TRYVIO- aprocitentan tablet, film coated
TRYVIO by
Drug Labeling and Warnings
TRYVIO by is a Prescription medication manufactured, distributed, or labeled by Idorsia Pharmaceuticals Ltd. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TRYVIO safely and effectively. See full prescribing information for TRYVIO.
TRYVIO™ (aprocitentan) tablets, for oral use
Initial U.S. Approval: 2024WARNING: EMBRYO–FETAL TOXICITY
See full prescribing information for complete boxed warning.
- Based on animal data TRYVIO may cause fetal harm if used by pregnant patients and is contraindicated in pregnancy. (4.1, 5.1, 8.1)
- For patients who can become pregnant, exclude pregnancy prior to initiation of treatment with TRYVIO. (2.2, 5.1, 8.3)
- Use effective contraception prior to initiation of treatment, during treatment, and for one month after stopping TRYVIO. (2.2, 4.1, 5.1, 8.3)
- When pregnancy is detected, discontinue TRYVIO as soon as possible (5.1)
RECENT MAJOR CHANGES
Boxed Warning 4/2025 Contraindications (4.1) 4/2025 Warnings and Precautions (5.1) 4/2025 INDICATIONS AND USAGE
TRYVIO is an endothelin receptor antagonist indicated for the treatment of hypertension in combination with other antihypertensive drugs, to lower blood pressure in adult patients who are not adequately controlled on other drugs. Lowering blood pressure reduces the risk of fatal and non-fatal cardiovascular events, primarily strokes and myocardial infarctions. (1)
DOSAGE AND ADMINISTRATION
- The recommended dosage of TRYVIO is 12.5 mg orally once daily, with or without food. (2.1)
DOSAGE FORMS AND STRENGTHS
Tablets: 12.5 mg (3)
WARNINGS AND PRECAUTIONS
- ERAs cause hepatotoxicity and liver failure. Measure serum aminotransferase levels and total bilirubin prior to initiation of treatment and repeat periodically during treatment and as clinically indicated. (5.2)
- Fluid retention may require intervention (5.3)
- Decreases in hemoglobin (5.4)
- Decreased sperm counts (5.5)
ADVERSE REACTIONS
Most common adverse reactions (more frequent than placebo and ≥ 2% in TRYVIO-treated patients) are edema/fluid retention and anemia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Idorsia Pharmaceuticals Ltd at 1-833-400-9611 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 9/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: EMBRYO-FETAL TOXICITY
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Pregnancy Testing in Females of Reproductive Potential
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Pregnancy
4.2 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
5.2 Hepatotoxicity
5.3 Fluid Retention
5.4 Hemoglobin Decrease
5.5 Decreased Sperm Counts
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: EMBRYO-FETAL TOXICITY
- TRYVIO is contraindicated for use during pregnancy because it may cause fetal harm if used by pregnant patients. Therefore in patients who can become pregnant, exclude pregnancy prior to initiation of TRYVIO.
- Advise use of effective contraception before the start of TRYVIO, during treatment and for one month after stopping treatment.
- When pregnancy is detected, discontinue TRYVIO as soon as possible [see Dosage and Administration (2.2), Contraindications (4.1), Warnings and Precautions (5.1), Use in Specific Populations (8.1)].
-
1 INDICATIONS AND USAGE
TRYVIO, in combination with other antihypertensive drugs, is indicated for the treatment of hypertension, to lower blood pressure (BP) in adult patients who are not adequately controlled on other drugs. Lowering BP reduces the risk of fatal and non-fatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes. There are no controlled trials demonstrating reduction of risk of these events with TRYVIO.
Control of high BP should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve BP goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program's Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is BP reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher BPs, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from BP reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower BP goal.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of TRYVIO is 12.5 mg orally once daily.
Swallow tablets whole. TRYVIO may be taken with or without food.
If a dose is missed, skip the missed dose and take the next dose at the regular time. Do not take two doses on the same day.
2.2 Pregnancy Testing in Females of Reproductive Potential
Exclude pregnancy before initiating treatment with TRYVIO in females of reproductive potential [see Boxed Warning, Contraindications (4.1), Warnings and Precautions (5.1), Use in Specific Populations (8.3)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
4.1 Pregnancy
Use of TRYVIO is contraindicated in patients who are pregnant [see Dosage and Administration (2.2), Warnings and Precautions (5.1) and Use in Specific Populations (8.1)].
