FASENRA- benralizumab injection, solution
FASENRA by
Drug Labeling and Warnings
FASENRA by is a Prescription medication manufactured, distributed, or labeled by AstraZeneca Pharmaceuticals LP, AstraZeneca PLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FASENRA® safely and effectively. See full prescribing information for FASENRA.
FASENRA (benralizumab) injection, for subcutaneous use
Initial U.S. Approval: 2017RECENT MAJOR CHANGES
Dosage and Administration (2) 10/2019
INDICATIONS AND USAGE
FASENRA is an interleukin-5 receptor alpha-directed cytolytic monoclonal antibody (IgG1, kappa) indicated for the add-on maintenance treatment of patients with severe asthma aged 12 years and older, and with an eosinophilic phenotype. (1)
Limitations of Use:
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Known hypersensitivity to benralizumab or excipients. (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity reactions: Hypersensitivity reactions (e.g., anaphylaxis, angioedema, urticaria, rash) have occurred after administration of FASENRA. Discontinue in the event of a hypersensitivity reaction. (5.1)
- Reduction in Corticosteroid Dosage: Do not discontinue systemic or inhaled corticosteroids abruptly upon initiation of therapy with FASENRA. Decrease corticosteroids gradually, if appropriate. (5.3)
- Parasitic (Helminth) Infection: Treat patients with pre-existing helminth infections before therapy with FASENRA. If patients become infected while receiving FASENRA and do not respond to anti-helminth treatment, discontinue FASENRA until the parasitic infection resolves. (5.4)
ADVERSE REACTIONS
Most common adverse reactions (incidence greater than or equal to 5%) include headache and pharyngitis. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
2.2 General Administration Instructions
2.3 Instructions for Administration of FASENRA Prefilled Syringe (Healthcare Providers)
2.4 Instructions for Administration of FASENRA PEN
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 Acute Asthma Symptoms or Deteriorating Disease
5.3 Reduction of Corticosteroid Dosage
5.4 Parasitic (Helminth) Infection
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
FASENRA is indicated for the add-on maintenance treatment of patients with severe asthma aged 12 years and older, and with an eosinophilic phenotype [see Clinical Studies (14)].
Limitations of use:
- FASENRA is not indicated for treatment of other eosinophilic conditions.
- FASENRA is not indicated for the relief of acute bronchospasm or status asthmaticus.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
FASENRA is for subcutaneous use only.
The recommended dose of FASENRA is 30 mg administered once every 4 weeks for the first 3 doses, and then once every 8 weeks thereafter by subcutaneous injection into the upper arm, thigh, or abdomen.
2.2 General Administration Instructions
FASENRA is intended for use under the guidance of a healthcare provider. In line with clinical practice, monitoring of patients after administration of biologic agents is recommended [see Warnings and Precautions (5.1)].
Administer FASENRA into the thigh or abdomen. The upper arm can also be used if a healthcare provider or caregiver administers the injection. Prior to administration, warm FASENRA by leaving carton at room temperature for about 30 minutes. Visually inspect FASENRA for particulate matter and discoloration prior to administration. FASENRA is clear to opalescent, colorless to slightly yellow, and may contain a few translucent or white to off-white particles. Do not use FASENRA if the liquid is cloudy, discolored, or if it contains large particles or foreign particulate matter.
Prefilled Syringe
The prefilled syringe is for administration by a healthcare provider.
Autoinjector (FASENRA PEN™)
FASENRA PEN is intended for administration by patients/caregivers. Patients/caregivers may inject after proper training in subcutaneous injection technique, and after the healthcare provider determines it is appropriate.
2.3 Instructions for Administration of FASENRA Prefilled Syringe (Healthcare Providers)
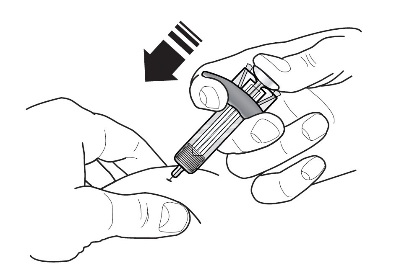
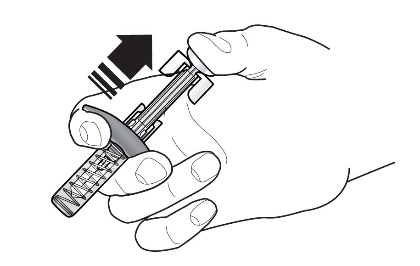
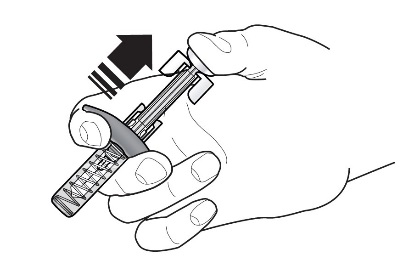
Refer to Figure 1 to identify the prefilled syringe components for use in the administration steps.
Figure 1
Do not touch the needle guard activation clips to prevent premature activation of the needle safety guard.
2.4 Instructions for Administration of FASENRA PEN
Refer to the FASENRA PEN ‘Instructions for Use’ for more detailed instructions on the preparation and administration of FASENRA PEN [See Instructions for Use]. A patient may self-inject or the patient caregiver may administer FASENRA PEN subcutaneously after the healthcare provider determines it is appropriate.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
FASENRA is contraindicated in patients who have known hypersensitivity to benralizumab or any of its excipients [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions (e.g., anaphylaxis, angioedema, urticaria, rash) have occurred following administration of FASENRA. These reactions generally occur within hours of administration, but in some instances have a delayed onset (i.e., days). In the event of a hypersensitivity reaction, FASENRA should be discontinued [see Contraindications (4)].
5.2 Acute Asthma Symptoms or Deteriorating Disease
FASENRA should not be used to treat acute asthma symptoms or acute exacerbations. Do not use FASENRA to treat acute bronchospasm or status asthmaticus. Patients should seek medical advice if their asthma remains uncontrolled or worsens after initiation of treatment with FASENRA.
5.3 Reduction of Corticosteroid Dosage
Do not discontinue systemic or inhaled corticosteroids abruptly upon initiation of therapy with FASENRA. Reductions in corticosteroid dose, if appropriate, should be gradual and performed under the direct supervision of a physician. Reduction in corticosteroid dose may be associated with systemic withdrawal symptoms and/or unmask conditions previously suppressed by systemic corticosteroid therapy.
5.4 Parasitic (Helminth) Infection
Eosinophils may be involved in the immunological response to some helminth infections. Patients with known helminth infections were excluded from participation in clinical trials. It is unknown if FASENRA will influence a patient’s response against helminth infections.
Treat patients with pre-existing helminth infections before initiating therapy with FASENRA. If patients become infected while receiving treatment with FASENRA and do not respond to anti-helminth treatment, discontinue treatment with FASENRA until infection resolves.
