SYEDA- drospirenone and ethinyl estradiol kit
Syeda by
Drug Labeling and Warnings
Syeda by is a Prescription medication manufactured, distributed, or labeled by Xiromed, LLC., XIROMED PHARMA ESPANA, S.L. . Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SyedaTM safely and effectively. See full prescribing information for SyedaTM.
SyedaTM (drospirenone and ethinyl estradiol) tablets, for oral use
Initial U.S. Approval: 2001WARNING: CIGARETTE SMOKING AND SERIOUS CARDIOVASCULAR EVENTS
See full prescribing information for complete boxed warning
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
SyedaTM (drospirenone and ethinyl estradiol tablets, USP) is an estrogen/progestin COC indicated for use by women to prevent pregnancy. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
SyedaTM (drospirenone and ethinyl estradiol tablets) consists of 28 film-coated, tablets in the following order (3):
- 21 yellow active tablets, each containing 3 mg drospirenone (DRSP) and 0.03 mg ethinyl estradiol (EE),
- 7 inert white tablets
CONTRAINDICATIONS
- Renal impairment (4)
- Adrenal insufficiency (4)
- A high risk of arterial or venous thrombotic diseases (4)
- Undiagnosed abnormal uterine bleeding (4)
- Breast cancer or other estrogen- or progestin-sensitive cancer (4)
- Liver tumors or liver disease (4)
- Pregnancy (4)
- Co-administration with Hepatits C drug combinations containing ombitasvir, paritaprevir/ritonavir, with or without dasabuvir (4)
WARNINGS AND PRECAUTIONS
- Vascular risks: Stop drospirenone and ethinyl estradiol if a thrombotic event occurs. Stop at least 4 weeks before and through 2 weeks after major surgery. Start no earlier than 4 weeks after delivery, in women who are not breastfeeding. (5.1)
- COCs containing DRSP may be associated with a higher risk of venous thromboembolism (VTE) than COCs containing levonorgestrel or some other progestins. Before initiating drospirenone and ethinyl estradiol in a new COC user or a woman who is switching from a contraceptive that does not contain DRSP, consider the risks and benefits of a DRSP-containing COC in light of her risk of a VTE. (5.1)
- Hyperkalemia: DRSP has anti-mineralocorticoid activity. Do not use in patients predisposed to hyperkalemia. Check serum potassium concentration during the first treatment cycle in women on long-term treatment with medications that may increase serum potassium concentration. (5.2, 7.1, 7.2)
- Liver disease: Discontinue drospirenone and ethinyl estradiol if jaundice occurs. (5.4)
- High blood pressure: Do not prescribe drospirenone and ethinyl estradiol for women with uncontrolled hypertension or hypertension with vascular disease. (5.5)
- Carbohydrate and lipid metabolic effects: Monitor prediabetic and diabetic women taking drospirenone and ethinyl estradiol. Consider an alternate contraceptive method for women with uncontrolled dyslipidemia. (5.7)
- Headache: Evaluate significant change in headaches and discontinue drospirenone and ethinyl estradiol if indicated. (5.8)
- Uterine bleeding: Evaluate irregular bleeding or amenorrhea. (5.9)
ADVERSE REACTIONS
The most frequent adverse reactions (≥ 2%) are premenstrual syndrome (13.2%), headache /migraine (10.7%), breast pain/tenderness/discomfort (8.3%), nausea/vomiting (4.5%), abdominal pain/tenderness/discomfort (2.3%), mood changes (2.3%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Xiromed, LLC. at 1-844-XIROMED (1-844-947-6633) or FDA at 1-800-FDA-1088 www.fda.gov/medwatch.
DRUG INTERACTIONS
Drugs or herbal products that induce certain enzymes (for example, CYP3A4) may decrease the effectiveness of COCs or increase breakthrough bleeding. Counsel patients to use a back-up or alternative method of contraception when enzyme inducers are used with COCs. (7.1)
USE IN SPECIFIC POPULATIONS
Nursing mothers: Not recommended; can decrease milk production. (8.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 12/2017
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: CIGARETTE SMOKING AND SERIOUS CARDIOVASCULAR EVENTS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 How to Take SyedaTM
2.2 How to Start SyedaTM
2.3 Advice in Case of Gastrointestinal Disturbances
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Thromboembolic Disorders and Other Vascular Problems
5.2 Hyperkalemia
5.3 Carcinoma of the Breasts and Reproductive Organs
5.4 Liver Disease
5.5 Risk of Liver Enzyme Elevations with Concomitant Hepatitis C Treatment
5.6 High Blood Pressure
5.7 Gallbladder Disease
5.8 Carbohydrate and Lipid Metabolic Effects
5.9 Headache
5.10 Bleeding Irregularities
5.11 COC Use Before or During Early Pregnancy
5.12 Depression
5.13 Interference with Laboratory Tests
5.14 Monitoring
5.15 Other Conditions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on Combined Oral Contraceptives
7.2 Effects of Combined Oral Contraceptives on Other Drugs
7.3 Concomitant Use of HCV Combination Therapy – Liver Enzyme Elevation
7.4 Interference with Laboratory Tests
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Renal Impairment
8.7 Patients with Hepatic Impairment
8.8 Race
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: CIGARETTE SMOKING AND SERIOUS CARDIOVASCULAR EVENTS
Cigarette smoking increases the risk of serious cardiovascular events from combination oral contraceptives (COC) use. This risk increases with age, particularly in women over 35 years of age, and with the number of cigarettes smoked. For this reason, COCs should not be used by women who are over 35 years of age and smoke [see CONTRAINDICATIONS (4)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 How to Take SyedaTM
Take one tablet by mouth at the same time every day. The failure rate may increase when pills are missed or taken incorrectly.
To achieve maximum contraceptive effectiveness, SyedaTM must be taken as directed, in the order directed on the blister pack. Single missed pills should be taken as soon as remembered.
2.2 How to Start SyedaTM
Instruct the patient to begin taking SyedaTM either on the first day of her menstrual period (Day 1 Start) or on the first Sunday after the onset of her menstrual period (Sunday Start).
Day 1 Start
During the first cycle of SyedaTM use, instruct the patient to take one yellow SyedaTM daily, beginning on Day 1 of her menstrual cycle. (The first day of menstruation is Day 1.) She should take one yellow SyedaTM daily for 21 consecutive days, followed by one white tablet daily on Days 22 through 28. SyedaTM should be taken in the order directed on the package at the same time each day, preferably after the evening meal or at bedtime with some liquid, as needed. SyedaTM can be taken without regard to meals. If SyedaTM is first taken later than the first day of the menstrual cycle, SyedaTM should not be considered effective as a contraceptive until after the first 7 consecutive days of product administration. Instruct the patient to use a non-hormonal contraceptive as back-up during the first 7 days. The possibility of ovulation and conception prior to initiation of medication should be considered.
Sunday Start
During the first cycle of SyedaTM use, instruct the patient to take one yellow SyedaTM daily, beginning on the first Sunday after the onset of her menstrual period. She should take one yellow SyedaTMdaily for 21 consecutive days, followed by one white tablet daily on Days 22 through 28. SyedaTM should be taken in the order directed on the package at the same time each day, preferably after the evening meal or at bedtime with some liquid, as needed. SyedaTM can be taken without regard to meals. SyedaTM should not be considered effective as a contraceptive until after the first 7 consecutive days of product administration. Instruct the patient to use a non-hormonal contraceptive as back-up during the first 7 days. The possibility of ovulation and conception prior to initiation of medication should be considered.
The patient should begin her next and all subsequent 28-day regimens of SyedaTM on the same day of the week that she began her first regimen, following the same schedule. She should begin taking her yellow tablets on the next day after ingestion of the last white tablet, regardless of whether or not a menstrual period has occurred or is still in progress. Anytime a subsequent cycle of SyedaTM is started later than the day following administration of the last white tablet, the patient should use another method of contraception until she has taken a yellow SyedaTM daily for seven consecutive days.
When switching from a different birth control pill
When switching from another birth control pill, SyedaTM should be started on the same day that a new pack of the previous oral contraceptive would have been started.
When switching from a method other than a birth control pill
When switching from a transdermal patch or vaginal ring, SyedaTMshould be started when the next application would have been due. When switching from an injection, SyedaTM should be started when the next dose would have been due. When switching from an intrauterine contraceptive or an implant, SyedaTMshould be started on the day of removal.
Withdrawal bleeding usually occurs within 3 days following the last yellow tablet. If spotting or breakthrough bleeding occurs while taking SyedaTM, instruct the patient to continue taking SyedaTM by the regimen described above. Counsel her that this type of bleeding is usually transient and without significance; however, advise her that if the bleeding is persistent or prolonged, she should consult her healthcare provider.
Although the occurrence of pregnancy is low if SyedaTM is taken according to directions, if withdrawal bleeding does not occur, consider the possibility of pregnancy. If the patient has not adhered to the prescribed dosing schedule (missed one or more active tablets or started taking them on a day later than she should have), consider the possibility of pregnancy at the time of the first missed period and take appropriate diagnostic measures. If the patient has adhered to the prescribed regimen and misses two consecutive periods, rule out pregnancy. Discontinue SyedaTM if pregnancy is confirmed.
The risk of pregnancy increases with each active yellow tablet missed. For additional patient instructions regarding missed pills, see the “WHAT TO DO IF YOU MISS PILLS” section in the FDA-Approved Patient Labeling. If breakthrough bleeding occurs following missed tablets, it will usually be transient and of no consequence. If the patient misses one or more white tablets, she should still be protected against pregnancy provided she begins taking a new cycle of yellow tablets on the proper day.
For postpartum women who do not breastfeed or after a second trimester abortion, start SyedaTM no earlier than 4 weeks postpartum due to the increased risk of thromboembolism. If the patient starts SyedaTM postpartum and has not yet had a period, evaluate for possible pregnancy, and instruct her to use an additional method of contraception until she has taken SyedaTM for 7 consecutive days.
-
3 DOSAGE FORMS AND STRENGTHS
SyedaTM (drospirenone and ethinyl estradiol tablets, USP) are available in blister packs.