4.2 Hypersensitivity
TRYVIO is contraindicated in patients who are hypersensitive to aprocitentan or any of its excipients [see Adverse Reactions (6.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
Based on data from animal reproduction studies with endothelin receptor antagonists (ERAs), TRYVIO may cause fetal harm when administered during pregnancy and is contraindicated for use in patients who are pregnant. The available human data for endothelin receptor antagonists do not establish the presence or absence of fetal harm related to the use of TRYVIO. Counsel patients who can become pregnant about the potential risk to a fetus. Obtain a pregnancy test prior to initiation of treatment with TRYVIO. Advise patients who can become pregnant to use effective contraception prior to initiation of treatment with TRYVIO, during treatment, and for one month after the final dose of TRYVIO. When pregnancy is detected, discontinue TRYVIO as soon as possible [see Dosage and Administration (2.2), Contraindications (4.1), Use in Specific Populations (8.1, 8.3)].
5.2 Hepatotoxicity
Elevations of aminotransferases and hepatotoxicity are known effects of ERAs, including TRYVIO. Elevations in alanine transaminase (ALT) or aspartate aminotransferase (AST) of greater than 5-fold upper limit of normal (ULN) were observed rarely in patients treated with aprocitentan in the clinical trial, including cases with positive rechallenge. There were no reports of patients with ALT and/or AST >3 × ULN and total bilirubin >2 × ULN or cases of liver failure observed in TRYVIO-treated patients in the clinical trials. To reduce the risk of potential serious hepatotoxicity, measure serum aminotransferase levels and total bilirubin prior to initiation of treatment and repeat during treatment periodically and as clinically indicated.
Do not initiate TRYVIO in patients with elevated aminotransferases (>3 × ULN) or moderate to severe hepatic impairment.
Advise patients with symptoms suggesting hepatotoxicity (nausea, vomiting, right upper quadrant pain, fatigue, anorexia, scleral icterus, jaundice, dark urine, fever, or itching) to immediately stop treatment with TRYVIO and seek medical attention.
If sustained, unexplained, clinically relevant aminotransferase elevations occur, or if elevations are accompanied by an increase in bilirubin >2 × ULN, or if clinical symptoms of hepatotoxicity occur, discontinue TRYVIO.
5.3 Fluid Retention
Fluid retention and peripheral edema are known effects of ERAs, including TRYVIO [see Adverse Reactions (6.1)]. Edema/fluid retention was reported in 9% of TRYVIO-treated patients compared with 18% of patients receiving aprocitentan 25 mg (twice the recommended dose) and 2% on placebo in the clinical trial, requiring additional diuretic use in some patients. Older age and chronic kidney disease are risk factors for edema/fluid retention with TRYVIO. TRYVIO has not been studied in patients with heart failure New York Heart Association stage III–IV, unstable cardiac function, or with NTproBNP ≥500 pg/mL. TRYVIO is not recommended in these patients.
Monitor for signs and symptoms of fluid retention, weight gain, and worsening heart failure. If clinically significant fluid retention develops, treat appropriately, and consider discontinuation of TRYVIO.
5.4 Hemoglobin Decrease
Decreases in hemoglobin concentration and hematocrit have occurred following administration of other ERAs and were observed in the clinical trial with TRYVIO. Hemoglobin decreases usually presented early, stabilized thereafter, and were reversible after discontinuation. A decrease in hemoglobin of >2 g/dL from baseline was observed in 7% of patients compared to 1% of placebo patients. A decrease to below 10.0 g/dL was observed in 3% of TRYVIO-treated patients compared to 0 patients taking placebo. Initiation of TRYVIO is not recommended in patients with severe anemia. Measure hemoglobin prior to initiation of treatment and periodically during treatment as clinically indicated [see Adverse Reactions (6.1)].
5.5 Decreased Sperm Counts
TRYVIO, like other ERAs, may have an adverse effect on spermatogenesis. Counsel men about potential effects on fertility [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
-
6 ADVERSE REACTIONS
Clinically significant adverse reactions that appear in other sections of the labeling include:
- Embryo-fetal toxicity [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Fluid retention [see Warnings and Precautions (5.3)]
- Hemoglobin decrease [see Warnings and Precautions (5.4)]
- Decreased sperm counts [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of TRYVIO was evaluated in a placebo-controlled phase 3 clinical study (PRECISION, NCT03541174) in adults with uncontrolled BP (systolic blood pressure [SBP] ≥140 mmHg) despite the use of at least three antihypertensive medications.
In this study, 724 patients received any dose of aprocitentan, with 633 patients treated for at least 26 weeks, 192 patients for at least 47 weeks, and 99 patients for at least 48 weeks.
