SEMPREX D- acrivastine and pseudoephedrine hydrochloride capsule
SEMPREX by
Drug Labeling and Warnings
SEMPREX by is a Prescription medication manufactured, distributed, or labeled by Endo Pharmaceuticals, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
SEMPREX-D Capsules (acrivastine and pseudoephedrine hydrochloride) are a fixed combination product formulated for oral administration. Acrivastine is an antihistamine and pseudoephedrine is a decongestant. Each capsule contains 8 mg acrivastine and 60 mg pseudoephedrine hydrochloride and the inactive ingredients: lactose, magnesium stearate and sodium starch glycolate. The green and white capsule shell consists of gelatin, D&C Yellow No. 10, FD&C Green No. 3, and titanium dioxide. The capsules may contain one or more parabens and are printed with edible black and white inks.
The chemical name of acrivastine is (E,E)-3-[6-[1-(4-methylphenyl)-3-(1-pyrrolidinyl)-1-propenyl]-2-pyridinyl]-2-propenoic acid; the molecular formula is C22H24N2O2. As an analog of triprolidine hydrochloride, acrivastine is classified as an alkylamine antihistamine. Acrivastine is an odorless, white to pale cream crystalline powder that is soluble in chloroform and alcohol and slightly soluble in water.
The chemical name of pseudoephedrine hydrochloride is [S-(R*,R*)]-α-[1-(methylamino)ethyl]benzenemethanol hydrochloride; the molecular formula is C10H15NOHCl. Pseudoephedrine is one of the naturally occurring dextrorotatory diastereoisomers of ephedrine and is classified as an indirect sympathomimetic amine. Pseudoephedrine hydrochloride occurs as odorless, fine white to off-white crystals or powder; the drug is soluble in water, alcohol and chloroform.
Structural formulae for the active ingredients of SEMPREX-D Capsules are as follows:
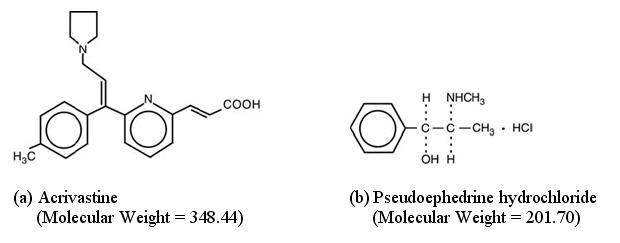
-
CLINICAL PHARMACOLOGY
Acrivastine, a structural analog of triprolidine hydrochloride, exhibits H1-antihistaminic activity in isolated tissues, animals, and humans, and has sedative effects in humans (see PRECAUTIONS). The propionic acid derivative of acrivastine is a metabolite in several animal species (as well as in man) and also exhibits H1-antihistaminic activity.
Pseudoephedrine hydrochloride is an indirect sympathomimetic agent; that is, it releases norepinephrine from adrenergic nerves.
In vitro test and in vivo studies in animals of acrivastine and pseudoephedrine in combination failed to demonstrate evidence of any beneficial or deleterious pharmacologic interaction between the two agents.
Pharmacokinetics And Metabolism
Acrivastine was absorbed rapidly from the combination capsule following oral administration and was as bioavailable as a solution of acrivastine. After administration of SEMPREX-D Capsules, maximum plasma acrivastine concentrations were achieved at 1.14 ± 0.23 hours. A mass balance study in 7 healthy volunteers showed that acrivastine is primarily eliminated by the kidneys. Over a 72-hour collection period, about 84% of the administered total radioactivity was recovered in urine and about 13% in feces, for a combined recovery of about 97%. Further, 67% of the administered radioactive dose was recovered in urine as the unchanged drug, 11% as the propionic acid metabolite, and 6% as other unknown metabolites.