Each blister pack contains 28 tablets in the following order:
- 21 active tablets are yellow colored, round , film-coated tablets, “SZ” and “U3” are debossed on opposite sides of the tablet and containing 3 mg drospirenone (DRSP) and 0.03 mg ethinyl estradiol (EE)
- 7 inert white tablets are white film-coated, round, ‘SZ” and J1 are debossed on opposite sides of the tablet
-
4 CONTRAINDICATIONS
Do not prescribe SyedaTM to women who are known to have the following:
- Renal Impairment
- Adrenal insufficiency
- A high risk of arterial or venous thrombotic diseases. Examples include women who are known to:
- Smoke, if over age 35 [see BOXED WARNING and WARNINGS AND PRECAUTIONS(5.1)]
- Have deep vein thorombosis or pulmonary embolism, now or in the past [see WARNINGS AND PRECAUTIONS (5.1)]
- Have cerebrovascular disease [see WARNINGS AND PRECAUTIONS (5.1)]
- Have coronary artery disease [see WARNINGS AND PRECAUTIONS (5.1)]
- Have thrombogenic valvular or thrombogenic rhythm diseases of the heart (for example, subacute bacterial endocarditis with valvular disease, or atrial fibrilation) [see WARNINGS AND PRECAUTIONS (5.1)]
- Have inherited or acquired hypercoagulopathies [see WARNINGS AND PRECAUTIONS (5.1)]
- Have uncontrolled hypertension [see WARNINGS AND PRECAUTIONS (5.5)]
- Have diabetes mellitus with vascular disease [see WARNINGS AND PRECAUTIONS (5.7)]
- Have headaches with focal neurological symptoms or have migrane headaches with or without aura if over age 35 [see WARNINGS AND PRECAUTIONS (5.8)]
- Undiagnosed abnormal uterine bleeding [see WARNINGS AND PRECAUTIONS (5.9)]
- Breast cancer or other estrogen- or progestin-sensitive cancer, now or in the past [see WARNINGS AND PRECAUTIONS (5.3)]
- Liver tumor (benign or malignant) or liver disease [see WARNINGS AND PRECAUTIONS (5.4) and USE IN SPECIFIC POPULATIONS (8.7)]
- Pregnancy, because there is no reason to use COCs during pregnancy [see WARNINGS AND PRECAUTIONS (5.10) and USE IN SPECIFIC POPULATIONS (8)]
- Use of Hepatits C drug combinations containing ombitasvir, paritaprevir/ritonavir, with or without dasabuvir due to the potential for ALT elevations [see WARNINGS AND PRECUTIONS (5.5) and DRUG INTERACTIONS (7.2)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Thromboembolic Disorders and Other Vascular Problems
Stop drospirenone and ethinyl estradiol if an arterial or venous thrombotic (VTE) event occurs.
Based on presently available information on drospirenone and ethinyl estradiol, DRSP-containing COCs may be associated with a higher risk of venous thromboembolism (VTE) than COCs containing the progestin levonorgestrel or some other progestins. Epidemiologic studies that compared the risk of VTE reported that the risk ranged from no increase to a three-fold increase. Before initiating use of drospirenone and ethinyl estradiol in a new COC user or a woman who is switching from a contraceptive that does not contain DRSP, consider the risks and benefits of a DRSP-containing COC in light of her risk of a VTE. Known risk factors for VTE include smoking, obesity, and family history of VTE, in addition to other factors that contraindicate use of COCs [see CONTRAINDICATIONS (4)].
A number of studies have compared the risk of VTE for users of drospirenone and ethinyl estradiol to the risk for users of other COCs, including COCs containing levonorgestrel. Those that were required or sponsored by regulatory agencies are summarized in Table 1.
Table 1: Estimates (Hazard Ratios) of Venous Thromboembolism Risk in Current Users of Drospirenone and Ethinyl Estradiol Compared to Users of Oral Contraceptives that Contain Other Progestins Epidemiologic Study (Author, Year of Publication) Population Studied
Comparator Product
(all are low-dose COCs; with ≤ 0.04 mg of EE)
Hazard Ratio (HR)
(95% CI)
i3 Ingenix
(Seeger 2007)
Initiators, including new users*
All COCs available in the US during the conduct of the study †
HR: 0.9
(0.5-1.6)
EURAS
(Dinger 2007)
Initiators, including new users*
All COC’s available in Europe during the conduct of the study‡
Levonorgestrel/EE
HR: 0.9
(0.6 -1.4)
HR: 1
(0.6-1.8)
“FDA-funded study” (2011)
New users*
All users
(i.e., initiation and continuing use of study combination hormonal contraception)
Other COCs available during the course of the study§
Levonorgestrel/0.03 mg EE
Other COCs available during the course of the study§
Levonorgestrel/0.03 mg EE
HR: 1.8
(1.3-2.4)
HR: 1.6
(1.1-2.2)
HR: (1.7)
(1.4-2.1)
HR: 1.5
(1.2-1.8)
*“New users” - no use of combination hormonal contraception for at least the prior 6 months
†Includes low-dose COCs containing the following progestins: norgestimate, norethindrone, levonorgestrel, desogestrel, norgestrel, medroxyprogesterone, or ethynodiol diacetate
‡Includes low-dose COCs containing the following progestins: levonorgestrel, desogestrel, dienogest, chlormadinone acetate, gestodene, cyproterone acetate, norgestimate, or norethindrone
§Includes low-dose COCs containing the following progestins: norgestimate, norethindrone, or levonorgestrel
In addition to these “regulatory studies,” other studies of various designs have been conducted. Overall, there are two prospective cohort studies (see Table 1): the US post-approval safety study Ingenix [Seeger 2007], the European post-approval safety study EURAS (European Active Surveillance Study) [Dinger 2007]. An extension of the EURAS study, the Long-Term Active Surveillance Study (LASS), did not enroll additional subjects, but continued to assess VTE risk. There are three retrospective cohort studies: one study in the US funded by the FDA (see Table 1), and two from Denmark [Lidegaard 2009, Lidegaard 2011]. There are two case-control studies: the Dutch MEGA study analysis [van Hylckama Vlieg 2009] and the German case-control study [Dinger 2010]. There are two nested case-control studies that evaluated the risk of non-fatal idiopathic VTE: the PharMetrics study [Jick 2011] and the GPRD study [Parkin 2011]. The results of all of these studies are presented in Figure 1.
Risk ratios displayed on logarithmic scale; risk ratio < 1 indicates a lower risk of VTE for DRSP, > 1 indicates an increased risk of VTE for DRSP.
*Comparator “Other COCs”, including LNG- containing COCs
‡LASS is an extension of the EURAS study
#Some adjustment factors are indicated by superscript letters: a) Current heavy smoking, b) hypertension, c) obesity, d) family history, e) age, f) BMI, g) duration of use, h) VTE history, i) period of inclusion, j) calendar year, k) education, l) length of use, m) parity, n) chronic disease, o) concomitant medication, p) smoking, q) duration of exposure, r) site
(References: Ingenix [Seeger 2007]1, EURAS (European Active Surveillance Study) [Dinger 2007]2, LASS (Long-Term Active Surveillance Study) [Dinger, unpublished document on file], FDA-funded study [Sidney 2011]3, Danish [Lidegaard 2009]4, Danish re-analysis [ Lidegaard 2011]5, MEGA study [van Hylckama Vlieg 2009]6, German Case-Control study [Dinger 2010]7, PharMetrics [Jick 2011]8, GPRD study [Parkin 2011]9)
Although the absolute VTE rates are increased for users of hormonal contraceptives compared to non-users, the rates during pregnancy are even greater, especially during the post-partum period (see Figure 2). The risk of VTE in women using COCs has been estimated to be 3 to 9 per 10,000 woman-years. The risk of VTE is highest during the first year of use. Data from a large, prospective cohort safety study of various COCs suggest that this increased risk, as compared to that in non-COC users, is greatest during the first 6 months of COC use. Data from this safety study indicate that the greatest risk of VTE is present after initially starting a COC or restarting (following a 4 week or greater pill-free interval) the same or a different COC.
The risk of thromboembolic disease due to oral contraceptives gradually disappears after COC use is discontinued.
Figure 2 shows the risk of developing a VTE for women who are not pregnant and do not use oral contraceptives, for women who use oral contraceptives, for pregnant women, and for women in the postpartum period. To put the risk of developing a VTE into perspective: If 10,000 women who are not pregnant and do not use oral contraceptives are followed for one year, between 1 and 5 of these women will develop a VTE.
If feasible, stop drospirenone and ethinyl estradiol at least 4 weeks before and through 2 weeks after major surgery or other surgeries known to have an elevated risk of thromboembolism.
Start drospirenone and ethinyl estradiol no earlier than 4 weeks after delivery, in women who are not breastfeeding. The risk of postpartum thromboembolism decreases after the third postpartum week, whereas the risk of ovulation increases after the third postpartum week.
Use of COCs also increases the risk of arterial thromboses such as strokes and myocardial infarctions, especially in women with other risk factors for these events.
COCs have been shown to increase both the relative and attributable risks of cerebrovascular events (thrombotic and hemorrhagic strokes), although, in general, the risk is greatest among older (>35 years of age), hypertensive women who also smoke. COCs also increase the risk for stroke in women with other underlying risk factors.
Oral contraceptives must be used with caution in women with cardiovascular disease risk factors.
Stop drospirenone and ethinyl estradiol if there is unexplained loss of vision, proptosis, diplopia, papilledema, or retinal vascular lesions. Evaluate for retinal vein thrombosis immediately. [see ADVERSE REACTIONS (6)]
5.2 Hyperkalemia
Drospirenone and ethinyl estradiol contains 3 mg of the progestin DRSP, which has anti-mineralocorticoid activity, including the potential for hyperkalemia in high-risk patients, comparable to a 25 mg dose of spironolactone. Drospirenone and ethinyl estradiol is contraindicated in patients with conditions that predispose to hyperkalemia (that is, renal impairment, hepatic impairment, and adrenal insufficiency). Women receiving daily, long-term treatment for chronic conditions or diseases with medications that may increase serum potassium concentration should have their serum potassium concentration checked during the first treatment cycle. Medications that may increase serum potassium concentration include ACE inhibitors, angiotensin–II receptor antagonists, potassium-sparing diuretics, potassium supplementation, heparin, aldosterone antagonists, and NSAIDs.
