolites formed by oxidation of the N-butyl side chain. These metabolites were detected in both urine and bile.
Excretion
Following oral administration of carbon-14 labeled bumetanide to healthy adults, 81% of the administered radioactivity was recovered in urine, with 45% excreted as unchanged drug. Biliary excretion accounted for 2% of the administered dose.
Specific Populations
Geriatric Patients
In a group of ten geriatric subjects between the ages of 65 and 73 years, total bumetanide clearance was significantly lower (1.8 ± 0.3 mL/min*kg) compared with younger subjects (2.9 ± 0.2 mL/min*kg) after a single oral bumetanide 0.5 mg dose. Maximum plasma concentrations were higher in geriatric subjects (16.9 ± 1.8 ng/mL) compared with younger subjects (10.3 ± 1.5 ng/mL). Urine flow rate and total excretion of sodium and potassium were increased less in the geriatric subjects compared with younger subjects, although potassium excretion and fractional sodium excretion were similar between the two age groups. Nonrenal clearance, bioavailability, and volume of distribution were not significantly different between the two groups.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Bumetanide was devoid of mutagenic activity in various strains of Salmonella typhimurium when tested in the presence or absence of an in vitro metabolic activation system. An 18-month study showed an increase in mammary adenomas of questionable significance in female rats receiving oral doses of 60 mg/kg/day (approximately 290 times a 2 mg/day human dose, based on BSA). A repeat study at the same doses failed to duplicate this finding.
Reproduction studies were performed to evaluate general reproductive performance and fertility in rats at oral dose levels of 10 mg/kg/day, 30 mg/kg/day, 60 mg/kg/day or 100 mg/kg/day. The pregnancy rate was slightly decreased in the treated animals; however, the differences were small and not statistically significant.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ENBUMYST is a clear, colorless solution, supplied as a 12-pack carton containing unit-dose nasal sprays as shown in Table 1. Each unit-dose nasal spray delivers 0.5 mg bumetanide.
Table 1: Available Packaging Configurations Description Package Configuration NDC 12-pack carton Two (2) 6-pack cartons containing twelve (12) blister packages each with a unit-dose nasal spray NDC: 84388-005-12 -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Administration
Advise patients on appropriate administration, including the number of devices to use [see Dosage and Administration (2)].
Lactation
Advise lactating women treated with ENBUMYST to monitor their breastfed infant for excessive urine output, dehydration, and lethargy [see Use in Specific Populations (8.2)].
Manufactured for Corstasis Therapeutics, 3535 Executive Terminal Drive, Henderson, NV 89042
Copyright © 2025 Corstasis Therapeutics. All rights reserved -
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: November 2025 PATIENT INFORMATION
ENBUMYST (En-byoo-mist)
(bumetanide nasal spray)What is the most important information I should know about ENBUMYST? - ENBUMYST is a diuretic that causes fluid loss and may lead to abnormalities with blood electrolytes and body energy systems (metabolic). Too much fluid loss can lead to dehydration and decreased blood volume and increase your risk of blood clots. Blood abnormalities may include decreased salts (electrolytes), and increased nitrogen, glucose and uric acid. The chance of getting these abnormalities is higher in people who are elderly, use higher doses, or who do not get enough electrolytes by mouth. Your healthcare provider should do blood tests to check for these abnormalities during treatment with ENBUMYST.
- You should not use ENBUMYST if you have severe electrolyte loss until it gets better.
- unusual thirst
- dizziness or lightheadedness
- weakness or tiredness
- fast heart rate
- dry mouth or tongue
- sudden pain, redness or swelling in your joints, especially your big toe (gout)
- pain, swelling, redness, or warmth in your leg
- shortness of breath
- fast breathing
- high blood sugar
- dark urine
- skin is pale, tight, cool or clammy
- muscle cramps
- confusion
- See "What are the possible side effects of ENBUMYST?" for more information about side effects.
What is ENBUMYST?
ENBUMYST is a prescription medicine used in adults to treat swelling caused by too much fluid in the body (edema) from congestive heart failure, liver disease and kidney disease, including a kidney problem called nephrotic syndrome.
It is not known if ENBUMYST is safe and effective in children.Who should not use ENBUMYST? Do not use ENBUMYST if you: - have very low or no urine production
- are allergic to bumetanide or to any of the ingredients in ENBUMYST. See the end of this Patient Information for a complete list of ingredients in ENBUMYST.