Acrivastine exhibits linear kinetics over dosages ranging from 2 to 32 mg t.i.d. The mean ± SD terminal half-life for acrivastine was 1.9 ± 0.3 hours following single oral doses and increased to 3.5 ± 1.9 hours at steady state. The terminal half-life for the propionic acid metabolite was 3.8 ± 1.4 hours. Because of the short half-lives of both acrivastine and its metabolites, accumulation in the plasma following multiple dosing is not expected.
The steady-state maximum acrivastine plasma concentration was 227 ± 47 ng/mL. The oral clearance, and apparent volume of distribution were 2.9 ± 0.7 mL/min/kg and 0.46 ± 0.05 L/kg, respectively, following a single oral dose; oral clearance did not change at steady state (2.86 ± 0.75 mL/min/kg). The apparent volume of distribution increased to 0.82 ± 0.6 L/kg to parallel the increase in the elimination half-life of the drug.
Acrivastine binding to human plasma proteins was 50% ± 2.0% and was concentration-independent over the range of 5 to 1000 ng/mL. The main binding protein was serum albumin although the drug was slightly bound to α-1-acid glycoprotein. No displacement interaction was observed between acrivastine and either phenytoin or theophylline. The binding of acrivastine was not affected by the presence of pseudoephedrine.
Pseudoephedrine hydrochloride was also rapidly absorbed from the combination capsule, and the capsule was as bioavailable as a solution of pseudoephedrine. Steady state maximum plasma concentration for pseudoephedrine was 498 ± 129 ng/mL. The terminal half-life, oral clearance and apparent volume of distribution were 6.2 ± 1.8 hours, 5.9 ± 1.7 mL/min/kg, and 3.0 ± 0.4 L/kg, respectively. Elimination of pseudoephedrine is primarily through the renal route as 55% to 75% of an administered dose appears unchanged in the urine. Pseudoephedrine elimination, however, is highly dependent upon urine pH; the plasma half-life decreased to about 4 hours at pH 5 and increased to 13 hours at pH 8.
Pseudoephedrine did not bind to human plasma proteins over the concentration range of 50 to 2000 ng/mL.
Acrivastine and pseudoephedrine do not influence the pharmacokinetics of the other drug when administered concomitantly.
Special Populations
A single dose pharmacokinetic study showed that the elimination half-lives of acrivastine, the propionic acid metabolite of acrivastine, and pseudoephedrine were prolonged in patients with chronic renal insufficiency. Compared to normal volunteers, the elimination half-life of acrivastine was about 50% increased in patients with mild renal insufficiency (creatinine clearance = 26 to 48 mL/min) and was increased by about 130% in patients with moderate (creatinine clearance = 12 to 17 mL/min) or severe (creatinine clearance 6 to 10 mL/min) renal insufficiency. Oral clearance of acrivastine was diminished by the same magnitude as the half-life was prolonged in each of the three renally impaired groups. The elimination half-life of the propionic acid metabolite of acrivastine was about 140% increased in patients with mild renal insufficiency and about 5 times increased in patients with moderate or severe renal insufficiency.
Compared to normal volunteers, the elimination half-life of pseudoephedrine was about 3 times increased in patients with mild renal insufficiency, about 7 times increased in patients with moderate renal insufficiency, and about 10 times increased in patients with severe renal insufficiency. Oral clearance of pseudoephedrine was diminished by about the same magnitude as the half-life was prolonged in each of the three renally impaired groups (see PRECAUTIONS, Use in Patients with Diminished Renal Function).
The total body load removed by dialysis is approximately 20%, 27% and 38% for acrivastine, the propionic acid metabolite of acrivastine, and pseudoephedrine, respectively, and therefore, a supplemental dose after a dialysis session is not required.