The most frequently reported adverse reactions to TRYVIO during the 4-week double-blind placebo-controlled treatment period (part 1) of the PRECISION study are presented in Table 1.
Table 1 Adverse reactions reported with a frequency of ≥2% in TRYVIO-treated patients and greater (≥1%) than in placebo-treated patients during the initial 4-week double-blind placebo-controlled treatment (part 1) Adverse Reaction 12.5 mg
N = 243
%Placebo
N = 242
%Edema/fluid retention 9.1 2.1 Anemia 3.7 0 Hypersensitivity Reactions
During the initial 4-week double-blind placebo-controlled treatment period (part 1), 0.8% of patients experienced an adverse reaction of hypersensitivity (i.e., rash, erythema, allergic edema) on TRYVIO compared to no reports in patients treated with placebo. One patient experienced allergic dermatitis requiring hospitalization while receiving aprocitentan 25 mg.
Laboratory Tests
Initiation of TRYVIO may cause an initial small decrease in estimated glomerular filtration rate (eGFR) that occurs within the first 6 weeks of starting therapy and then stabilizes.
In the initial 4-week double-blind treatment period, TRYVIO 12.5 mg caused a mean decrease of about 0.8 g/dL in hemoglobin compared to no change in the placebo patients.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on animal reproduction studies with other ERAs, TRYVIO can cause embryo-fetal toxicity, including birth defects and fetal death when administered to a pregnant patient and is contraindicated during pregnancy [see Contraindications (4.1)]. Available data from post-marketing reports and published literature over decades of use with endothelin receptor antagonists in the same class as TRYVIO have not identified an increased risk of fetal harm; however, these data are limited. Methodological limitations of these post-marketing reports and published literature include lack of a control group; limited information regarding dose, duration, and timing of drug exposure; and missing data. These limitations preclude establishing a reliable estimate of the risk of adverse fetal and neonatal outcomes with maternal endothelin receptor antagonist use. Administration of macitentan, where approximately ≥50% of total exposure was to aprocitentan, was teratogenic in rats and rabbits at all doses tested (see Data). Available data from reports of pregnancy in clinical trials with TRYVIO are insufficient to rule out a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. Advise pregnant patients of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In embryo-fetal development toxicity studies in pregnant rats and rabbits given macitentan (for which aprocitentan is a major metabolite) during the period of major organogenesis, cardiovascular and mandibular arch fusion malformations were observed at all doses studied. The lowest doses in rats and rabbits produced aprocitentan exposures that were equivalent to and 15-fold, respectively, the clinical exposures at the maximum recommended human dose (MRHD) based on area under the curve (AUC).
In pre- and post-natal development studies, female rats given macitentan (for which aprocitentan is a major metabolite) from late pregnancy through lactation showed reduced pup survival and impairment of the male fertility of the offspring at all doses. The lowest dose produced aprocitentan exposures approximately 2-fold the clinical exposures at the MRHD based on AUC.
8.2 Lactation
Risk Summary
There are no data on the presence of aprocitentan in human milk, the effects on the breastfed infant, or the effect on milk production. In rats, aprocitentan was excreted into milk during lactation (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the potential for serious adverse reactions in breastfed infants, advise women not to breastfeed during treatment with TRYVIO.
8.3 Females and Males of Reproductive Potential
Based on data from animal reproductive toxicity studies with other ERAs, TRYVIO can cause fetal harm, including birth defects and fetal death, when administered to a pregnant patient and is contraindicated during pregnancy [see Contraindications (4.1), Use in Specific Populations (8.1)].
Pregnancy TestingVerify that patients who can become pregnant, are not pregnant prior to initiating TRYVIO. The patient should contact their physician immediately if onset of menses is delayed or pregnancy is suspected. If the pregnancy test is positive, the physician and patient should discuss the risks to the patient, the pregnancy, and the fetus [see Warnings and Precautions (5.1), Dosage and Administration (2.2), Contraindications (4.1)].
ContraceptionPatients using TRYVIO who can become pregnant should use effective contraception prior to initiation of treatment, during treatment, and for one month after discontinuation of treatment with TRYVIO to prevent pregnancy [see Warnings and Precautions (5.1)].
InfertilityOther ERAs have shown an adverse effect on spermatogenesis in humans and/or animals. TRYVIO, like other ERAs, may impair fertility in males of reproductive potential. It is not known whether effects on fertility would be reversible [see Warnings and Precautions (5.5), Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and efficacy of TRYVIO in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of subjects in the PRECISION study of TRYVIO, 321 (44%) were 65 years and older, while 72 (10%) were 75 years and older. Edema/fluid retention was more common in these patients than younger patients [see Warnings and Precautions (5.3)].
