TRANDOLAPRIL AND VERAPAMIL HYDROCHLORIDE ER- trandolapril and verapamil hydrochloride tablet, film coated, extended release
Trandolapril and Verapamil Hydrochloride by
Drug Labeling and Warnings
Trandolapril and Verapamil Hydrochloride by is a Prescription medication manufactured, distributed, or labeled by Greenstone LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
BOXED WARNING
(What is this?)
WARNING: FETAL TOXICITY
- When pregnancy is detected, discontinue trandolapril/verapamil hydrochloride ER tablets as soon as possible.
- Drugs that act directly on the renin-angiotensin system can cause injury and death to the developing fetus(see WARNINGS: Fetal Toxicity).
-
DESCRIPTION
Trandolapril/verapamil hydrochloride ER tablets combine a slow release formulation of a calcium channel blocker, verapamil hydrochloride, and an immediate release formulation of an angiotensin converting enzyme inhibitor, trandolapril.
Verapamil Component
Verapamil hydrochloride is chemically described as benzeneacetonitrile, α[3-[[2-(3,4-dimethoxyphenyl)ethyl]methylamino]propyl]-3, 4-dimethoxy-α-(1-methylethyl) hydrochloride. Its empirical formula is C27H38N2O4 HCl and its structural formula is:
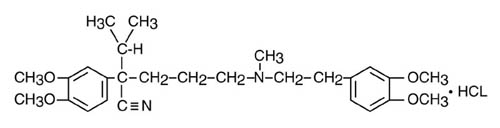
Verapamil hydrochloride is an almost white crystalline powder, with a molecular weight of 491.08. It is soluble in water, chloroform, and methanol. It is practically free of odor, with a bitter taste.
Trandolapril Component
Trandolapril is the ethyl ester prodrug of a nonsulfhydryl angiotensin converting enzyme (ACE) inhibitor, trandolaprilat. It is chemically described as (2S,3aR,7aS)-1-[(S)-N-[(S)-1-Carboxy-3-phenylpropyl]alanyl] hexahydro-2-indolinecarboxylic acid, 1-ethyl ester. Its empirical formula is C24 H34 N2O5 and its structural formula is:
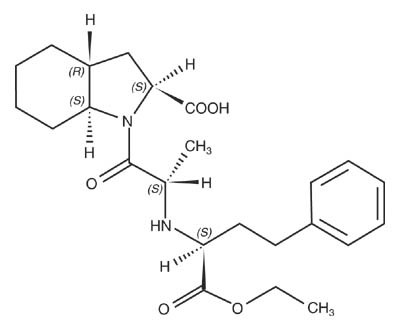
Trandolapril is a white or almost white powder with a molecular weight of 430.54. It is soluble (>100 mg/mL) in chloroform, dichloromethane, and methanol.
Trandolapril/verapamil hydrochloride ER tablets are formulated for oral administration, containing verapamil hydrochloride as a controlled release formulation and trandolapril as an immediate release formulation. The tablet strengths are trandolapril 2 mg/verapamil hydrochloride ER 180 mg, trandolapril 2 mg/verapamil hydrochloride ER 240 mg, and trandolapril 4 mg/verapamil hydrochloride ER 240 mg. The tablets also contain the following ingredients: corn starch, dioctyl sodium sulfosuccinate, ethanol, hydroxypropyl cellulose, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol, povidone, purified water, silicon dioxide, sodium alginate, sodium stearyl fumarate, synthetic iron oxides, talc, and titanium dioxide.
-
CLINICAL PHARMACOLOGY
Verapamil hydrochloride and trandolapril have been used individually and in combination for the treatment of hypertension. For the four dosing strengths, the antihypertensive effect of the combination is approximately additive to the individual components.
Verapamil Component
Verapamil is a calcium channel blocker that exerts its pharmacologic effects by modulating the influx of ionic calcium across the cell membrane of the arterial smooth muscle as well as in conductile and contractile myocardial cells. Verapamil exerts antihypertensive effects by decreasing systemic vascular resistance, usually without orthostatic decreases in blood pressure or reflex tachycardia. During isometric or dynamic exercise, verapamil does not alter systolic cardiac function in patients with normal ventricular function. Verapamil does not alter total serum calcium levels.
Trandolapril Component
Trandolapril is de-esterified to its diacid metabolite, trandolaprilat. Both inhibit angiotensin-converting enzyme (ACE) in human subjects and in animals. Trandolaprilat is about 8 times more potent than trandolapril. ACE is a peptidyl dipeptidase that catalyzes the conversion of angiotensin I to the vasoconstrictor, angiotensin II. Angiotensin II also stimulates aldosterone secretion by the adrenal cortex.
Inhibition of ACE results in decreased plasma angiotensin II, which leads to decreased vasopressor activity and to decreased aldosterone secretion. The latter decrease may result in a small increase of serum potassium. In controlled clinical trials, treatment with trandolapril/verapamil hydrochloride ER tablets resulted in mean increases in potassium of 0.1 mEq/L (see PRECAUTIONS). Removal of angiotensin II negative feedback on renin secretion leads to increased plasma renin activity (PRA).
ACE is identical to kininase II, an enzyme that degrades bradykinin. Whether increased levels of bradykinin, a potent vasodepressor peptide, play a role in the therapeutic effect of trandolapril/verapamil hydrochloride ER tablets remains to be elucidated.
While the mechanism through which trandolapril lowers blood pressure is believed to be primarily suppression of the renin-angiotensin-aldosterone system, trandolapril has an antihypertensive effect even in patients with low renin hypertension. Trandolapril is an effective antihypertensive in all races studied. Both black patients (usually a predominantly low renin group) and non-black patients respond to 2 to 4 mg of trandolapril.
Pharmacokinetics and Metabolism
Trandolapril/verapamil Hydrochloride ER Tablets
Following a single oral dose of trandolapril/verapamil hydrochloride ER tablets in healthy subjects, peak plasma concentrations are reached within 0.5-2 hours for trandolapril and within 4-15 hours for verapamil. Peak plasma concentrations of the active desmethyl metabolite of verapamil, norverapamil, are reached within 5-15 hours. Cleavage of the ester group converts trandolapril to its active diacid metabolite, trandolaprilat, which reaches peak plasma concentrations within 2-12 hours. The pharmacokinetics of trandolapril and trandolaprilat are not altered when trandolapril is administered in combination with verapamil, compared to monotherapy.
The AUC and Cmax for both verapamil and norverapamil are increased when 240 mg of controlled release verapamil is administered concomitantly with 4 mg trandolapril. The increase in Cmax is 54 and 30% and the AUC is increased by 65 and 32% for verapamil and norverapamil, respectively. Administration of trandolapril/verapamil hydrochloride ER 4/240 tablets (4 mg trandolapril and 240 mg verapamil hydrochloride ER) with a high-fat meal does not alter the bioavailability of trandolapril whereas verapamil peak concentrations and area under the curve (AUC) decrease 37% and 28%, respectively. Food thus decreases verapamil bioavailability and the time to peak plasma concentration for both verapamil and norverapamil are delayed by approximately 7 hours. Both optical isomers of verapamil are similarly affected.
The elimination half life of trandolapril is about 6 hours. At steady state, the effective half-life of trandolaprilat is 22.5 hours. Like all ACE inhibitors, trandolaprilat also has a prolonged terminal elimination phase, involving a small fraction of administered drug, probably representing binding to plasma and tissue ACE.
The terminal half-life of verapamil is 6-11 hours. Steady-state plasma concentrations of the two components are achieved after about a week of once-daily dosing of trandolapril/verapamil hydrochloride ER tablets. At steady-state, plasma concentrations of verapamil and trandolaprilat are up to two-fold higher than those observed after a single oral trandolapril/verapamil hydrochloride ER tablets dose.