Based on a multiple dose cross study comparison, the apparent volume of distribution for acrivastine was 44% lower in elderly (n = 36, 65-75 yr) than in young volunteers (n = 16, 19-33 yr). This difference could be attributed to the decrease in total body water that occurs with aging. Despite this difference, no appreciable differences in plasma acrivastine concentrations were seen in the elderly compared to the young, and no appreciable accumulation of acrivastine occurred in plasma at steady-state. The elimination half-life for pseudoephedrine was 18% longer in elderly (7.9 hours) than in younger subjects (6.7 hours), presumably due to the decline in average renal function that occurs with aging. Despite this difference, clearance of pseudoephedrine was not appreciably different in elderly and younger subjects. Elderly patients can therefore be given the same dosage as younger patients. SEMPREX-D Capsules are not recommended, however, in patients with renal impairment (see PRECAUTIONS, Use in Patients with Diminished Renal Functionand Geriatric Use).
The effect of age and sex on the pharmacokinetic parameters of acrivastine and pseudoephedrine was determined in 93 healthy volunteers who participated in various studies. All of the 93 volunteers were Caucasian (81 males and 12 females); 57 were between the ages of 18 and 38 years and 36 were between the ages of 65 and 75 years. There were no age- or sex-related differences in the pharmacokinetic parameters of either acrivastine or pseudoephedrine.
The effect of race on acrivastine and pseudoephedrine pharmacokinetics was examined by screening data obtained from 1035 patients, age 12 to 71 years, who participated in the eight safety and efficacy studies. No race-related differences were observed in the pharmacokinetics of either acrivastine or pseudoephedrine.
-
CLINICAL STUDIES
In healthy volunteers, histamine-induced wheal and flare areas were significantly reduced relative to placebo at 30 minutes after administration of a single dose of acrivastine 8 mg. Maximum reductions of wheal and flare occurred by 1 to 2 hours and significant reductions relative to placebo persisted for up to 6 hours after a single oral dose of acrivastine 8 mg. No additional reductions of wheal and flare were observed following single doses of acrivastine up to 24 mg. The exact correlation between responses on skin testing and clinical efficacy is not established.
Five randomized, placebo- and/or active-controlled trials compared SEMPREX-D with its acrivastine and pseudoephedrine components for the symptomatic relief of seasonal allergic rhinitis. In these studies, 696 patients received four daily doses of acrivastine 8 mg plus pseudoephedrine hydrochloride 60 mg (i.e., SEMPREX-D Capsules or bioequivalent formulations administered concurrently) or the same doses of the components for 14 days. The combination reduced the intensity of sneezing, rhinorrhea, pruritus, and lacrimation more than pseudoephedrine and reduced the intensity of nasal congestion more than acrivastine, demonstrating a contribution of each of the components. The onset of antihistaminic and nasal decongestant actions occurred within one or two hours after the first dose of SEMPREX-D Capsules. Somnolence occurred in about 12% of patients given SEMPREX-D compared with about 6% on placebo.
-
INDICATIONS AND USAGE
SEMPREX-D Capsules are indicated for relief of symptoms associated with seasonal allergic rhinitis such as sneezing, rhinorrhea, pruritus, lacrimation, and nasal congestion. SEMPREX-D Capsules should be administered when both the antihistaminic activity of acrivastine and the nasal decongestant activity of pseudoephedrine are desired (see CLINICAL PHARMACOLOGY). The efficacy of SEMPREX-D Capsules beyond 14 days of continuous treatment in patients with seasonal allergic rhinitis has not been adequately investigated in clinical trials.
SEMPREX-D Capsules have not been adequately studied for effectiveness in relieving the symptoms of the common cold.
-
CONTRAINDICATIONS
SEMPREX-D Capsules are contraindicated in patients with a known sensitivity to acrivastine, other alkylamine antihistamines (e.g., triprolidine), pseudoephedrine, other sympathomimetic amines (e.g., phenylpropanolamine), or to any other components of the formulation. SEMPREX-D Capsules are contraindicated in patients with severe hypertension or severe coronary artery disease. SEMPREX-D Capsules are contraindicated in patients taking monoamine oxidase (MAO) inhibitors and for 14 days after stopping use of an MAO inhibitor (see PRECAUTIONS, Drug Interactions).
