JEVTANA- cabazitaxel kit
Jevtana by
Drug Labeling and Warnings
Jevtana by is a Prescription medication manufactured, distributed, or labeled by Sanofi-Aventis U.S. LLC, Sanofi Winthrop Industrie, Sanofi-Aventis Deutschland GmbH. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use JEVTANA safely and effectively. See full prescribing information for JEVTANA.
JEVTANA® (cabazitaxel) injection, for intravenous use
Initial U.S. Approval: 2010WARNING: NEUTROPENIA AND HYPERSENSITIVITY
See full prescribing information for complete boxed warning.
- Neutropenic deaths have been reported. Obtain frequent blood counts to monitor for neutropenia. JEVTANA is contraindicated in patients with neutrophil counts of ≤1,500 cells/mm3. Primary prophylaxis with G-CSF is recommended in patients with high-risk clinical features. (4, 5.1, 5.2)
- Severe hypersensitivity can occur and may include generalized rash/erythema, hypotension and bronchospasm. Discontinue JEVTANA immediately if severe reactions occur and administer appropriate therapy. (2.1, 5.2)
- Contraindicated if history of severe hypersensitivity reactions to cabazitaxel or to drugs formulated with polysorbate 80. (4)
RECENT MAJOR CHANGES
Warnings and Precautions (5.9) 01/2020 INDICATIONS AND USAGE
JEVTANA is a microtubule inhibitor indicated in combination with prednisone for treatment of patients with metastatic castration-resistant prostate cancer previously treated with a docetaxel-containing treatment regimen. (1)
DOSAGE AND ADMINISTRATION
Recommended Dose: JEVTANA 20 mg/m2 administered every three weeks as a one-hour intravenous infusion in combination with oral prednisone 10 mg administered daily throughout JEVTANA treatment. (2.1)
A dose of 25 mg/m2 can be used in select patients at the discretion of the treating healthcare provider. (2.1, 5.1, 5.2, 6.1, 14)
- JEVTANA requires two dilutions prior to administration. (2.5)
- Use the entire contents of the accompanying diluent to achieve a concentration of 10 mg/mL JEVTANA. (2.5)
- PVC equipment should not be used. (2.5)
-
Premedication Regimen: Administer intravenously 30 minutes before each dose of JEVTANA:
- Antihistamine (dexchlorpheniramine 5 mg or diphenhydramine 25 mg or equivalent antihistamine)
- Corticosteroid (dexamethasone 8 mg or equivalent steroid)
- H2 antagonist (ranitidine 50 mg or equivalent H2 antagonist) (2.1)
- Dosage Modifications: See full prescribing information (2.2, 2.3, 2.4)
DOSAGE FORMS AND STRENGTHS
- Single-dose vial 60 mg/1.5 mL, supplied with diluent (5.7 mL) for JEVTANA (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Bone marrow suppression (particularly neutropenia) and its clinical consequences (febrile neutropenia, neutropenic infections, and death): Monitor blood counts frequently to determine if dosage modification or initiation of G-CSF is needed. Primary prophylaxis with G-CSF is recommended in patients with high-risk clinical features. Closely monitor patients with hemoglobin <10 g/dL. (2.2, 4, 5.1)
- Increased toxicities in elderly patients: Patients ≥65 years of age were more likely to experience fatal outcomes and certain adverse reactions, including neutropenia and febrile neutropenia. Monitor closely. (5.2, 8.5)
- Hypersensitivity: Severe hypersensitivity reactions can occur. Premedicate with corticosteroids and H2 antagonists. Discontinue infusion immediately if hypersensitivity is observed and treat as indicated. (4, 5.3)
- Gastrointestinal disorders: Nausea, vomiting, and diarrhea may occur. Mortality related to diarrhea has been reported. Rehydrate and treat with antiemetics and antidiarrheals as needed. If experiencing Grade ≥3 diarrhea, dosage should be modified. (2.2) Deaths have occurred due to gastrointestinal hemorrhage, perforation and neutropenic enterocolitis. Delay or discontinue JEVTANA and treat as indicated. (5.4)
- Renal failure, including cases with fatal outcomes, has been reported. Identify cause and manage aggressively. (5.5)
- Urinary disorders including cystitis: Cystitis, radiation cystitis, and hematuria may occur. Monitor patients who previously received pelvic radiation for signs and symptoms of cystitis. Interrupt or discontinue JEVTANA and provide medical or surgical supportive care, as needed, in patients experiencing severe hemorrhagic cystitis. (5.6)
- Respiratory disorders: Interstitial pneumonia/pneumonitis, interstitial lung disease and acute respiratory distress syndrome, including fatal outcomes, have been reported. Delay or discontinue JEVTANA and treat as indicated. (5.7)
- Hepatic impairment: Administer JEVTANA at a dose of 20 mg/m2 in patients with mild hepatic impairment. Administer JEVTANA at a dose of 15 mg/m2 in patients with moderate hepatic impairment. (2.3, 5.8)
- Embryo-fetal toxicity: JEVTANA can cause fetal harm and loss of pregnancy. Advise males with female partners of reproductive potential to use effective contraception. (5.9, 8.1, 8.3)
ADVERSE REACTIONS
Most common all grades adverse reactions and laboratory abnormalities (≥10%) with JEVTANA 20 mg/m2 or 25 mg/m2 are neutropenia, anemia, leukopenia, thrombocytopenia, diarrhea, fatigue, nausea, vomiting, constipation, asthenia, abdominal pain, hematuria, back pain, anorexia, peripheral neuropathy, pyrexia, dyspnea, dysgeusia, cough, arthralgia, and alopecia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact sanofi-aventis U.S. LLC at 1-800-633-1610 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: NEUTROPENIA AND HYPERSENSITIVITY
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
2.2 Dose Modifications for Adverse Reactions
2.3 Dose Modifications for Hepatic Impairment
2.4 Dose Modifications for Use with Strong CYP3A Inhibitors
2.5 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Bone Marrow Suppression
5.2 Increased Toxicities in Elderly Patients
5.3 Hypersensitivity Reactions
5.4 Gastrointestinal Adverse Reactions
5.5 Renal Failure
5.6 Urinary Disorders Including Cystitis
5.7 Respiratory Disorders
5.8 Use in Patients with Hepatic Impairment
5.9 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 CYP3A Inhibitors
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 TROPIC Trial (JEVTANA + prednisone compared to mitoxantrone)
14.2 PROSELICA Trial (comparison of two doses of JEVTANA)
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
16.3 Handling and Disposal
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: NEUTROPENIA AND HYPERSENSITIVITY
Neutropenia: Neutropenic deaths have been reported. Monitor for neutropenia with frequent blood cell counts. JEVTANA is contraindicated in patients with neutrophil counts of ≤1,500 cells/mm3. Primary prophylaxis with G-CSF is recommended in patients with high-risk clinical features [see Contraindications (4) and Warnings and Precautions (5.1, 5.2)].
Severe hypersensitivity: Severe hypersensitivity reactions can occur and may include generalized rash/erythema, hypotension and bronchospasm. Severe hypersensitivity reactions require immediate discontinuation of the JEVTANA infusion and administration of appropriate therapy. Patients should receive premedication. JEVTANA is contraindicated in patients who have a history of severe hypersensitivity reactions to cabazitaxel or to other drugs formulated with polysorbate 80 [see Dosage and Administration (2.1), Contraindications (4), and Warnings and Precautions (5.3)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The recommended dose of JEVTANA is based on calculation of the Body Surface Area (BSA), and is 20 mg/m2 administered as a one-hour intravenous infusion every three weeks in combination with oral prednisone 10 mg administered daily throughout JEVTANA treatment.
A dose of 25 mg/m2 can be used in select patients at the discretion of the treating healthcare provider [see Warnings and Precautions (5.1, 5.2), Adverse Reactions (6.1), and Clinical Studies (14)].
Premedicate at least 30 minutes prior to each dose of JEVTANA with the following intravenous medications to reduce the risk and/or severity of hypersensitivity [see Warnings and Precautions (5.3)]:
- antihistamine (dexchlorpheniramine 5 mg, or diphenhydramine 25 mg or equivalent antihistamine),
- corticosteroid (dexamethasone 8 mg or equivalent steroid),
- H2 antagonist (ranitidine 50 mg or equivalent H2 antagonist).
Antiemetic prophylaxis is recommended and can be given orally or intravenously as needed [see Warnings and Precautions (5.3)].
JEVTANA injection single-dose vial requires two dilutions prior to administration [see Dosage and Administration (2.5)].
2.2 Dose Modifications for Adverse Reactions
Reduce or discontinue JEVTANA dosing for adverse reactions as described in Table 1.
Table 1: Recommended Dosage Modifications for Adverse Reactions in Patients Treated with JEVTANA Toxicity Dosage Modification Prolonged grade ≥3 neutropenia (greater than 1 week) despite appropriate medication including granulocyte-colony stimulating factor (G-CSF) Delay treatment until neutrophil count is >1,500 cells/mm3, then reduce dosage of JEVTANA by one dose level. Use G-CSF for secondary prophylaxis. Febrile neutropenia or neutropenic infection Delay treatment until improvement or resolution, and until neutrophil count is >1,500 cells/mm3, then reduce dosage of JEVTANA by one dose level. Use G-CSF for secondary prophylaxis. Grade ≥3 diarrhea or persisting diarrhea despite appropriate medication, fluid and electrolytes replacement Delay treatment until improvement or resolution, then reduce dosage of JEVTANA by one dose level. Grade 2 peripheral neuropathy Delay treatment until improvement or resolution, then reduce dosage of JEVTANA by one dose level. Grade ≥3 peripheral neuropathy Discontinue JEVTANA. Patients at a 20 mg/m2 dose who require dose reduction should decrease dosage of JEVTANA to 15 mg/m2 [see Adverse Reactions (6.1)].