The pharmacokinetics of verapamil and trandolaprilat are significantly different in the elderly (≥65 years) than in younger subjects. The bioavailability of verapamil and norverapamil are increased by 87% and 77%, respectively, and that of trandolapril by approximately 35% in the elderly. AUCs are approximately 80% and 35% higher, respectively.
Verapamil Component
With the immediate release formulation, more than 90% of the orally administered dose is absorbed with peak plasma concentrations of verapamil observed 1 to 2 hours after dosing. A delayed rate but similar extent of absorption is observed for the sustained release formulation when compared to the immediate release formulation. Because of the rapid biotransformation of verapamil during its first pass through the portal circulation, absolute bioavailability ranges from 20% to 35%. A nonlinear correlation exists between verapamil dose and plasma concentrations.
In early dose titration with verapamil, a relationship exists between plasma concentrations of verapamil and prolongation of the PR interval. However, during chronic administration, this relationship may disappear. No relationship has been established between the plasma concentration of verapamil and reduction in blood pressure.
In healthy subjects, orally administered verapamil undergoes extensive metabolism in the liver. Twelve metabolites have been identified in plasma; all except norverapamil are present in trace amounts only. Approximately 70% of an administered dose is excreted as metabolites in the urine and 16% or more in the feces within 5 days. Urinary excretion of unchanged drug is about 3% to 4% of the dose. Verapamil is approximately 90% bound to plasma proteins.
In patients with hepatic insufficiency, verapamil clearance is decreased about 30% and the elimination half-life is prolonged up to 14 to 16 hours (see PRECAUTIONS). In patients with liver dysfunction, a dosage adjustment may be required. In the elderly (≥65 years), verapamil clearance is reduced resulting in increases in elimination half-life.
Trandolapril Component
Following oral administration of trandolapril, the absolute bioavailability of trandolapril is approximately 10% as trandolapril and 70% as trandolaprilat. Plasma concentrations of trandolaprilat but not trandolapril increase in proportion with dose. Plasma concentrations of trandolaprilat decline in a triphasic manner. The more prolonged terminal elimination phase probably represents a small fraction of dose saturably bound to ACE.
After an oral radiolabeled dose of trandolapril, excretion of trandolapril and metabolites account for 33% of the dose in the urine and about 66% in the feces. Less than 1% of the dose is excreted in the urine as unchanged drug. Serum protein binding of trandolapril is about 80%, and is independent of concentration. Binding of trandolaprilat is concentration-dependent, varying from 65% at 1000 ng/mL to 94% at 0.1 ng/mL, indicating saturation of binding with increasing concentration.
Compared to normal subjects, the plasma concentrations of trandolapril and trandolaprilat are approximately 2-fold greater and renal clearance is reduced by about 85% in patients with creatinine clearance below 30 mL/min and in patients on hemodialysis. Dosage adjustment is recommended in renally impaired patients (see DOSAGE AND ADMINISTRATION).
Following oral administration in patients with mild to moderate alcoholic cirrhosis, plasma concentrations of trandolapril and trandolaprilat were, respectively, 9-fold and 2-fold greater than in normal subjects, but inhibition of ACE activity was not affected. Lower doses should be considered in patients with hepatic insufficiency (see DOSAGE AND ADMINISTRATION).
Pharmacodynamics
Trandolapril/verapamil Hydrochloride ER Tablets
Verapamil does not interfere with ACE inhibition by trandolapril. Trandolapril does not alter the effect of verapamil on intra-cardiac conduction.
Verapamil Component
Verapamil dilates the main coronary arteries and coronary arterioles, both in normal and ischemic regions, and is a potent inhibitor of coronary artery spasm. This property increases myocardial oxygen delivery in patients with coronary artery spasm, and is responsible for the effectiveness of verapamil in vasospastic (Prinzmetal's or variant) as well as unstable angina at rest.
Verapamil regularly reduces the total systemic resistance (afterload) by dilating peripheral arterioles. By decreasing the influx of calcium, verapamil prolongs the effective refractory period within the AV node and slows AV conduction in a rate-related manner.
Normal sinus rhythm is usually not affected, but in patients with sick sinus syndrome, verapamil may interfere with sinus node impulse generation and may induce sinus arrest or sinoatrial block. Atrioventricular block can occur in patients without preexisting conduction defects (see WARNINGS).
Verapamil does not alter the normal atrial action potential or intraventricular conduction time, but depresses amplitude, velocity of depolarization and conduction in depressed atrial fibers. Verapamil may shorten the antegrade effective refractory period of accessory bypass tracts. Acceleration of ventricular rate and/or ventricular fibrillation has been reported in patients with atrial flutter or atrial fibrillation and a coexisting accessory AV pathway following administration of verapamil (see WARNINGS).
Hemodynamics and Myocardial Metabolism: Verapamil reduces afterload and myocardial contractility. Improved left ventricular diastolic function in patients with idiopathic hypertrophic subaortic stenosis (IHSS) and those with coronary heart disease has also been observed with verapamil therapy. In most patients, including those with organic cardiac disease, the negative inotropic action of verapamil is countered by a reduction of afterload and cardiac index is usually not reduced. However, in patients with severe left ventricular dysfunction (e.g., pulmonary wedge pressure about 20 mmHg or ejection fraction less than 30%), or in patients taking beta-adrenergic blocking agents or other cardio-depressant drugs, deterioration of ventricular function may occur (see PRECAUTIONS - Drug Interactions).
Pulmonary Function: Verapamil does not induce bronchoconstriction and hence, does not impair ventilatory function.
Trandolapril Component
After a single 2 mg dose of trandolapril, inhibition of ACE activity reaches a maximum (70-85%) at 4 hours with about 10% decline at 24 hours. Eight days after dosing, ACE inhibition is still 40%.
Four placebo-controlled dose response studies were conducted using once daily oral dosing of trandolapril in doses from 0.25 to 16 mg per day in 827 black and non-black patients with mild to moderate hypertension. The minimal effective once daily dose was 1.0 mg in non-black patients and 2.0 mg in black patients. Further decreases in trough supine diastolic blood pressure were obtained in non-black patients with higher doses, and no further response was seen with doses above 4 mg (up to 16 mg). The antihypertensive effect diminished somewhat at the end of the dosing interval.
During chronic therapy, the maximum reduction in blood pressure with any dose is achieved within one week. Following 6 weeks of monotherapy in placebo-controlled trials in patients with mild to moderate hypertension, once daily doses of 2 to 4 mg lowered supine or standing systolic/diastolic blood pressure 24 hours after dosing by an average 7-10/4-5 mmHg below placebo responses in non-black patients. Once daily doses of 2 to 4 mg lowered blood pressures 4-6/3-4 mmHg below placebo responses in black patients.
-
CLINICAL STUDIES
In controlled clinical trials, once daily doses of trandolapril/verapamil hydrochloride ER tablets, trandolapril 4 mg/verapamil HCl ER 240 mg or trandolapril 2 mg/verapamil HCl ER 180 mg, decreased placebo-corrected seated pressure (systolic/diastolic) 24 hours after dosing by about 7-12/6-8 mmHg. Each of the components of trandolapril/verapamil hydrochloride ER tablets added to the antihypertensive effect. Treatment effects were consistent across age groups (<65, ≥65 years), and gender (male, female).
Blood pressure reductions were significantly greater for the trandolapril/verapamil hydrochloride ER tablets 4/240 combination than for either of the components used alone.
The antihypertensive effects of trandolapril/verapamil hydrochloride ER tablets have continued during therapy for at least 1 year.
-
INDICATIONS AND USAGE
Trandolapril/verapamil hydrochloride ER tablets are indicated for the treatment of hypertension.
This fixed combination drug is not indicated for the initial therapy of hypertension (see DOSAGE AND ADMINISTRATION).