-
WARNINGS
SEMPREX-D Capsules should be used with caution in patients with hypertension, diabetes mellitus, ischemic heart disease, increased intraocular pressure, hyperthyroidism, prostatic hypertrophy, stenosing peptic ulcer, or pyloroduodenal obstruction. Overdose of sympathomimetic amines may produce CNS stimulation with convulsions or cardiovascular collapse with accompanying hypotension. The elderly are more likely to have adverse reactions to sympathomimetic amines.
-
PRECAUTIONS
General
Acrivastine is sedating in some patients. In controlled clinical trials, somnolence (i.e., drowsiness, sedation, sleepiness) was more common with SEMPREX-D Capsules (by an average of 6%) than with placebo (see ADVERSE EXPERIENCES).
Patients should be advised to assess their individual responses to SEMPREX-D Capsules before engaging in any activity requiring mental alertness, such as driving a motor vehicle or operating machinery. Concurrent use of SEMPREX-D Capsules with alcohol or other CNS depressants may cause additional reductions in alertness and impairment of CNS performance and should be avoided (see PRECAUTIONS, Drug Interactions).
Use in Patients with Diminished Renal Function
Acrivastine and pseudoephedrine are excreted primarily through the kidney. Both compounds therefore accumulate in patients with impaired renal function. Due to the differential effects of renal failure on the serum half-life and clearance of acrivastine and pseudoephedrine, use of SEMPREX-D Capsules, a fixed combination product, in patients with renal impairment (creatinine clearance ≤ 48 mL/min) is not recommended (see OVERDOSAGE and CLINICAL PHARMACOLOGY).
Information to Patients
Patients taking SEMPREX-D Capsules should receive the following information. SEMPREX-D Capsules are prescribed to reduce symptoms associated with seasonal allergic rhinitis. Patients should be instructed to take SEMPREX-D Capsules only as prescribed and not to exceed the prescribed dose. Patients should be advised against the concurrent use of SEMPREX-D with over-the-counter antihistamines and decongestants. Patients who are or may become pregnant should be told that this product should be used in pregnancy or during lactation only if the potential benefit justifies the potential risks to the fetus or nursing infant. Due to the risk of hypertensive crisis, patients should be instructed not to take SEMPREX-D Capsules (acrivastine and pseudoephedrine hydrochloride) if they are presently taking a monoamine oxidase inhibitor or for 14 days after stopping use of an MAO inhibitor. Patients should be advised to assess their individual responses to SEMPREX-D Capsules before engaging in any activity requiring mental alertness, such as driving a car or operating machinery. Patients should be advised that the concurrent use of SEMPREX-D Capsules with alcohol and other CNS depressants may lead to additional reductions in alertness and impairment of CNS performance and should be avoided.
Drug Interactions
MAO inhibitors and beta-adrenergic agonists increase the effects of sympathomimetic amines. Concomitant use of sympathomimetic amines with MAO inhibitors can result in a hypertensive crisis (see CONTRAINDICATIONS). Because MAO inhibitors are long-acting, SEMPREX-D Capsules should not be taken with an MAO inhibitor or for 14 days after stopping use of an MAO inhibitor.
Because of their pseudoephedrine content, SEMPREX-D Capsules may reduce the antihypertensive effects of drugs that interfere with sympathetic activity. Care should be taken in the administration of SEMPREX-D Capsules concomitantly with other sympathomimetic amines because the combined effects on the cardiovascular system may be harmful to the patient.
Concomitant administration of SEMPREX-D Capsules with alcohol and other CNS depressants may result in additional reductions in alertness and impairment of CNS performance and should be avoided.