Patients at a 25 mg/m2 dose who require dose reduction should decrease dosage of JEVTANA to 20 mg/m2. One additional dose reduction to 15 mg/m2 may be considered [see Adverse Reactions (6.1)].
2.3 Dose Modifications for Hepatic Impairment
- Mild hepatic impairment (total bilirubin >1 to ≤1.5 × Upper Limit of Normal (ULN) or AST >1.5 × ULN): Administer JEVTANA at a dose of 20 mg/m2.
- Moderate hepatic impairment (total bilirubin >1.5 to ≤3 × ULN and AST = any): Administer JEVTANA at a dose of 15 mg/m2 based on tolerability data in these patients; however, the efficacy of this dose is unknown.
- Severe hepatic impairment (total bilirubin >3 × ULN): JEVTANA is contraindicated in patients with severe hepatic impairment [see Warning and Precautions (5.8) and Clinical Pharmacology (12.3)].
2.4 Dose Modifications for Use with Strong CYP3A Inhibitors
Concomitant drugs that are strong CYP3A inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, voriconazole) may increase plasma concentrations of cabazitaxel. Avoid the coadministration of JEVTANA with these drugs. If patients require coadministration of a strong CYP3A inhibitor, consider a 25% JEVTANA dose reduction [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
2.5 Preparation and Administration
JEVTANA is a cytotoxic anticancer drug. Follow applicable special handling and disposal procedures [see References (15)]. If JEVTANA first diluted solution, or second (final) dilution for intravenous infusion should come into contact with the skin or mucous, immediately and thoroughly wash with soap and water.
Do not use PVC infusion containers or polyurethane infusions sets for preparation and administration of JEVTANA infusion solution.
JEVTANA should not be mixed with any other drugs.
Preparation
Read this entire section carefully before mixing and diluting. JEVTANA requires two dilutions prior to administration. Follow the preparation instructions provided below, as improper preparation may lead to overdose [see Overdosage (10)].
Note: Both the JEVTANA injection and the diluent vials contain an overfill to compensate for liquid loss during preparation. This overfill ensures that after dilution with the entire contents of the accompanying diluent, there is an initial diluted solution containing 10 mg/mL JEVTANA.
Inspect the JEVTANA injection and supplied diluent vials. The JEVTANA injection is a clear yellow to brownish-yellow viscous solution.
Step 1 – first dilution
Each vial of JEVTANA (cabazitaxel) 60 mg/1.5 mL must first be mixed with the entire contents of supplied diluent. Once reconstituted, the resultant solution contains 10 mg/mL of JEVTANA.
When transferring the diluent, direct the needle onto the inside wall of JEVTANA vial and inject slowly to limit foaming. Remove the syringe and needle and gently mix the initial diluted solution by repeated inversions for at least 45 seconds to assure full mixing of the drug and diluent. Do not shake.
Let the solution stand for a few minutes to allow any foam to dissipate, and check that the solution is homogeneous and contains no visible particulate matter. It is not required that all foam dissipate prior to continuing the preparation process.
The resulting initial diluted JEVTANA solution (cabazitaxel 10 mg/mL) requires further dilution before administration. The second dilution should be done immediately (within 30 minutes) to obtain the final infusion as detailed in Step 2.
Step 2 – second (final) dilution
Withdraw the recommended dose from the JEVTANA solution containing 10 mg/mL as prepared in Step 1 using a calibrated syringe and further dilute into a sterile 250 mL PVC-free container of either 0.9% sodium chloride solution or 5% dextrose solution for infusion. If a dose greater than 65 mg of JEVTANA is required, use a larger volume of the infusion vehicle so that a concentration of 0.26 mg/mL JEVTANA is not exceeded. The concentration of the JEVTANA final infusion solution should be between 0.10 mg/mL and 0.26 mg/mL.
Remove the syringe and thoroughly mix the final infusion solution by gently inverting the bag or bottle.
As the final infusion solution is supersaturated, it may crystallize over time. Do not use if this occurs and discard.
Fully prepared JEVTANA infusion solution (in either 0.9% sodium chloride solution or 5% dextrose solution) should be used within 8 hours at ambient temperature (including the one-hour infusion), or for a total of 24 hours (including the one-hour infusion) under the refrigerated conditions.
Discard any unused portion.
Administration
Inspect visually for particulate matter, any crystals and discoloration prior to administration. If the JEVTANA first diluted solution or second (final) infusion solution is not clear or appears to have precipitation, it should be discarded.
Use an in-line filter of 0.22 micrometer nominal pore size (also referred to as 0.2 micrometer) during administration.
The final JEVTANA infusion solution should be administered intravenously as a one-hour infusion at room temperature.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
JEVTANA is contraindicated in patients with:
- neutrophil counts of ≤1,500/mm3 [see Warnings and Precautions (5.1)]
- history of severe hypersensitivity reactions to cabazitaxel or to other drugs formulated with polysorbate 80 [see Warnings and Precautions (5.3)]
- severe hepatic impairment (total bilirubin >3 × ULN) [see Warnings and Precautions (5.8)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Bone Marrow Suppression
JEVTANA is contraindicated in patients with neutrophils ≤1,500/mm3 [see Contraindications (4)]. Closely monitor patients with hemoglobin <10 g/dL.
Bone marrow suppression manifested as neutropenia, anemia, thrombocytopenia and/or pancytopenia may occur. Neutropenic deaths have been reported.
In a randomized trial (TROPIC) in previously treated patients with metastatic castration-resistant prostate cancer, five patients (1.3%) died from infection (sepsis or septic shock). All had grade 4 neutropenia and one had febrile neutropenia. One additional patient's death was attributed to neutropenia without a documented infection. Twenty-two (6%) patients discontinued JEVTANA treatment due to neutropenia, febrile neutropenia, infection, or sepsis. The most common adverse reaction leading to treatment discontinuation in the JEVTANA group was neutropenia (2%). Grade 3–4 neutropenia has been observed in 82% of patients treated with JEVTANA in the randomized trial.
In a randomized trial (PROSELICA) comparing two doses of JEVTANA in previously treated metastatic castration-resistant prostate cancer, 8 patients (1%) on the 20 mg/m2 arm and 15 patients (3%) on the 25 mg/m2 arm died from infection; of these, 4 deaths on the 20 mg/m2 arm and 8 deaths on the 25 mg/m2 arm occurred within the first 30 days of treatment.
Fewer patients receiving JEVTANA 20 mg/m2 were reported to have infectious adverse reactions. Grade 1–4 infections were experienced by 160 patients (28%) on the 20 mg/m2 arm and 227 patients (38%) on the 25 mg/m2 arm. Grade 3–4 infections were experienced by 57 patients (10%) on the 20 mg/m2 arm and 120 patients (20%) on the 25 mg/m2 arm. Noninferiority for overall survival was demonstrated between these two arms [see Clinical Studies (14)].
Based on guidelines for the use of G-CSF and the adverse reactions profile of JEVTANA, primary prophylaxis with G-CSF is recommended in patients with high-risk clinical features (older patients, poor performance status, previous episodes of febrile neutropenia, extensive prior radiation ports, poor nutritional status, or other serious comorbidities) that predispose them to increased complications from prolonged neutropenia. The effectiveness of primary prophylaxis with G-CSF in patients receiving JEVTANA has not been studied. Therapeutic use of G-CSF and secondary prophylaxis should be considered in all patients at increased risk for neutropenia complications.
Monitoring of complete blood counts is essential on a weekly basis during cycle 1 and before each treatment cycle thereafter so that the dose can be adjusted, if needed [see Dosage and Administration (2.2)].
5.2 Increased Toxicities in Elderly Patients
In a randomized trial (TROPIC), 2% of patients (3/131) <65 years of age and 6% (15/240) ≥65 years of age died of causes other than disease progression within 30 days of the last JEVTANA dose. Patients ≥65 years of age are more likely to experience certain adverse reactions, including neutropenia and febrile neutropenia. The incidence of the following grade 3–4 adverse reactions were higher in patients ≥65 years of age compared to younger patients; neutropenia (87% vs 74%), and febrile neutropenia (8% vs 6%).
In a randomized clinical trial (PROSELICA) comparing two doses of JEVTANA, deaths due to infection within 30 days of starting JEVTANA occurred in 0.7% (4/580) patients on the 20 mg/m2 arm and 1.3% (8/595) patients on the 25 mg/m2 arm; all of these patients were >60 years of age.
In PROSELICA, on the 20 mg/m2 arm, 3% (5/178) of patients <65 years of age and 2% (9/402) ≥65 years of age died of causes other than disease progression within 30 days of the last JEVTANA dose. On the 25 mg/m2 arm, 2% (3/175) patients <65 years of age and 5% (20/420) ≥65 years of age died of causes other than disease progression within 30 days of the last JEVTANA dose [see Adverse Reactions (6) and Use in Specific Populations (8.5)].
5.3 Hypersensitivity Reactions
Hypersensitivity reactions may occur within a few minutes following the initiation of the infusion of JEVTANA, thus facilities and equipment for the treatment of hypotension and bronchospasm should be available. Severe hypersensitivity reactions can occur and may include generalized rash/erythema, hypotension and bronchospasm.
Premedicate all patients prior to the initiation of the infusion of JEVTANA [see Dosage and Administration (2.1)]. Observe patients closely for hypersensitivity reactions, especially during the first and second infusions. Severe hypersensitivity reactions require immediate discontinuation of the JEVTANA infusion and appropriate therapy. JEVTANA is contraindicated in patients with a history of severe hypersensitivity reactions to cabazitaxel or to other drugs formulated with polysorbate 80 [see Contraindications (4)].