In using trandolapril/verapamil hydrochloride ER tablets, consideration should be given to the fact that an angiotensin converting enzyme inhibitor, captopril, has caused agranulocytosis, particularly in patients with renal impairment or collagen vascular disease, and that available data are insufficient to show that trandolapril does not have similar risk (see WARNINGS - Neutropenia/Agranulocytosis).
-
CONTRAINDICATIONS
Trandolapril/verapamil hydrochloride ER tablets are contraindicated in patients who are hypersensitive to any ACE inhibitor or verapamil.
Because of the verapamil component, trandolapril/verapamil hydrochloride ER tablets are contraindicated in:
- Severe left ventricular dysfunction (see WARNINGS).
- Hypotension (systolic pressure less than 90 mmHg) or cardiogenic shock.
- Sick sinus syndrome (except in patients with a functioning artificial ventricular pacemaker).
- Second- or third-degree AV block (except in patients with a functioning artificial ventricular pacemaker).
- Patients with atrial flutter or atrial fibrillation and an accessory bypass tract (e.g. Wolff-Parkinson-White, Lown-Ganong-Levine syndromes) (see WARNINGS).
- Patients taking flibanserin (see PRECAUTIONS, Drug Interactions).
Because of the trandolapril component, trandolapril/verapamil hydrochloride ER tablets are contraindicated in patients with a history of angioedema related to previous treatment with an angiotensin converting enzyme (ACE) inhibitor.
Do not co-administer aliskiren with trandolapril/verapamil hydrochloride ER tablets in patients with diabetes (see PRECAUTIONS, Drug Interactions).
Trandolapril/verapamil hydrochloride ER tablets are contraindicated in combination with a neprilysin inhibitor (e.g., sacubitril). Do not administer trandolapril/verapamil hydrochloride ER tablets within 36 hours of switching to or from sacubitril/valsartan, a neprilysin inhibitor (see WARNINGS).
-
WARNINGS
Verapamil Component
Verapamil has a negative inotropic effect which, in most patients, is compensated by its afterload reduction (decreased systemic vascular resistance) properties without a net impairment of ventricular performance. In clinical experience with 4,954 patients, 87 (1.8%) developed congestive heart failure or pulmonary edema. Verapamil should be avoided in patients with severe left ventricular dysfunction (e.g., ejection fraction less than 30%, pulmonary wedge pressure above 20 mmHg, or severe symptoms of cardiac failure) and in patients with any degree of ventricular dysfunction if they are receiving a beta adrenergic blocker (see PRECAUTIONS - Drug Interactions). Patients with milder ventricular dysfunction should, if possible, be controlled with optimum doses of digitalis and/or diuretics before verapamil treatment (Note interactions with digoxin under: PRECAUTIONS).
Trandolapril Component
Trandolapril, as an ACE inhibitor, may cause excessive hypotension in patients with congestive heart failure (see WARNINGS - Hypotension).
Hypotension
Verapamil Component
Occasionally, the pharmacologic action of verapamil may produce a decrease in blood pressure below normal levels which may result in dizziness or symptomatic hypotension.
Trandolapril Component
Trandolapril can cause symptomatic hypotension. Like other ACE inhibitors, trandolapril has only rarely been associated with symptomatic hypotension in uncomplicated hypertensive patients. Symptomatic hypotension is most likely to occur in patients who are salt- or volume-depleted as a result of prolonged treatment with diuretics, dietary salt restriction, dialysis, diarrhea, or vomiting. Volume and/or salt depletion should be corrected before initiating treatment with trandolapril (see PRECAUTIONS - Drug Interactions and ADVERSE REACTIONS).
In controlled studies, hypotension was observed in 0.6% of patients receiving any combination of trandolapril and verapamil HCl ER.
In patients with concomitant congestive heart failure, with or without associated renal insufficiency, ACE inhibitor therapy may cause excessive hypotension, which may be associated with oliguria or azotemia, and, rarely, with acute renal failure and death (see DOSAGE AND ADMINISTRATION).
If symptomatic hypotension occurs, the patient should be placed in the supine position and, if necessary, normal saline may be administered intravenously. A transient hypotensive response is not a contraindication to further doses; however, lower doses of verapamil HCl ER and/or trandolapril or reduced concomitant diuretic therapy should be considered.
Elevated Liver Enzymes/Hepatic Failure
Verapamil Component
Elevations of transaminases with and without concomitant elevations in alkaline phosphatase and bilirubin have been reported. Such elevations have sometimes been transient and may disappear even in the face of continued verapamil treatment. Several cases of hepatocellular injury related to verapamil have been proven by rechallenge; half of these had clinical symptoms (malaise, fever, and/or right upper quadrant pain) in addition to elevations of SGOT, SGPT, and alkaline phosphatase.
Trandolapril Component
ACE inhibitors rarely have been associated with a syndrome of cholestatic jaundice, fulminant hepatic necrosis, and death. The mechanism of this syndrome is not understood. Patients receiving ACE inhibitors who develop jaundice should discontinue the ACE inhibitor and receive appropriate medical follow-up.
Liver abnormalities were noted in 3.2% of patients taking any of several combinations of trandolapril/verapamil doses. Periodic monitoring of liver function in patients taking trandolapril/verapamil hydrochloride ER tablets is therefore prudent.
Accessory Bypass Tract (Wolff-Parkinson-White or Lown-Ganong-Levine Syndromes)
Verapamil Component
Some patients with paroxysmal and/or chronic atrial fibrillation or atrial flutter and a coexisting accessory AV pathway have developed increased antegrade conduction across the accessory pathway bypassing the AV node, producing a very rapid ventricular response or ventricular fibrillation after receiving intravenous verapamil (or digitalis). Although a risk of this occurring with oral verapamil has not been established, such patients receiving oral verapamil may be at risk and its use in these patients is contraindicated (see CONTRAINDICATIONS).
Treatment is usually DC-cardioversion. Cardioversion has been used safely and effectively after oral verapamil.
Atrioventricular Block
Verapamil Component
The effect of verapamil on AV conduction and the SA node may lead to asymptomatic first-degree AV block and transient bradycardia, sometimes accompanied by nodal escape rhythms. PR interval prolongation is correlated with verapamil plasma concentrations, especially during the early titration phases of therapy. Higher degrees of AV block, however, were infrequently (0.8%) observed. Marked first-degree block or progressive development to second- or third-degree AV block requires a reduction in dosage or, in rare instances, discontinuation of verapamil HCl and institution of appropriate therapy depending upon the clinical situation.
Patients with Hypertrophic Cardiomyopathy (IHSS)
Verapamil Component
In 120 patients with hypertrophic cardiomyopathy (most of them refractory or intolerant to propranolol) who received therapy with verapamil at doses up to 720 mg/day, a variety of serious adverse effects were seen. Three patients died in pulmonary edema; all had severe left ventricular outflow obstruction and a past history of left ventricular dysfunction. Eight other patients had pulmonary edema and/or severe hypotension; abnormally high (over 20 mmHg) capillary wedge pressure and a marked left ventricular outflow obstruction were present in most of these patients. Sinus bradycardia occurred in 11% of the patients, second-degree AV block in 4% and sinus arrest in 2%. It must be appreciated that this group of patients had a serious disease with a high mortality rate. Most adverse effects responded well to dose reduction and only rarely did verapamil have to be discontinued.
Anaphylactoid and Possibly Related Reactions
Presumably because angiotensin-converting enzyme inhibitors affect the metabolism of eicosanoids and polypeptides, including endogenous bradykinin, patients receiving ACE inhibitors, including trandolapril may be subject to a variety of adverse reactions, some of them serious.