Consider monitoring serum potassium concentration in high-risk patients who take a strong CYP3A4 inhibitor long-term and concomitantly. Strong CYP3A4 inhibitors include azole antifungals (e.g. ketoconazole, itraconazole, voriconazole), HIV/HCV protease inhibitors (e.g., indinavir, boceprevir), and clarithromycin [see CLINICAL PHARMACOLOGY (12.3)].
5.3 Carcinoma of the Breasts and Reproductive Organs
Women who currently have or have had breast cancer should not use drospirenone and ethinyl estradiol because breast cancer is a hormonally-sensitive tumor.
There is substantial evidence that COCs do not increase the incidence of breast cancer. Although some past studies have suggested that COCs might increase the incidence of breast cancer, more recent studies have not confirmed such findings.
Some studies suggest that COCs are associated with an increase in the risk of cervical cancer or intraepithelial neoplasia. However, there is controversy about the extent to which these findings may be due to differences in sexual behavior and other factors.
5.4 Liver Disease
Discontinue drospirenone and ethinyl estradiol if jaundice develops. Steroid hormones may be poorly metabolized in patients with impaired liver function. Acute or chronic disturbances of liver function may necessitate the discontinuation of COC use until markers of liver function return to normal and COC causation has been excluded.
Hepatic adenomas are associated with COC use. An estimate of the attributable risk is 3.3 cases/100,000 COC users. Rupture of hepatic adenomas may cause death through intra-abdominal hemorrhage.
Studies have shown an increased risk of developing hepatocellular carcinoma in long-term (>8 years) COC users. However, the attributable risk of liver cancers in COC users is less than one case per million users.
Oral contraceptive-related cholestasis may occur in women with a history of pregnancy-related cholestasis. Women with a history of COC-related cholestasis may have the condition recur with subsequent COC use.
5.5 Risk of Liver Enzyme Elevations with Concomitant Hepatitis C Treatment
During clinical trials with the Hepatitis C combination drug regimen that contains ombitasvir/paritaprevir/ritonavir, with or without dasabuvir, ALT elevations greater than 5 times the upper limit of normal (ULN), including some cases greater than 20 times the ULN, were significantly more frequent in women using ethinyl estradiol-containing medications, such as COCs. Discontinue drospirenone and ethinyl estradiol prior to starting therapy with the combination drug regimen ombitasvir/paritaprevir/ritonavir, with or without dasabuvir [see CONTRAINDICATIONS (4)]. Drospirenone and ethinyl estradiol can be restarted approximately 2 weeks following completion of treatment with the Hepatitis C combination drug regimen.
5.6 High Blood Pressure
For women with well-controlled hypertension, monitor blood pressure and stop drospirenone and ethinyl estradiol if blood pressure rises significantly. Women with uncontrolled hypertension or hypertension with vascular disease should not use COCs.
An increase in blood pressure has been reported in women taking COCs, and this increase is more likely in older women and with extended duration of use. The incidence of hypertension increases with increasing concentration of progestin.
5.7 Gallbladder Disease
Studies suggest a small increased relative risk of developing gallbladder disease among COC users.
5.8 Carbohydrate and Lipid Metabolic Effects
Carefully monitor prediabetic and diabetic women who are taking drospirenone and ethinyl estradiol. COCs may decrease glucose tolerance in a dose-related fashion.
Consider alternative contraception for women with uncontrolled dyslipidemia. A small proportion of women will have adverse lipid changes while on COCs.
Women with hypertriglyceridemia, or a family history thereof, may be at an increased risk of pancreatitis when using COCs.
5.9 Headache
If a woman taking drospirenone and ethinyl estradiol develops new headaches that are recurrent, persistent, or severe, evaluate the cause and discontinue drospirenone and ethinyl estradiol if indicated.
An increase in frequency or severity of migraine during COC use (which may be prodromal of a cerebrovascular event) may be a reason for immediate discontinuation of the COC.
5.10 Bleeding Irregularities
Unscheduled (breakthrough or intracyclic) bleeding and spotting sometimes occur in patients on COCs, especially during the first three months of use. If bleeding persists or occurs after previously regular cycles, check for causes such as pregnancy or malignancy. If pathology and pregnancy are excluded, bleeding irregularities may resolve over time or with a change to a different COC.
Data from ten contraceptive efficacy clinical trials (N=2,467) show that the percent of women who took drospirenone and ethinyl estradiol and experienced unscheduled bleeding decreased over time from 12% at cycle 2 to 6% (cycle 13). A total of 24 subjects out of 2,837 in the drospirenone and ethinyl estradiol trials (<1%) discontinued due to bleeding complaints. These are described as metrorrhagia, vaginal hemorrhage, menorrhagia, abnormal withdrawal bleeding, and menometrorrhagia.
The average duration of scheduled bleeding episodes in the majority of subjects (86% to 88%) was 4 to 7 days. Women who use drospirenone and ethinyl estradiol may experience absence of withdrawal bleeding, even if they are not pregnant. Based on subject diaries from contraceptive efficacy trials, during cycles 2 to 13, 1 to 11% of women per cycle experienced no withdrawal bleeding. Some women may encounter post-pill amenorrhea or oligomenorrhea, especially when such a condition was pre-existent.
If withdrawal bleeding does not occur, consider the possibility of pregnancy. If the patient has not adhered to the prescribed dosing schedule (missed one or more active tablets or started taking them on a day later than she should have), consider the possibility of pregnancy at the time of the first missed period and take appropriate diagnostic measures. If the patient has adhered to the prescribed regimen and misses two consecutive periods, rule out pregnancy.
5.11 COC Use Before or During Early Pregnancy
Extensive epidemiological studies have revealed no increased risk of birth defects in women who have used oral contraceptives prior to pregnancy. Studies also do not suggest a teratogenic effect when COCs are taken inadvertently during early pregnancy, particularly in so far as cardiac anomalies and limb-reduction defects are concerned.
The administration of oral contraceptives to induce withdrawal bleeding should not be used as a test for pregnancy [see USE IN SPECIFIC POPULATIONS (8.1)].
5.12 Depression
Women with a history of depression should be carefully observed and drospirenone and ethinyl estradiol discontinued if depression recurs to a serious degree.
5.13 Interference with Laboratory Tests
The use of COCs may change the results of some laboratory tests, such as coagulation factors, lipids, glucose tolerance, and binding proteins. Women on thyroid hormone replacement therapy may need increased doses of thyroid hormone because serum concentrations of thyroid-binding globulin increase with use of COCs [see DRUG INTERACTIONS (7.2)].
DRSP causes an increase in plasma renin activity and plasma aldosterone induced by its mild anti-mineralocorticoid activity.
5.14 Monitoring
A woman who is taking COCs should have a yearly visit with her healthcare provider for a blood pressure check and for other indicated healthcare.
5.15 Other Conditions
In women with hereditary angioedema, exogenous estrogens may induce or exacerbate symptoms of angioedema. Chloasma may occasionally occur, especially in women with a history of chloasma gravidarum. Women with a tendency to chloasma should avoid exposure to the sun or ultraviolet radiation while taking COCs.
-
6 ADVERSE REACTIONS
The following serious adverse reactions with the use of COCs are discussed elsewhere in the labeling:
- Serious cardiovascular events and stroke [see BOXED WARNING AND WARNINGS AND PRECAUTIONS (5.1)]
- Vascular events [see WARNINGS AND PRECAUTIONS (5.1)]
- Liver disease [see WARNINGS AND PRECAUTIONS (5.4)]
- Adverse reactions commonly reported by COC users are:
- Irregular uterine bleeding
- Nausea
- Breast tenderness
- Headache
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in practice.
The data provided reflect the experience with the use of drospirenone and ethinyl estradiol (3 mg DRSP/0.03 mg EE) in the adequate and well-controlled studies for contraception (N=2,837). The US pivotal clinical study (N=326) was a multicenter, open-label trial in healthy women aged 18 to 35 who were treated for up to 13 cycles. The second pivotal study (N=442)was a multicenter, randomized, open-label comparative European study of drospirenone and ethinyl estradiol vs. 0.150 mg desogestrel/0.03 mg EE conducted in healthy women aged 17 to 40 who were treated for up to 26 cycles.
The most common adverse reactions (≥ 2% of users) were: premenstrual syndrome (13.2%), headache/migraine (10.7%), breast pain/tenderness/discomfort (8.3%), nausea/vomiting (4.5%) abdominal pain/discomfort/tenderness (2.3%) and mood changes (depression, depressed mood, irritability, mood swings, mood altered and affect lability (2.3%).
Adverse Reactions (≥ 1%) Leading to Study Discontinuation
Of 2,837 women, 6.7% discontinued from the clinical trials due to an adverse reaction; the most frequent adverse reaction leading to discontinuation was headache/migraine (1.5%).
Serious Adverse Reactions
Depression, pulmonary embolism, toxic skin eruption, and uterine leiomyoma.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of drospirenone and ethinyl estradiol. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Adverse reactions, including fatalities, are grouped into System Organ Classes and ordered by frequency.
Vascular disorders: Venous and arterial thromboembolic events (including pulmonary emboli, deep vein thrombosis, intracardiac thrombosis, intracranial venous sinus thrombosis, sagittal sinus thrombosis, retinal vein occlusion, myocardial infarction and stroke), hypertension
Hepatobiliary disorders: Gallbladder disease
Immune system disorders: Hypersensitivity
Metabolism and nutrition disorders: Hyperkalemia
Skin and subcutaneous tissue disorders: Chloasma
-
7 DRUG INTERACTIONS
Consult the labeling of all concurrently-used drugs to obtain further information about interactions with hormonal contraceptives or the potential for enzyme alterations.
7.1 Effects of Other Drugs on Combined Oral Contraceptives
Substances diminishing the efficacy of COCs:
Drugs or herbal products that induce certain enzymes, including cytochrome P450 3A4 (CYP3A4), may decrease the effectiveness of COCs or increase breakthrough bleeding. Some drugs or herbal products that may decrease the effectiveness of hormonal contraceptives include phenytoin, barbiturates, carbamazepine, bosentan, felbamate, griseofulvin, oxcarbazepine, rifampin, topiramate and products containing St. John’s wort. Interactions between oral contraceptives and other drugs may lead to breakthrough bleeding and/or contraceptive failure. Counsel women to use an alternative method of contraception or a back-up method when enzyme inducers are used with COCs, and to continue back-up contraception for 28 days after discontinuing the enzyme inducer to ensure contraceptive reliability.