-
6 ADVERSE REACTIONS
The following adverse reactions are described in greater detail in other sections:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Across Trials 1, 2, and 3, 1,808 patients received at least 1 dose of FASENRA [see Clinical Studies (14)]. The data described below reflect exposure to FASENRA in 1,663 patients, including 1,556 exposed for at least 24 weeks and 1,387 exposed for at least 48 weeks. The safety exposure for FASENRA is derived from two Phase 3 placebo-controlled studies (Trials 1 and 2) from 48 weeks duration [FASENRA every 4 weeks (n=841), FASENRA every 4 weeks for 3 doses, then every 8 weeks (n=822), and placebo (n=847)]. While a dosing regimen of FASENRA every 4 weeks was included in clinical trials, FASENRA administered every 4 weeks for 3 doses, then every 8 weeks thereafter is the recommended dose [see Dosage and Administration (2.1)]. The population studied was 12 to 75 years of age, of which 64% were female and 79% were white.
Adverse reactions that occurred at greater than or equal to 3% incidence are shown in Table 1.
Table 1. Adverse Reactions with FASENRA with Greater than or Equal to 3% Incidence in Patients with Asthma (Trials 1 and 2) - * Pharyngitis was defined by the following terms: ‘Pharyngitis’, ‘Pharyngitis bacterial’, ‘Viral pharyngitis’, ‘Pharyngitis streptococcal’.
- † Hypersensitivity Reactions were defined by the following terms: ‘Urticaria’, ‘Urticaria papular’, and ‘Rash’ [see Warnings and Precautions (5.1)].
Adverse Reactions
FASENRA
(N=822)
%
Placebo
(N=847)
%
Headache
8
6
Pyrexia
3
2
Pharyngitis*
5
3
Hypersensitivity reactions†
3
3
28-Week Trial
Adverse reactions from Trial 3 with 28 weeks of treatment with FASENRA (n=73) or placebo (n=75) in which the incidence was more common in FASENRA than placebo include headache (8.2% compared to 5.3%, respectively) and pyrexia (2.7% compared to 1.3%, respectively) [see Clinical Studies (14)]. The frequencies for the remaining adverse reactions with FASENRA were similar to placebo.
Injection site reactions
In Trials 1 and 2, injection site reactions (e.g., pain, erythema, pruritus, papule) occurred at a rate of 2.2% in patients treated with FASENRA compared with 1.9% in patients treated with placebo.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to benralizumab in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Overall, treatment-emergent anti-drug antibody response developed in 13% of patients treated with FASENRA at the recommended dosing regimen during the 48 to 56 week treatment period. A total of 12% of patients treated with FASENRA developed neutralizing antibodies. Anti-benralizumab antibodies were associated with increased clearance of benralizumab and increased blood eosinophil levels in patients with high anti-drug antibody titers compared to antibody negative patients. No evidence of an association of anti-drug antibodies with efficacy or safety was observed.
The data reflect the percentage of patients whose test results were positive for antibodies to benralizumab in specific assays.
6.3 Postmarketing Experience
In addition to adverse reactions reported from clinical trials, the following adverse reactions have been identified during post approval use of FASENRA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These events have been chosen for inclusion due to either their seriousness, frequency of reporting, or causal connection to FASENRA or a combination of these factors.
Immune System Disorders: Hypersensitivity reactions, including anaphylaxis.
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to FASENRA during pregnancy. Healthcare providers can enroll patients or encourage patients to enroll themselves by calling 1-877-311-8972 or visiting mothertobaby.org/Fasenra.
Risk Summary
The data on pregnancy exposure from the clinical trials are insufficient to inform on drug-associated risk. Monoclonal antibodies such as benralizumab are transported across the placenta during the third trimester of pregnancy; therefore, potential effects on a fetus are likely to be greater during the third trimester of pregnancy. In a prenatal and postnatal development study conducted in cynomolgus monkeys, there was no evidence of fetal harm with IV administration of benralizumab throughout pregnancy at doses that produced exposures up to approximately 310 times the exposure at the maximum recommended human dose (MRHD) of 30 mg SC [see Data].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk:
In women with poorly or moderately controlled asthma, evidence demonstrates that there is an increased risk of preeclampsia in the mother and prematurity, low birth weight, and small for gestational age in the neonate. The level of asthma control should be closely monitored in pregnant women and treatment adjusted as necessary to maintain optimal control.
Data
Animal Data
In a prenatal and postnatal development study, pregnant cynomolgus monkeys received benralizumab from beginning on GD20 to GD22 (dependent on pregnancy determination), on GD35, once every 14 days thereafter throughout the gestation period and 1-month postpartum (maximum 14 doses) at doses that produced exposures up to approximately 310 times that achieved with the MRHD (on an AUC basis with maternal IV doses up to 30 mg/kg once every 2 weeks). Benralizumab did not elicit adverse effects on fetal or neonatal growth (including immune function) up to 6.5 months after birth. There was no evidence of treatment-related external, visceral, or skeletal malformations. Benralizumab was not teratogenic in cynomolgus monkeys. Benralizumab crossed the placenta in cynomolgus monkeys. Benralizumab concentrations were approximately equal in mothers and infants on postpartum day 7, but were lower in infants at later time points. Eosinophil counts were suppressed in infant monkeys with gradual recovery by 6 months postpartum; however, recovery of eosinophil counts was not observed for one infant monkey during this period.
8.2 Lactation
Risk Summary
There is no information regarding the presence of benralizumab in human or animal milk, and the effects of benralizumab on the breast fed infant and on milk production are not known. However, benralizumab is a humanized monoclonal antibody (IgG1/κ-class), and immunoglobulin G (IgG) is present in human milk in small amounts. If benralizumab is transferred into human milk, the effects of local exposure in the gastrointestinal tract and potential limited systemic exposure in the infant to benralizumab are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for benralizumab and any potential adverse effects on the breast-fed child from benralizumab or from the underlying maternal condition.
8.4 Pediatric Use
There were 108 adolescents aged 12 to 17 with asthma enrolled in the Phase 3 exacerbation trials (Trial 1: n=53, Trial 2: n=55). Of these, 46 received placebo, 40 received FASENRA every 4 weeks for 3 doses, followed by every 8 weeks thereafter, and 22 received FASENRA every 4 weeks. Patients were required to have a history of 2 or more asthma exacerbations requiring oral or systemic corticosteroid treatment in the past 12 months and reduced lung function at baseline (pre-bronchodilator FEV1<90%) despite regular treatment with medium or high dose ICS and LABA with or without OCS or other controller therapy. The pharmacokinetics of benralizumab in adolescents 12 to 17 years of age were consistent with adults based on population pharmacokinetic analysis and the reduction in blood eosinophil counts was similar to that observed in adults following the same FASENRA treatment. The adverse event profile in adolescents was generally similar to the overall population in the Phase 3 studies [see Adverse Reactions (6.1)]. The safety and efficacy in patients younger than 12 years of age has not been established.
8.5 Geriatric Use
Of the total number of patients in clinical trials of benralizumab, 13% (n=320) were 65 and over, while 0.4% (n=9) were 75 and over. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
-
10 OVERDOSAGE
Doses up to 200 mg were administered subcutaneously in clinical trials to patients with eosinophilic disease without evidence of dose-related toxicities.