Angioedema
Angioedema of the face, extremities, lips, tongue, glottis, and larynx has been reported in patients treated with ACE inhibitors including trandolapril. Symptoms suggestive of angioedema or facial edema occurred in 0.13% of trandolapril-treated patients. Two of the four cases were life-threatening and resolved without treatment or with medication (corticosteroids). Angioedema associated with laryngeal edema can be fatal. If laryngeal stridor or angioedema of the face, tongue or glottis occurs, treatment with trandolapril/verapamil hydrochloride ER tablets should be discontinued immediately, the patient treated in accordance with accepted medical care and carefully observed until the swelling disappears. In instances where swelling is confined to the face and lips, the condition generally resolves without treatment; antihistamines may be useful in relieving symptoms. Where there is involvement of the tongue, glottis, or larynx, likely to cause airway obstruction, emergency therapy, including but not limited to subcutaneous epinephrine solution 1:1,000 (0.3 to 0.5 mL) should be promptly administered (see PRECAUTIONS and ADVERSE REACTIONS).
Patients receiving coadministration of an ACE inhibitor with an mTOR (mammalian target of rapamycin) inhibitor (e.g., temsirolimus, sirolimus, everolimus) or a neprilysin inhibitor (e.g., sacubitril) may be at increased risk for angioedema.
Anaphylactoid Reactions During Desensitization
Two patients undergoing desensitizing treatment with hymenoptera venom while receiving ACE inhibitors sustained life-threatening anaphylactoid reactions. In the same patients, these reactions did not occur when ACE inhibitors were temporarily withheld, but they reappeared when the ACE inhibitors were inadvertently readministered.
Anaphylactoid Reactions During Membrane Exposure
Anaphylactoid reactions have been reported in patients dialyzed with high-flux membranes and treated concomitantly with an ACE inhibitor. Anaphylactoid reactions have also been reported in patients undergoing low-density lipoprotein apheresis with dextran sulfate absorption.
Neutropenia/Agranulocytosis
Trandolapril Component
Another ACE inhibitor, captopril, has been shown to cause agranulocytosis and bone marrow depression rarely in patients with uncomplicated hypertension, but more frequently in patients with renal impairment, especially if they also have a collagen-vascular disease such as systemic lupus erythematosus or scleroderma. Available data from clinical trials of trandolapril or trandolapril/verapamil hydrochloride ER tablets are insufficient to show that trandolapril does not cause agranulocytosis at similar rates. As with other ACE inhibitors, periodic monitoring of white blood cell counts in patients with collagen-vascular disease and/or renal disease should be considered.
Fetal Toxicity
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue trandolapril/verapamil hydrochloride ER tablets as soon as possible. These adverse outcomes are usually associated with use of these drugs in the second and third trimester of pregnancy. Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin-angiotensin system from other antihypertensive agents. Appropriate management of maternal hypertension during pregnancy is important to optimize outcomes for both mother and fetus.
In the unusual case that there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system for a particular patient, apprise the mother of the potential risk to the fetus. Perform serial ultrasound examinations to assess the intra-amniotic environment. If oligohydramnios is observed, discontinue trandolapril/verapamil hydrochloride ER tablets, unless it is considered lifesaving for the mother. Fetal testing may be appropriate, based on the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury. Closely observe infants with histories of in utero exposure to trandolapril/verapamil hydrochloride ER tablets for hypotension, oliguria, and hyperkalemia (see PRECAUTIONS - Pediatric Use).
Doses of 0.8 mg/kg/day (9.4 mg/m2/day) in rabbits, 1000 mg/kg/day (7000 mg/m2/day) in rats, and 25 mg/kg/day (295 mg/m2/day) in cynomolgus monkeys did not produce teratogenic effects. These doses represent 10 and 3 times (rabbits), 1250 and 2564 times (rats), and 312 and 108 times (monkeys) the maximum projected human dose of 4 mg based on body-weight and body-surface-area, respectively assuming a 50 kg woman.
Trandolapril in doses of 0.8 mg/kg/day in rabbits, 100.0 mg/kg/day in rats, and 25 mg/kg/day in cynomolgus monkeys (10, 1250, and 312 times the maximum projected human dose, respectively, assuming a 50 kg woman) did not produce teratogenic effects.
-
PRECAUTIONS
Use in Patients with Impaired Hepatic Function
Trandolapril/verapamil hydrochloride ER tablets have not been evaluated in subjects with impaired hepatic function.
Verapamil Component
Since verapamil is highly metabolized by the liver, it should be administered cautiously to patients with impaired hepatic function. Severe liver dysfunction prolongs the elimination half-life of immediate release verapamil to about 14 to 16 hours; hence, approximately 30% of the dose given to patients with normal liver function should be administered to these patients.
Careful monitoring for abnormal prolongation of the PR interval or other signs of excessive pharmacologic effects (see OVERDOSAGE) should be carried out.
Use in Patients with Impaired Renal Function
Trandolapril/verapamil hydrochloride ER tablets have not been evaluated in patients with impaired renal function.
Verapamil Component
About 70% of an administered dose of verapamil is excreted as metabolites in the urine. Verapamil is not removed by hemodialysis. Until further data are available, verapamil should be administered cautiously to patients with impaired renal function. These patients should be carefully monitored for abnormal prolongation of the PR interval or other signs of overdosage (see OVERDOSAGE).
Trandolapril Component
As a consequence of inhibiting the renin-angiotensin-aldosterone system, changes in renal function may be anticipated in susceptible individuals. In patients with severe heart failure whose renal function may depend on the activity of the renin-angiotensin-aldosterone system, treatment with ACE inhibitors, including trandolapril, may be associated with oliguria and/or progressive azotemia and rarely with acute renal failure and/or death.
In hypertensive patients with unilateral or bilateral renal artery stenosis, increases in blood urea nitrogen and serum creatinine have been observed in some patients following ACE inhibitor therapy. These increases were almost always reversible upon discontinuation of the ACE inhibitor and/or diuretic therapy. In such patients, renal function should be monitored during the first few weeks of therapy.
Some hypertensive patients with no apparent pre-existing renal vascular disease have developed increases in blood urea and serum creatinine, usually minor and transient, especially when ACE inhibitors have been given concomitantly with a diuretic. This is more likely to occur in patients with pre-existing renal impairment. Dosage reduction and/or discontinuation of any diuretic and/or the ACE inhibitor may be required.
Evaluation of hypertensive patients should always include assessment of renal function (see DOSAGE AND ADMINISTRATION).
Use in Patients with Attenuated (Decreased) Neuromuscular Transmission
Verapamil Component
It has been reported that verapamil decreases neuromuscular transmission in patients with Duchenne's muscular dystrophy, and that verapamil prolongs recovery from the neuromuscular blocking agent vecuronium. It may be necessary to decrease the dosage of verapamil when it is administered to patients with attenuated neuromuscular transmission (see PRECAUTIONS - Surgery/Anesthesia).
Hyperkalemia and Potassium-sparing Diuretics
Trandolapril Component
In clinical trials, hyperkalemia (serum potassium > 6.00 mEq/L) occurred in approximately 0.4 percent of hypertensive patients receiving trandolapril and in 0.8% of patients receiving a dose of trandolapril (0.5-8 mg) in combination with a dose of verapamil SR (120-240 mg). In most cases, elevated serum potassium levels were isolated values, which resolved despite continued therapy. None of these patients were discontinued from the trials because of hyperkalemia. Risk factors for the development of hyperkalemia include renal insufficiency, diabetes mellitus, and the concomitant use of potassium-sparing diuretics, potassium supplements, and/or potassium-containing salt substitutes, which should be used cautiously, if at all, with trandolapril (see PRECAUTIONS - Drug Interactions).
Cough
Presumably due to the inhibition of the degradation of endogenous bradykinin, persistent nonproductive cough has been reported with all ACE inhibitors, always resolving after discontinuation of therapy. ACE inhibitor-induced cough should be considered in the differential diagnosis of cough. In controlled trials of trandolapril, cough was present in 2% of trandolapril patients and 0% of patients given placebo. There was no evidence of a relationship to dose.