No dose adjustment is required in patients over the age of 65 years [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
TRYVIO is not recommended in patients with kidney failure (eGFR <15 mL/min) or on dialysis. The effect of kidney failure (eGFR <15 mL/min) or dialysis on aprocitentan pharmacokinetics is unknown [see Clinical Pharmacology (12.3)]. Patients with renal impairment are at increased risk of edema/fluid retention [see Warnings and Precautions (5.3)].
No dose adjustment is required in patients with mild to severe renal impairment (eGFR ≥15 mL/min).
8.7 Hepatic Impairment
TRYVIO is not recommended in patients with moderate and severe hepatic impairment (Child-Pugh class B and C) because these patients may be at increased risk for poor outcomes from hepatotoxicity.
No dose adjustment is required in patients with mild hepatic impairment (Child-Pugh class A) [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
TRYVIO has been administered as a single dose of up to 600 mg, and as multiple doses of up to 100 mg daily, to healthy subjects (48 and 8 times the recommended dose, respectively). Adverse events of headache, nasal congestion, nausea, and upper respiratory tract infection were observed. In the event of an overdose, standard supportive measures should be taken, as required. Dialysis is unlikely to be effective because aprocitentan is highly protein-bound. Consult a Certified Poison Control Center for the most up-to-date information on the management of overdosage (1-800-222-1222 or www.poison.org).
-
11 DESCRIPTION
TRYVIO (aprocitentan) is an endothelin receptor antagonist. The chemical name of aprocitentan is N-[5-(4-bromophenyl)-6- [2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-sulfamide. It has a molecular formula of C16H14Br2N6O4S and a molecular weight of 546.2 g/mol.
The structural formula is:
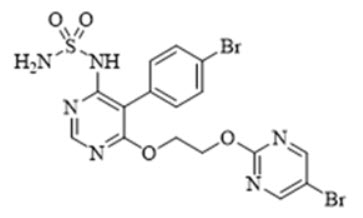
Aprocitentan is a white to off-white powder that is insoluble in water.
TRYVIO is available as film-coated 12.5 mg strength tablets for oral administration. The inactive ingredients in TRYVIO are croscarmellose sodium, hydroxypropyl cellulose, lactose monohydrate, magnesium stearate, and microcrystalline cellulose.
The film coating contains the following inactive ingredients: hydroxypropyl cellulose, iron oxide black, iron oxide red, iron oxide yellow, polyvinyl alcohol, silica colloidal hydrated, talc, titanium dioxide, and triethyl citrate.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Aprocitentan is an ERA that inhibits the binding of endothelin (ET)-1 to ETA and ETB receptors.
ET-1, via its receptors (ETA and ETB), mediates a variety of deleterious effects such as vasoconstriction, fibrosis, cell proliferation, and inflammation. In hypertension, ET-1 can cause endothelial dysfunction, vascular hypertrophy and remodeling, sympathetic activation, and increased aldosterone synthesis.
12.2 Pharmacodynamics
Aprocitentan exposure-response relationships and the time course of pharmacodynamic response are not fully characterized.
12.3 Pharmacokinetics
The aprocitentan mean (%CV) Cmax is approximately 1.3 mcg/mL (19) following a single 25 mg dose (twice the recommended dose). The mean (%CV) aprocitentan AUC to the dosing interval (AUC0-tau) is approximately 23 mcg⸱h/mL (17) following a single 25 mg dose. Aprocitentan plasma concentrations increased in a dose-proportional manner following once-daily administration of 5, 25, and 100 mg (0.4 times the recommended dose to 8 times the recommended dose). Aprocitentan steady state is reached by Day 8 with approximately 3-fold accumulation following once daily administration. Aprocitentan is primarily unchanged in plasma following oral TRYVIO administration.
Absorption
The absolute oral bioavailability of aprocitentan is unknown. The time to reach Cmax is between 4 and 5 hours after administration of 25 mg aprocitentan (twice the recommended dose).
Effect of Food
No clinically significant differences in aprocitentan pharmacokinetics were observed following administration of a high-fat, high-calorie meal (approximately 150, 250, and 500–600 calories from protein, carbohydrate, and fat, respectively) in healthy subjects.
Distribution
The apparent volume of distribution of aprocitentan is approximately 20 L. Aprocitentan is >99% bound to plasma proteins, primarily albumin. Protein binding is not affected by renal or hepatic impairment. The aprocitentan blood-to-plasma ratio is 0.63.