Substances increasing the plasma concentrations of COCs:
Co-administration of atorvastatin and certain COCs containing EE increase AUC values for EE by approximately 20%. Ascorbic acid and acetaminophen may increase plasma EE concentrations, possibly by inhibition of conjugation.
Concomitant administration of moderate or strong CYP3A4 inhibitors such as azole antifungals (e.g., ketoconazole, itraconazole, voriconazole, fluconazole), verapamil, macrolides (e.g., clarithromycin, erythromycin), diltiazem, and grapefruit juice can increase the plasma concentrations of the estrogen or the progestin or both. In a clinical drug-drug interaction study conducted in premenopausal women, once daily co-administration of DRSP 3 mg/EE 0.02 mg containing tablets with strong CYP3A4 inhibitor, ketoconazole 200 mg twice daily for 10 days resulted in a moderate increase of DRSP systemic exposure. The exposure of EE was increased mildly [see WARNINGS AND PRECAUTIONS (5.2) and CLINICAL PHARMACOLGY (12.3)].
Human immunodeficiency virus (HIV)/Hepatitis C virus (HCV) protease inhibitors and non-nucleoside reverse transcriptase inhibitors
Significant changes (increase or decrease) in the plasma concentrations of estrogen and progestin have been noted in some cases of co-administration with HIV/HCV protease inhibitors or with non-nucleoside reverse transcriptase inhibitors.
Antibiotics
There have been reports of pregnancy while taking hormonal contraceptives and antibiotics, but clinical pharmacokinetic studies have not shown consistent effects of antibiotics on plasma concentrations of synthetic steroids.
7.2 Effects of Combined Oral Contraceptives on Other Drugs
COCs containing EE may inhibit the metabolism of other compounds. COCs have been shown to significantly decrease plasma concentrations of lamotrigine, likely due to induction of lamotrigine glucuronidation. This may reduce seizure control; therefore, dosage adjustments of lamotrigine may be necessary. Consult the labeling of the concurrently-used drug to obtain further information about interactions with COCs or the potential for enzyme alterations.
COCs Increasing the Plasma Concentrations of CYP450 Enzymes
In clinical studies, administration of a hormonal contraceptive containing EE did not lead to any increase or only to a weak increase in plasma concentrations of CYP3A4 substrates (e.g., midazolam) while plasma concentrations of CYP2C19 substrates (e.g., omeprazole and voriconazole) and CYP1A2 substrates (e.g., theophylline and tizanidine) can have a weak or moderate increase.
Clinical studies did not indicate an inhibitory potential of DRSP towards human CYP enzymes at clinically relevant concentrations [see CLINICAL PHARMACOLOGY (12.3)].
Women on thyroid hormone replacement therapy may need increased doses of thyroid hormone because serum concentration of thyroid-binding globulin increases with use of COCs.
Potential to Increase Serum Potassium Concentration
There is a potential for an increase in serum potassium concentration in women taking drospirenone and ethinyl estradiol with other drugs that may increase serum potassium concentration [see WARNINGS AND PRECAUTIONS (5.2) AND CLINICAL PHARMACOLOGY (12.3)].
7.3 Concomitant Use of HCV Combination Therapy – Liver Enzyme Elevation
Do not co-administer drospirenone and ethinyl with HCV drug combinations containing ombitasvir/paritaprevir/ritonavir, with or without dasabuvir, due to potential for ALT elevations [see WARNINGS AND PRECAUTIONS (5.5)].
7.4 Interference with Laboratory Tests
The use of contraceptive steroids may influence the results of certain laboratory tests, such as coagulation factors, lipids, glucose tolerance, and binding proteins. DRSP causes an increase in plasma renin activity and plasma aldosterone induced by its mild anti-mineralocorticoid activity [see WARNINGS AND PRECAUTIONS (5.12) AND DRUG INTERACTIONS (7.2)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
There is little or no increased risk of birth defects in women who inadvertently use COCs during early pregnancy. Epidemiologic studies and meta-analyses have not found an increased risk of genital or non-genital birth defects (including cardiac anomalies and limb-reduction defects) following exposure to low dose COCs prior to conception or during early pregnancy.
The administration of COCs to induce withdrawal bleeding should not be used as a test for pregnancy. COCs should not be used during pregnancy to treat threatened or habitual abortion.
Women who do not breastfeed may start COCs no earlier than four weeks postpartum.
8.3 Nursing Mothers
When possible, advise the nursing mother to use other forms of contraception until she has weaned her child. Estrogen-containing COCs can reduce milk production in breastfeeding mothers. This is less likely to occur once breastfeeding is well-established; however, it can occur at any time in some women. Small amounts of oral contraceptive steroids and/or metabolites are present in breast milk.
After oral administration of drospirenone and ethinyl estradiol, about 0.02% of the DRSP dose was excreted into the breast milk of postpartum women within 24 hours. This results in a maximal daily dose of about 0.003 mg DRSP in an infant.
8.4 Pediatric Use
Safety and efficacy of drospirenone and ethinyl estradiol has been established in women of reproductive age. Efficacy is expected to be the same for postpubertal adolescents under the age of 18 and for users 18 years and older. Use of this product before menarche is not indicated.
8.5 Geriatric Use
Drospirenone and ethinyl estradiol has not been studied in postmenopausal women and is not indicated in this population.
8.6 Patients with Renal Impairment
Drospirenone and ethinyl estradiol is contraindicated in patients with renal impairment [see CONTRAINDICATIONS (4) and WARNINGS AND PRECAUTIONS (5.2)].
In subjects with creatinine clearance (CLcr) of 50 to 79 mL/min, serum DRSP concentrations were comparable to those in a control group with CLcr ≥ 80 mL/min. In subjects with CLcr of 30 to 49 mL/min, serum DRSP concentrations were on average 37% higher than those in the control group. In addition, there is a potential to develop hyperkalemia in subjects with renal impairment whose serum potassium is in the upper reference range, and who are concomitantly using potassium sparing drugs [see CLINICAL PHARMACOLOGY (12.3)].
8.7 Patients with Hepatic Impairment
Drospirenone and ethinyl estradiol is contraindicated in patients with hepatic disease [see CONTRAINDICATIONS (4) AND WARNINGS AND PRECAUTIONS (5.4)]. The mean exposure to DRSP in women with moderate liver impairment is approximately three times higher than the exposure in women with normal liver function. Drospirenone and ethinyl estradiol has not been studied in women with severe hepatic impairment.
8.8 Race
No clinically significant difference was observed between the pharmacokinetics of DRSP or EE in Japanese versus Caucasian women [see CLINICAL PHARMACOLOGY (12.3)].
-
10 OVERDOSAGE
There have been no reports of serious ill effects from overdose, including ingestion by children. Overdosage may cause withdrawal bleeding in females and nausea.
DRSP is a spironolactone analogue which has anti-mineralocorticoid properties. Serum concentration of potassium and sodium, and evidence of metabolic acidosis, should be monitored in cases of overdose.
-
11 DESCRIPTION
SyedaTM (drospirenone and ethinyl estradiol tablets) provide an oral contraceptive regimen consisting of 28 film-coated tablets that contain the ingredients specified for each tablet below:
- 21 yellow tablets each containing 3 mg drospirenone and 0.03 mg ethinyl estradiol
- 7 inert white tablets
The inactive ingredients are corn starch, crospovidone (Polyplasdone XL), crosspovidone (Polyplasdone XL-10), lactose fast flo, macrogol/PEG 3350, magnesium stearate vegetable (kemilub em-f-v), polysorbate 80 (tween 80), polyvinyl alcohol-part. hydrolyzed, povidone k-30 (kollidon K-30), pregelatinized starch (sepistab ST200), talc and titanium dioxide. In addition, each active tablet contains yellow iron oxide.
Drospirenone (6R,7R,8R,9S,10R,13S,14S,15S,16S,17S)-1,3',4',6,6a,7,8,9,10,11,12,13, 14,15,15a,16-hexadecahydro-10,13-dimethylspiro-[17H-dicyclopropa-[6,7:15,16] cyclopenta[a]phenanthrene-17,2'(5H)-furan]-3,5'(2H)-dione) is a synthetic progestational compound and has a molecular weight of 366.5 and a molecular formula of C24H30O3.
Ethinyl estradiol (19-nor-17α-pregna 1,3,5(10)-triene-20-yne-3,17-diol) is a synthetic estrogenic compound and has a molecular weight of 296.4 and a molecular formula of C20H24O2.
The structural formulas are as follows:
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
COCs lower the risk of becoming pregnant primarily by suppressing ovulation. Other possible mechanisms may include cervical mucus changes that inhibit sperm penetration and endometrial changes that reduce the likelihood of implantation.
12.2 Pharmacodynamics
Drospirenone is a spironolactone analogue with anti-mineralocorticoid activity. The estrogen in SyedaTM is ethinyl estradiol (EE).
No specific pharmacodynamic studies were conducted with drospirenone and ethinyl estradiol
12.3 Pharmacokinetics
Absorption
The absolute bioavailability of DRSP from a single entity tablet is about 76%. The absolute bioavailability of EE is approximately 40% as a result of presystemic conjugation and first-pass metabolism. The absolute bioavailability of drospirenone and ethinyl estradiol, which is a combination tablet of DRSP and EE, has not been evaluated. Serum concentrations of DRSP and EE reached peak levels within 1 to 2 hours after administration of drospirenone and ethinyl estradiol.
The pharmacokinetics of DRSP are dose proportional following single doses ranging from 1 to 10 mg. Following daily dosing of drospirenone and ethinyl estradiol, steady state DRSP concentrations were observed after 8 days. There was about 2 to 3 fold accumulation in serum Cmax and AUC (0-24h) values of DRSP following multiple dose administration of drospirenone and ethinyl estradiol (see Table 2).
For EE, steady-state conditions are reported during the second half of a treatment cycle. Following daily administration of drospirenone and ethinyl estradiol serum Cmax and AUC (0-24h) values of EE accumulate by a factor of about 1.5 to 2 (see Table 2).