Before using ENBUMYST, tell your healthcare provider about all your medical conditions, including if you: - have low levels of salts (electrolytes) in your blood, or are on a low-salt diet
- have diarrhea
- have diabetes
- have nasal problems including a runny nose, nasal congestion, nasal polyps, history of injury such as a broken nose, or any past nasal surgery
- are pregnant or plan to become pregnant. It is not known if ENBUMYST will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if ENBUMYST passes into your breast milk. If you use ENBUMYST and breastfeed, watch your baby for signs of urinating more than usual, dehydration, or unusual sleepiness, drowsiness, or sluggishness.
ENBUMYST and other medicines may affect each other, causing side effects, and may affect the way each other works.Especially tell your healthcare provider if you take or use: - other nasal sprays
- water pills (diuretics)
- lithium
- probenecid
- indomethacin
- antibiotic medicines called aminoglycosides
- medicines to treat high blood pressure
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. How should I use ENBUMYST? - Read the Instructions for Use that come with ENBUMYST for detailed information about how to use the device.
- Use ENBUMYST exactly as your healthcare provider tells you to. Talk to your healthcare provider or pharmacist if you have questions about how to use ENBUMYST.
- ENBUMYST is for use in the nose only.
- Each ENBUMYST device has 1 spray of medicine and cannot be reused. Do not test or prime (pre-spray) the device.
- ENBUMYST comes in a carton with 12 devices. Use the following number of nasal spray devices for your prescribed dose:
Prescribed Dose Number of Nasal Spray Devices 0.5 mg 1 device 1 mg 2 devices 1.5 mg 3 devices 2 mg 4 devices - Your healthcare provider will prescribe the dose of ENBUMYST that is right for you.
- ENBUMYST is for short term use only for a single treatment course (episode) of edema.
- If you use too much ENBUMYST call your healthcare provider right away.
What are the possible side effects of ENBUMYST? ENBUMYST may cause serious side effects, including: - See "What is the most important information I should know about ENBUMYST?"
- Worsening kidney function. Your healthcare provider may stop treatment if your kidney function gets worse.
- Ear damage (ototoxicity) including ringing in the ears (tinnitus) or hearing loss, especially in people with severe kidney problems, taking higher than recommended doses, with low levels of protein in the blood, and when used with certain other medicines.
The most common side effects of ENBUMYST include: - low levels of fluid in the body
- headache
- muscle cramps
- dizziness
- low blood pressure
- nausea
- abnormal brain functioning in people with certain liver problems
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of ENBUMYST.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store ENBUMYST? - Store ENBUMYST at room temperature between 59°F to 77°F (15°C to 25°C).
- Do not freeze.
Keep ENBUMYST and all medicines out of the reach of children. General information about the safe and effective use of ENBUMYST.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ENBUMYST for a condition for which it was not prescribed. Do not give ENBUMYST to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ENBUMYST that is written for health professionals.What are the ingredients in ENBUMYST?
Active Ingredient: bumetanide
Inactive Ingredients: benzyl alcohol (0.5 mg per 0.1 mL spray), carboxymethylcellulose sodium, mannitol, potassium hydroxide (pH modifier), hydrochloric acid (to adjust pH) and water for injection.
Manufactured for Corstasis Therapeutics, 3535 Executive Terminal Drive, Henderson, NV 89042
Copyright © 2025 Corstasis Therapeutics. All rights reserved.
For more information about ENBUMYST, call 1-877-300-5339 or visit ENBUMYST.com. -
PRINCIPAL DISPLAY PANEL - 0.5 mg Device Blister Pack Carton
NDC: 84388-005-06
Rx OnlyEnbumyst™
(bumetanide nasal spray)
0.5 mg per deviceFor Use in the Nose Only
6 Unit-dose Nasal Spray Devices
Dispense in this sealed carton

-
INGREDIENTS AND APPEARANCE
ENBUMYST
bumetanide sprayProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 84388-005 Route of Administration NASAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength bumetanide (UNII: 0Y2S3XUQ5H) (bumetanide - UNII:0Y2S3XUQ5H) bumetanide 0.5 mg in 0.1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 84388-005-06 6 in 1 CARTON 09/12/2025 1 1 in 1 BLISTER PACK 1 0.1 mL in 1 VIAL, SINGLE-DOSE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 2 NDC: 84388-005-12 12 in 1 CARTON 09/12/2025 2 1 in 1 BLISTER PACK 2 0.1 mL in 1 VIAL, SINGLE-DOSE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219500 09/12/2025 Labeler - Corstasis USA LLC (119325696)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.