Elimination
The aprocitentan effective half-life is approximately 41 hours, and the apparent clearance is approximately 0.3 L/h.
Metabolism
Aprocitentan is primarily metabolized by UGT1A1- and UGT2B7-mediated N-glucosidation and non-enzymatic hydrolysis.
Excretion
After a single dose of radiolabeled aprocitentan, approximately 52% of the dose was eliminated via urine (0.2% unchanged) and 25% via feces (6.8% unchanged).
Specific Populations
No clinically significant differences in the pharmacokinetics of aprocitentan were observed based on age (18–84 years), sex, race/ethnicity, body weight (44–196 kg), between patients and healthy subjects, mild to severe renal impairment (eGFR ≥15 mL/min), or mild to moderate hepatic impairment (Child-Pugh class A to B). The effect of kidney failure (eGFR <15 mL/min), dialysis, or severe hepatic impairment (Child-Pugh class C) on aprocitentan pharmacokinetics is unknown.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
No clinically significant differences in the pharmacokinetics of the following drugs were observed when used concomitantly with aprocitentan: midazolam (CYP3A4 substrate), rosuvastatin (breast cancer resistance protein [BCRP] substrate) or oral contraceptive (ethinyl estradiol and levonorgestrel).
In Vitro Studies
UDP-glucuronosyltransferase (UGT) inducers: Concomitant administration of aprocitentan with UGT inducers may decrease aprocitentan exposure.
CYP450 enzymes: Aprocitentan inhibits CYP3A4 and all members of the CYP2C family, but did not inhibit CYP1A2, CYP2A6, CYP2B6, CYP2D6, and CYP2E1. Aprocitentan is an inducer of CYP3A4 but did not induce CYP1A2 or CYP2C9.
Transporter systems: Aprocitentan is a substrate of P-glycoprotein (P-gp) and BCRP. However, inhibitors of these transporters are not anticipated to influence the PK of aprocitentan. Aprocitentan is an inhibitor of BCRP, bile salt export pump (BSEP), and sodium taurocholate co-transporting polypeptide (NTCP), but does not inhibit P-gp, organic cationic transporter (OCT)1, OCT2, human multi-drug and toxin compound extrusion (MATE)1, or MATE2K. Aprocitentan does not inhibit organic anion transporter (OAT)1, OAT3, OATP1B1, or OATP1B3 at therapeutic concentrations.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two-year carcinogenicity studies with macitentan (for which aprocitentan is a major metabolite) did not identify any carcinogenic potential at doses up to 100 mg/kg/day and 250 mg/kg/day in mice and rats, which produced aprocitentan exposures approximately 30-fold and 11-fold, respectively, the clinical aprocitentan exposure at the MRHD based on AUC.
Mutagenesis
Aprocitentan did not induce mutagenicity or genotoxicity in a standard battery of in vitro and in vivo assays that included a bacterial reverse mutation assay, a chromosome aberration test in human lymphocytes, and an in vivo bone marrow micronucleus test in rats.
Impairment of Fertility
In a fertility study in male rats given aprocitentan for 15 weeks at doses up to 250 mg/kg/day, no effect on fertility or spermatogenesis was observed at 52-fold the clinical exposure at the MRHD based on AUC. In repeated dose toxicity studies, treatment with aprocitentan resulted in testicular tubular degeneration/atrophy in male rats and dogs at high doses of 250 mg/kg/day and 25 mg/kg/day, respectively, which represents approximately 41- and 52-fold the clinical exposure at the MRHD, based on AUC, respectively. The testicular toxicity was not evident in male rats and dogs at 50 mg/kg/day and 5 mg/kg/day, respectively, which represents approximately 14- and 10-fold the clinical exposure at the MRHD based on AUC.
In female rats given aprocitentan prior to mating, minimally increased pre-implantation loss was observed at doses ≥50 mg/kg/day, which represent ≥23-fold the clinical exposure at the MRHD based on AUC. No impact on fertility was observed at 10 mg/kg/day, which represents 5-fold the clinical exposure at the MRHD based on AUC.
-
14 CLINICAL STUDIES
The efficacy of TRYVIO (aprocitentan) was evaluated in a multipart, phase 3 multicenter study (PRECISION, NCT03541174) in adults with SBP ≥140 mmHg who were prescribed at least three antihypertensive medications. The trial included a placebo run-in period, which was followed by three parts as described below. Prior to the placebo run-in period, all patients were switched to standard background antihypertensive therapy consisting of an angiotensin receptor blocker, a calcium channel blocker, and a diuretic, which was continued throughout the study. Patients with concomitant use of beta-blockers continued this treatment throughout the study.