There is no specific treatment for an overdose with benralizumab. If overdose occurs, the patient should be treated supportively with appropriate monitoring as necessary.
-
11 DESCRIPTION
Benralizumab is a humanized monoclonal antibody (IgG1/κ-class) selective for interleukin-5 receptor alpha subunit (IL-5Rα). Benralizumab is produced in Chinese hamster ovary cells by recombinant DNA technology. Benralizumab has a molecular weight of approximately 150 kDa.
FASENRA (benralizumab) injection is a sterile, preservative-free, clear to opalescent, colorless to slightly yellow solution for subcutaneous injection. Since benralizumab is a protein, a few translucent or white to off-white particles may be present in the solution. Each single-dose prefilled syringe or single-dose autoinjector delivers 1 mL containing 30 mg benralizumab, L-histidine (1.4 mg); L-histidine hydrochloride monohydrate (2.3 mg); polysorbate 20 (0.06 mg); α,α‑trehalose dihydrate (95 mg); and Water for Injection, USP. The single-dose prefilled syringe or single-dose autoinjector contains a 1 mL glass syringe with a staked 29 gauge ½ inch stainless steel needle.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Benralizumab is a humanized afucosylated, monoclonal antibody (IgG1, kappa) that directly binds to the alpha subunit of the human interleukin-5 receptor (IL-5Rα) with a dissociation constant of 11 pM. The IL-5 receptor is expressed on the surface of eosinophils and basophils. In an in vitro setting, the absence of fucose in the Fc domain of benralizumab facilitates binding (45.5 nM) to FcɣRIII receptors on immune effector cells, such as natural killer (NK) cells, leading to apoptosis of eosinophils and basophils through antibody-dependent cell-mediated cytotoxicity (ADCC).
Inflammation is an important component in the pathogenesis of asthma. Multiple cell types (e.g., mast cells, eosinophils, neutrophils, macrophages, lymphocytes) and mediators (e.g., histamine, eicosanoids, leukotrienes, cytokines) are involved in inflammation. Benralizumab, by binding to the IL-5Rα chain, reduces eosinophils through ADCC; however, the mechanism of benralizumab action in asthma has not been definitively established.
12.2 Pharmacodynamics
In the 52-week Phase 2 dose-ranging trial, asthma patients received 1 of 3 doses of benralizumab [2 mg (n=81), 20 mg (n=81), or 100 mg (n=222)] or placebo (n=222). All doses were administered every 4 weeks for the first 3 doses, followed by every 8 weeks thereafter. Median blood eosinophil levels at baseline were 310, 280, 190 and 190 cells/μL in the 2, 20, and 100 mg benralizumab and placebo groups, respectively. Dose-dependent reductions in blood eosinophils were observed. At the time of the last dose (Week 40), median blood eosinophil counts were 100, 50, 40, 170 cells/μL in the 2, 20, and 100 mg benralizumab and placebo groups, respectively.
A reduction in blood eosinophil counts was observed 24 hours post dosing in a Phase 2 trial.
In Trials 1 and 2, following SC administration of benralizumab at the recommended dose blood eosinophils were reduced to a median absolute blood eosinophil count of 0 cells/μL [see Clinical Studies (14)]. This magnitude of reduction was seen at the first observed time point, 4 weeks of treatment, and was maintained throughout the treatment period.
Treatment with benralizumab was also associated with reductions in blood basophils, which was consistently observed across all clinical studies. In the Phase 2 dose-ranging trial, blood basophil counts were measured by flow cytometry. Median blood basophil counts were 45, 52, 46, and 40 cells/µL in the 2 mg, 20 mg and 100 mg benralizumab and placebo groups, respectively. At 52 weeks (12 weeks after the last dose), median blood basophil counts were 42, 18, 17, and 46 cells/µL in the 2 mg, 20 mg and 100 mg benralizumab and placebo groups, respectively.
12.3 Pharmacokinetics
The pharmacokinetics of benralizumab was approximately dose-proportional in patients with asthma following subcutaneous administration over a dose range of 20 to 200 mg.
Absorption
Following subcutaneous administration to patients with asthma, the absorption half-life was approximately 3.5 days. Based on population pharmacokinetic analysis, the estimated absolute bioavailability was approximately 59% and there was no clinically relevant difference in relative bioavailability in the administration to the abdomen, thigh, or arm.
Distribution:
Based on population pharmacokinetic analysis, central and peripheral volume of distribution of benralizumab was 3.1 L and 2.5 L, respectively, for a 70 kg individual.
Metabolism:
Benralizumab is a humanized IgG1 monoclonal antibody that is degraded by proteolytic enzymes widely distributed in the body and not restricted to hepatic tissue.
Elimination:
From population pharmacokinetic analysis, benralizumab exhibited linear pharmacokinetics and no evidence of target receptor-mediated clearance pathway. The estimated typical systemic clearance (CL) for benralizumab was 0.29 L/d for a subject weighing 70kg. Following subcutaneous administration, the elimination half-life was approximately 15.5 days.
Specific populations:
Age:
Based on population pharmacokinetic analysis, age did not affect benralizumab clearance.
Gender, Race:
A population pharmacokinetics analysis indicated that there was no significant effect of gender and race on benralizumab clearance.
Renal impairment:
No formal clinical studies have been conducted to investigate the effect of renal impairment on benralizumab. Based on population pharmacokinetic analysis, benralizumab clearance was comparable in subjects with creatinine clearance values between 30 and 80 mL/min and patients with normal renal function. There are limited data available in subjects with creatinine clearance values less than 30 mL/min; however, benralizumab is not cleared renally.
Hepatic impairment:
No formal clinical studies have been conducted to investigate the effect of hepatic impairment on benralizumab. IgG monoclonal antibodies are not primarily cleared via hepatic pathway; change in hepatic function is not expected to influence benralizumab clearance. Based on population pharmacokinetic analysis, baseline hepatic function biomarkers (ALT, AST, and bilirubin) had no clinically relevant effect on benralizumab clearance.
Drug-Drug Interaction:
No formal drug-drug interaction studies have been conducted.
Cytochrome P450 enzymes, efflux pumps and protein-binding mechanisms are not involved in the clearance of benralizumab. There is no evidence of IL-5Rα expression on hepatocytes and eosinophil depletion does not produce chronic systemic alterations of proinflammatory cytokines.
An effect of benralizumab on the pharmacokinetics of co-administered medications is not expected. Based on the population analysis, commonly co-administered medications had no effect on benralizumab clearance in patients with asthma.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been performed to evaluate the carcinogenic potential of benralizumab. Published literature using animal models suggests that IL-5 and eosinophils are part of an early inflammatory reaction at the site of tumorigenesis and can promote tumor rejection. However, other reports indicate that eosinophil infiltration into tumors can promote tumor growth. Therefore, the malignancy risk in humans from an antibody that binds to IL-5Rα such as benralizumab is unknown.