Table 2 Mean Pharmacokinetic Parameters Of Drospirenone And Ethinyl Estradiol (DRSP 3 mg and EE 0.03 mg ) DRSP
Mean (%CV) ValuesCycle / Day
No. of Subjects
Cmax(ng/mL)
Tmax(h)
AUC(0-24h) (ngh/mL)
t1/2 (h)
1/1
12
36.9 (13)
1.7 (47)
288 (25)
NA
1/21
12
87.5 (59)
1.7 (20)
827 (23)
30.9 (44)
6/21
12
84.2 (19)
1.8 (19)
930 (19)
32.5 (38)
9/21
12
81.3 (19)
1.6 (38)
957 (23)
31.4 (39)
13/21
12
78.7 (18)
1.6 (26)
968 (24)
31.1 (36)
EE
Mean (%CV) ValuesCycle / Day
No. of Subjects
Cmax(pg/mL)
Tmax(h)
AUC(0-24h) (pgh/mL)
t1/2 (h)
1/1
11
53.5 (43)
1.9 (45)
280 (87)
NA
1/21
11
92.1 (35)
1.5 (40)
461 (94)
NA
6/21
11
99.1 (45)
1.5 (47)
346 (74)
NA
9/21
11
87 (43)
1.5 (42)
485 (92)
NA
13/21
10
90.5 (45)
1.6 (38)
469 (83)
NA
N/A – Not available
Food Effect
The rate of absorption of DRSP and EE following single administration of a formulation similar to drospirenone and ethinyl estradiol was slower under fed (high fat meal) conditions with the serum Cmax being reduced about 40% for both components. The extent of absorption of DRSP, however, remained unchanged. In contrast, the extent of absorption of EE was reduced by about 20% under fed conditions.
Distribution
DRSP and EE serum concentrations decline in two phases. The apparent volume of distribution of DRSP is approximately 4 L/kg and that of EE is reported to be approximately 4 to 5 L/kg.
DRSP does not bind to sex hormone binding globulin (SHBG) or corticosteroid binding globulin (CBG) but binds about 97% to other serum proteins. Multiple dosing over 3 cycles resulted in no change in the free fraction (as measured at trough concentrations). EE is reported to be highly but non-specifically bound to serum albumin (approximately 98.5 %) and induces an increase in the serum concentrations of both SHBG and CBG. EE induced effects on SHBG and CBG were not affected by variation of the DRSP dosage in the range of 2 to 3 mg.
Metabolism
The two main metabolites of DRSP found in human plasma were identified to be the acid form of DRSP generated by opening of the lactone ring and the 4,5-dihydrodrospirenone-3-sulfate, formed by reduction and subsequent sulfation. These metabolites were shown not to be pharmacologically active. Drospirenone is also subject to oxidative metabolism catalyzed by CYP3A4.
EE has been reported to be subject to significant gut and hepatic first-pass metabolism. Metabolism of EE and its oxidative metabolites occur primarily by conjugation with glucuronide or sulfate.
CYP3A4 in the liver is responsible for the 2-hydroxylation which is the major oxidative reaction. The 2-hydroxy metabolite is further transformed by methylation and glucuronidation prior to urinary and fecal excretion.
Excretion
DRSP serum concentrations are characterized by a terminal disposition phase half-life of approximately 30 hours after both single and multiple dose regimens. Excretion of DRSP was nearly complete after ten days and amounts excreted were slightly higher in feces compared to urine. DRSP was extensively metabolized and only trace amounts of unchanged DRSP were excreted in urine and feces. At least 20 different metabolites were observed in urine and feces. About 38 to 47% of the metabolites in urine were glucuronide and sulfate conjugates. In feces, about 17 to 20% of the metabolites were excreted as glucuronides and sulfates.
For EE the terminal disposition phase half-life has been reported to be approximately 24 hours. EE is not excreted unchanged. EE is excreted in the urine and feces as glucuronide and sulfate conjugates and undergoes enterohepatic circulation.
Use in Specific Populations
Pediatric Use:
Safety and efficacy of drospirenone and ethinyl estradiol has been established in women of reproductive age. Efficacy is expected to be the same for postpubertal adolescents under the age of 18 and for users 18 years and older. Use of this product before menarche is not indicated.
Geriatric Use:
Drospirenone and ethinyl estradiol has not been studied in postmenopausal women and is not indicated in this population.
Race:
No clinically significant difference was observed between the pharmacokinetics of DRSP or EE in Japanese versus Caucasian women (age 25 to 35) when 3 mg DRSP/0.02 mg EE was administered daily for 21 days. Other ethnic groups have not been specifically studied.
Renal Impairment:
Drospirenone and ethinyl estradiol is contraindicated in patients with renal impairment.
The effect of renal impairment on the pharmacokinetics of DRSP (3 mg daily for 14 days) and the effect of DRSP on serum potassium concentrations were investigated in three separate groups of female subjects (n=28, age 30 to 65). All subjects were on a low potassium diet. During the study, 7 subjects continued the use of potassium-sparing drugs for the treatment of their underlying illness. On the 14th day (steady-state) of DRSP treatment, the serum DRSP concentrations in the group with CLcr of 50 to 79 mL/min were comparable to those in a the control group with CLcr ≥ 80 mL/min. The serum DRSP concentrations were on average 37% higher in the group with CLcr of 30 to 49 mL/min compared to those in the control group. DRSP treatment did not show any clinically significant effect on serum potassium concentration. Although hyperkalemia was not observed in the study, in five of the seven subjects who continued use of potassium sparing drugs during the study, mean serum potassium concentrations increased by up to 0.33 mEq/L. [See CONTRAINDICATIONS (4) and WARNINGS AND PRECAUTIONS (5.2).]
Hepatic Impairment
Drospirenone and ethinyl estradiol is contraindicated in patients with hepatic disease.
The mean exposure to DRSP in women with moderate liver impairment is approximately three times higher than the exposure in women with normal liver function. Drospirenone and ethinyl estradiol has not been studied in women with severe hepatic impairment [see CONTRAINDICATIONS (4) and WARNINGS AND PRECAUTIONS (5.4)].
Drug Interactions
Consult the labeling of all concurrently used drugs to obtain further information about interactions with oral contraceptives or the potential for enzyme alterations.
Effects of Other Drugs on Combined Oral Contraceptives
Substances diminishing the efficacy of COCs
Drugs or herbal products that induce certain enzymes, including CYP3A4, may decrease the effectiveness of COCs or increase breakthrough bleeding.
Substances increasing the plasma concentrations of COCs
Co-administration of atorvastatin and certain COCs containing ethinyl estradiol increase AUC values for ethinyl estradiol by approximately 20%. Ascorbic acid and acetaminophen may increase plasma ethinyl estradiol concentrations, possibly by inhibition of conjugation. In a clinical drug-drug interaction study conducted in 20 premenopausal women, co-administration of a DRSP (3 mg)/EE (0.02 mg) COC with the strong CYP3A4 inhibitor ketoconazole (200 mg twice daily) for 10 days increased the AUC(0-24h) of DRSP and EE by 2.68-fold (90% CI: 2.44, 2.95) and 1.40-fold (90% CI: 1.31, 1.49), respectively. The increases in Cmax were 1.97-fold (90% CI: 1.79, 2.17) and 1.39-fold (90% CI: 1.28, 1.52) for DRSP and EE, respectively. Although no clinically relevant effects on safety or laboratory parameters including serum potassium were observed, this study only assessed subjects for 10 days. The clinical impact for a patient taking a DRSP-containing COC concomitantly with chronic use of a CYP3A4/5 inhibitor is unknown [see WARNINGS AND PRECAUTIONS (5.2)].
HIV/HCV protease inhibitors and non-nucleoside reverse transcriptase inhibitors
Significant changes (increase or decrease) in the plasma concentrations of estrogen and progestin have been noted in some cases of co-administration with HIV/HCV protease inhibitors or with non-nucleoside reverse transcriptase inhibitors.
Antibiotics
There have been reports of pregnancy while taking hormonal contraceptives and antibiotics, but clinical pharmacokinetic studies have not shown consistent effects of antibiotics on plasma concentrations of synthetic steroids.
Effects of Combined Oral Contraceptives on Other Drugs
COCs containing ethinyl estradiol may inhibit the metabolism of other compounds. COCs have been shown to significantly decrease plasma concentrations of lamotrigine, likely due to induction of lamotrigine glucuronidation. This may reduce seizure control; therefore, dosage adjustments of lamotrigine may be necessary. Consult the labeling of the concurrently-used drug to obtain further information about interactions with COCs or the potential for enzyme alterations.
In vitro, EE is a reversible inhibitor of CYP2C19, CYP1A1 and CYP1A2 as well as a mechanism-based inhibitor of CYP3A4/5, CYP2C8, and CYP2J2. Metabolism of DRSP and potential effects of DRSP on hepatic CYP enzymes have been investigated in in vitro and in vivo studies. In in vitro studies DRSP did not affect turnover of model substrates of CYP1A2 and CYP2D6, but had an inhibitory influence on the turnover of model substrates of CYP1A1, CYP2C9, CYP2C19, and CYP3A4, with CYP2C19 being the most sensitive enzyme. The potential effect of DRSP on CYP2C19 activity was investigated in a clinical pharmacokinetic study using omeprazole as a marker substrate. In the study with 24 postmenopausal women [including 12 women with homozygous (wild type) CYP2C19 genotype and 12 women with heterozygous CYP2C19 genotype] the daily oral administration of 3 mg DRSP for 14 days did not affect the oral clearance of omeprazole (40 mg, single oral dose) and the CYP2C19 product 5-hydroxy omeprazole. Furthermore, no significant effect of DRSP on the systemic clearance of the CYP3A4 product omeprazole sulfone was found. These results demonstrate that DRSP did not inhibit CYP2C19 and CYP3A4 in vivo.
Two additional clinical drug-drug interaction studies using simvastatin and midazolam as marker substrates for CYP3A4 were each performed in 24 healthy postmenopausal women. The results of these studies demonstrated that pharmacokinetics of the CYP3A4 substrates were not influenced by steady state DRSP concentrations achieved after administration of 3 mg DRSP/day.
Women on thyroid hormone replacement therapy may need increased doses of thyroid hormone because serum concentration of thyroid-binding globulin increases with use of COCs.