Surgery/anesthesia
Trandolapril Component
In patients undergoing major surgery or during anesthesia with agents that produce hypotension, trandolapril will block angiotensin II formation secondary to compensatory renin release. If hypotension occurs and is considered to be due to this mechanism, it can be corrected by volume expansion (see PRECAUTIONS - Use in Patients with Attenuated (Decreased) Neuromuscular Transmission).
Drug Interactions
In vitro metabolic studies indicate that verapamil is metabolized by cytochrome P450 including CYP3A4, CYP1A2, CYP2C8, CYP2C9 and CYP2C18. Verapamil has been shown to be an inhibitor of CYP3A4 enzymes and P-glycoprotein (P-gp).
Clinically significant interactions have been reported with inhibitors of CYP3A4 (e.g. erythromycin, ritonavir) causing elevation of plasma levels of verapamil while inducers of CYP3A4 (e.g. rifampin) have caused a lowering of plasma levels of verapamil. Therefore, patients receiving inhibitors or inducers of the cytochrome P450 system should be monitored for drug interactions.
Concurrent use of verapamil increases exposure to ivabradine and may exacerbate bradycardia and conduction disturbances. Avoid co-administration of verapamil and ivabradine.
Clinical use of verapamil in digitalized patients has shown the combination to be well tolerated if digoxin doses are properly adjusted. Chronic verapamil treatment can increase serum digoxin levels by 50 to 75% during the first week of therapy, and this can result in digoxin toxicity. In patients with hepatic cirrhosis, the influence of verapamil on digoxin kinetics is magnified. Verapamil may reduce total body clearance and extrarenal clearance of digitoxin by 27% and 29%, respectively. Maintenance digoxin doses should be reduced when verapamil is administered, and the patient should be carefully monitored to avoid over- or under-digitalization. Whenever overdigitalization is suspected, the daily dose of digoxin should be reduced or temporarily discontinued. Upon discontinuation of any verapamil-containing regime including trandolapril/verapamil hydrochloride ER tablets, the patient should be reassessed to avoid underdigitalization. No clinically significant pharmacokinetic interaction has been found between trandolapril (or its metabolites) and digoxin.
Verapamil Component
Increased sensitivity to the effects of lithium (neurotoxicity) has been reported during concomitant verapamil-lithium therapy with either no change or an increase in serum lithium levels. Increased serum lithium levels and symptoms of lithium toxicity have been reported in patients receiving concomitant lithium and ACE inhibitor therapy. Trandolapril/verapamil hydrochloride ER tablets and lithium should be coadministered with caution, and frequent monitoring of serum lithium levels is recommended. If a diuretic is also used, the risk of lithium toxicity may be increased.
Hypotension, bradyarrhythmias, and lactic acidosis have been observed in patients receiving concurrent clarithromycin.
Hypotension, bradyarrhythmias, and lactic acidosis have been observed in patients receiving concurrent erythromycin ethylsuccinate.
The interaction between cimetidine and chronically administered verapamil has not been studied. Variable results on clearance have been obtained in acute studies of healthy volunteers; clearance of verapamil was either reduced or unchanged. No clinically significant pharmacokinetic interaction has been found between trandolapril (or its metabolites) and cimetidine.
Verapamil Component
Data on possible interactions between verapamil and disopyramide phosphate are not available. Therefore, disopyramide should not be administered within 48 hours before or 24 hours after verapamil administration.
A study of healthy volunteers showed that the concomitant administration of flecainide and verapamil may have additive effects on myocardial contractility, AV conduction, and repolarization. Concomitant therapy with flecainide and verapamil may result in additive negative inotropic effect and prolongation of atrioventricular conduction.
In a small number of patients with hypertrophic cardiomyopathy (IHSS), concomitant use of verapamil and quinidine resulted in significant hypotension. Until further data are obtained, combined therapy of verapamil and quinidine in patients with hypertrophic cardiomyopathy should probably be avoided.
The electrophysiological effects of quinidine and verapamil on AV conduction were studied in 8 patients. Verapamil significantly counteracted the effects of quinidine on AV conduction. There has been a report of increased quinidine levels during verapamil therapy.
Antihypertensive Agents
Concomitant use of trandolapril/verapamil hydrochloride ER tablets with other antihypertensive agents including diuretics, vasodilators, beta-adrenergic blockers, and alpha-antagonists may result in additive hypotensive effects. There are reports that verapamil may result in higher concentrations of the alpha-agonists prazosin and terazosin.
Dual Blockade of the Renin-Angiotensin System (RAS)
Dual blockade of the RAS with angiotensin receptor blockers, ACE inhibitors, or aliskiren is associated with increased risks of hypotension, hyperkalemia, and changes in renal function (including acute renal failure) compared to monotherapy. Most patients receiving the combination of two RAS inhibitors do not obtain any additional benefit compared to monotherapy. In general, avoid combined use of RAS inhibitors. Closely monitor blood pressure, renal function and electrolytes in patients on trandolapril/verapamil hydrochloride ER tablets and other agents that affect the RAS.
Do not co-administer aliskiren with trandolapril/verapamil hydrochloride ER tablets in patients with diabetes. Avoid use of aliskiren with trandolapril/verapamil hydrochloride ER tablets in patients with renal impairment (GFR <60 ml/min).
Verapamil Component
Concomitant therapy with beta-adrenergic blockers and verapamil may result in additive negative effects on heart rate, atrioventricular conduction, and/or cardiac contractility. Drug interaction studies have indicated that the maximum concentrations of metoprolol and propanolol are increased after the administration of verapamil. The use of verapamil in combination with a beta-adrenergic blocker should be used only with caution, and close monitoring.
Asymptomatic bradycardia (36 beats/min) with a wandering atrial pacemaker has been observed in a patient receiving concomitant timolol (a beta-adrenergic blocker) eyedrops and oral verapamil.
Trandolapril Component
As with other ACE inhibitors, patients on diuretics, especially those on recently instituted diuretic therapy, may occasionally experience an excessive reduction of blood pressure after initiation of therapy with trandolapril/verapamil hydrochloride ER tablets. The possibility of exacerbation of hypotensive effects with trandolapril/verapamil hydrochloride ER tablets may be minimized by either discontinuing the diuretic or cautiously increasing salt intake prior to initiation of treatment with trandolapril/verapamil hydrochloride ER tablets. If it is not possible to discontinue the diuretic, the starting dose of trandolapril/verapamil hydrochloride ER tablets should be reduced (see DOSAGE AND ADMINISTRATION). No clinically significant pharmacokinetic interaction has been found between trandolapril (or its metabolites) and furosemide.
Trandolapril Component
Trandolapril can attenuate potassium loss caused by thiazide diuretics and increase serum potassium when used alone. Use of potassium-sparing diuretics (spironolactone, triamterene, or amiloride), potassium supplements, or potassium-containing salt substitutes concomitantly with ACE inhibitors can increase the risk of hyperkalemia. If concomitant use of such agents is indicated, they should be used with caution and with appropriate monitoring of serum potassium (see PRECAUTIONS).
HMG-CoA Reductase Inhibitors (“Statins”)
The use of HMG-CoA reductase inhibitors that are CYP3A4 substrates in combination with verapamil has been associated with reports of myopathy/rhabdomyolysis.
Co-administration of multiple doses of 10 mg of verapamil with 80 mg simvastatin resulted in exposure to simvastatin 2.5-fold that following simvastatin alone. Limit the dose of simvastatin in patients on verapamil to 10 mg daily. Limit the daily dose of lovastatin to 40 mg. Lower starting and maintenance doses of other CYP3A4 substrates (e.g., atorvastatin) may be required as verapamil may increase the plasma concentration of these drugs.
Non-Steroidal Anti-Inflammatory Agents including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, co-administration of NSAIDs, including selective COX-2 inhibitors, with ACE inhibitors, including trandolapril, may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically in patients receiving trandolapril and NSAID therapy.