5.4 Gastrointestinal Adverse Reactions
Nausea, vomiting and severe diarrhea, at times, may occur. Deaths related to diarrhea and electrolyte imbalance occurred in the randomized clinical trials. Intensive measures may be required for severe diarrhea and electrolyte imbalance. Antiemetic prophylaxis is recommended. Treat patients with rehydration, antidiarrheal or antiemetic medications as needed. Treatment delay or dosage reduction may be necessary if patients experience Grade ≥3 diarrhea [see Dosage and Administration (2.2)].
Gastrointestinal (GI) hemorrhage and perforation, ileus, enterocolitis, neutropenic enterocolitis, including fatal outcome, have been reported in patients treated with JEVTANA [see Adverse Reactions (6.2)]. Risk may be increased with neutropenia, age, steroid use, concomitant use of NSAIDs, antiplatelet therapy or anticoagulants, and patients with a prior history of pelvic radiotherapy, adhesions, ulceration and GI bleeding.
Abdominal pain and tenderness, fever, persistent constipation, diarrhea, with or without neutropenia, may be early manifestations of serious gastrointestinal toxicity and should be evaluated and treated promptly. JEVTANA treatment delay or discontinuation may be necessary.
The incidence of gastrointestinal adverse reactions is greater in the patients who have received prior radiation. In PROSELICA, diarrhea was reported in 41% (297/732) of patients who had received prior radiation and in 27% (118/443) of patients without prior radiation. Of the patients who had previously received radiation, more patients on the 25 mg/m2 arm reported diarrhea, compared to patients on the 20 mg/m2 arm.
5.5 Renal Failure
In the randomized clinical trial (TROPIC), renal failure of any grade occurred in 4% of the patients being treated with JEVTANA, including four cases with fatal outcome. Most cases occurred in association with sepsis, dehydration, or obstructive uropathy [see Adverse Reactions (6.1)]. Some deaths due to renal failure did not have a clear etiology. Appropriate measures should be taken to identify causes of renal failure and treat aggressively.
5.6 Urinary Disorders Including Cystitis
Cystitis, radiation cystitis, and hematuria, including that requiring hospitalization, has been reported with JEVTANA in patients who previously received pelvic radiation [see Adverse Reactions (6.2)]. In PROSELICA, cystitis and radiation cystitis were reported in 1.2% and 1.5% of patients who received prior radiation, respectively. Hematuria was reported in 19.4% of patients who received prior radiation and in 14.4% of patients who did not receive prior radiation. Cystitis from radiation recall may occur late in treatment with JEVTANA. Monitor patients who previously received pelvic radiation for signs and symptoms of cystitis while on JEVTANA. Interrupt or discontinue JEVTANA in patients experiencing severe hemorrhagic cystitis. Medical and/or surgical supportive treatment may be required to treat severe hemorrhagic cystitis.
5.7 Respiratory Disorders
Interstitial pneumonia/pneumonitis, interstitial lung disease and acute respiratory distress syndrome have been reported and may be associated with fatal outcome [see Adverse Reactions (6.2)]. Patients with underlying lung disease may be at higher risk for these events. Acute respiratory distress syndrome may occur in the setting of infection.
Interrupt JEVTANA if new or worsening pulmonary symptoms develop. Closely monitor, promptly investigate, and appropriately treat patients receiving JEVTANA. Consider discontinuation. The benefit of resuming JEVTANA treatment must be carefully evaluated.
5.8 Use in Patients with Hepatic Impairment
Cabazitaxel is extensively metabolized in the liver.
JEVTANA is contraindicated in patients with severe hepatic impairment (total bilirubin >3 × ULN) [see Contraindications (4)]. Dose should be reduced for patients with mild (total bilirubin >1 to ≤1.5 × ULN or AST >1.5 × ULN) and moderate (total bilirubin >1.5 to ≤3.0 × ULN and any AST) hepatic impairment, based on tolerability data in these patients [see Dosage and Administration (2.3) and Use in Specific Populations (8.7)]. Administration of JEVTANA to patients with mild and moderate hepatic impairment should be undertaken with caution and close monitoring of safety.
5.9 Embryo-Fetal Toxicity
Based on findings in animal reproduction studies and its mechanism of action, JEVTANA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data in pregnant women to inform the drug-associated risk. In animal reproduction studies, intravenous administration of cabazitaxel in pregnant rats during organogenesis caused embryonic and fetal death at doses lower than the maximum recommended human dose (approximately 0.06 times the Cmax in patients at the recommended human dose). Advise males with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of JEVTANA [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in another section of the label:
- Bone Marrow Suppression [see Warnings and Precautions (5.1)]
- Increased Toxicities in Elderly Patients [see Warnings and Precautions (5.2)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Gastrointestinal Adverse Reactions [see Warnings and Precautions (5.4)]
- Renal Failure [see Warnings and Precautions (5.5)]
- Urinary Disorders Including Cystitis [see Warnings and Precautions (5.6)]
- Respiratory Disorders [see Warnings and Precautions (5.7)]
- Use in Patients with Hepatic Impairment [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other trials and may not reflect the rates observed in clinical practice.
TROPIC Trial (JEVTANA + prednisone compared to mitoxantrone)
The safety of JEVTANA in combination with prednisone was evaluated in 371 patients with metastatic castration-resistant prostate cancer treated in the randomized TROPIC trial, compared to mitoxantrone plus prednisone.
Deaths due to causes other than disease progression within 30 days of last study drug dose were reported in 18 (5%) JEVTANA-treated patients and 3 (<1%) mitoxantrone-treated patients. The most common fatal adverse reactions in JEVTANA-treated patients were infections (n=5) and renal failure (n=4). The majority (4 of 5 patients) of fatal infection-related adverse reactions occurred after a single dose of JEVTANA. Other fatal adverse reactions in JEVTANA-treated patients included ventricular fibrillation, cerebral hemorrhage, and dyspnea.
The most common (≥10%) grade 1–4 adverse reactions were anemia, leukopenia, neutropenia, thrombocytopenia, diarrhea, fatigue, nausea, vomiting, constipation, asthenia, abdominal pain, hematuria, back pain, anorexia, peripheral neuropathy, pyrexia, dyspnea, dysgeusia, cough, arthralgia, and alopecia.
The most common (≥5%) grade 3–4 adverse reactions in patients who received JEVTANA were neutropenia, leukopenia, anemia, febrile neutropenia, diarrhea, fatigue, and asthenia.
Treatment discontinuations due to adverse drug reactions occurred in 18% of patients who received JEVTANA and 8% of patients who received mitoxantrone. The most common adverse reactions leading to treatment discontinuation in the JEVTANA group were neutropenia and renal failure. Dose reductions were reported in 12% of JEVTANA-treated patients and 4% of mitoxantrone-treated patients. Dose delays were reported in 28% of JEVTANA-treated patients and 15% of mitoxantrone-treated patients.
Table 2: Incidence of Adverse Reactions* and Hematologic Abnormalities in ≥5% of Patients Receiving JEVTANA in Combination with Prednisone or Mitoxantrone in Combination with Prednisone in TROPIC JEVTANA 25 mg/m2 every 3 weeks with prednisone 10 mg daily
n=371Mitoxantrone 12 mg/m2 every 3 weeks with prednisone 10 mg daily
n=371Grade 1–4
n (%)Grade 3–4
n (%)Grade 1–4
n (%)Grade 3–4
n (%)- * Graded using NCI CTCAE version 3.
- † Based on laboratory values, JEVTANA: n=369, mitoxantrone: n=370.
- ‡ Includes atrial fibrillation, atrial flutter, atrial tachycardia, atrioventricular block complete, bradycardia, palpitations, supraventricular tachycardia, tachyarrhythmia, and tachycardia.
- § Includes abdominal discomfort, abdominal pain lower, abdominal pain upper, abdominal tenderness, and GI pain.
- ¶ Includes gastroesophageal reflux disease and reflux gastritis.
- # Includes urinary tract infection enterococcal and urinary tract infection fungal.
- Þ Includes peripheral motor neuropathy and peripheral sensory neuropathy.
Any Adverse Reaction Blood and Lymphatic System Disorders Neutropenia† 347 (94%) 303 (82%) 325 (87%) 215 (58%) Febrile Neutropenia 27 (7%) 27 (7%) 5 (1%) 5 (1%) Anemia† 361 (98%) 39 (11%) 302 (82%) 18 (5%) Leukopenia† 355 (96%) 253 (69%) 343 (93%) 157 (42%) Thrombocytopenia† 176 (48%) 15 (4%) 160 (43%) 6 (2%) Cardiac Disorders Arrhythmia‡ 18 (5%) 4 (1%) 6 (2%) 1 (<1%) Gastrointestinal Disorders Diarrhea 173 (47%) 23 (6%) 39 (11%) 1 (<1%) Nausea 127 (34%) 7 (2%) 85 (23%) 1 (<1%) Vomiting 83 (22%) 6 (2%) 38 (10%) 0 Constipation 76 (20%) 4 (1%) 57 (15%) 2 (<1%) Abdominal Pain§ 64 (17%) 7 (2%) 23 (6%) 0 Dyspepsia¶ 36 (10%) 0 9 (2%) 0 General Disorders and Administration Site Conditions Fatigue 136 (37%) 18 (5%) 102 (27%) 11 (3%) Asthenia 76 (20%) 17 (5%) 46 (12%) 9 (2%) Pyrexia 45 (12%) 4 (1%) 23 (6%) 1 (<1%) Peripheral Edema 34 (9%) 2 (<1%) 34 (9%) 2 (<1%) Mucosal Inflammation 22 (6%) 1 (<1%) 10 (3%) 1 (<1%) Pain 20 (5%) 4 (1%) 18 (5%) 7 (2%) Infections and Infestations Urinary Tract Infection# 29 (8%) 6 (2%) 12 (3%) 4 (1%) Investigations Weight Decreased 32 (9%) 0 28 (8%) 1 (<1%) Metabolism and Nutrition Disorders Anorexia 59 (16%) 3 (<1%) 39 (11%) 3 (<1%) Dehydration 18 (5%) 8 (2%) 10 (3%) 3 (<1%) Musculoskeletal and Connective Tissue Disorders Back Pain 60 (16%) 14 (4%) 45 (12%) 11 (3%) Arthralgia 39 (11%) 4 (1%) 31 (8%) 4 (1%) Muscle Spasms 27 (7%) 0 10 (3%) 0 Nervous System Disorders Peripheral NeuropathyÞ 50 (13%) 3 (<1%) 12 (3%) 3 (<1%) Dysgeusia 41 (11%) 0 15 (4%) 0 Dizziness 30 (8%) 0 21 (6%) 2 (<1%) Headache 28 (8%) 0 19 (5%) 0 Renal and Urinary Tract Disorders Hematuria 62 (17%) 7 (2%) 13 (4%) 1 (<1%) Dysuria 25 (7%) 0 5 (1%) 0 Respiratory, Thoracic and Mediastinal Disorders Dyspnea 43 (12%) 4 (1%) 16 (4%) 2 (<1%) Cough 40 (11%) 0 22 (6%) 0 Skin and Subcutaneous Tissue Disorders Alopecia 37 (10%) 0 18 (5%) 0 Vascular Disorders Hypotension 20 (5%) 2 (<1 %) 9 (2%) 1 (<1%) Median Duration of Treatment 6 cycles 4 cycles PROSELICA Trial (comparison of two doses of JEVTANA)
In a noninferiority, multicenter, randomized, open-label study (PROSELICA), 1175 patients with metastatic castration-resistant prostate cancer, previously treated with a docetaxel-containing regimen, were treated with either JEVTANA 25 mg/m2 (n=595) or the 20 mg/m2 (n=580) dose.