No formal drug interaction studies between SEMPREX-D Capsules and other possibly co-administered drugs have been performed.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with the combination of acrivastine and pseudoephedrine have not been performed. Oral doses of acrivastine alone at levels up to 40 mg/kg/day (236 mg/m2/day or 10 times the recommended human daily dose) for 20 to 22 months in rats and up to 250 mg/kg/day (750 mg/m2/day or 32 times the recommended human daily dose) for 20 to 24 months in mice revealed no evidence of carcinogenic potential. No evidence of mutagenicity (with or without metabolic activation) was observed in the Ames Salmonella mutagenicity assay or in the L5178Y/tk+/- mouse lymphoma assay. In an in vitro cytogenetic study performed in cultured human lymphocytes, acrivastine induced structural chromosomal abnormalities in the absence of metabolic activation, but not in its presence. In an in vivo cytogenetic study in rats given single oral doses of acrivastine up to 1000 mg/kg (5900 mg/m2 or 249 times the recommended human daily dose) there were no structural chromosomal alterations.
Reproduction-fertility studies in rats given acrivastine alone at levels up to 200 mg/kg/day (1180 mg/m2/day or 50 times the recommended human daily dose) had no effect on male or female fertility. Similarly, no effect on fertility was seen in male rats given acrivastine 20 mg/kg/day and pseudoephedrine 100 mg/kg/ day (118 and 590 mg/m2/day or 5 and 3 times the recommended human daily doses, respectively) or in female rats given acrivastine 4 mg/kg/day and pseudoephedrine 20 mg/kg/day (23.6 and 118 mg/m2/day or 1 and 0.7 times the recommended human daily doses, respectively).
Pregnancy
Teratogenic Effects
No evidence of teratogenicity was seen in rats and rabbits given acrivastine 1000 and 400 mg/kg/day, respectively (5900 and 4720 mg/m2/day or 249 and 200 times the recommended human daily dose). No evidence of teratogenicity was seen in rats given a combination of acrivastine 30 mg/kg/day and pseudoephedrine 150 mg/kg/day (177 and 885 mg/m2/day or 8 and 5 times the recommended human daily dose, respectively). Similarly, no evidence of teratogenicity was observed in rabbits given acrivastine 20 mg/kg/day and pseudoephedrine 100 mg/kg/day (236 and 1180 mg/m2/day or 10 and 7 times the recommended human daily doses, respectively). There are, however, no adequate and well-controlled studies in pregnant women. Because animal teratology studies are not always predictive of human responses, SEMPREX-D Capsules should be used during pregnancy only if the potential benefit justifies the potential risks to the fetus.
Nonteratogenic Effects
In a perinatal-postnatal study in rats, acrivastine given alone at levels up to 500 mg/kg/day (2950 mg/m2/day or 124 times the recommended human daily dose) was associated with maternal and neonatal mortality at the maximum dose level. Neonatal survival was decreased in rats given a combination of acrivastine 20 mg/kg/day and pseudoephedrine 100 mg/ kg/day (118 and 590 mg/m2/day or 5 and 3 times the human dose, respectively).
Nursing Mothers
It is not known whether acrivastine is excreted in human milk; pseudoephedrine is excreted in human milk. SEMPREX-D Capsules should only be used in nursing mothers when the potential benefit justifies the potential risks to the nursing infant.
Pediatric Use
Safety and effectiveness of SEMPREX-D Capsules in pediatric patients under the age of 12 years have not been established.
Geriatric Use
Of the total number of subjects in clinical studies of SEMPREX-D, 349 were 60 years of age or older and 53 were 70 years of age and older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. Antihistamines, however, as a pharmaceutical class, are more likely to cause dizziness, sedation, bladder-neck obstruction, and hypotension in elderly patients. The elderly are also more likely to have adverse reactions to sympathomimetics such as pseudoephedrine (see CLINICAL PHARMACOLOGY and WARNINGS).
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. Use of SEMPREX-D in patients with renal impairment (creatinine clearance ≤ 48 mL/min) is not recommended (see PRECAUTIONS, Use in Patients with Diminished Renal Function).