Male and female fertility were unaffected based upon no adverse histopathological findings in the reproductive organs from cynomolgus monkeys treated with benralizumab for 9 months at IV doses up to 25 mg/kg or at SC doses of up to 30 mg/kg once every 2 weeks (approximately 400 and 270 times the MRHD on an AUC basis).
-
14 CLINICAL STUDIES
The asthma development program for FASENRA included one 52-week dose ranging exacerbation trial (NCT01238861) three confirmatory trials, (Trial 1 [NCT01928771], Trial 2 [NCT01914757], Trial 3 [NCT02075255]) and one 12-week lung function trial (NCT02322775).
Dose-Ranging Trial
The Phase 2 randomized, double-blind, placebo-controlled, 52-week dose-ranging trial, enrolled 609 asthmatic patients 18 years of age and older. Patients were treated with benralizumab 2 mg, 20 mg, or 100 mg or placebo administered subcutaneously every 4 weeks for 3 doses followed by every 8 weeks. The primary endpoint was the annual exacerbation rate and forced expiratory volume in 1 second (FEV1) and ACQ-6 were key secondary endpoints. Patients were required to have a history of 2 or more asthma exacerbations (but no more than 6 exacerbations) requiring systemic corticosteroid treatment in the past 12 months, ACQ-6 score of 1.5 at least twice during screening, and reduced morning lung function at screening [pre-bronchodilator FEV1 below 90%] despite treatment with medium- or high-dose ICS plus LABA. Patients were stratified by eosinophilic status. The annual exacerbation rate reduction for patients receiving benralizumab 2 mg, 20 mg, and 100 mg were -12% (80% CI: -52, 18), 34% (80% CI: 6, 54), 29% (80% CI: 10, 44), respectively, compared to placebo (rate 0.56).
Results from this trial and exposure-response modelling of exacerbation rate reduction supported the evaluation of benralizumab 30 mg in the subsequent trials [see Clinical Pharmacology (12.2 and 12.3)]. FASENRA is not approved at 2 mg, 20 mg, or 100 mg doses, and should only be administered at the recommended dose of 30 mg [see Dosage and Administration (2.1)].
Confirmatory Trials
Trial 1 and Trial 2, were randomized, double-blind, parallel-group, placebo-controlled, exacerbation trials in patients 12 years of age and older and 48 and 56 weeks in duration, respectively. The trials randomized a total of 2510 patients. Patients were required to have a history of 2 or more asthma exacerbations requiring oral or systemic corticosteroid treatment in the past 12 months, ACQ-6 score of 1.5 or more at screening, and reduced lung function at baseline [pre-bronchodilator FEV1 below 80% in adults, and below 90% in adolescents] despite regular treatment with high dose inhaled corticosteroid (ICS) (Trial 1) or with medium or high dose ICS (Trial 2) plus a long-acting beta agonist (LABA) with or without oral corticosteroids (OCS) and additional asthma controller medications. Patients were stratified by geography, age, and blood eosinophils count (≥300 cells/μL or <300 cells/μL). FASENRA administered once every 4 weeks for the first 3 doses, and then every 4 or 8 weeks thereafter as add-on to background treatment was evaluated compared to placebo.
All subjects continued their background asthma therapy throughout the duration of the trials.
Trial 3 was a randomized, double-blind, parallel-group, OCS reduction trial in 220 asthma patients. Patients were required treatment with daily OCS (7.5 to 40 mg per day) in addition to regular use of high-dose ICS and LABA with or without additional controller(s). The trial included an 8-week run-in period during which the OCS was titrated to the minimum effective dose without losing asthma control. For the purposes of the OCS dose titration, asthma control was assessed by the investigator based on a patient’s FEV1, peak expiratory flow, nighttime awakenings, short-acting bronchodilator rescue medication use or any other symptoms that would require an increase in OCS dose. Baseline median OCS dose was similar across all treatment groups. Patients were required to have blood eosinophil counts greater than or equal to 150 cells/μL and a history of at least one exacerbation in the past 12 months. The baseline median OCS dose was 10 mg (range: 8 to 40 mg) for all 3 treatment groups (placebo, FASENRA every 4 weeks, and FASENRA every 4 weeks for the first 3 doses, and then once every 8 weeks).
While 2 dosing regimens were studied in Trials 1, 2, and 3, the recommended dosing regimen is 30 mg FASENRA administered every 4 weeks for the first 3 doses, then every 8 weeks thereafter [see Dosage and Administration (2.1)].
Table 2. Demographics and Baseline Characteristics of Asthma Trials Total Population Trial 1
(N=1204)
Trial 2
(N=1306)
Trial 3
(N=220)
Mean age (yr)
49
49
51
Female (%)
66
62
61
White (%)
73
84
93
Duration of asthma, median (yr)
15
16
12
Never smoked (%)
80
78
79
Mean baseline FEV1 pre-bronchodilator (L)
1.67
1.76
1.85
Mean baseline % predicted FEV1
57
58
60
Mean post-SABA FEV1/FVC (%)
66
65
62
Mean baseline eosinophil count (cells/μL)
472
472
575
Mean number of exacerbations in previous year
3
3
3
Exacerbations
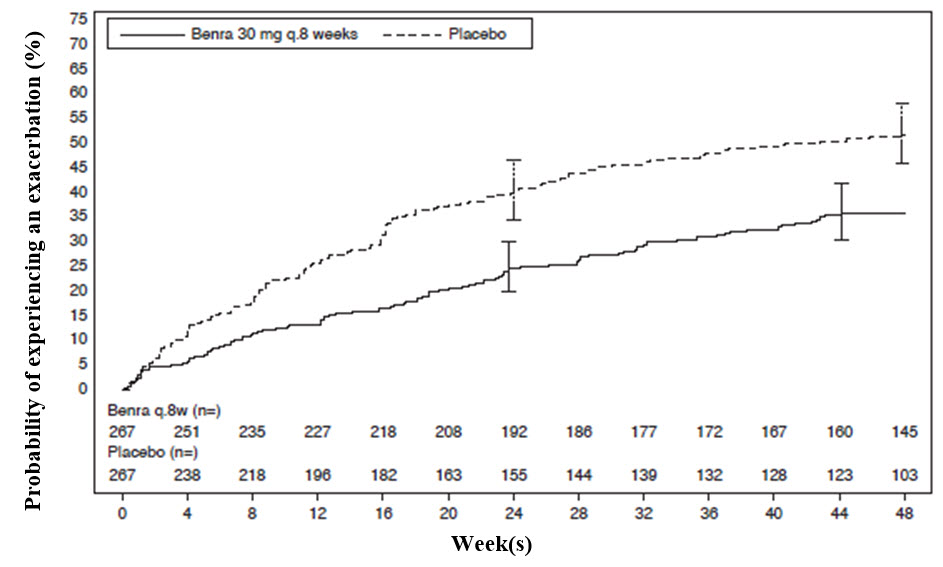
The primary endpoint for Trials 1 and 2 was the rate of asthma exacerbations in patients with baseline blood eosinophil counts of greater than or equal to 300 cells/μL who were taking high-dose ICS and LABA. Asthma exacerbation was defined as a worsening of asthma requiring use of oral/systemic corticosteroids for at least 3 days, and/or emergency department visits requiring use of oral/systemic corticosteroids and/or hospitalization. For patients on maintenance oral corticosteroids, an asthma exacerbation requiring oral corticosteroids was defined as a temporary increase in stable oral/systemic corticosteroids for at least 3 days or a single depo-injectable dose of corticosteroids. In Trial 1, 35% of patients receiving FASENRA experienced an asthma exacerbation compared to 51% on placebo. In Trial 2, 40% of patients receiving FASENRA experienced an asthma exacerbation compared to 51% on placebo (Table 3).