Interactions With Drugs That Have the Potential to Increase Serum Potassium Concentration:
There is a potential for an increase in serum potassium concentration in women taking drospirenone and ethinyl estradiol with other drugs that may increase serum potassium concentration [see WARNINGS AND PRECAUTIONS (5.2)].
A drug-drug interaction study of DRSP 3 mg/estradiol (E2) 1 mg versus placebo was performed in 24 mildly hypertensive postmenopausal women taking enalapril maleate 10 mg twice daily. Potassium concentrations were obtained every other day for a total of 2 weeks in all subjects. Mean serum potassium concentrations in the DRSP/E2 treatment group relative to baseline were 0.22 mEq/L higher than those in the placebo group. Serum potassium concentrations also were measured at multiple time points over 24 hours at baseline and on Day 14. On Day 14, the ratios for serum potassium Cmax and AUC in the DRSP/E2 group to those in the placebo group were 0.955 (90% CI: 0.914, 0.999) and 1.010 (90% CI: 0.944, 1.08), respectively. No patient in either treatment group developed hyperkalemia (serum potassium concentrations > 5.5 mEq/L).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 24 month oral carcinogenicity study in mice dosed with 10 mg/kg/day DRSP alone or 1 + 0.01, 3 + 0.03 and 10 + 0.1 mg/kg/day of DRSP and EE, 0.1 to 2 times the exposure (AUC of DRSP) of women taking a contraceptive dose, there was an increase in carcinomas of the harderian gland in the group that received the high dose of DRSP alone. In a similar study in rats given 10 mg/kg/day DRSP alone or 0.3 + 0.003, 3 + 0.03 and 10 + 0.1 mg/kg/day DRSP and EE, 0.8 to 10 times the exposure of women taking a contraceptive dose, there was an increased incidence of benign and total (benign and malignant) adrenal gland pheochromocytomas in the group receiving the high dose of DRSP. Mutagenesis studies for DRSP were conducted in vivo and in vitro and no evidence of mutagenic activity was observed.
-
14 CLINICAL STUDIES
In the clinical efficacy studies of up to 2 years duration, 2,629 subjects completed 33,160 cycles of use without any other contraception. The mean age of the subjects was 25.5 ± 4.7 years. The age range was 16 to 37 years. The racial demographic was: 83% Caucasian, 1% Hispanic, 1% Black, <1% Asian, <1% other, <1% missing data, 14% not inquired and <1% unspecified. Pregnancy rates in the clinical trials were less than one per 100 woman-years of use.
-
15 REFERENCES
- 1. Seeger, J.D., Loughlin, J., Eng, P.M., Clifford, C.R., Cutone, J., and Walker, A.M. (2007). Risk of thromboembolism in women taking ethinylestradiol/drospirenone and other oral contraceptives. Obstet Gynecol 110, 587-593.
- 2. Dinger, J.C., Heinemann, L.A., and Kuhl-Habich, D. (2007). The safety of a drospirenone-containing oral contraceptive: final results from the European Active Surveillance Study on oral contraceptives based on 142,475 women-years of observation. Contraception 75, 344-354.
- 3. Combined hormonal contraceptives (CHCs) and the risk of cardiovascular endpoints. Sidney, S. (primary author) http://www.fda.gov/downloads/Drugs/DrugSafety/UCM277384.pdf, accessed Oct 27, 2011.
- 4. Lidegaard, O., Lokkegaard, E., Svendsen, A.L., and Agger, C. (2009). Hormonal contraception and risk of venous thromboembolism: national follow-up study. BMJ 339, b2890.
- 5. Lidegaard, O., Nielsen, L.H., Skovlund, C.W., Skjeldestad, F.E., and Lokkegaard, E. (2011). Risk of venous thromboembolism from use of oral contraceptives containing different progestogens and oestrogen doses: Danish cohort study, 2001-9. BMJ 343, d6423.
- 6. van Hylckama Vlieg, A., Helmerhorst, F.M., Vandenbroucke, J.P., Doggen, C.J., and Rosendaal, F.R. (2009). The venous thrombotic risk of oral contraceptives, effects of oestrogen dose and progestogen type: results of the MEGA case-control study. BMJ 339, b2921.
- 7. Dinger, J., Assmann, A., Mohner, S., and Minh, T.D. (2010). Risk of venous thromboembolism and the use of dienogest- and drospirenone-containing oral contraceptives: results from a German case-control study. J Fam Plann Reprod Health Care 36, 123-129.
- 8. Jick, S.S., and Hernandez, R.K. (2011). Risk of non-fatal venous thromboembolism in women using oral contraceptives containing drospirenone compared with women using oral contraceptives containing levonorgestrel: case-control study using United States claims data. BMJ 342, d2151.
- 9. Parkin, L., Sharples, K., Hernandez, R.K., and Jick, S.S. (2011). Risk of venous thromboembolism in users of oral contraceptives containing drospirenone or levonorgestrel: nested case-control study based on UK General Practice Research Database. BMJ 342, d2139.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
SyedaTM tablets (drospirenone and ethinyl estradiol tablets, USP) are (active tablets), round, yellow colored, film coated tablets, SZ and U3 are debossed on opposite sides of the tablet.
Inert/Placebo, are round, white film coated tablets, SZ and J1 are debossed on opposite sides of the tablet. NDC: 70700-115-85, one box containing 3 individual unit cartons
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
- Counsel patients that cigarette smoking increases the risk of serious cardiovascular events from COC use, and that women who are over 35 years old and smoke should not use COCs.
- Counsel patients that the increased risk of VTE compared to non-users of COCs is greatest after initially starting a COC or restarting (following a 4-week or greater pill-free interval) the same or a different COC.
- Counsel patients about the information regarding the risk of VTE with DRSP-containing COCs compared to COCs that contain levonorgestrel or some other progestins.
- Counsel patients that SyedaTM does not protect against HIV infection (AIDS) and other sexually transmitted diseases.
- Counsel patients on Warnings and Precautions associated with COCs.
- Counsel patients that SyedaTM contains DRSP. Drospirenone may increase potassium. Patients should be advised to inform their healthcare provider if they have kidney, liver or adrenal disease because the use of SyedaTM in the presence of these conditions could cause serious heart and health problems. They should also inform their healthcare provider if they are currently on daily, long-term treatment (NSAIDs, potassium-sparing diuretics, potassium supplementation, ACE inhibitors, angiotensin-II receptor antagonists, heparin or aldosterone antagonists) for a chronic condition or taking strong CYP3A4 inhibitors.
- Inform patients that SyedaTMis not indicated during pregnancy. If pregnancy occurs during treatment with SyedaTM, instruct the patient to stop further intake.
- Counsel patients to take one tablet daily by mouth at the same time every day. Instruct patients what to do in the event pills are missed. See “What to Do if You Miss Pills” section in FDA-Approved Patient Labeling.
- Counsel patients to use a back-up or alternative method of contraception when enzyme inducers are used with COCs.
- Counsel patients who are breastfeeding or who desire to breastfeed that COCs may reduce breast milk production. This is less likely to occur if breastfeeding is well established.
- Counsel any patient who starts COCs postpartum, and who has not yet had a period, to use an additional method of contraception until she has taken a yellow tablet for 7 consecutive days.
- Counsel patients that amenorrhea may occur. Rule out pregnancy in the event of amenorrhea in two or more consecutive cycles.
Manufactured by Laboratorios Leon Farma S.A., Spain
For Xiromed LLC, Florham Park, NJ 07932
-
FDA Approved Patient Labeling
Guide for Using SyedaTM
SyedaTM
(Drospirenone and Ethinyl Estradiol Tablets, USP)
3 mg and 0.03 mg
WARNING TO WOMEN WHO SMOKE
Do not use SyedaTM if you smoke cigarettes and are over 35 years old. Smoking increases your risk of serious cardiovascular side effects (heart and blood vessel problems) from birth control pills, including death from heart attack, blood clots or stroke. This risk increases with age and the number of cigarettes you smoke.
Birth control pills help to lower the chances of becoming pregnant when taken as directed. They do not protect against HIV infection (AIDS) and other sexually transmitted diseases.
What is SyedaTM?
SyedaTM is a birth control pill. It contains two female hormones, a synthetic estrogen called ethinyl estradiol and a progestin called drospirenone.
The progestin drospirenone may increase potassium. Therefore, you should not take SyedaTM if you have kidney, liver or adrenal disease because this could cause serious heart and health problems. Other drugs may also increase potassium. If you are currently on daily, long-term treatment for a chronic condition with any of the medications below, you should consult your healthcare provider about whether SyedaTM is right for you, and during the first month that you take SyedaTM, you should have a blood test to check your potassium level.
- NSAIDs (ibuprofen [Motrin, Advil], naproxen [Aleve and others] when taken long-term and daily for treatment of arthritis or other problems)
- Potassium-sparing diuretics (spironolactone and others)
- Potassium supplementation
- ACE inhibitors (Capoten, Vasotec, Zestril and others)
- Angiotensin-II receptor antagonists (Cozaar, Diovan, Avapro and others)
- Heparin
- Aldosterone antagonists
How Well Does SyedaTMWork?
Your chance of getting pregnant depends on how well you follow the directions for taking your birth control pills. The better you follow the directions, the less chance you have of getting pregnant.
Based on the results of two clinical studies, about 1 woman out of 100 women may get pregnant during the first year they use SyedaTM.
The following chart shows the chance of getting pregnant for women who use different methods of birth control. Each box on the chart contains a list of birth control methods that are similar in effectiveness. The most effective methods are at the top of the chart. The box on the bottom of the chart shows the chance of getting pregnant for women who do not use birth control and are trying to get pregnant.
How Do I Take SyedaTM?
- 1. Be sure to read these directions before you start taking your pills or anytime you are not sure what to do.
- 2.
The right way to take the pill is to take one pill every day at the same time in the order directed on the package. Preferably, take the pill after the evening meal or at bedtime, with some liquid, as needed. SyedaTM can be taken without regard to meals.
If you miss pills you could get pregnant. This includes starting the pack late. The more pills you miss, the more likely you are to get pregnant. See "WHAT TO DO IF YOU MISS PILLS” below. - 3.
Many women have spotting or light bleeding at unexpected times, or may feel sick to their stomach during the first 1 to 3 packs of pills.