Deaths within 30 days of last study drug dose were reported in 22 (3.8%) patients in the 20 mg/m2 and 32 (5.4%) patients in the 25 mg/m2 arm. The most common fatal adverse reactions in JEVTANA-treated patients were related to infections, and these occurred more commonly on the 25 mg/m2 arm (n=15) than on the 20 mg/m2 arm (n=8). Other fatal adverse reactions in JEVTANA-treated patients included cerebral hemorrhage, respiratory failure, paralytic ileus, diarrhea, acute pulmonary edema, disseminated intravascular coagulation, renal failure, sudden death, cardiac arrest, ischemic stroke, diverticular perforation, and cardiorenal syndrome.
Grade 1–4 adverse reactions occurring ≥5% more commonly in patients on the 25 mg/m2 versus 20 mg/m2 arms were leukopenia, neutropenia, thrombocytopenia, febrile neutropenia, decreased appetite, nausea, diarrhea, asthenia, and hematuria.
Grade 3–4 adverse reactions occurring ≥5% more commonly in patients on the 25 mg/m2 versus 20 mg/m2 arms were leukopenia, neutropenia, and febrile neutropenia.
Treatment discontinuations due to adverse drug reactions occurred in 17% of patients in the 20 mg/m2 group and 20% of patients in the 25 mg/m2 group. The most common adverse reactions leading to treatment discontinuation were fatigue and hematuria. The patients in the 20 mg/m2 group received a median of 6 cycles (median duration of 18 weeks), while patients in the 25 mg/m2 group received a median of 7 cycles (median duration of 21 weeks). In the 25 mg/m2 group, 128 patients (22%) had a dose reduced from 25 to 20 mg/m2, 19 patients (3%) had a dose reduced from 20 to 15 mg/m2 and 1 patient (0.2%) had a dose reduced from 15 to 12 mg/m2. In the 20 mg/m2 group, 58 patients (10%) had a dose reduced from 20 to 15 mg/m2, and 9 patients (2%) had a dose reduced from 15 to 12 mg/m2.
Table 3: Incidence of Adverse Reactions* in ≥5% of Patients Receiving JEVTANA 20 mg/m2 or 25 mg/m2 in Combination with Prednisone in PROSELICA JEVTANA 20 mg/m2 every 3 weeks with prednisone 10 mg daily
n=580JEVTANA 25 mg/m2 every 3 weeks with prednisone 10 mg daily
n=595Primary System Organ Class
Preferred TermGrade 1–4
n (%)Grade 3–4
n (%)Grade 1–4
n (%)Grade 3–4
n (%)- * Grade from NCI CTCAE version 4.03.
- † Based on adverse event reporting.
- ‡ Includes urinary tract infection staphylococcal, urinary tract infection bacterial, urinary tract infection fungal, and urosepsis.
- § Includes neutropenic sepsis.
Blood and Lymphatic System Disorders Febrile Neutropenia 12 (2%) 12 (2%) 55 (9%) 55 (9%) Neutropenia† 18 (3%) 14 (2%) 65 (11%) 57 (10%) Infections and Infestations Urinary tract infection‡ 43 (7%) 12 (2%) 66 (11%) 14 (2%) Neutropenic infection§ 15 (3%) 13 (2%) 42 (7%) 36 (6%) Metabolism and Nutrition Disorders Decreased appetite 76 (13%) 4 (0.7%) 110 (19%) 7 (1%) Nervous System Disorders Dysgeusia 41 (7%) 0 63 (11%) 0 Peripheral sensory neuropathy 38 (7%) 0 63 (11%) 4 (0.7%) Dizziness 24 (4%) 0 32 (5%) 0 Headache 29 (5%) 1 (0.2%) 24 (4%) 1 (0.2%) Respiratory, Thoracic and Mediastinal Disorders Dyspnea 30 (5%) 5 (0.9%) 46 (8%) 4 (0.7%) Cough 34 (6%) 0 35 (6%) 0 Gastrointestinal Disorders Diarrhea 178 (31%) 8 (1%) 237 (40%) 24 (4%) Nausea 142 (25%) 4 (0.7%) 191 (32%) 7 (1%) Vomiting 84 (15%) 7 (1.2%) 108 (18 %) 8 (1%) Constipation 102 (18%) 2 (0.3%) 107 (18%) 4 (0.7%) Abdominal pain 34 (6%) 3 (0.5%) 52 (9%) 7 (1%) Stomatitis 27 (5%) 0 30 (5%) 2 (0.3%) Skin and Subcutaneous Tissue Disorders Alopecia 15 (3%) 0 36 (6.1%) 0 Musculoskeletal and Connective Tissue Disorders Back pain 64 (11%) 5 (0.9%) 83 (14%) 7 (1%) Bone pain 46 (8%) 10 (2%) 50 (8%) 13 (2 %) Arthralgia 49 (8%) 3 (0.5%) 41 (7%) 5 (0.8%) Pain in extremity 30 (5%) 1 (0.2%) 41 (7%) 3 (0.5%) Renal and Urinary Disorders Hematuria 82 (14%) 11 (2%) 124 (21%) 25 (4%) Dysuria 31 (5%) 2 (0.3%) 24 (4%) 0 General Disorders and Administration Site Conditions Fatigue 143 (25%) 15 (3%) 161 (27%) 22 (4%) Asthenia 89 (15%) 11 (2%) 117 (20%) 12 (2%) Edema peripheral 39 (7%) 1 (0.2%) 53 (9%) 1 (0.2%) Pyrexia 27 (5%) 1 (0.2%) 38 (6 %) 1 (0.2%) Investigations Weight decreased 24 (4%) 1 (0.2%) 44 (7%) 0 Injury, Poisoning and Procedural Complications Wrong technique in drug usage process 2 (0.3%) 0 32 (5%) 0 Table 4: Incidence of Hematologic Laboratory Abnormalities in Patients Receiving JEVTANA 20 mg/m2 or 25 mg/m2 in Combination with Prednisone in Study PROSELICA Laboratory Abnormality JEVTANA 20 mg/m2 every 3 weeks with prednisone 10 mg daily
n=577JEVTANA 25 mg/m2 every 3 weeks with prednisone 10 mg daily
n=590Grade 1–4
n (%)Grade 3–4
n (%)Grade 1–4
n (%)Grade 3–4
n (%)Neutropenia 384 (67%) 241 (42%) 522 (89%) 432 (73%) Anemia 576 (99.8%) 57 (10%) 588 (99.7%) 81 (14%) Leukopenia 461 (80%) 167 (29%) 560 (95%) 351 (60%) Thrombocytopenia 202 (35%) 15 (3%) 251 (43%) 25 (4%) Hematuria
In study TROPIC, adverse reactions of hematuria, including those requiring medical intervention, were more common in JEVTANA-treated patients. The incidence of grade ≥2 hematuria was 6% in JEVTANA-treated patients and 2% in mitoxantrone-treated patients. Other factors associated with hematuria were well-balanced between arms and do not account for the increased rate of hematuria on the JEVTANA arm.
In study PROSELICA, hematuria of all grades was observed in 18% of patients overall.
6.2 Postmarketing Experience
The following adverse reactions have been identified from clinical trials and/or postmarketing surveillance. Because they are reported from a population of unknown size, precise estimates of frequency cannot be made.
Gastrointestinal: Gastritis, intestinal obstruction.
Respiratory: Interstitial pneumonia/pneumonitis, interstitial lung disease and acute respiratory distress syndrome.
Renal and urinary disorders: Radiation recall hemorrhagic cystitis.
-
7 DRUG INTERACTIONS
7.1 CYP3A Inhibitors
Cabazitaxel is primarily metabolized through CYP3A [see Clinical Pharmacology (12.3)]. Strong CYP3A inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, voriconazole) may increase plasma concentrations of cabazitaxel. Avoid the coadministration of JEVTANA with strong CYP3A inhibitors. If patients require coadministration of a strong CYP3A inhibitor, consider a 25% JEVTANA dose reduction [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The safety and efficacy of JEVTANA have not been established in females. There are no human data on the use of JEVTANA in pregnant women to inform the drug-associated risk. In animal reproduction studies, intravenous administration of cabazitaxel in pregnant rats during organogenesis caused embryonic and fetal death at doses lower than the maximum recommended human dose [see Data].