Table 3. Rate of Exacerbations, Trial 1 and 2 (ITT Population)* Trial Treatment Exacerbations per year - * Baseline blood eosinophil counts of greater than or equal to 300 cells/μL and taking high-dose ICS
- † FASENRA 30 mg administered every 4 weeks for the first 3 doses, and every 8 weeks thereafter
Rate
Difference
Rate Ratio (95% CI)
All exacerbations
Trial 1
FASENRA† (n=267)
0.74
-0.78
0.49 (0.37, 0.64)
Placebo (n=267)
1.52
--
--
Trial 2
FASENRA† (n=239)
0.73
-0.29
0.72 (0.54, 0.95)
Placebo (n=248)
1.01
--
--
Exacerbations requiring hospitalization/emergency room visit
Trial 1
FASENRA† (n=267)
0.09
-0.16
0.37 (0.20, 0.67)
Placebo (n=267)
0.25
--
--
Trial 2
FASENRA† (n=239)
0.12
0.02
1.23 (0.64, 2.35)
Placebo (n=248)
0.10
--
--
Exacerbations requiring hospitalization
Trial 1
FASENRA† (n=267)
0.07
-0.07
0.48 (0.22, 1.03)
Placebo (n=267)
0.14
--
--
Trial 2
FASENRA† (n=239)
0.07
0.02
1.48 (0.65, 3.37)
Placebo (n=248)
0.05
--
--
The time to first exacerbation was longer for the patients receiving FASENRA compared with placebo in Trial 1 (Figure 2). Similar findings were seen in Trial 2.
Figure 2. Kaplan-Meier Cumulative Incidence Curves for Time to First Exacerbation, Trial 1
Subgroup analyses from Trials 1 and 2 identified patients with a higher prior exacerbation history and baseline blood eosinophil count as potential predictors of improved treatment response. Reductions in exacerbation rates were observed irrespective of baseline peripheral eosinophil counts; however, patients with a baseline blood eosinophil count ≥300 cells/μL showed a numerically greater response than those with counts <300 cells/μL. In both trials patients with a history of 3 or more exacerbations within the 12 months prior to FASENRA randomization showed a numerically greater exacerbation response than those with fewer prior exacerbations.
Oral Corticosteroid Reduction
Trial 3 evaluated the effect of FASENRA on reducing the use of maintenance oral corticosteroids. The primary endpoint was percent reduction from baseline of the final OCS dose during Weeks 24 to 28, while maintaining asthma control (see definition of asthma control in trial description). Compared to placebo, patients receiving FASENRA achieved greater reductions in daily maintenance oral corticosteroid dose, while maintaining asthma control. The median percent reduction in daily OCS dose from baseline was 75% in patients receiving FASENRA (95% CI: 60, 88) compared to 25% in patients receiving placebo (95% CI: 0, 33). Reductions of 50% or higher in the OCS dose were observed in 48 (66%) patients receiving FASENRA compared to those receiving placebo 28 (37%). The proportion of patients with a mean final dose less than or equal to 5 mg at Weeks 24 to 28 was 59% for FASENRA and 33% for placebo (odds ratio 2.74, 95% CI: 1.41, 5.31). Only patients with an optimized baseline OCS dose of 12.5 mg or less were eligible to achieve a 100% reduction in OCS dose during the study. Of those patients, 52% (22 of 42) receiving FASENRA and 19% (8 of 42) on placebo achieved a 100% reduction in OCS dose. Exacerbations resulting in hospitalization and/or ER visit were also assessed as a secondary endpoint. In this 28-week trial, patients receiving FASENRA had 1 event while those on placebo had 14 events (annualized rate 0.02 and 0.32, respectively; rate ratio of 0.07, 95% CI: 0.01, 0.63).
Lung Function
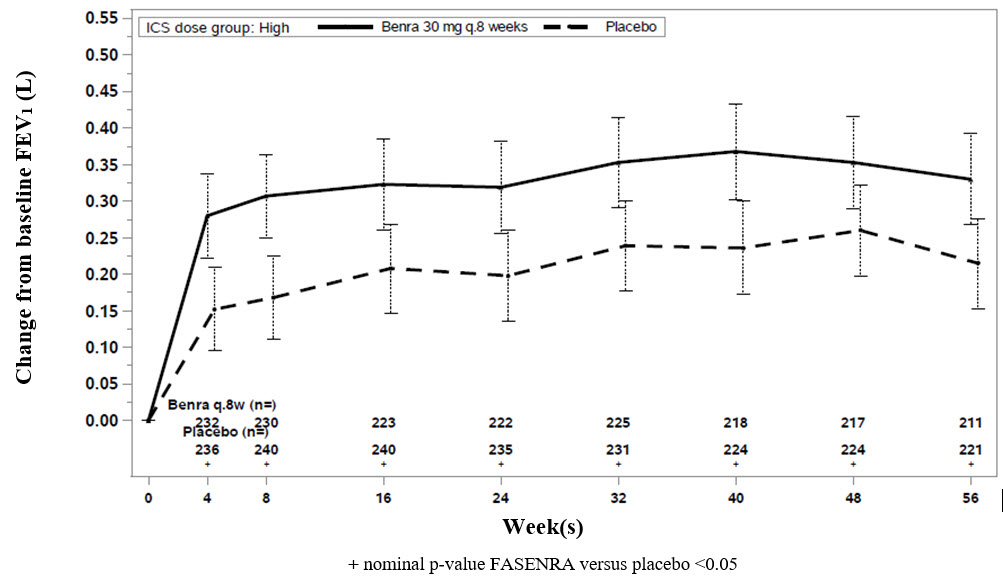
Change from baseline in mean FEV1 was assessed in Trials 1, 2, and 3 as a secondary endpoint. Compared with placebo, FASENRA provided consistent improvements over time in the mean change from baseline in FEV1 (Figure 3 and Table 4).
Figure 3. Mean Change from Baseline in Pre-Bronchodilator FEV1 (L), Trial 2
Table 4. Change from Baseline in Mean Pre-Bronchodilator FEV1 (L) at End of Trial* - * Week 48 in Trial 1, Week 56 in Trial 2, Week 28 in Trial 3.