If you do have spotting or light bleeding or feel sick to your stomach, do not stop taking the pill. The problem will usually go away. If it does not go away, check with your healthcare provider. - 4.
Missing pills can also cause spotting or light bleeding, even when you make up these missed pills.
On the days you take two pills, to make up for missed pills, you could also feel a little sick to your stomach. - 5.
If you have vomiting (within 3 to 4 hours after you take your pill), you should follow the instructions for "WHAT TO DO IF YOU MISS PILLS." If you have diarrhea or if you take certain medicines, including some antibiotics and some herbal products such as St. John's Wort, your pills may not work as well.
Use a back-up method (such as condoms and spermicides) until you check with your healthcare provider. - 6. If you have trouble remembering to take the pill, talk to your healthcare provider about how to make pill-taking easier or about using another method of birth control.
- 7. If you have any questions or are unsure about the information in this leaflet, call your healthcare provider.
Before You Start Taking Your Pills
1. Decide What Time of Day You Want to Take Your Pill
It is important to take SyedaTM in the order directed on the package at the same time every day, preferably after the evening meal or at bedtime, with some liquid, as needed. SyedaTM can be taken without regard to meals.
2. Look at Your Pill Pack – It has 28 Pills
The SyedaTM pill pack has 21 yellow pills (with hormones) to be taken for three weeks, followed by 7 white pills (without hormones) to be taken for one week.
3. Also look for:
a) Where on the pack to start taking pills,
b) In what order to take the pills (follow the arrows)
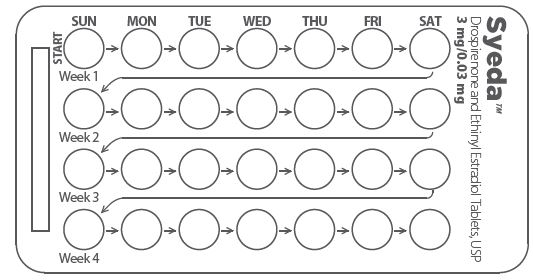
4. Be sure you have ready at all times (a) another kind of birth control (such as condoms and spermicides) to use as a back-up in case you miss pills, and (b) an extra, full pill pack.
When To Start the First Pack of Pills
You have a choice for which day to start taking your first pack of pills. Decide with your healthcare provider which is the best day for you. Pick a time of day which will be easy to remember.
Day 1 Start:
- 1. Take the first yellow pill of the pack during the first 24 hours of your period.
- 2. You will not need to use a back-up method of birth control, because you are starting the Pill at the beginning of your period. However, if you start SyedaTM later than the first day of your period, you should use another method of birth control (such as a condom and spermicide) as a back-up method until you have taken 7 yellow pills.
Sunday Start:
- 1. Take the first yellow pill of the pack on the Sunday after your period starts, even if you are still bleeding. If your period begins on Sunday, start the pack that same day.
- 2. Use another method of birth control (such as a condom and spermicide) as a back-up method if you have sex anytime from the Sunday you start your first pack until the next Sunday (7 days). This also applies if you start SyedaTM after having been pregnant and you have not had a period since your pregnancy.
When You Switch From a Different Birth Control Pill
When switching from another birth control pill, SyedaTM should be started on the same day that a new pack of the previous birth control pill would have been started.
When You Switch From Another Type of Birth Control Method
When switching from a transdermal patch or vaginal ring, SyedaTM should be started when the next application would have been due. When switching from an injection, SyedaTM should be started when the next dose would have been due. When switching from an intrauterine contraceptive or an implant, SyedaTM should be started on the day of removal.
What to Do During the Month
- 1.
Take one pill at the same time every day until the pack is empty.
Do not skip pills even if you are spotting or bleeding between monthly periods or feel sick to your stomach (nausea).
Do not skip pills even if you do not have sex very often. - 2. When you finish a pack of pills, start the next pack on the day after your last white pill. Do not wait any days between packs.
What to Do if You Miss Pills
If you miss 1 yellow pill of your pack:
- 1. Take it as soon as you remember. Take the next pill at your regular time. This means you may take two pills in one day.
- 2. You do not need to use a back-up birth control method if you have sex.
If you miss 2 yellow pills in a row in Week 1 or Week 2 of your pack:
- 1. Take two pills on the day you remember and two pills the next day.
- 2. Then take one pill a day until you finish the pack.
- 3. You could become pregnant if you have sex in the 7 days after you restart your pills. You must use another birth control method (such as a condom and spermicide) as a back-up for those 7 days.
If you miss 2 yellow pills in a row in Week 3 or Week 4 of your pack:
- 1.
If you are a Day 1 Starter:
Throw out the rest of the pill pack and start a new pack that same day. - 2.
If you are a Sunday Starter:
Keep taking one pill every day until Sunday. On Sunday, throw out the rest of the pack and start a new pack of pills that same day. - 3. You could become pregnant if you have sex in the 7 days after you restart your pills. You must use another birth control method (such as a condom and spermicide) as a back-up for those 7 days.
- 4. You may not have your period this month but this is expected. However, if you miss your period two months in a row, call your healthcare provider because you might be pregnant.
If you miss 3 or more yellow pills in a row during any week:
- 1.
If you are a Day 1 Starter:
Throw out the rest of the pill pack and start a new pack that same day. - 2.
If you are a Sunday Starter:
Keep taking 1 pill every day until Sunday. On Sunday, throw out the rest of the pack and start a new pack of pills that same day. - 3. You could become pregnant if you have sex in the 7 days after you restart your pills. You must use another birth control method (such as condoms and spermicides) as a back-up for those 7 days.
- 4. You may not have your period this month but this is expected. However, if you miss your period two months in a row, call your healthcare provider because you might be pregnant.
If you miss any of the 7 white pills in Week 4:
Throw away the pills you missed.
Keep taking one pill each day until the pack is empty.
You do not need a back-up method.
Finally, if you are still not sure what to do about the pills you have missed:
Use a back-up method (such as condoms and spermicides) anytime you have sex.
Contact your healthcare provider and continue taking one active yellow pill each day until otherwise directed.
WHO SHOULD NOT TAKE SYEDATM?
Your healthcare provider will not give you SyedaTM if you:
- Ever had blood clots in your legs (deep vein thrombosis), lungs (pulmonary embolism), or eyes (retinal thrombosis)
- Ever had a stroke
- Ever had a heart attack
- Have certain heart valve problems or heart rhythm abnormalities that can cause blood clots to form in the heart
- Have an inherited problem with your blood that makes it clot more than normal
- Have high blood pressure that medicine can’t control
- Have diabetes with kidney, eye, nerve, or blood vessel damage
- Ever had certain kinds of severe migraine headaches with aura, numbness, weakness or changes in vision
- Ever had breast cancer or any cancer that is sensitive to female hormones
- Have liver disease, including liver tumors
- Take any Hepatitis C drug combination containing ombitasvir/paritaprevir/ritonavir, with or without dasabuvir. This may increase levels of the liver enzyme “alanine aminotransferase” (ALT) in the blood.
- Have kidney disease
- Have adrenal disease
Also, do not take birth control pills if you:
- Smoke and are over 35 years old
- Are or suspect you are pregnant
Birth control pills may not be a good choice for you if you have ever had jaundice (yellowing of the skin or eyes) caused by pregnancy (also called cholestasis of pregnancy).
Tell your healthcare provider if you have ever had any of the above conditions (your healthcare provider can recommend another method of birth control).
What Else Should I Know about Taking SyedaTM?
Birth control pills do not protect you against any sexually transmitted disease, including HIV, the virus that causes AIDS.
Do not skip any pills, even if you do not have sex often.
If you miss a period, you could be pregnant. However, some women miss periods or have light periods on birth control pills, even when they are not pregnant. Contact your healthcare provider for advice if you:
- Think you are pregnant
- Miss one period and have not taken your birth control pills on time every day
- Miss two periods in a row
Birth control pills should not be taken during pregnancy. However, birth control pills taken by accident during pregnancy are not known to cause birth defects.
Due to an increased risk of blood clots, you should stop SyedaTM at least four weeks before you have major surgery and not restart it until at least two weeks after the surgery.
If you are breastfeeding, consider another birth control method until you are ready to stop breastfeeding. Birth control pills that contain estrogen, like SyedaTM, may decrease the amount of milk you make. A small amount of the pill's hormones pass into breast milk.
If you have vomiting or diarrhea, your birth control pills may not work as well. Take another pill if you vomit within 3 to 4 hours after taking your pill, or use another birth control method, like condoms and a spermicide, until you check with your healthcare provider.
If you are scheduled for any laboratory tests, tell your doctor you are taking birth-control pills. Certain blood tests may be affected by birth-control pills.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements.
SyedaTM may affect the way other medicines work, and other medicines may affect how well SyedaTM works. Know the medicines you take.
Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
What are the Most Serious Risks of Taking Birth Control Pills?
Like pregnancy, birth control pills increase the risk of serious blood clots (see following graph), especially in women who have other risk factors, such as smoking, obesity, or age greater than 35. This increased risk is highest when you first start taking birth control pills and when you restart the same or different birth control pills after not using them for a month or more. . Women who use birth control pills with drospirenone (like Syeda™) may have a higher risk of getting a blood clot. Some studies reported that the risk of blood clots was higher for women who use birth control pills that contain drospirenone than for women who use birth control pills that do not contain drospirenone.
Talk with your healthcare provider about your risk of getting a blood clot before deciding which birth control pill is right for you.
It is possible to die or be permanently disabled from a problem caused by a blood clot, such as a heart attack or a stroke. Some examples of serious clots are blood clots in the:
- Legs (deep vein thrombosis or DVT)
- Lungs (pulmonary embolus or PE)
- Eyes (loss of eyesight)
- Heart (heart attack)
- Brain (stroke)
To put the risk of developing a blood clot into perspective: If 10,000 women who are not pregnant and do not use birth control pills are followed for one year, between 1 and 5 of these women will develop a blood clot. The figure below shows the likelihood of developing a serious blood clot for women who are not pregnant and do not use birth control pills, for women who use birth control pills, for pregnant women, and for women in the first 12 weeks after delivering a baby.