-
ADVERSE EXPERIENCES
Information on the incidence of adverse events in clinical investigations conducted in the United States was obtained from 33 controlled and 15 uncontrolled clinical studies in which 2499 patients received acrivastine and 2631 patients received acrivastine plus pseudoephedrine hydrochloride for treatment periods ranging from one day to one year. The majority of patients in clinical trials were exposed to acrivastine or acrivastine plus pseudoephedrine for less than 90 days. Acrivastine dosage ranged from 3 to 96 mg/day; 1336 patients received dosages equal to or greater than acrivastine 24 mg/day. Acrivastine plus pseudoephedrine hydrochloride dosages ranged from acrivastine 8 to 48 mg/day plus pseudoephedrine hydrochloride 60 to 240 mg/day. A total of 2335 patients received three or four daily doses of acrivastine 8 mg plus pseudoephedrine hydrochloride 60 mg.
In controlled clinical trials, only 12 spontaneously elicited adverse events were reported with frequencies greater than 1% in the acrivastine plus pseudoephedrine hydrochloride treatment group (see Table 1).
TABLE 1 ADVERSE EVENTS REPORTED IN CLINICAL TRIALS* (PERCENT OF PATIENTS REPORTING)† - * Includes all events regardless of casual relationship to treatment.
- † Includes all adverse events with a reported frequency of > 1% for the acrivastine plus pseudoephedrine treatment group.
- ‡ SEMPREX-D demonstrates a statistically higher frequency of events than placebo, p ≤ 0.05.
Controlled Studies Placebo
(N = 1767)
Acrivastine
(N = 1935)Pseudoephedrine
(N = 887)Acrivastine plus Pseudoephedrine
(N = 1650)CNS Somnolence‡ 6 12 8 12 Headache 18 19 19 19 Dizziness 2 3 3 3 Nervousness‡ 1 2 4 3 Insomnia‡ 1 1 6 4 MISCELLANEOUS Nausea 2 3 3 2 Dry Mouth‡ 2 3 5 7 Asthenia 2 3 2 2 Dyspepsia 1 1 2 2 Pharyngitis 2 1 1 3 Cough Increase 1 2 1 2 Dysmenorrhea 1 2 3 2 The nature and overall frequencies of adverse events from international clinical trials (35 studies involving approximately 1600 patients) were similar to the results obtained in the US studies.
Post-marketing clinical experience reports with acrivastine and acrivastine plus pseudoephedrine have included rare serious hypersensitivity reactions manifested by anaphylaxis, angioedema, bronchospasm, and erythema multiforme. No deaths associated with use of acrivastine or acrivastine plus pseudoephedrine have been reported.
Pseudoephedrine may cause ephedrine-like reactions such as tachycardia, palpitations, headache, dizziness, or nausea (see WARNINGS and OVERDOSAGE).
Serious skin reactions, including acute generalized exanthematous pustulosis (AGEP), have been reported with pseudoephedrine-containing products.
-
OVERDOSAGE
There have been no reports of overdosage with Semprex-D Capsules. In the clinical trial program and in international post-marketing experience, there have been two reported overdoses with acrivastine. Doses were 72 mg and 322 mg. Both patients recovered without sequelae. Adverse events included trembling, stridor, loss of consciousness and possible convulsions in the first patient and somnolence in the second.
Since acrivastine and pseudoephedrine have pharmacologically different actions, it is difficult to predict how an individual will respond to overdosage with SEMPREX-D Capsules. However, acute overdosage with SEMPREX-D Capsules may produce clinical signs of either CNS stimulation or depression. Overdosage of sympathomimetics has been associated with the following events: fear, anxiety, tenseness, restlessness, tremor, weakness, pallor, respiratory difficulty, dysuria, insomnia, hallucinations, convulsions, CNS depression, arrhythmias, and cardiovascular collapse with hypotension. Treatment for overdosage with SEMPREX-D Capsules should follow general symptomatic and supportive principles.