The antihypertensive effect of ACE inhibitors, including trandolapril may be attenuated by NSAIDs.
Patients taking concomitant neprilysin inhibitors (e.g., sacubitril) may be at increased risk for angioedema (see WARNINGS).
Use of a moderate CYP3A4 inhibitor such as verapamil with flibanserin significantly increases flibanserin concentrations, which can lead to severe hypotension and syncope. Concomitant use is contraindicated (see CONTRAINDICATIONS). Discontinue trandolapril/verapamil hydrochloride ER tablets at least 2 weeks prior to starting flibanserin. Do not administer trandolapril/verapamil hydrochloride ER tablets within 2 days of discontinuing flibanserin.
Other (Verapamil Component)
Verapamil has been given concomitantly with short- and long-acting nitrates without any undesirable drug interactions. The pharmacologic profile of both drugs and the clinical experience suggest beneficial interactions.
Verapamil may increase carbamazepine concentrations during combined therapy. This may produce carbamazepine side effects such as diplopia, headache, ataxia, or dizziness.
Therapy with rifampin may markedly reduce oral verapamil bioavailability. There have been reports that erythromycin and telithromycin may increase concentrations of verapamil.
Tranquilizers/ Anti-depressants
Due to metabolism via the CYP enzyme system, there have been reports that verapamil may increase the concentrations of buspirone, midazolam, almotriptan and imipramine.
Colchicine is a substrate for both CYP3A and the efflux transporter, P-gp. Verapamil is known to inhibit CYP3A and P-gp. When verapamil and colchicine are administered together, the potential inhibition of P-gp and/or CYP3A by verapamil may lead to increased exposure to colchicine (see PRECAUTIONS - Drug Interactions).
Verapamil, a P-gp inhibitor, increases exposure to dabigatran (a thrombin inhibitor) when administered concomitantly; however, no dose adjustment of dabigatran is required when administered with verapamil.
Concentrations of verapamil may be increased by the concomitant administration of protease inhibitors such as ritonavir, and reduced by the concomitant administration of sulfinpyrazone, or St John’s Wort.
Concentrations of doxorubicin may be increased by the administration of verapamil.
There have been reports that verapamil may elevate the concentrations of the oral anti-diabetic glyburide.
Animal experiments have shown that inhalation anesthetics depress cardiovascular activity by decreasing the inward movement of calcium ions. When used concomitantly, inhalation anesthetics and calcium antagonists, such as verapamil, should be titrated carefully to avoid excessive cardiovascular depression.
Gold
Nitritoid reactions (symptoms include facial flushing, nausea, vomiting and hypotension) have been reported rarely in patients on therapy with injectable gold (sodium aurothiomalate) and concomitant ACE inhibitor therapy including trandolapril/verapamil hydrochloride ER tablets.
Other (Trandolapril Component)
No clinically significant pharmacokinetic interaction has been found between trandolapril (or its metabolites) and nifedipine.
The anticoagulant effect of warfarin was not significantly changed by trandolapril.
Mammalian Target of Rapamycin (mTOR) Inhibitors
Patients taking concomitant mTOR inhibitor (e.g., temsirolimus, sirolimus, everolimus) therapy may be at increased risk for angioedema (see WARNINGS - Angioedema).
Carcinogenesis, Mutagenesis, Impairment of Fertility
Verapamil Component
An 18-month toxicity study in rats, at a low multiple (6 fold) of the maximum recommended human dose, and not the maximum tolerated dose, did not suggest a tumorigenic potential. There was no evidence of a carcinogenic potential of verapamil administered in the diet of rats for two years at doses of 10, 35, and 120 mg/kg per day or approximately 1x, 3.5x, and 12x, respectively, the maximum recommended human daily dose (480 mg per day or 9.6 mg/kg/day).
Verapamil was not mutagenic in the Ames test in 5 test strains at 3 mg per plate, with or without metabolic activation.
Studies in female rats at daily dietary doses up to 5.5 times (55 mg/kg/day) the maximum recommended human dose did not show impaired fertility. Effects on male fertility have not been determined.
Long-term studies were conducted with oral trandolapril administered by gavage to mice (78 weeks) and rats (104 and 106 weeks). No evidence of carcinogenic potential was seen in mice dosed up to 25 mg/kg/day (85 mg/m2/day) or rats dosed up to 8 mg/kg/day (60 mg/m2/day). These doses are 313 and 32 times (mice), and 100 and 23 times (rats) the maximum recommended human daily dose (MRHDD) of 4 mg based on body-weight and body-surface-area, respectively assuming a 50 kg individual. The genotoxic potential of trandolapril was evaluated in the microbial mutagenicity (Ames) test, the point mutation and chromosome aberration assays in Chinese hamster V79 cells, and the micronucleus test in mice. There was no evidence of mutagenic or clastogenic potential in these in vitro and in vivo assays.
Reproduction studies in rats did not show any impairment of fertility at doses up to 100 mg/kg/day (710 mg/m2/day) of trandolapril, or 1250 and 260 times the MRHDD on the basis of body-weight and body-surface-area, respectively.
Pregnancy
Female patients of childbearing age should be told about the consequences of exposure to trandolapril/verapamil hydrochloride ER tablets during pregnancy (see WARNINGS). Discuss treatment options with women planning to become pregnant. Patients should be asked to report pregnancies to their physicians as soon as possible.
Nursing Mothers
Verapamil is excreted in human milk. Radiolabeled trandolapril or its metabolites are secreted in rat milk. Trandolapril/verapamil hydrochloride ER tablets should not be administered to nursing mothers.
Geriatric Use
In placebo-controlled studies, where 23% of patients receiving trandolapril/verapamil hydrochloride ER tablets were 65 years and older, and 2.4% were 75 years and older, no overall differences in effectiveness or safety were observed between these patients and younger patients. However, greater sensitivity of some older individual patients cannot be ruled out.
Pediatric Use
Neonates with a history of in utero exposure to trandolapril/verapamil hydrochloride ER tablets:
If oliguria or hypotension occurs, direct attention toward support of blood pressure and renal perfusion. Exchange transfusions or dialysis may be required as a means of reversing hypotension and/or substituting for disordered renal function.
The safety and effectiveness of trandolapril/verapamil hydrochloride ER tablets in children below the age of 18 have not been established.
Animal Pharmacology and/or Animal Toxicology
In chronic animal toxicology studies, verapamil caused lenticular and/or suture line changes at 30 mg/kg/day or greater and frank cataracts at 62.5 mg/kg/day or greater in the beagle dog but not the rat. Development of cataracts due to verapamil has not been reported in man.
-
ADVERSE REACTIONS
Trandolapril/verapamil hydrochloride ER tablets have been evaluated in over 1,957 subjects and patients. Of these, 541 patients, including 23% elderly patients, participated in U.S. controlled clinical trials, and 251 were studied in foreign controlled clinical trials. In clinical trials with trandolapril/verapamil hydrochloride ER tablets, no adverse experiences peculiar to this combination drug have been observed. Adverse experiences that have occurred have been limited to those that have been previously reported with verapamil or trandolapril. Trandolapril/verapamil hydrochloride ER tablets have been evaluated for long-term safety in 272 patients treated for 1 year or more. Adverse experiences were usually mild and transient.
Discontinuation of therapy because of adverse events in U.S. placebo-controlled hypertension studies was required in 2.6% and 1.9% of patients treated with trandolapril/verapamil hydrochloride ER tablets and placebo, respectively.
Adverse experiences occurring in 1% or more of the 541 patients in placebo-controlled hypertension trials who were treated with a range of trandolapril (0.5-8 mg) and verapamil (120-240 mg) combinations are shown below.