Data
Animal data
In an early embryonic developmental toxicity study in rats, cabazitaxel was administered intravenously for 15 days prior to mating through Day 6 of pregnancy, which resulted in an increase in pre-implantation loss at 0.2 mg/kg/day and an increase in early resorptions at ≥0.1 mg/kg/day (approximately 0.06 and 0.02 times the Cmax in patients at the recommended human dose, respectively).
In an embryo-fetal developmental toxicity study in rats, cabazitaxel caused maternal and embryo-fetal toxicity consisting of increased postimplantation loss, embryolethality, and fetal deaths when administered intravenously at a dose of 0.16 mg/kg/day (approximately 0.06 times the Cmax in patients at the recommended human dose). Decreased mean fetal birthweight associated with delays in skeletal ossification was observed at doses ≥0.08 mg/kg. Cabazitaxel crossed the placenta barrier within 24 hours of a single intravenous administration of 0.08 mg/kg to pregnant rats at gestational day 17. A dose of 0.08 mg/kg in rats resulted in a Cmax approximately 0.02 times that observed in patients at the recommended human dose. Administration of cabazitaxel did not result in fetal abnormalities in rats or rabbits at exposure levels significantly lower than the expected human exposures.
8.2 Lactation
Risk Summary
The safety and efficacy of JEVTANA have not been established in females. There is no information available on the presence of cabazitaxel in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. Cabazitaxel or cabazitaxel metabolites are excreted in maternal milk of lactating rats [see Data].
Data
Animal data
In a milk excretion study, radioactivity related to cabazitaxel was detected in the stomachs of nursing pups within 2 hours of a single intravenous administration of cabazitaxel to lactating rats at a dose of 0.08 mg/kg (approximately 0.02 times the Cmax in patients at the recommended human dose). This was detectable 24 hours post dose. Approximately 1.5% of the dose delivered to the mother was calculated to be delivered in the maternal milk.
8.3 Females and Males of Reproductive Potential
Contraception
Males
Based on findings in animal reproduction studies, advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the final dose of JEVTANA [see Use in Specific Populations (8.1)].
Infertility
Males
Based on animal toxicology studies, JEVTANA may impair human fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of JEVTANA in pediatric patients have not been established.
JEVTANA was evaluated in 39 pediatric patients (ages 3 to 18 years) receiving prophylactic G-CSF. The maximum tolerated dose (MTD) was 30 mg/m2 intravenously over 1 hour on Day 1 of a 21 day cycle in pediatric patients with solid tumors based on the dose-limiting toxicity (DLT) of febrile neutropenia. No objective responses were observed in 11 patients with refractory high grade glioma (HGG) or diffuse intrinsic pontine glioma (DIPG). One patient had a partial response among the 9 patients with ependymoma.
Infusion related/hypersensitivity reactions were seen in 10 patients (26%). Three patients experienced serious adverse events of anaphylactic reaction. The incidence of infusion related/hypersensitivity reactions decreased with steroid pre-medication. The most frequent treatment-emergent adverse events were similar to those reported in adults.
Based on the population pharmacokinetics analysis conducted with data from 31 pediatric patients with cancer (ages 3 to 18 years), the clearances by body surface area were comparable to those in adults.
8.5 Geriatric Use
In the TROPIC study, of the 371 patients with prostate cancer treated with JEVTANA every three weeks plus prednisone, 240 patients (64.7%) were 65 years of age and over, while 70 patients (18.9%) were 75 years of age and over. No overall differences in effectiveness were observed between patients ≥65 years of age and younger patients. Elderly patients (≥65 years of age) may be more likely to experience certain adverse reactions. The incidence of death due to causes other than disease progression within 30 days of the last cabazitaxel dose were higher in patients who were 65 years of age or greater compared to younger patients [see Warnings and Precautions (5.2)]. The incidence of grade 3–4 neutropenia and febrile neutropenia were higher in patients who were 65 years of age or greater compared to younger patients. The following grade 1–4 adverse reactions were reported at rates ≥5% higher in patients 65 years of age or older compared to younger patients: fatigue (40% vs 30%), neutropenia (97% vs 89%), asthenia (24% vs 15%), pyrexia (15% vs 8%), dizziness (10% vs 5%), urinary tract infection (10% vs 3%), and dehydration (7% vs 2%), respectively.
In the PROSELICA study, the grade 1–4 adverse reactions reported at rates of at least 5% higher in patients 65 years of age or older compared to younger patients were diarrhea (43% vs 33%), fatigue (30% vs 19%), asthenia (22% vs 13%), constipation (20% vs 13%), clinical neutropenia (13% vs 6%), febrile neutropenia (11% vs 5%), and dyspnea (10% vs 3%).
Based on a population pharmacokinetic analysis, no significant difference was observed in the pharmacokinetics of cabazitaxel between patients <65 years (n=100) and older (n=70).
8.6 Renal Impairment
No dose adjustment is necessary in patients with renal impairment not requiring hemodialysis. Patients presenting with end-stage renal disease (creatinine clearance CLCR <15 mL/min/1.73 m2), should be monitored carefully during treatment [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Cabazitaxel is extensively metabolized in the liver. Patients with mild hepatic impairment (total bilirubin >1 to ≤1.5 × ULN or AST >1.5 × ULN) should have JEVTANA dose of 20 mg/m2. Administration of cabazitaxel to patients with mild hepatic impairment should be undertaken with caution and close monitoring of safety [see Clinical Pharmacology (12.3)]. The maximum tolerated dose in patients with moderate hepatic impairment (total bilirubin >1.5 to ≤3.0 × ULN and AST = any) was 15 mg/m2, however, the efficacy at this dose level was unknown. JEVTANA is contraindicated in patients with severe hepatic impairment (total bilirubin >3 × ULN) [see Contraindications (4)].
-
10 OVERDOSAGE
There is no known antidote for JEVTANA overdose. Overdose has resulted from improper preparation [see Dosage and Administration (2.5)]. Read the entire section Dosage and Administration (2) carefully before mixing or diluting. Complications of overdose include exacerbation of adverse reactions such as bone marrow suppression and gastrointestinal disorders. Overdose has led to fatal outcome.
In case of overdose, the patient should be kept in a specialized unit where vital signs, chemistry and particular functions can be closely monitored. Patients should receive therapeutic G-CSF as soon as possible after discovery of overdose. Other appropriate symptomatic measures should be taken, as needed.
-
11 DESCRIPTION
JEVTANA (cabazitaxel) injection is an antineoplastic agent belonging to the taxane class that is for intravenous use. It is prepared by semi-synthesis with a precursor extracted from yew needles.
The chemical name of cabazitaxel is (2α,5β,7β,10β,13α)-4-acetoxy-13-({(2R,3S)-3-[(tertbutoxycarbonyl) amino]-2-hydroxy-3-phenylpropanoyl}oxy)-1-hydroxy-7,10-dimethoxy-9-oxo-5,20-epoxytax-11-en-2-yl benzoate – propan-2-one (1:1).
Cabazitaxel has the following structural formula:
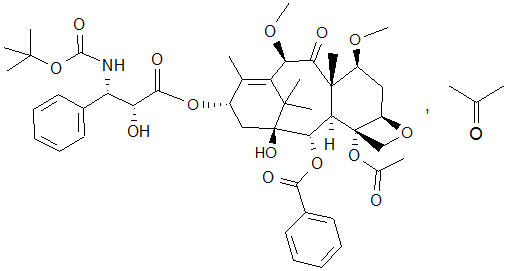
Cabazitaxel is a white to almost-white powder with a molecular formula of C45H57NO14C3H6O and a molecular weight of 894.01 (for the acetone solvate) / 835.93 (for the solvent free). It is lipophilic, practically insoluble in water and soluble in alcohol.
JEVTANA (cabazitaxel) injection 60 mg/1.5 mL is a sterile, non-pyrogenic, clear yellow to brownish-yellow viscous solution and is available in single-dose vials containing 60 mg cabazitaxel (anhydrous and solvent free) and 1.56 g polysorbate 80.
Each mL contains 40 mg cabazitaxel (anhydrous) and 1.04 g polysorbate 80.
DILUENT for JEVTANA is a clear, colorless, sterile, and non-pyrogenic solution containing 13% (w/w) ethanol in water for injection, approximately 5.7 mL.
JEVTANA requires two dilutions prior to intravenous infusion. JEVTANA injection should be diluted only with the supplied DILUENT for JEVTANA, followed by dilution in either 0.9% sodium chloride solution or 5% dextrose solution.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Cabazitaxel is a microtubule inhibitor. Cabazitaxel binds to tubulin and promotes its assembly into microtubules while simultaneously inhibiting disassembly. This leads to the stabilization of microtubules, which results in the inhibition of mitotic and interphase cellular functions.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of cabazitaxel following a single dose of 25 mg/m2 administered by intravenous infusion on QTc interval was evaluated in 94 patients with solid tumors. No large changes in the mean QT interval (i.e., >20 ms) from baseline based on Fridericia correction method were detected. However, a small increase in the mean QTc interval (i.e., <10 ms) cannot be excluded due to study design limitations.
12.3 Pharmacokinetics
A population pharmacokinetic analysis was conducted in 170 patients with solid tumors at doses ranging from 10 to 30 mg/m2 weekly or every three weeks.
Absorption
Based on the population pharmacokinetic analysis, after an intravenous dose of cabazitaxel 25 mg/m2 every three weeks, the mean Cmax in patients with metastatic prostate cancer was 226 ng/mL (CV 107%) and was reached at the end of the one-hour infusion (Tmax). The mean AUC in patients with metastatic prostate cancer was 991 ng∙h/mL (CV 34%).