Trial
Difference from Placebo in Mean Change from Pre-Bronchodilator Baseline FEV1 (L)
(95% CI)1
0.159 (0.068, 0.249)
2
0.116 (0.028, 0.204)
3
0.112 (-0.033, 0.258)
Sub group analyses also showed greater improvements in FEV1 in patients with higher baseline blood eosinophil counts and more frequent prior exacerbation history.
The clinical development program for FASENRA also included a 12-week, randomized, double-blind, placebo-controlled lung function trial conducted in 211 patients with mild to moderate asthma. Patients were treated with placebo or benralizumab 30 mg SC every 4 weeks for 3 doses. Lung function, as measured by the change from baseline in FEV1 at Week 12 was improved in the benralizumab treatment group compared to placebo.
Patient Reported Outcomes
The Asthma Control Questionnaire-6 (ACQ-6) and Standardized Asthma Quality of Life Questionnaire for 12 Years and Older (AQLQ(S)+12) were assessed in Trials 1, 2 and 3. The responder rate for both measures was defined as improvement in score of 0.5 or more as threshold at the end of Trials 1, 2, and 3 (48, 56, and 28 weeks, respectively). In Trial 1, the ACQ-6 responder rate for FASENRA was 60% vs 50% placebo (odds ratio 1.55; 95% CI: 1.09, 2.19). In Trial 2, the ACQ-6 responder rate for the FASENRA was 63% vs 59% placebo (odds ratio 1.16; 95% CI: 0.80, 1.68). In Trial 1, the responder rate for AQLQ(S)+12 for FASENRA was 57% vs 49% placebo (odds ratio 1.42; 95% CI: 0.99, 2.02), and in Trial 2, 60% FASENRA vs 59% placebo (odds ratio of 1.03; 95% CI: 0.70,1.51). Similar results were seen in Trial 3.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
FASENRA (benralizumab) injection is a sterile, preservative-free, clear to opalescent, colorless to slightly yellow solution and may contain a few translucent or white to off-white particles, for subcutaneous injection supplied as a single-dose prefilled syringe or single-dose autoinjector. The prefilled syringe (including stopper and cap) and autoinjector are not made with natural rubber latex.
FASENRA is available as:
-
- Single-Dose Prefilled Syringe
Carton contains one 30 mg/mL single-dose prefilled syringe: NDC: 0310-1730-30
-
- Single-Dose Autoinjector FASENRA PEN
Carton contains one 30 mg/mL single-dose autoinjector: NDC: 0310-1830-30
16.2 Storage and Handling
Store refrigerated at 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
If needed, the prefilled syringe and autoinjector may be stored at room temperature up to 77°F (25°C) for a maximum of 14 days in the original carton to protect from light. Once removed from the refrigerator and brought to room temperature (up to 77°F [25°C]), the prefilled syringe and autoinjector must be used within 14 days or discarded.
Do not freeze. Do not shake. Do not expose to heat.
-
-
17 PATIENT COUNSELING INFORMATION
Advise the patients and/or caregivers to read the FDA-approved patient labeling (Patient Information and Instructions for Use for FASENRA PEN) before the patient starts using FASENRA and each time the prescription is renewed as there may be new information they need to know.
Provide proper training to patients and/or caregivers on proper subcutaneous injection technique using the FASENRA PEN, including aseptic technique, and the preparation and administration of FASENRA PEN prior to use. Advise patients to follow sharps disposal recommendations [see Instructions for Use].
Hypersensitivity Reactions
Inform patients that hypersensitivity reactions (e.g., anaphylaxis, angioedema, urticaria, rash) have occurred after administration of FASENRA. These reactions generally occurred within hours of FASENRA administration, but in some instances had a delayed onset (i.e., days). Instruct patients to contact their healthcare provider if they experience symptoms of an allergic reaction [see Warnings and Precautions (5.1)].
Not for Acute Symptoms or Deteriorating Disease
Inform patients that FASENRA does not treat acute asthma symptoms or acute exacerbations. Inform patients to seek medical advice if their asthma remains uncontrolled or worsens after initiation of treatment with FASENRA [see Warnings and Precautions (5.2)].
Reduction of Corticosteroid Dosage
Inform patients to not discontinue systemic or inhaled corticosteroids except under the direct supervision of a physician. Inform patients that reduction in corticosteroid dose may be associated with systemic withdrawal symptoms and/or unmask conditions previously suppressed by systemic corticosteroid therapy [see Warnings and Precautions (5.3)].
Pregnancy Exposure Registry
Inform women there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to FASENRA during pregnancy and that they can enroll in the Pregnancy Exposure Registry by calling 1-877-311-8972 or by visiting mothertobaby.org/Fasenra[see Use in Specific Populations (8.1)].
Manufactured by
AstraZeneca AB
Södertälje, Sweden SE-15185
US License No. 2059
Distributed by
AstraZeneca Pharmaceuticals LP,
Wilmington, DE 19850
FASENRA is a trademark of the AstraZeneca group of companies.
©AstraZeneca 2019
-
PATIENT PACKAGE INSERT
Patient Information
FASENRA® (fas-en-rah)
(benralizumab)
injection, for subcutaneous use
What is FASENRA?
FASENRA is a prescription medicine used with other asthma medicines for the maintenance treatment of asthma in people 12 years and older whose asthma is not controlled with their current asthma medicines. FASENRA helps prevent severe asthma attacks (exacerbations) and may improve your breathing. Medicines such as FASENRA reduce blood eosinophils. Eosinophils are a type of white blood cell that may contribute to your asthma.
- FASENRA is not used to treat other problems caused by eosinophils.
- FASENRA is not used to treat sudden breathing problems. Tell your healthcare provider if your asthma does not get better or if it gets worse after you start treatment with FASENRA.
It is not known if FASENRA is safe and effective in children under 12 years of age.
Do not use FASENRA if you are allergic to benralizumab or any of the ingredients in FASENRA. See the end of this leaflet for a complete list of ingredients in FASENRA.
Before using FASENRA, tell your healthcare provider about all of your medical conditions, including if you:
- are taking oral or inhaled corticosteroid medicines. Do not stop taking your corticosteroid medicines unless instructed by your healthcare provider. This may cause other symptoms that were controlled by the corticosteroid medicine to come back.
- have a parasitic (helminth) infection.
-
are pregnant or plan to become pregnant. It is not known if FASENRA will harm your unborn baby. Tell your healthcare provider if you become pregnant during your treatment with FASENRA.
- o Pregnancy Registry. There is a pregnancy registry for women who use FASENRA while pregnant. The purpose of the registry is to collect information about the health of you and your baby. You can talk to your healthcare provider about how to take part in this registry or you can get more information and register by calling 1‑877-311-8972 or go to mothertobaby.org/Fasenra.
- are breastfeeding or plan to breastfeed. It is not known if FASENRA passes into your breast milk. You and your healthcare provider should decide if you will use FASENRA and breastfeed. Talk to your healthcare provider about the best way to feed your baby if you use FASENRA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Do not stop taking your other asthma medicines unless your healthcare provider tells you to.
How will I use FASENRA?
- FASENRA is injected under your skin (subcutaneously) one time every 4 weeks for the first 3 doses, and then every 8 weeks.