Following the 4-week placebo run-in period, 730 patients were randomized equally to aprocitentan at either 12.5 mg, 25 mg, or placebo once daily during the initial 4-week double-blind (DB) treatment period (part 1). At the end of 4 weeks, all patients entered the single-blind treatment period (part 2) where they received 25 mg aprocitentan once daily for 32 weeks. At the end of the 32 weeks, patients were re-randomized to receive either 25 mg aprocitentan or placebo, once daily, during a 12-week DB-withdrawal period (part 3).
The primary efficacy endpoint was the change in sitting SBP (SiSBP) from baseline to Week 4 during part 1, measured at trough by unattended automated office blood pressure (uAOBP).
The key secondary endpoint was the change in SiSBP measured at trough by uAOBP from Week 36 (i.e., prior to randomized withdrawal to 25 mg aprocitentan or placebo in part 3) to Week 40.
Patients had a mean age of 62 years (range 24 to 84 years) and 60% were male. Patients were White (83%), African American (11%) or Asian (5%). Approximately 10% were Hispanic. The mean body mass index (BMI) was 34 kg/m2 (range 18 to 64 kg/m2). At baseline, 19% of patients had an eGFR 30–59 mL/min/1.73 m2 and 3% had an eGFR 15–29 mL/min/1.73 m2. At baseline, 24% of patients had a urine albumin-to-creatinine ratio (UACR) of 30–300 mg/g and 13% had a UACR >300 mg/g. Approximately 54% of patients had a medical history of diabetes mellitus, 31% ischemic heart disease, and 20% congestive heart failure. At baseline, 63% of patients reported taking four or more antihypertensive medications.
BP reductions compared to placebo based on uAOBP measurements at trough are shown in Table 2. TRYVIO 12.5 mg was statistically superior to placebo in reducing SiSBP at Week 4 (part 1). The treatment effect was consistent for sitting diastolic BP (SiDBP) (Table 2).
Table 2 Reduction in sitting trough BP (mmHg) at Week 4 of DB treatment Difference to placebo Treatment group N Baseline* Mean LS Mean LS Mean p-value BP = blood pressure; CL = confidence limits; DB = double-blind; LS Mean = least squares mean; SiDBP = sitting diastolic blood pressure; SiSBP = sitting systolic blood pressure. - * Observed baseline value.
- † Statistically significant at the 2.5% level as prespecified in the testing strategy.
SiSBP (primary endpoint) LS Mean
(97.5% CL)LS Mean
(97.5% CL)12.5 mg 243 153.2 −15.4 (−17.5, −13.3) −3.8 (−6.8, −0.8) 0.0043† Placebo 244 153.3 −11.6 (−13.7, −9.5) – – SiDBP LS Mean
(97.5% CL)LS Mean
(97.5% CL)12.5 mg 243 87.9 −10.4 (−11.7, −9.1) −4.0 (−5.8, −2.1) − Placebo 244 87.1 −6.4 (−7.8, −5.1) – – The persistence of the BP-lowering effect of TRYVIO was demonstrated in part 3 of the trial, in which patients on aprocitentan were re-randomized to placebo or 25 mg aprocitentan following a period during which all patients were treated with 25 mg. In patients re-randomized to placebo, the mean SiSBP increased, whereas in patients re-randomized to 25 mg aprocitentan the mean effect on SiSBP was maintained and was statistically superior to placebo at Week 40. The treatment effect was consistent for SiDBP.
Most of the BP-lowering effect occurred within the first two weeks of treatment with TRYVIO.
TRYVIO is not approved for use at a 25 mg dose. The 25 mg dose has not demonstrated a meaningful improvement in blood pressure reduction as compared to the 12.5 mg dose and had an increased risk of edema/fluid retention [see Warnings and Precautions (5.3)].
TRYVIO's BP-lowering effect appeared consistent among subgroups defined by age, sex, race, BMI, baseline eGFR, baseline UACR, medical history of diabetes, and between BP measurement methodologies (uAOBP and ambulatory BP measurements).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
TRYVIO tablets are available as:
- 12.5 mg: yellow to orange round, film-coated tablet, debossed with "AN" on one side and plain on the other side.
- – NDC: 80491-8012-8, each blister contains 10 tablets
- – NDC: 80491-8012-3, each bottle contains 30 tablets and has a child-resistant closure
16.2 Storage and Handling
Store at 20ºC to 25ºC (68ºF to 77ºF); excursions permitted from 15ºC to 30ºC (59ºF to 86ºF) [see USP Controlled Room Temperature]. Store in the original package. Dispense to patient in original container only. Replace cap securely each time after opening. Do not discard desiccant. Protect from light and moisture.