No major deviation from the dose proportionality was observed from 10 to 30 mg/m2 in patients with advanced solid tumors.
Distribution
The volume of distribution (Vss) was 4,864 L (2,643 L/m2 for a patient with a median BSA of 1.84 m2) at steady state.
In vitro, the binding of cabazitaxel to human serum proteins was 89% to 92% and was not saturable up to 50,000 ng/mL, which covers the maximum concentration observed in clinical trials. Cabazitaxel is mainly bound to human serum albumin (82%) and lipoproteins (88% for HDL, 70% for LDL, and 56% for VLDL). The in vitro blood-to-plasma concentration ratio in human blood ranged from 0.90 to 0.99, indicating that cabazitaxel was equally distributed between blood and plasma.
Metabolism
Cabazitaxel is extensively metabolized in the liver (>95%), mainly by the CYP3A4/5 isoenzyme (80% to 90%), and to a lesser extent by CYP2C8. Cabazitaxel is the main circulating moiety in human plasma. Seven metabolites were detected in plasma (including the 3 active metabolites issued from O-demethylation), with the main one accounting for 5% of cabazitaxel exposure. Around 20 metabolites of cabazitaxel are excreted into human urine and feces.
Elimination
After a one-hour intravenous infusion [14C]-cabazitaxel 25 mg/m2, approximately 80% of the administered dose was eliminated within 2 weeks. Cabazitaxel is mainly excreted in the feces as numerous metabolites (76% of the dose); while renal excretion of cabazitaxel and metabolites account for 3.7% of the dose (2.3% as unchanged drug in urine).
Based on the population pharmacokinetic analysis, cabazitaxel has a plasma clearance of 48.5 L/h (CV 39%; 26.4 L/h/m2 for a patient with a median BSA of 1.84 m2) in patients with metastatic prostate cancer. Following a one-hour intravenous infusion, plasma concentrations of cabazitaxel can be described by a three-compartment pharmacokinetic model with α-, β-, and γ- half-lives of 4 minutes, 2 hours, and 95 hours, respectively.
Renal Impairment
Cabazitaxel is minimally excreted via the kidney. A population pharmacokinetic analysis carried out in 170 patients including 14 patients with moderate renal impairment (30 mL/min ≤CLCR <50 mL/min) and 59 patients with mild renal impairment (50 mL/min ≤CLCR <80 mL/min) showed that mild to moderate renal impairment did not have meaningful effects on the pharmacokinetics of cabazitaxel. This was confirmed by a dedicated comparative pharmacokinetic study in patients with solid tumors with normal renal function (n=8, CLCR >80 mL/min/1.73 m2), or moderate (n=8, 30 mL/min/1.73 m2 ≤CLCR <50 mL/min/1.73 m2) and severe (n=9, CLCR <30 mL/min/1.73 m2) renal impairment, who received several cycles of cabazitaxel in single IV infusion up to 25 mg/m2. Limited pharmacokinetic data were available in patients with end-stage renal disease (n=2, CLCR <15 mL/min/1.73 m2).
Hepatic Impairment
Cabazitaxel is extensively metabolized in the liver.
A dedicated study in 43 cancer patients with hepatic impairment showed no influence of mild (total bilirubin >1 to ≤1.5 × ULN or AST >1.5 × ULN) or moderate (total bilirubin >1.5 to ≤3.0 × ULN) hepatic impairment on cabazitaxel pharmacokinetics. The maximum tolerated dose (MTD) of cabazitaxel was 20 and 15 mg/m2, respectively.
In 3 patients with severe hepatic impairment (total bilirubin >3 × ULN), a 39% decrease in clearance was observed when compared to patients with mild hepatic impairment (ratio=0.61, 90% CI: 0.36–1.05), indicating some effect of severe hepatic impairment on cabazitaxel pharmacokinetics. The MTD of cabazitaxel in patients with severe hepatic impairment was not established. Based on safety and tolerability data, cabazitaxel dose should be maintained at 20 mg/m2 in patients with mild hepatic impairment and reduced to 15 mg/m2 in patients with moderate hepatic impairment [see Warnings and Precautions (5.8) and Use in Specific Populations (8.7)]. Cabazitaxel is contraindicated in patients with severe hepatic impairment [see Contraindications (4) and Use in Specific Populations (8.7)].
Drug Interactions
A drug interaction study of JEVTANA in 23 patients with advanced cancers has shown that repeated administration of ketoconazole (400 mg orally once daily), a strong CYP3A inhibitor, increased the exposure to cabazitaxel (5 mg/m2 intravenous) by 25%.
A drug interaction study of JEVTANA in 13 patients with advanced cancers has shown that repeated administration of aprepitant (125 or 80 mg once daily), a moderate CYP3A inhibitor, did not modify the exposure to cabazitaxel (15 mg/m2 intravenous).
A drug interaction study of JEVTANA in 21 patients with advanced cancers has shown that repeated administration of rifampin (600 mg once daily), a strong CYP3A inducer, decreased the exposure to cabazitaxel (15 mg/m2 intravenous) by 17%.
A drug interaction study of JEVTANA in 11 patients with advanced cancers has shown that cabazitaxel (25 mg/m2 administered as a single 1-hour infusion) did not modify the exposure to midazolam, a probe substrate of CYP3A.
Prednisone or prednisolone administered at 10 mg daily did not affect the pharmacokinetics of cabazitaxel.
Based on in vitro studies, the potential for cabazitaxel to inhibit drugs that are substrates of other CYP isoenzymes (1A2, -2B6, -2C9, -2C8, -2C19, -2E1, -2D6, and CYP3A4/5) is low. In addition, cabazitaxel did not induce CYP isozymes (-1A, -2C9 and -3A) in vitro.
In vitro, cabazitaxel did not inhibit the multidrug-resistance protein 1 (MRP1), 2 (MRP2) or organic cation transporter (OCT1). In vitro, cabazitaxel inhibited P-gp, BRCP, and organic anion transporting polypeptides (OATP1B1, OATP1B3). However the in vivo risk of cabazitaxel inhibiting MRPs, OCT1, P-gp, BCRP, OATP1B1 or OATP1B3 is low at the dose of 25 mg/m2.
In vitro, cabazitaxel is a substrate of P-gp, but not a substrate of MRP1, MRP2, BCRP, OCT1, OATP1B1 or OATP1B3.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been performed to evaluate the carcinogenic potential of cabazitaxel.
Cabazitaxel was positive for clastogenesis in the in vivo micronucleus test, inducing an increase of micronuclei in rats at doses ≥0.5 mg/kg. Cabazitaxel increased numerical aberrations with or without metabolic activation in an in vitro test in human lymphocytes though no induction of structural aberrations was observed. Cabazitaxel did not induce mutations in the bacterial reverse mutation (Ames) test. The positive in vivo genotoxicity findings are consistent with the pharmacological activity of the compound (inhibition of tubulin depolymerization).
In a fertility study performed in female rats at cabazitaxel doses of 0.05, 0.1, or 0.2 mg/kg/day there was no effect of administration of the drug on mating behavior or the ability to become pregnant. In repeat-dose toxicology studies in rats with intravenous cabazitaxel administration once every three weeks for up to 6 months, atrophy of the uterus was observed at the 5 mg/kg dose level (approximately the AUC in patients with cancer at the recommended human dose) along with necrosis of the corpora lutea at doses ≥1 mg/kg (approximately 0.2 times the AUC at the clinically recommended human dose).
In a fertility study in male rats, cabazitaxel did not affect mating performances or fertility at doses of 0.05, 0.1, or 0.2 mg/kg/day. In repeat-dose toxicology studies with intravenous cabazitaxel administration once every three weeks for up to 9 months, degeneration of seminal vesicle and seminiferous tubule atrophy in the testis were observed in rats at a dose of 1 mg/kg (approximately 0.2 times the AUC in patients at the recommended human dose), and minimal testicular degeneration (minimal epithelial single cell necrosis in epididymis) was observed in dogs treated at a dose of 0.5 mg/kg (approximately 0.1 times the AUC in patients at the recommended human dose).
-
14 CLINICAL STUDIES
14.1 TROPIC Trial (JEVTANA + prednisone compared to mitoxantrone)
The efficacy and safety of JEVTANA in combination with prednisone were evaluated in a randomized, open-label, international, multi-center study in patients with metastatic castration-resistant prostate cancer previously treated with a docetaxel-containing treatment regimen (TROPIC, NCT00417079).
A total of 755 patients were randomized to receive either JEVTANA 25 mg/m2 intravenously every 3 weeks for a maximum of 10 cycles with prednisone 10 mg orally daily (n=378), or to receive mitoxantrone 12 mg/m2 intravenously every 3 weeks for 10 cycles with prednisone 10 mg orally daily (n=377) for a maximum of 10 cycles.
This study included patients over 18 years of age with hormone-refractory metastatic prostate cancer either measurable by RECIST criteria or non-measurable disease with rising PSA levels or appearance of new lesions, and ECOG (Eastern Cooperative Oncology Group) performance status 0–2. Patients had to have neutrophils >1,500 cells/mm3, platelets >100,000 cells/mm3, hemoglobin >10 g/dL, creatinine <1.5 × upper limit of normal (ULN), total bilirubin <1 × ULN, AST <1.5 × ULN, and ALT <1.5 × ULN. Patients with a history of congestive heart failure, or myocardial infarction within the last 6 months, or patients with uncontrolled cardiac arrhythmias, angina pectoris, and/or hypertension were not included in the study.
Demographics, including age, race, and ECOG performance status (0–2) were balanced between the treatment arms. The median age was 68 years (range 46–92) and the racial distribution for all groups was 83.9% Caucasian, 6.9% Asian, 5.3% Black, and 4% Others in the JEVTANA group.