- FASENRA comes in a single dose prefilled syringe and in a single dose autoinjector.
- A healthcare provider will inject FASENRA using the single-dose prefilled syringe.
- If your healthcare provider decides that you or a caregiver can give the injection of FASENRA, you or your caregiver should receive training on the right way to prepare and give the injection using the FASENRA PEN.
- Do not try to inject FASENRA until you have been shown the right way by your healthcare provider. See the detailed “Instructions for Use” that comes with FASENRA PEN for information on how to prepare and inject FASENRA.
- If you miss a dose of FASENRA, call your healthcare provider.
What are the possible side effects of FASENRA?
FASENRA may cause serious side effects, including:
- allergic (hypersensitivity) reactions, including anaphylaxis. Serious allergic reactions can happen after you get your FASENRA injection. Allergic reactions can sometimes happen hours or days after you get your injection. Tell your healthcare provider or get emergency help right away if you have any of the following symptoms of an allergic reaction:
- o swelling of your face, mouth and tongue
- o breathing problems
- o fainting, dizziness, feeling lightheaded (low blood pressure)
- o rash
- o hives
The most common side effects of FASENRA include headache and sore throat.
These are not all the possible side effects of FASENRA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store FASENRA?
- Store FASENRA in the refrigerator between 36°F to 46°F (2°C to 8°C).
- FASENRA may be stored at room temperature between 68°F to 77°F (20°C to 25°C) for up to 14 days.
- Once removed from the refrigerator and brought to room temperature FASENRA must be used within 14 days or thrown away.
- Store FASENRA in the original carton until you are ready to use it to protect it from light.
- Do not freeze FASENRA. Do not use FASENRA that has been frozen.
- Do not expose FASENRA to heat.
- Do not use FASENRA past the expiration date.
- Keep FASENRA and all medicines out of the reach of children.
General information about the safe and effective use of FASENRA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use FASENRA for a condition for which it was not prescribed. Do not give FASENRA to other people, even if they have the same symptoms you have. It may harm them.
You can ask your doctor or pharmacist for information about FASENRA that is written for health providers.
What are the ingredients in FASENRA?
Active ingredient: benralizumab
Inactive ingredients: L-histidine, L-histidine hydrochloride monohydrate, polysorbate 20, α,α-trehalose dihydrate, and Water for Injection
Manufactured by: AstraZeneca AB, Södertälje, Sweden SE-15185
U.S. License Number 2059
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
FASENRA is a trademark of the AstraZeneca group of companies.
©AstraZeneca 2019
For more information, go to https://www.FASENRA.com or call 1-800-236-9933.
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: October 2019
-
INSTRUCTIONS FOR USE
Instructions for Use
FASENRA PEN™ (fas-en-rah)
(benralizumab)
for Subcutaneous Injection
Single-dose Autoinjector
Before using your FASENRA PEN, your healthcare provider should show you or your caregiver how to use it the right way.
Read this Instructions for Use before you start using your FASENRA PEN and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
If you or your caregiver have any questions, talk to your healthcare provider.
Important information:
- Store FASENRA in a refrigerator between 36°F to 46°F (2°C to 8°C) in its original carton until you are ready to use it.
- FASENRA may be kept at room temperature between 68°F to 77°F (20°C to 25°C) for a maximum of 14 days.
- Once removed from the refrigerator and brought to room temperature, FASENRA must be used within 14 days or thrown away (disposed of).
Do not use your FASENRA PEN if:
- it has been frozen
- it has been dropped or damaged
- the security seal on the carton has been broken
- the expiration date (EXP) has passed
Do not:
- shake your FASENRA PEN
- share or reuse your FASENRA PEN
- expose your FASENRA PEN to heat
If any of these happen, throw away the FASENRA PEN in a puncture-resistant sharps disposal container and use a new FASENRA PEN.
Each FASENRA PEN contains 1 dose of FASENRA that is for one time use only.
Keep FASENRA and all medicines out of the sight and reach of children.
Your FASENRA PEN
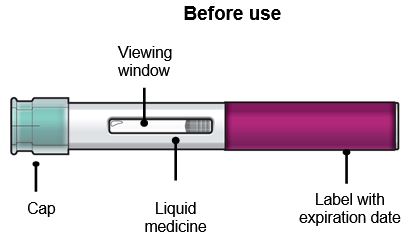
Do not remove the cap until you have reached Step 6 of these instructions and are ready to inject FASENRA.
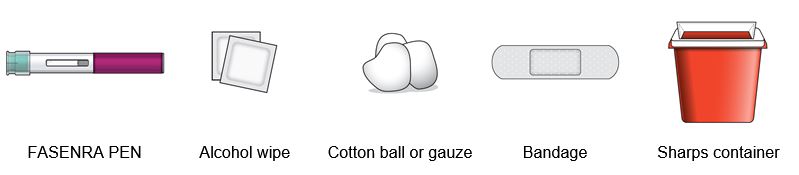
Step 1 – Gather supplies
- 1 FASENRA PEN from the refrigerator
- 1 alcohol wipe
- 1 cotton ball or gauze
- 1 bandage
- 1 puncture-resistant sharps disposal container. See Step 10 for instructions on how to throw away (dispose of) the used FASENRA PEN safely.
Step 2 – Prepare to use your FASENRA PEN
Check the expiration date (EXP). Do not use if the expiration date has passed.
Let FASENRA warm up at room temperature between 68°F to 77°F (20°C to 25°C) for about 30 minutes before giving the injection.
Do not warm the FASENRA PEN in any other way. For example, do not warm it in a microwave or hot water, or put it near other heat sources.
Use FASENRA within 14 days of removing from the refrigerator. After 14 days, throw away the FASENRA PEN.
Do not remove the cap until you have reached Step 6.

Step 3 – Check the liquid
Look at the liquid in the FASENRA PEN through the viewing window. The liquid should be clear and colorless to slightly yellow. It may contain small white particles.
Do not inject FASENRA if the liquid is cloudy, discolored, or contains large particles.
You may see small air bubbles in the liquid. This is normal. You do not need to do anything about it.
Step 4 – Choose the injection site
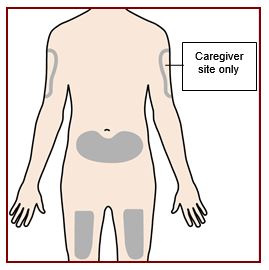
If you are giving yourself the injection, the recommended injection site is the front of your thigh or the lower part of your stomach (abdomen).
A caregiver may inject you in the upper-arm, thigh, or abdomen. Do not try to inject yourself in the arm.
For each injection, choose a different site that is at least 1-inch (3-cm) away from where you last injected.
Do not inject:
- into the 2-inch (5-cm) area around your belly-button
- where the skin is tender, bruised, scaly or hard
- into scars or damaged skin
- through clothing
Step 5 – Clean the injection site
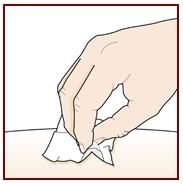
Wash your hands well with soap and water.