ADVERSE EVENTS OCCURRING in ≥ 1% of TRANDOLAPRIL/VERAPAMIL HYDROCHLORIDE ER TABLETS PATIENTS IN U.S. PLACEBO-CONTROLLED TRIALS TRANDOLAPRIL/VERAPAMIL HYDROCHLORIDE ER TABLETS
(N = 541)
% Incidence
(% Discontinuance)PLACEBO
(N = 206)
% Incidence
(% Discontinuance)AV Block First Degree 3.9 (0.2) 0.5 (0.0) Bradycardia 1.8 (0.0) 0.0 (0.0) Bronchitis 1.5 (0.0) 0.5 (0.0) Chest Pain 2.2 (0.0) 1.0 (0.0) Constipation 3.3 (0.0) 1.0 (0.0) Cough 4.6 (0.0) 2.4 (0.0) Diarrhea 1.5 (0.2) 1.0 (0.0) Dizziness 3.1 (0.0) 1.9 (0.5) Dyspnea 1.3 (0.4) 0.0 (0.0) Edema 1.3 (0.0) 2.4 (0.0) Fatigue 2.8 (0.4) 2.4 (0.0) Headache(s)+ 8.9 (0.0) 9.7 (0.5) Increased Liver Enzymes* 2.8 (0.2) 1.0 (0.0) Nausea 1.5 (0.2) 0.5 (0.0) Pain Extremity(ies) 1.1 (0.2) 0.5 (0.0) Pain Back+ 2.2 (0.0) 2.4 (0.0) Pain Joint(s) 1.7 (0.0) 1.0 (0.0) Upper Respiratory Tract Infection(s)+ 5.4 (0.0) 7.8 (0.0) Upper Respiratory Tract Congestion+ 2.4 (0.0) 3.4 (0.0) * Also includes increase in SGPT, SGOT, Alkaline Phosphatase
+ Incidence of adverse events is higher in Placebo group than trandolapril/verapamil hydrochloride ER tablets patientsOther clinical adverse experiences possibly, probably, or definitely related to drug treatment occurring in 0.3% or more of patients treated with trandolapril/verapamil combinations with or without concomitant diuretic in controlled or uncontrolled trials (N = 990) and less frequent, clinically significant events (in italics) include the following:
Cardiovascular
Angina, AV block second degree, bundle branch block, edema, flushing, hypotension, myocardial infarction , palpitations, premature ventricular contractions, nonspecific ST-T changes, near syncope, tachycardia.
Angioedema
Angioedema has been reported in 3 (0.15%) patients receiving trandolapril/verapamil hydrochloride ER tablets in U.S. and foreign studies (N = 1,957). Angioedema associated with laryngeal edema may be fatal. If angioedema of the face, extremities, lips, tongue, glottis, and/or larynx occurs, treatment with trandolapril/verapamil hydrochloride ER tablets should be discontinued and appropriate therapy instituted immediately (see WARNINGS).
Hypotension
(See WARNINGS). In hypertensive patients, hypotension occurred in 0.6% and near syncope occurred in 0.1%. Hypotension or syncope was a cause for discontinuation of therapy in 0.4% of hypertensive patients.
Treatment of Acute Cardiovascular Adverse Reactions
The frequency of cardiovascular adverse reactions which require therapy is rare, hence, experience with their treatment is limited. Whenever severe hypotension or complete AV block occur following oral administration of trandolapril/verapamil hydrochloride ER tablets (verapamil component), the appropriate emergency measures should be applied immediately, e.g., intravenously administered isoproterenol HCl, levarterenol bitartrate, atropine (all in the usual doses), or calcium gluconate (10% solution). In patients with hypertrophic cardiomyopathy (IHSS), alpha-adrenergic agents (phenylephrine, metaraminol bitartrate or methoxamine) should be used to maintain blood pressure, and isoproterenol and levarterenol should be avoided. If further support is necessary, inotropic agents (dopamine or dobutamine) may be administered. Actual treatment and dosage should depend on the severity and the clinical situation and the judgment and experience of the treating physician.
Other
Other adverse experiences (in addition to those in table and listed above) that have been reported with the individual components are listed below.
Verapamil Component
(See WARNINGS). CHF/pulmonary edema, AV block 3°, atrioventricular dissociation, claudication, purpura (vasculitis), syncope.
Gingival hyperplasia. Reversible, (upon discontinuation of verapamil) nonobstructive, paralytic ileus has been infrequently reported in association with the use of verapamil.
Clinical Laboratory Test Findings
Renal Function Tests
Increases in creatinine and blood urea nitrogen levels occurred in 1.1 percent and 0.3 percent, respectively, of patients receiving trandolapril/verapamil hydrochloride ER tablets with or without hydrochlorothiazide therapy. None of these increases required discontinuation of treatment. Increases in these laboratory values are more likely to occur in patients with renal insufficiency or those pretreated with a diuretic and, based on experience with other ACE inhibitors, would be expected to be especially likely in patients with renal artery stenosis (see PRECAUTIONS and WARNINGS).
Liver Function Tests
Elevations of liver enzymes (SGOT, SGPT, LDH, and alkaline phosphatase) and/or serum bilirubin occurred. Discontinuation for elevated liver enzymes occurred in 0.9 percent of patients (see WARNINGS).
Post Marketing Experience
There has been a single postmarketing report of paralysis (tetraparesis) associated with the combined use of verapamil and colchicine. This may have been caused by colchicine crossing the blood-brain barrier due to CYP3A4 and P-gp inhibition by verapamil. Combined use of verapamil and colchicine is not recommended (see PRECAUTIONS - Drug Interactions).
-
OVERDOSAGE
No specific information is available on the treatment of overdosage with trandolapril/verapamil hydrochloride ER tablets.
Verapamil Component
Overdose with verapamil may lead to pronounced hypotension, bradycardia, and conduction system abnormalities (e.g., junctional rhythm with AV dissociation and high degree AV block, including asystole). Other symptoms secondary to hypoperfusion (e.g., metabolic acidosis, hyperglycemia, hyperkalemia, renal dysfunction, and convulsions) may be evident.
Treat all verapamil overdoses as serious and maintain observation for at least 48 hours, preferably under continuous hospital care. Delayed pharmacodynamic consequences may occur with the sustained release formulation. Verapamil is known to decrease gastrointestinal transit time. In cases of overdose, tablets of ISOPTIN SR have occasionally been reported to form concretions within the stomach or intestines. These concretions have not been visible on plain radiographs of the abdomen, and no medical means of gastrointestinal emptying is of proven efficacy in removing them. Endoscopy might reasonably be considered in cases of overdose when symptoms are unusually prolonged. Verapamil cannot be removed by hemodialysis.
Treatment of overdosage should be supportive. Beta adrenergic stimulation or parenteral administration of calcium solutions may increase calcium ion flux across the slow channel, and have been used effectively in treatment of deliberate overdosage with verapamil. The following measures may be considered:
Trandolapril Component
The oral LD50 of trandolapril in mice was 4875 mg/kg in males and 3990 mg/kg in females. In rats, an oral dose of 5000 mg/kg caused low mortality (1 male out of 5; 0 females). In dogs, an oral dose of 1000 mg/kg did not cause mortality and abnormal clinical signs were not observed.
In humans, the most likely clinical manifestation would be symptoms attributable to severe hypotension. Laboratory determinations of serum levels of trandolapril and its metabolites are not widely available, and such determinations have, in any event, no established role in the management of trandolapril overdose. No data are available to suggest that physiological maneuvers (e.g., maneuvers to change pH of the urine) might accelerate elimination of trandolapril and its metabolites. It is not known if trandolapril or trandolaprilat can be usefully removed from the body by hemodialysis.
Angiotensin II could presumably serve as a specific antagonist antidote in the setting of trandolapril overdose, but angiotensin II is essentially unavailable outside of scattered research facilities. Because the hypotensive effect of trandolapril is achieved through vasodilation and effective hypovolemia, it is reasonable to treat trandolapril overdose by infusion of normal saline solution.