Efficacy results for the JEVTANA arm versus the control arm are summarized in Table 5 and Figure 1.
Table 5: Efficacy of JEVTANA in TROPIC in the Treatment of Patients with Metastatic Castration-Resistant Prostate Cancer (intent-to-treat analysis) JEVTANA + Prednisone
n=378Mitoxantrone + Prednisone
n=377- * Hazard ratio estimated using Cox model; a hazard ratio of less than 1 favors JEVTANA.
Overall Survival Number of deaths (%) 234 (61.9 %) 279 (74.0%) Median survival (month) (95% CI) 15.1 (14.1–16.3) 12.7 (11.6–13.7) Hazard Ratio* (95% CI) 0.70 (0.59–0.83) p-value <0.0001 Figure 1: Kaplan-Meier Overall Survival Curves (TROPIC)
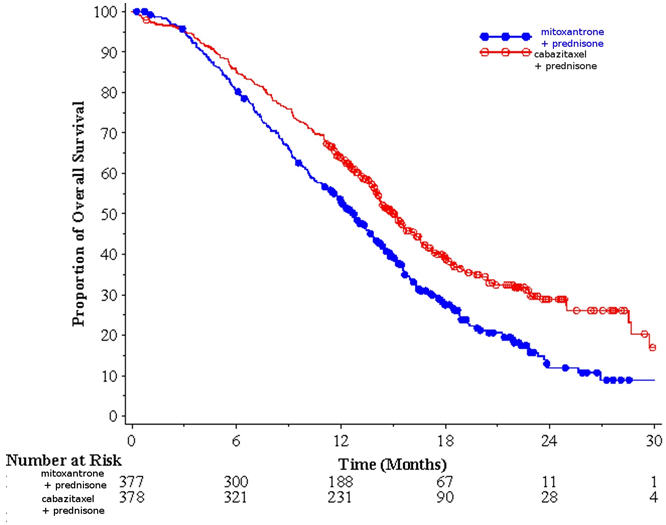
Investigator-assessed tumor response of 14.4% (95% CI: 9.6–19.3) was higher for patients in the JEVTANA arm compared to 4.4% (95% CI: 1.6–7.2) for patients in the mitoxantrone arm, p=0.0005.
14.2 PROSELICA Trial (comparison of two doses of JEVTANA)
In a noninferiority, multicenter, randomized, open-label study (PROSELICA, NCT01308580), 1200 patients with metastatic castration-resistant prostate cancer, previously treated with a docetaxel-containing regimen, were randomized to receive either JEVTANA 25 mg/m2 (n=602) or 20 mg/m2 (n=598) dose. Overall survival (OS) was the major efficacy outcome.
Demographics, including age, race, and ECOG performance status (0–2) were balanced between the treatment arms. The median age was 68 years (range 45–89) and the racial distribution for all groups was 87% Caucasian, 6.9% Asian, 2.3% Black, and 3.8% Others in the JEVTANA 20 mg/m2 group. The median age was 69 years (range 45–88) and the racial distribution for all groups was 88.7% Caucasian, 6.6% Asian, 1.8% Black, and 2.8% Others in the JEVTANA 25 mg/m2 group.
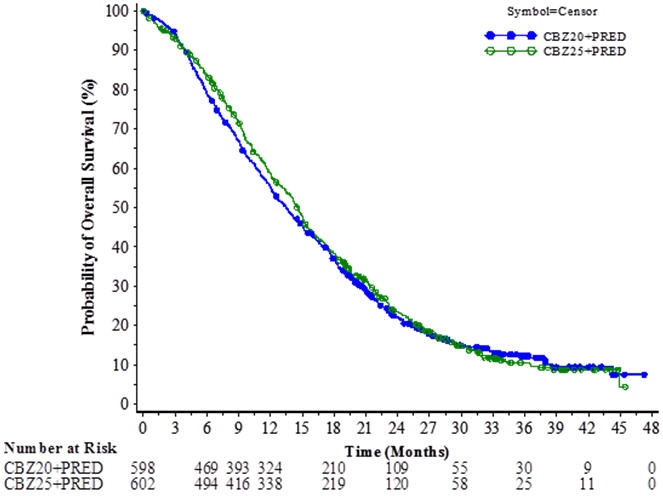
The study demonstrated noninferiority in overall survival (OS) of JEVTANA 20 mg/m2 in comparison with JEVTANA 25 mg/m2 in an intent-to-treat population (see Table 6 and Figure 2). Based on the per-protocol population, the estimated median OS was 15.1 months on JEVTANA 20 mg/m2 and 15.9 months on JEVTANA 25 mg/m2, the observed hazard ratio (HR) of OS was 1.042 (97.78% CI: 0.886, 1.224). Among the subgroup analyses intended for assessing the heterogeneity, no notable difference in OS was observed on the JEVTANA 25 mg/m2 arm compared to the JEVTANA 20 mg/m2 arm in subgroups based on the stratification factors of ECOG performance status score, measurability of disease, or region.
Table 6: Overall Survival in PROSELICA for JEVTANA 20 mg/m2 versus JEVTANA 25 mg/m2 (intent-to-treat analysis) CBZ20+PRED
n=598CBZ25+PRED
n=602CBZ20=Cabazitaxel 20 mg/m2, CBZ25=Cabazitaxel 25 mg/m2, PRED=Prednisone/Prednisolone. CI=confidence interval. - * Hazard ratio is estimated using a Cox Proportional Hazards regression model. A hazard ratio <1 indicates a lower risk of death for Cabazitaxel 20 mg/m2 with respect to 25 mg/m2.
- † Adjusted for interim OS analyses. The noninferiority margin is 1.214.
Overall Survival Number of deaths, n (%) 497 (83.1 %) 501 (83.2%) Median survival (95% CI) (months) 13.4 (12.2 to 14.9) 14.5 (13.5 to 15.3) Hazard Ratio* (97.78% CI†) 1.024 (0.886, 1.184) Figure 2: Kaplan-Meier Overall Survival Curves (intent-to-treat population) (PROSELICA)
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
JEVTANA is supplied as a kit consisting of the following:
- One single-dose vial of JEVTANA (cabazitaxel) injection: a clear yellow to brownish-yellow viscous solution of 60 mg/1.5 mL in a clear glass vial with a grey rubber closure, aluminum cap, and light green plastic flip-off cap
- One single-dose vial of Diluent for JEVTANA: a clear colorless solution of 13% (w/w) ethanol in water for injection in a clear glass vial with a grey rubber closure, gold-color aluminum cap, and colorless plastic flip-off cap.
Both items are in a blister pack in one carton.
NDC: 0024-5824-11
16.2 Storage
JEVTANA injection and Diluent for JEVTANA:
Store at 25°C (77°F); excursions permitted between 15°C–30°C (59°F–86°F).
Do not refrigerate.
16.3 Handling and Disposal
JEVTANA is a cytotoxic anticancer drug. Follow applicable special handling and disposable procedures [see References (15)].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Hypersensitivity Reactions
Educate patients about the risk of potential hypersensitivity associated with JEVTANA. Confirm patients do not have a history of severe hypersensitivity reactions to cabazitaxel or to other drugs formulated with polysorbate 80. Instruct patients to immediately report signs of a hypersensitivity reaction [see Contraindications (4) and Warnings and Precautions (5.3)].
Bone Marrow Suppression
Inform patients that JEVTANA decreases blood count such as white blood cells, platelets and red blood cells. Thus, it is important that periodic assessment of their blood count be performed to detect the development of neutropenia, thrombocytopenia, anemia, and/or pancytopenia [see Contraindications (4) and Warnings and Precautions (5.1)]. Instruct patients to monitor their temperature frequently and immediately report any occurrence of fever to their healthcare provider.
Increased Toxicities in Elderly Patients
Inform elderly patients that certain side effects may be more frequent or severe [see Warnings and Precautions (5.2) and Use in Specific Populations (8.5)].
Importance of Prednisone
Explain that it is important to take the oral prednisone as prescribed. Instruct patients to report if they were not compliant with oral corticosteroid regimen [see Dosage and Administration (2.1)].
Infections, Dehydration, Renal Failure
Explain to patients that severe and fatal infections, dehydration, and renal failure have been associated with cabazitaxel exposure. Patients should immediately report fever, significant vomiting or diarrhea, decreased urinary output, and hematuria to their healthcare provider [see Warnings and Precautions (5.1, 5.4, 5.5)].
Urinary Disorders Including Cystitis
Inform patients that hematuria may occur during treatment with JEVTANA. Inform patients that previously received pelvic radiation that cystitis and radiation cystitis may occur during treatment with JEVTANA. Advise patients to report any occurrence of hematuria, or any signs and symptoms of cystitis or radiation cystitis, to their healthcare provider [see Warnings and Precautions (5.6)].
Respiratory Disorders
Explain to patients that severe and fatal interstitial pneumonia/pneumonitis, interstitial lung disease and acute respiratory distress syndrome have occurred with JEVTANA. Instruct patients to immediately report new or worsening pulmonary symptoms to their healthcare provider [see Warnings and Precautions (5.7)].
Drug Interactions
Inform patients about the risk of drug interactions and the importance of providing a list of prescription and non-prescription drugs to their healthcare provider [see Drug Interactions (7.1)].
Embryo-Fetal Toxicity
Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of JEVTANA [see Use in Specific Populations (8.3)].
Infertility
Advise male patients that JEVTANA may impair fertility [see Use in Specific Populations (8.3)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
Patient Information
JEVTANA® (JEV-ta-na)
(cabazitaxel)
InjectionThis Patient Information has been approved by the U.S. Food and Drug Administration. Revised: March 2020 What is the most important information I should know about JEVTANA?