Clean the injection site with an alcohol wipe in a circular motion. Let it air dry.
Do not touch the cleaned area before injecting.
Do not fan or blow on the cleaned area.
Step 6 – Pull off the cap
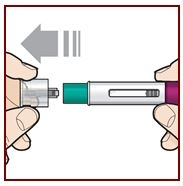
Hold the FASENRA PEN with 1 hand. Carefully pull the cap straight off with your other hand.
Put the cap aside to throw away later.
The green needle guard is now exposed. It is there to prevent you from touching the needle.
Do not try to touch the needle or push on the needle guard with your finger.
Do not try to put the cap back on the FASENRA PEN. You could cause the
injection to happen too soon or damage the needle.
Complete the following steps right away after removing the cap.
Step 7 – Inject FASENRA
Follow your healthcare providers instructions on how to inject. You can either gently pinch at the injection site or give the injection without pinching the skin.
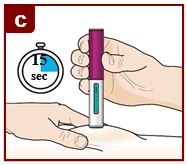
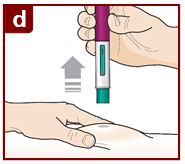
Inject FASENRA by following the steps in figures a, b, c, and d.
Hold the FASENRA PEN in place for the entire injection.
Do not change the position of the FASENRA PEN after the injection has started.
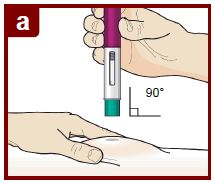
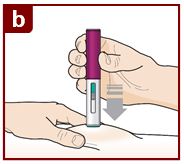
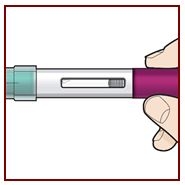
Position the FASENRAPEN at the injection site.
Place the needle guard of the FASENRA PEN flat against your skin (90 degree angle).
Make sure you can see the viewing window.
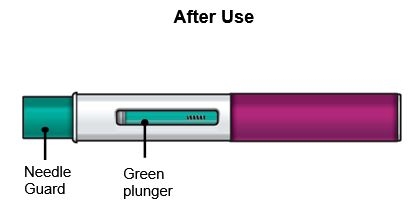
Press down firmly.
You will hear a click. A ‘click’ tells you the injection has started. The green plunger will move down in the viewing window during the injection.
Hold down firmly for 15 seconds.
You will hear a second ‘click’. The second click tells you the injection has finished. The green plunger will fill the viewing window.
Lift the FASENRAPEN straight up.
The needle guard will slide down and lock into place over the needle.
Step 8 – Check the viewing window
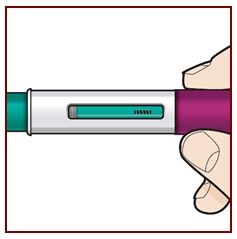
Check the viewing window to make sure all the liquid has been injected.
If the green plunger does not fill the viewing window, you may not have received the full dose. If this happens or if you have any other concerns, call your healthcare provider.
- Before Injection


After Injection
Step 9 – Check the injection site
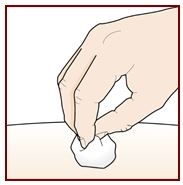
There may be a small amount of blood or liquid where you injected. This is normal.
Gently hold pressure over your skin with a cotton ball or gauze until the bleeding stops.
Do not rub the injection site.
If needed, cover the injection site with a small bandage.
Step 10 – Dispose of the used FASENRAPEN safely
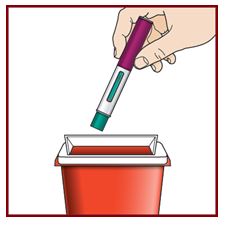
- Each FASENRA PEN contains a single dose of FASENRA and cannot be reused.
- Put your used FASENRA PEN in a FDA-cleared sharps disposal container right away after use.
Do not throw away the FASENRA PEN in your household trash.
Throw away the cap and other used supplies in your household trash.
Disposal guidelines
If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the area that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal.
Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
Do not recycle your used sharps disposal container.
For more information go to www.FasenraPen.com or call 1-800-236-9933. If you still have questions, call your healthcare provider.
Manufactured by: AstraZeneca AB, Södertälje, Sweden SE-15185
U.S. License Number 2059
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
FASENRA is a trademark of the AstraZeneca group of companies.
©AstraZeneca 2019
This Instructions for Use has been approved by the U.S. Food and Drug Administration
Issued: October 2019
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 0310-1730-30 Rx only
FASENRA®
(benralizumab) Injection
For Subcutaneous Injection Only
Store the prefilled syringe refrigerated at 36°F - 46°F (2°C - 8°C) in original carton to protect from light.
Do not shake, freeze, or expose to heat.
30 mg/mL
1 single-dose prefilled syringe
Discard unused portion.
AstraZeneca
-
Package/Label Display Panel
NDC: 0310-1830-30 Rx only
FASENRA PEN™
(benralizumab) Injection
For Subcutaneous Injection Only
Store the FASENRA PEN refrigerated at 36°F - 46°F (2°C - 8°C) in original carton to protect from light.
Do not shake, freeze, or expose to heat.
30 mg/mL
1 single-dose prefilled autoinjector
Discard unused portion.
AstraZeneca
-
INGREDIENTS AND APPEARANCE
FASENRA
benralizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-1730 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BENRALIZUMAB (UNII: 71492GE1FX) (BENRALIZUMAB - UNII:71492GE1FX) BENRALIZUMAB 30 mg in 1 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 1.4 mg in 1 mL HISTIDINE MONOHYDROCHLORIDE (UNII: 1D5Q932XM6) 2.3 mg in 1 mL TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) 95 mg in 1 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.06 mg in 1 mL WATER (UNII: 059QF0KO0R) 910 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-1730-30 1 in 1 CARTON 11/14/2017 1 1 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC: 0310-1730-85 1 in 1 CARTON 11/14/2017 2 1 mL in 1 SYRINGE, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761070 11/14/2017 FASENRA
benralizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-1830 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BENRALIZUMAB (UNII: 71492GE1FX) (BENRALIZUMAB - UNII:71492GE1FX) BENRALIZUMAB 30 mg in 1 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 1.4 mg in 1 mL HISTIDINE MONOHYDROCHLORIDE (UNII: 1D5Q932XM6) 2.3 mg in 1 mL TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) 95 mg in 1 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.06 mg in 1 mL WATER (UNII: 059QF0KO0R) 910 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-1830-30 1 in 1 CARTON 10/04/2019 1 1 mL in 1 CARTRIDGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC: 0310-1830-85 1 in 1 CARTON 10/04/2019 2 1 mL in 1 CARTRIDGE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761070 10/04/2019 Labeler - AstraZeneca Pharmaceuticals LP (054743190) Registrant - AstraZeneca PLC (230790719)
Trademark Results [FASENRA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 FASENRA 98375565 not registered Live/Pending |
AstraZeneca AB 2024-01-25 |
 FASENRA 87128498 5612391 Live/Registered |
AstraZeneca AB 2016-08-05 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.