-
DOSAGE AND ADMINISTRATION
The recommended usual dosage range of trandolapril for hypertension is 1 to 4 mg per day administered in a single dose or two divided doses. The recommended usual dosage range of Isoptin-SR for hypertension is 120 to 480 mg per day administered in a single dose or two divided doses.
The hazards (see WARNINGS) of trandolapril are generally independent of dose; those of verapamil are a mixture of dose-dependent phenomena (primarily dizziness, AV block, constipation) and dose-independent phenomena, the former much more common than the latter. Therapy with any combination of trandolapril and verapamil will thus be associated with both sets of dose-independent hazards. The dose-dependent side effects of verapamil have not been shown to be decreased by the addition of trandolapril nor vice versa.
Rarely, the dose-independent hazards of trandolapril are serious. To minimize dose-independent hazards, it is usually appropriate to begin therapy with trandolapril/verapamil hydrochloride ER tablets only after a patient has either (a) failed to achieve the desired antihypertensive effect with one or the other monotherapy at its respective maximally recommended dose and shortest dosing interval, or (b) the dose of one or the other monotherapy cannot be increased further because of dose-limiting side effects.
Clinical trials with trandolapril/verapamil hydrochloride ER tablets have explored only once-a-day doses. The antihypertensive effect and or adverse effects of adding 4 mg of trandolapril once-a-day to a dose of 240 mg Isoptin-SR administered twice-a-day has not been studied, nor have the effects of adding as little of 180 mg Isoptin-SR to 2 mg trandolapril administered twice-a-day been evaluated. Over the dose range of Isoptin-SR 120 to 240 mg once-a-day and trandolapril 0.5 to 8 mg once-a-day, the effects of the combination increase with increasing doses of either component.
Replacement Therapy
For convenience, patients receiving trandolapril (up to 8 mg) and verapamil (up to 240 mg) in separate tablets, administered once-a-day, may instead wish to receive tablets of trandolapril/verapamil hydrochloride ER tablets containing the same component doses.
Trandolapril/verapamil hydrochloride ER tablets should be administered with food.
-
HOW SUPPLIED
Trandolapril/verapamil hydrochloride ER 2/180 mg tablets are supplied as pink, oval, film-coated tablets containing 2 mg trandolapril in an immediate release form and 180 mg verapamil hydrochloride in a sustained release form. The tablet is debossed with a triangle and 182 on one side and plain on the other side.
NDC 59762-0142-1 - bottles of 100
Trandolapril/verapamil hydrochloride ER 2/240 mg tablets are supplied as gold, oval, film-coated tablets containing 2 mg trandolapril in an immediate release form and 240 mg verapamil hydrochloride in a sustained release form. The tablet is debossed with a triangle and 242 on one side and plain on the other side.
NDC 59762-0132-1 - bottles of 100
Trandolapril/verapamil hydrochloride ER 4/240 mg tablets are supplied as reddish-brown, oval, film-coated tablets containing 4 mg trandolapril in an immediate release form and 240 mg verapamil hydrochloride in a sustained release form. The tablet is debossed with a triangle and 244 on one side and plain on the other side.
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
TRANDOLAPRIL AND VERAPAMIL HYDROCHLORIDE ER
trandolapril and verapamil hydrochloride tablet, film coated, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59762-0142 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TRANDOLAPRIL (UNII: 1T0N3G9CRC) (TRANDOLAPRILAT - UNII:RR6866VL0O) TRANDOLAPRIL 2 mg VERAPAMIL HYDROCHLORIDE (UNII: V3888OEY5R) (VERAPAMIL - UNII:CJ0O37KU29) VERAPAMIL HYDROCHLORIDE 180 mg Inactive Ingredients Ingredient Name Strength ALCOHOL (UNII: 3K9958V90M) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) MAGNESIUM STEARATE (UNII: 70097M6I30) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM ALGINATE (UNII: C269C4G2ZQ) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) DOCUSATE SODIUM (UNII: F05Q2T2JA0) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) STARCH, CORN (UNII: O8232NY3SJ) FERRIC OXIDE RED (UNII: 1K09F3G675) WATER (UNII: 059QF0KO0R) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) Product Characteristics Color PINK Score no score Shape OVAL Size 18mm Flavor Imprint Code 182 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59762-0142-1 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/25/2015 08/14/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020591 02/25/2015 08/14/2021 TRANDOLAPRIL AND VERAPAMIL HYDROCHLORIDE ER
trandolapril and verapamil hydrochloride tablet, film coated, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59762-0152 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TRANDOLAPRIL (UNII: 1T0N3G9CRC) (TRANDOLAPRILAT - UNII:RR6866VL0O) TRANDOLAPRIL 1 mg VERAPAMIL HYDROCHLORIDE (UNII: V3888OEY5R) (VERAPAMIL - UNII:CJ0O37KU29) VERAPAMIL HYDROCHLORIDE 240 mg Inactive Ingredients Ingredient Name Strength ALCOHOL (UNII: 3K9958V90M) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) WATER (UNII: 059QF0KO0R) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM ALGINATE (UNII: C269C4G2ZQ) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) DOCUSATE SODIUM (UNII: F05Q2T2JA0) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) STARCH, CORN (UNII: O8232NY3SJ) Product Characteristics Color WHITE Score no score Shape OVAL Size 19mm Flavor Imprint Code 241 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59762-0152-1 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/25/2015 02/28/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020591 02/25/2015 02/28/2018 TRANDOLAPRIL AND VERAPAMIL HYDROCHLORIDE ER
trandolapril and verapamil hydrochloride tablet, film coated, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59762-0132 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TRANDOLAPRIL (UNII: 1T0N3G9CRC) (TRANDOLAPRILAT - UNII:RR6866VL0O) TRANDOLAPRIL 2 mg VERAPAMIL HYDROCHLORIDE (UNII: V3888OEY5R) (VERAPAMIL - UNII:CJ0O37KU29) VERAPAMIL HYDROCHLORIDE 240 mg Inactive Ingredients Ingredient Name Strength ALCOHOL (UNII: 3K9958V90M) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) WATER (UNII: 059QF0KO0R) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM ALGINATE (UNII: C269C4G2ZQ) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) DOCUSATE SODIUM (UNII: F05Q2T2JA0) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) STARCH, CORN (UNII: O8232NY3SJ) Product Characteristics Color YELLOW (gold) Score no score Shape OVAL Size 19mm Flavor Imprint Code 242 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59762-0132-1 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/25/2015 05/28/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020591 02/25/2015 05/28/2021 TRANDOLAPRIL AND VERAPAMIL HYDROCHLORIDE ER
trandolapril and verapamil hydrochloride tablet, film coated, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59762-0122 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TRANDOLAPRIL (UNII: 1T0N3G9CRC) (TRANDOLAPRILAT - UNII:RR6866VL0O) TRANDOLAPRIL 4 mg VERAPAMIL HYDROCHLORIDE (UNII: V3888OEY5R) (VERAPAMIL - UNII:CJ0O37KU29) VERAPAMIL HYDROCHLORIDE 240 mg Inactive Ingredients Ingredient Name Strength ALCOHOL (UNII: 3K9958V90M) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) STARCH, CORN (UNII: O8232NY3SJ) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) MAGNESIUM STEARATE (UNII: 70097M6I30) WATER (UNII: 059QF0KO0R) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM ALGINATE (UNII: C269C4G2ZQ) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE RED (UNII: 1K09F3G675) DOCUSATE SODIUM (UNII: F05Q2T2JA0) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) Product Characteristics Color RED (reddish-brown) Score no score Shape OVAL Size 19mm Flavor Imprint Code 244 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59762-0122-1 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/25/2015 08/18/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020591 02/25/2015 08/18/2021 Labeler - Greenstone LLC (825560733)



© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.