- 12.5 mg: yellow to orange round, film-coated tablet, debossed with "AN" on one side and plain on the other side.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Embryo-Fetal Toxicity
Counsel patients who can become pregnant to use effective methods of contraception before treatment with TRYVIO, during treatment with TRYVIO, and for one month after treatment discontinuation. Patients who can become pregnant should have a negative pregnancy test prior to initiation of TRYVIO, [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1, 8.3)].
Patients should be instructed to contact their healthcare provider if they suspect they may be pregnant. Patients should seek additional contraceptive advice from a gynecologist or similar expert as needed.
Educate and counsel patients who can become pregnant on the use of emergency contraception in the event of unprotected sex or contraceptive failure.
Advise pre-pubertal females and/or their guardian(s) to report any changes in their reproductive status immediately to their prescriber.
Lactation
Advise females not to breastfeed during treatment with TRYVIO [see Use in Specific Populations (8.2)].
Hepatotoxicity
Educate patients on signs of hepatotoxicity. Advise patients that they should contact their healthcare provider if they have unexplained nausea, vomiting, right upper quadrant pain, fatigue, anorexia, jaundice, dark urine, fever, or itching.
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration.
IDRSTRMG03272025Issued: X/2025 MEDICATION GUIDE
TRYVIO™ (try-vee-oh)
(aprocitentan)
tablets, for oral useRead this Medication Guide before you start taking TRYVIO and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your healthcare provider about your medical condition or your treatment. What is the most important information I should know about TRYVIO?
TRYVIO may cause serious side effects, including:
Serious birth defects.- TRYVIO may cause serious birth defects if taken during pregnancy.
- Females who can become pregnant should not be pregnant when they start taking TRYVIO or become pregnant during treatment with TRYVIO or for 1 month after stopping treatment with TRYVIO.
-
Females who can become pregnant should have a negative pregnancy test before starting treatment with TRYVIO.
- Females who can become pregnant are females who:
- have entered puberty, even if they have not started their menstrual period, and
- have a uterus, and
- have not gone through menopause. Menopause means that you have not had a menstrual period for at least 12 months for natural reasons, or that you have had your ovaries removed.
- Females who cannot become pregnant are females who:
- have not yet entered puberty, or
- do not have a uterus, or
- have gone through menopause. Menopause means that you have not had a menstrual period for at least 12 months for natural reasons, or that you have had your ovaries removed, or
- are infertile for other medical reasons and this infertility is permanent and cannot be reversed.
- Females who can become pregnant are females who:
-
Females who can become pregnant should use effective birth control before starting treatment with TRYVIO, during treatment with TRYVIO, and for 1 month after stopping TRYVIO because the medicine may still be in your body.
- Talk with your healthcare provider or gynecologist (a healthcare provider who specializes in female reproduction) to find out about options for effective birth control that you may use to prevent pregnancy during treatment with TRYVIO.
- If you decide that you want to change the form of birth control that you use, talk with your healthcare provider or gynecologist to be sure that you choose another acceptable form of birth control.
- Do not have unprotected sex. Talk to your healthcare provider or pharmacist right away if you have unprotected sex or if you think your birth control has failed. Your healthcare provider may talk with you about using emergency birth control.
- Tell your healthcare provider right away if you miss a menstrual period or you think you might be pregnant.
See "What are the possible side effects of TRYVIO?" for more information about side effects. What is TRYVIO?
TRYVIO is a prescription medicine used to treat high blood pressure (hypertension) in adults who are taking other high blood pressure medicines and whose blood pressure is not well controlled.
It is not known if TRYVIO is safe and effective in children.Who should not take TRYVIO?
Do not take TRYVIO if you are:- pregnant or currently trying to become pregnant. See "What is the most important information I should know about TRYVIO?".
- allergic to aprocitentan or any of the ingredients in TRYVIO. See the end of this Medication Guide for a complete list of ingredients in TRYVIO.
Before taking TRYVIO, tell your healthcare provider about all of your medical conditions, including if you: - have liver problems
- have heart failure
- have anemia
- have kidney problems or get dialysis
- are pregnant or plan to become pregnant during treatment with TRYVIO. TRYVIO can cause serious birth defects. See "What is the most important information I should know about TRYVIO?"
- are breastfeeding or plan to breastfeed. It is not known if TRYVIO passes into your breastmilk. Do not breastfeed if you take TRYVIO. Talk to your healthcare provider about the best way to feed your baby if you take TRYVIO.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. How should I take TRYVIO? - Take TRYVIO exactly how your healthcare provider tells you to take it and do not stop taking TRYVIO unless your healthcare provider tells you to.