In a placebo-controlled, double-blind clinical trial in 18 healthy male subjects, single doses of acrivastine up to 400 mg (50 times the recommended antihistaminic dose) produced only a weak vagolytic effect, manifested as an increase in heart rate, and did not cause cardiac repolarization delays (i.e., increased QTc). Daily doses of acrivastine up to 2400 mg (75 times the recommended antihistamine dose) in an uncontrolled study in 38 cancer patients produced a 15-beats-per-minute increase in mean heart rate and occasional episodes of nausea and vomiting. The effects of acrivastine plus pseudoephedrine at single or multiple doses higher than the recommended daily dose of SEMPREX-D Capsules (i.e., 32 mg acrivastine plus 240 mg pseudoephedrine) on heart rate and cardiac repolarization have not been investigated in clinical trials.
The mean LD50 (single, oral dose) of acrivastine is greater than 4000 mg/kg (23600 mg/m2 or 1000 times the recommended human daily dose) in rats and greater than 1200 mg/kg (3600 mg/m2 or 153 times the recommended human daily dose) in mice. The mean LD50 (single, oral dose) of pseudoephedrine hydrochloride is 2206 mg/kg (13015 mg/m2 or 73 times the recommended human daily dose) in rats and 726 mg/kg (2178 mg/m2 or 12 times the recommended human daily dose) in mice. The toxic and lethal concentrations of acrivastine and pseudoephedrine in human biologic fluids are not known. Based upon pharmacokinetic screening data from clinical trials, the maximum plasma acrivastine concentration after dosing with acrivastine 8 mg was 393 ng/mL and the maximum plasma pseudoephedrine concentration after dosing with pseudoephedrine hydrochloride 60 mg was 1308 ng/mL.
- DOSAGE AND ADMINISTRATION
-
HOW SUPPLIED
SEMPREX-D Capsules (dark green opaque cap and white opaque body) contain acrivastine 8 mg and pseudoephedrine hydrochloride 60 mg. The cap is printed with “404” in white ink, and the body is printed with “SEMPREX-D” in black ink.
NDC: 52244-404-10 Bottle of 100
-
SPL UNCLASSIFIED SECTION
To report suspected adverse reactions, contact Endo Pharmaceuticals Inc. at 1-800-462-3636.
Distributed by:
Endo Pharmaceuticals Inc.
Malvern, PA 19355SEMPREX is a registered trademark of Endo International plc or one of its affiliates.
©2019 Endo Pharmaceuticals Inc. All rights reserved.
Revised: 01/2019
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
SEMPREX D
acrivastine and pseudoephedrine hydrochloride capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 52244-404 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ACRIVASTINE (UNII: A20F9XAI7W) (ACRIVASTINE - UNII:A20F9XAI7W) ACRIVASTINE 8 mg PSEUDOEPHEDRINE HYDROCHLORIDE (UNII: 6V9V2RYJ8N) (PSEUDOEPHEDRINE - UNII:7CUC9DDI9F) PSEUDOEPHEDRINE HYDROCHLORIDE 60 mg Inactive Ingredients Ingredient Name Strength LACTOSE, UNSPECIFIED FORM (UNII: J2B2A4N98G) MAGNESIUM STEARATE (UNII: 70097M6I30) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) GELATIN (UNII: 2G86QN327L) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C GREEN NO. 3 (UNII: 3P3ONR6O1S) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color GREEN (dark green opaque cap and white opaque body) Score no score Shape CAPSULE Size 16mm Flavor Imprint Code 404;SEMPREX;D Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 52244-404-10 100 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 01/26/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA019806 01/26/2012 Labeler - Endo Pharmaceuticals, Inc. (178074951)

Trademark Results [SEMPREX]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 SEMPREX 78429596 3023249 Live/Registered |
ACTIENT PHARMACEUTICALS LLC 2004-06-03 |
 SEMPREX 74338456 1879087 Dead/Cancelled |
Burroughs Wellcome Co. 1992-12-09 |
 SEMPREX 73514851 1421747 Live/Registered |
SEMPREX CORPORATION 1984-12-24 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.