JEVTANA may cause serious side effects including:-
Low white blood cells. Low white blood cells can cause you to get serious infections, and may lead to death. Men who are 65 years or older may be more likely to have these problems. Your healthcare provider:
- will do blood tests regularly to check your white blood cell counts during your treatment with JEVTANA.
- may lower your dose of JEVTANA, change how often you receive it, or stop JEVTANA until your healthcare provider decides that you have enough white blood cells.
- may prescribe a medicine for you called G-CSF, to help prevent complications if your white blood cell count is too low.
Tell your healthcare provider right away if you have any of these symptoms of infection during treatment with JEVTANA:- fever. Take your temperature often during treatment with JEVTANA.
- cough
- burning on urination
- muscle aches
Also, tell your healthcare provider if you have any diarrhea during the time that your white blood cell count is low. Your healthcare provider may prescribe treatment for you as needed. -
Severe allergic reactions. Severe allergic reactions can happen within a few minutes after your infusion of JEVTANA starts, especially during the first and second infusions. Your healthcare provider should prescribe medicines before each infusion to help prevent severe allergic reactions.
Tell your healthcare provider or nurse right away if you have any of these symptoms of a severe allergic reaction during or soon after an infusion of JEVTANA:
- rash or itching
- feeling dizzy or faint
- chest or throat tightness
- skin redness
- breathing problems
- swelling of your face
-
Severe stomach and intestine (gastrointestinal) problems.
-
JEVTANA can cause severe vomiting and diarrhea, which may lead to death. Severe vomiting and diarrhea with JEVTANA can lead to loss of too much body fluid (dehydration), or too much of your body salts (electrolytes). Death has happened from having severe diarrhea and losing too much body fluid or body salts with JEVTANA. You may need to go to a hospital for treatment. Your healthcare provider will prescribe medicines to prevent or treat vomiting and diarrhea, as needed, with JEVTANA.
Tell your healthcare provider right away if you develop vomiting or diarrhea or if your symptoms get worse or do not get better. -
JEVTANA can cause a leak in the stomach or intestine, intestinal blockage, infection, and bleeding in the stomach or intestine, which may lead to death.
Tell your healthcare provider if you develop any of these symptoms:- severe stomach-area (abdomen) pain
- constipation
- fever
- blood in your stool, or changes in the color of your stool
-
JEVTANA can cause severe vomiting and diarrhea, which may lead to death. Severe vomiting and diarrhea with JEVTANA can lead to loss of too much body fluid (dehydration), or too much of your body salts (electrolytes). Death has happened from having severe diarrhea and losing too much body fluid or body salts with JEVTANA. You may need to go to a hospital for treatment. Your healthcare provider will prescribe medicines to prevent or treat vomiting and diarrhea, as needed, with JEVTANA.
-
Kidney failure. Kidney failure may happen with JEVTANA, because of severe infection, loss of too much body fluid (dehydration), and other reasons, which may lead to death. Your healthcare provider will check you for this problem and treat you if needed.
Tell your healthcare provider if you develop these signs or symptoms:- swelling of your face or body
- decrease in the amount of urine that your body makes each day
- blood in your urine
-
Lung or breathing problems. Lung or breathing problems may happen with JEVTANA and may lead to death. Men who have lung disease before receiving JEVTANA may have a higher risk for developing lung or breathing problems with JEVTANA treatment. Your healthcare provider will check you for this problem and treat you if needed.
Tell your healthcare provider right away if you develop any new or worsening symptoms, including trouble breathing, shortness of breath, chest pain, cough or fever.
What is JEVTANA?
JEVTANA is a prescription medicine used with the steroid medicine prednisone. JEVTANA is used to treat men with castration-resistant prostate cancer (prostate cancer that is resistant to medical or surgical treatments that lower testosterone) that has spread to other parts of the body, and that has worsened (progressed) after treatment with other medicines that included docetaxel.
It is not known if JEVTANA is safe and effective in children.Who should not receive JEVTANA?
Do not receive JEVTANA if:- your white blood cell (neutrophil count) is too low
- you have had a severe allergic reaction to cabazitaxel or other medicines that contain polysorbate 80. Ask your healthcare provider if you are not sure.
- you have severe liver problems
Before receiving JEVTANA, tell your healthcare provider about all your medical conditions, including if you: - are over the age of 65
- had allergic reactions in the past
- have kidney or liver problems
- have lung problems
- are pregnant or plan to become pregnant. JEVTANA can cause harm to your unborn baby and loss of pregnancy (miscarriage).
- are a male with a female partner who is able to become pregnant. Males should use effective birth control (contraception) during treatment with JEVTANA and for 3 months after the last dose of JEVTANA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
JEVTANA can interact with many other medicines. Do not take any new medicines without asking your healthcare provider first. Your healthcare provider will tell you if it is safe to take the new medicine with JEVTANA.How will I receive JEVTANA? - JEVTANA will be given to you by an intravenous (IV) infusion into your vein.
- Your treatment will take about 1 hour.
- JEVTANA is usually given every 3 weeks. Your healthcare provider will decide how often you will receive JEVTANA.
- Your healthcare provider will also prescribe another medicine called prednisone for you to take by mouth every day during treatment with JEVTANA.
- Your healthcare provider will tell you how and when to take your prednisone.
- It is important that you take prednisone exactly as prescribed by your healthcare provider. If you forget to take your prednisone, or do not take it on schedule, make sure to tell your healthcare provider or nurse.
- Before each infusion of JEVTANA, you may receive other medicines to prevent or treat side effects.
What are the possible side effects of JEVTANA?
JEVTANA may cause serious side effects including:- See "What is the most important information I should know about JEVTANA?"
- Inflammation of the bladder and blood in the urine. Blood in the urine is common with JEVTANA, but it can also sometimes be severe. Some people who have had pelvic radiation in the past may develop inflammation of the bladder and blood in the urine that is severe enough that they need to be hospitalized for medical treatment or surgery. Your healthcare provider will check you for these problems during treatment with JEVTANA. Your healthcare provider may stop your treatment with JEVTANA for a short time, or permanently, if you develop inflammation of the bladder and bleeding that is severe.
- Low red blood cell count (anemia). Low red blood cell count is common with JEVTANA, but can sometimes also be serious. Your healthcare provider will regularly check your red blood cell count. Symptoms of anemia include shortness of breath and tiredness.
- Low blood platelet count. Low platelet count is common with JEVTANA, but can sometimes also be serious. Tell your healthcare provider if you have any unusual bruising or bleeding.
- diarrhea
- tiredness
- nausea
- vomiting
- constipation
- weakness
- stomach (abdominal) pain
- back pain
- decreased appetite
- shortness of breath
- hair loss
- cough
JEVTANA may cause fertility problems in males. This may affect your ability to father a child. Talk to your healthcare provider if you have concerns about fertility.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of JEVTANA. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of JEVTANA
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about JEVTANA that is written for health professionals.What are the ingredients in JEVTANA?
Active ingredient: cabazitaxel
Inactive ingredient: polysorbate 80
Manufactured by: sanofi-aventis U.S. LLC, Bridgewater, NJ 08807, A SANOFI COMPANY
JEVTANA is a registered trademark of sanofi-aventis ©2020 sanofi-aventis U.S. LLC
For more information, go to www.sanofi-aventis.us or call 1-800-633-1610. -
Low white blood cells. Low white blood cells can cause you to get serious infections, and may lead to death. Men who are 65 years or older may be more likely to have these problems. Your healthcare provider:
-
PRINCIPAL DISPLAY PANEL - Kit Carton
NDC: 0024-5824-11
JEVTANA®
(cabazitaxel)
Injection60 mg/1.5 mL Before First Dilution*
This carton contains: 1 JEVTANA vial and 1 Diluent vial
*Requires two dilutions before administration-See back panel for details
FOR INTRAVENOUS INFUSION ONLY AFTER SECOND DILUTIONCYTOTOXIC AGENT
RX ONLYSANOFI

-
INGREDIENTS AND APPEARANCE
JEVTANA
cabazitaxel kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0024-5824 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0024-5824-11 1 in 1 CARTON 06/17/2010 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, GLASS 5.7 mL Part 2 1 VIAL, GLASS 5.7 mL Part 1 of 2 JEVTANA
cabazitaxel injection, solution, concentrateProduct Information Item Code (Source) NDC: 0024-5823 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength cabazitaxel (UNII: 51F690397J) (cabazitaxel - UNII:51F690397J) cabazitaxel 60 mg in 1.5 mL Inactive Ingredients Ingredient Name Strength Polysorbate 80 (UNII: 6OZP39ZG8H) 1.56 g in 1.5 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0024-5823-15 5.7 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA201023 06/17/2010 Part 2 of 2 DILUENT
alcohol and water injectionProduct Information Item Code (Source) NDC: 0024-5822 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength alcohol (UNII: 3K9958V90M) water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0024-5822-01 5.7 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA201023 06/17/2010 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA201023 06/17/2010 Labeler - Sanofi-Aventis U.S. LLC (824676584) Establishment Name Address ID/FEI Business Operations Sanofi Chimie 291592785 ANALYSIS(0024-5824) , API MANUFACTURE(0024-5824) Establishment Name Address ID/FEI Business Operations Sanofi-Aventis Deutschland GmbH 313218430 ANALYSIS(0024-5824) , LABEL(0024-5824) , MANUFACTURE(0024-5824) , PACK(0024-5824)
Trademark Results [Jevtana]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 JEVTANA 78779621 3790268 Dead/Cancelled |
SANOFI-AVENTIS 2005-12-22 |
 JEVTANA 78532860 3032226 Live/Registered |
SANOFI 2004-12-15 |
 JEVTANA 77925080 4148244 Dead/Cancelled |
sanofi-aventis 2010-02-01 |
 JEVTANA 77804650 3874581 Live/Registered |
SANOFI 2009-08-14 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.