- Swallow TRYVIO tablets whole.
- Take TRYVIO with or without food.
- If you take too much TRYVIO, contact your healthcare provider or Poison Control Center at 1-800-222-1222 or www.poison.org, or go to the nearest hospital emergency room right away.
- If you miss a dose of TRYVIO, skip the missed dose and take the next dose at your regularly scheduled time. Do not take 2 doses in the same day to make up for a missed dose.
What are the possible side effects of TRYVIO?
TRYVIO may cause serious side effects, including:- Serious birth defects. See "What is the most important information I should know about TRYVIO?"
- Liver problems. TRYVIO may cause liver problems. Your healthcare provider should do blood tests to check your liver before starting treatment and as needed during treatment with TRYVIO. Tell your healthcare provider if you have any of the following symptoms of liver problems during treatment with TRYVIO:
- nausea or vomiting
- pain in the upper right stomach
- tiredness
- loss of appetite
- yellowing of your skin or whites of your eyes
- dark urine
- fever
- itching
- Fluid retention. Fluid retention and swelling are common during treatment with TRYVIO and can be serious. Tell your healthcare provider right away if you have any unusual weight gain, trouble breathing, or swelling of your ankles or legs. Your healthcare provider may treat you with other medicines (diuretics) if you develop fluid retention or swelling.
- Low red blood cell levels (anemia). Anemia is common during treatment with TRYVIO and can be serious. Your healthcare provider will do blood tests to check your red blood cells before starting and as needed during treatment with TRYVIO.
- Decreased sperm count. TRYVIO may cause decreased sperm counts in males and may affect the ability to father a child. Tell your healthcare provider if being able to have children is important to you.
Your healthcare provider may stop treatment with TRYVIO if you develop certain side effects. Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of TRYVIO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store TRYVIO? - Store TRYVIO tablets at room temperature, between 68°F to 77°F (20°C and 25°C).
- Keep TRYVIO tablets in the original container.
- Tightly close the TRYVIO bottle cap each time after opening.
- The TRYVIO bottle contains a desiccant packet to help keep your tablets dry (protect them from moisture). Do not throw away the desiccant packet and keep it in the bottle.
- Protect TRYVIO from light and moisture.
- Keep TRYVIO and all medicines out of the reach of children.
General information about the safe and effective use of TRYVIO.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use TRYVIO for a condition for which it was not prescribed. Do not give TRYVIO to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TRYVIO that is written for health professionals.What are the ingredients in TRYVIO?
Active ingredient: aprocitentan
Inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, lactose monohydrate, magnesium stearate, and microcrystalline cellulose. Film coating: hydroxypropyl cellulose, iron oxide black, iron oxide red, iron oxide yellow, polyvinyl alcohol, silica colloidal hydrated, talc, titanium dioxide, and triethyl citrate.
Distributed by:
Idorsia Pharmaceuticals US Inc.
One Radnor Corporate Center, Suite 101
100 Matsonford Rd.
Radnor, PA 19087
For more information call 1-833-400-9611 or visit www.TRYVIO.com. -
PRINCIPAL DISPLAY PANEL - 30 Tablet Bottle Carton
NDC: 80491-8012-3
TRYVIO™
(aprocitentan)
tablets12.5 mg
Attention: Dispense the enclosed
Medication Guide to each patient.Rx only
30 film-coated tablets
idorsia
-
INGREDIENTS AND APPEARANCE
TRYVIO
aprocitentan tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 80491-8012 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength APROCITENTAN (UNII: MZI81HV01P) (APROCITENTAN - UNII:MZI81HV01P) APROCITENTAN 12.5 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) HYDRATED SILICA (UNII: Y6O7T4G8P9) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIETHYL CITRATE (UNII: 8Z96QXD6UM) Product Characteristics Color YELLOW (yellow to orange) Score no score Shape ROUND Size 6mm Flavor Imprint Code AN Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 80491-8012-3 1 in 1 CARTON 10/01/2024 1 30 in 1 BOTTLE; Type 0: Not a Combination Product 2 NDC: 80491-8012-8 1 in 1 CARTON 03/20/2024 2 NDC: 80491-8012-7 10 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA217686 03/20/2024 Labeler - Idorsia Pharmaceuticals Ltd (480176487)
Trademark Results [TRYVIO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TRYVIO 97131670 not registered Live/Pending |
JOHNSON & JOHNSON 2021-11-18 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.