A few women who take birth control pills may get:
- High blood pressure
- Gallbladder problems
- Rare cancerous or noncancerous liver tumors
All of these events are uncommon in healthy women.
Call your healthcare provider right away if you have:
- Persistent leg pain
- Sudden shortness of breath
- Sudden blindness, partial or complete
- Severe pain in your chest
- Sudden, severe headache unlike your usual headaches
- Weakness or numbness in an arm or leg, or trouble speaking
- Yellowing of the skin or eyeballs
What are the Common Side Effects of Birth Control Pills?
The most common side effects of birth control pills are:
- Spotting or bleeding between menstrual periods
- Nausea
- Breast tenderness
- Headache
These side effects are usually mild and usually disappear with time.
Less common side effects are:
- Acne
- Less sexual desire
- Bloating or fluid retention
- Blotchy darkening of the skin, especially on the face
- High blood sugar, especially in women who already have diabetes
- High fat (cholesterol; triglyceride) levels in the blood
- Depression, especially if you have had depression in the past. Call your healthcare provider immediately if you have any thoughts of harming yourself.
- Problems tolerating contact lenses
- Weight changes
This is not a complete list of possible side effects. Talk to your healthcare provider if you develop any side effects that concern you. You may report side effects to Xiromed LLC at 1-844-XIROMED (1-844-947-6633) or the FDA at 1-800-FDA-1088.
No serious problems have been reported from a birth control pill overdose, even when accidentally taken by children.
Do Birth Control Pills Cause Cancer?
Birth control pills do not seem to cause breast cancer. However, if you have breast cancer now, or have had it in the past, do not use birth control pills because some breast cancers are sensitive to hormones.
Women who use birth control pills may have a slightly higher chance of getting cervical cancer. However, this may be due to other reasons such as having more sexual partners.
What Should I Know about My Period when Taking SyedaTM?
Irregular vaginal bleeding or spotting may occur while you are taking SyedaTM. Irregular bleeding may vary from slight staining between menstrual periods to breakthrough bleeding, which is a flow much like a regular period. Irregular bleeding occurs most often during the first few months of oral contraceptive use, but may also occur after you have been taking the pill for some time. Such bleeding may be temporary and usually does not indicate any serious problems. It is important to continue taking your pills on schedule. If the bleeding occurs in more than one cycle, is unusually heavy, or lasts for more than a few days, call your healthcare provider.
Some women may not have a menstrual period but this should not be cause for alarm as long as you have taken the pills regularly on time.
What if I Miss My Scheduled Period when Taking SyedaTM?
It is not uncommon to miss your period. However, if you miss two periods in a row or miss one period when you have not taken your birth control pills regularly on time, call your healthcare provider. Also notify your healthcare provider if you have symptoms of pregnancy such as morning sickness or unusual breast tenderness. It is important that your healthcare provider checks you to find out if you are pregnant. Stop taking SyedaTM if you are pregnant.
What if I Want to Become Pregnant?
You may stop taking the pill whenever you wish. Consider a visit with your healthcare provider for a pre-pregnancy checkup before you stop taking the pill.
General Advice about SyedaTM
Your healthcare provider prescribed SyedaTM for you. Please do not share SyedaTM with anyone else. Keep SyedaTM out of the reach of children.
If you have concerns or questions, ask your healthcare provider. You may also ask your healthcare provider for a more detailed label written for medical professionals.
SyedaTM is a trademark of Xiromed, LLC.
The other brands listed are the registered trademarks of their respective owners and are not trademarks of Xiromed, LLC.
Manufactured by Laboratorios Leon Farma S.A., Spain
For Xiromed LLC, Florham Park , NJ 07932
Rev. December 2017
PI-115-00
-
Principal Display Panel
NDC: 70700-115-85
3 Units (3(x 28 Tablet blister cards)
Syeda TM
(Drospirenone and Ethinyl Estradiol Tablets)3 mg/0.03 mg
Rx Only
-
INGREDIENTS AND APPEARANCE
SYEDA
drospirenone and ethinyl estradiol kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 70700-115 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 70700-115-85 3 in 1 CARTON 03/05/2018 1 NDC: 70700-115-84 1 in 1 CARTON 1 1 in 1 BLISTER PACK Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 21 Part 2 7 Part 1 of 2 SYEDA
drospirenone and ethinyl estradiol tablet, film coatedProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DROSPIRENONE (UNII: N295J34A25) (DROSPIRENONE - UNII:N295J34A25) DROSPIRENONE 3 mg ETHINYL ESTRADIOL (UNII: 423D2T571U) (ETHINYL ESTRADIOL - UNII:423D2T571U) ETHINYL ESTRADIOL 0.03 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) CROSPOVIDONE (UNII: 68401960MK) LACTOSE (UNII: J2B2A4N98G) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYSORBATE 80 (UNII: 6OZP39ZG8H) POLYVINYL ALCOHOL (UNII: 532B59J990) POVIDONE K30 (UNII: U725QWY32X) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color YELLOW Score no score Shape ROUND Size 6mm Flavor Imprint Code SZ;U3 Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090114 12/01/2017 Part 2 of 2 SYEDA
inert tablet, film coatedProduct Information Route of Administration ORAL Inactive Ingredients Ingredient Name Strength CROSPOVIDONE (UNII: 68401960MK) STARCH, CORN (UNII: O8232NY3SJ) LACTOSE (UNII: J2B2A4N98G) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYSORBATE 80 (UNII: 6OZP39ZG8H) POLYVINYL ALCOHOL (UNII: 532B59J990) POVIDONE K30 (UNII: U725QWY32X) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color WHITE Score no score Shape ROUND Size 6mm Flavor Imprint Code SZ;J1 Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090114 12/01/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090114 12/01/2017 Labeler - Xiromed, LLC. (080228637) Registrant - XIROMED PHARMA ESPANA, S.L. (468835741)
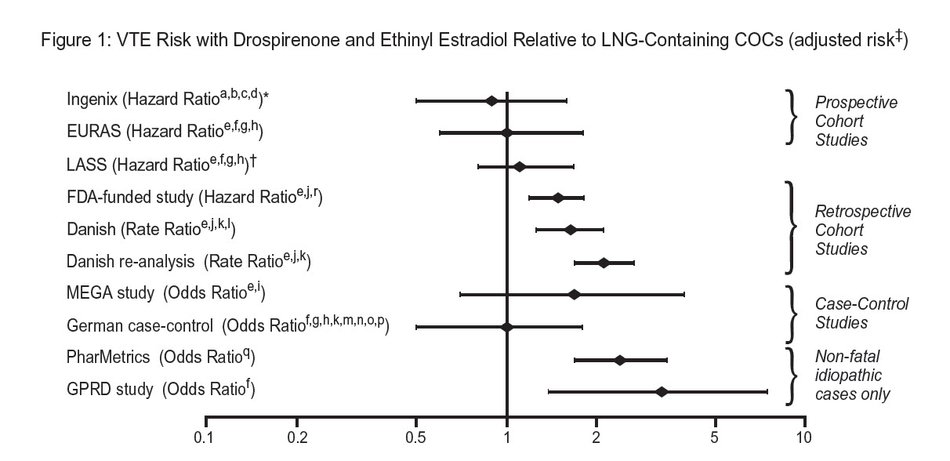
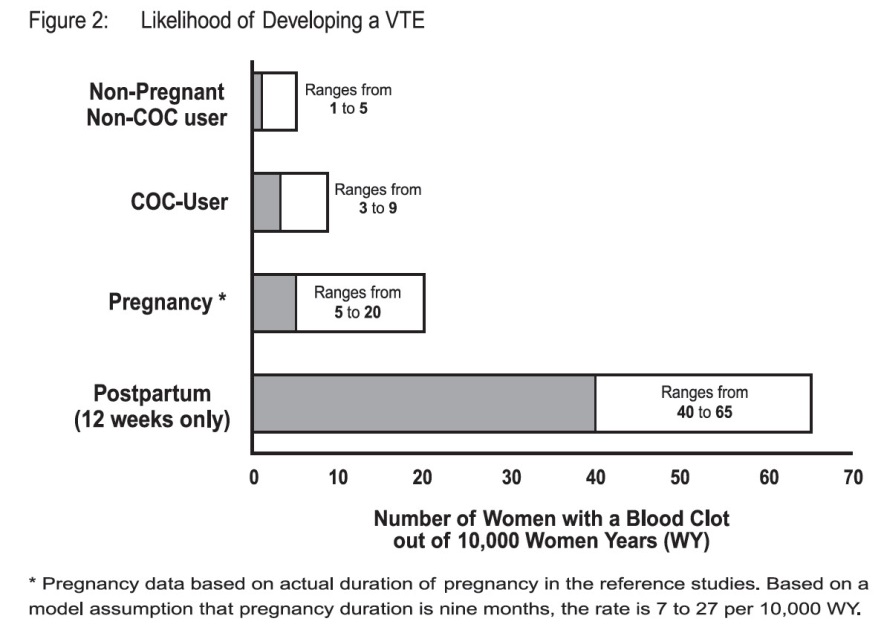
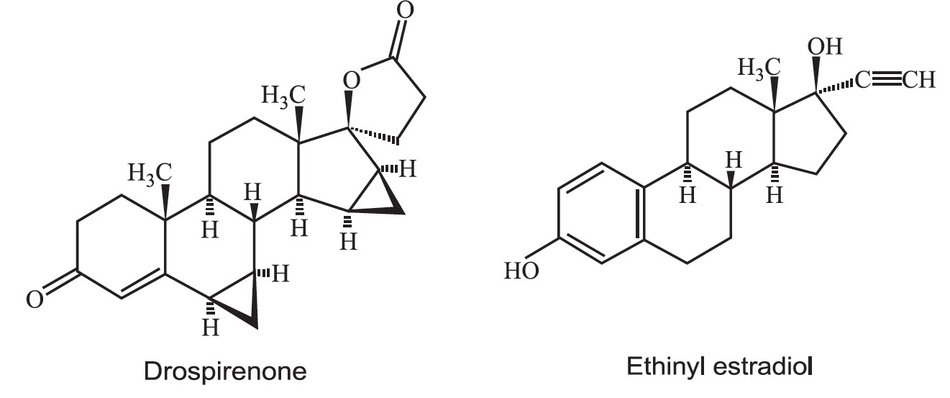
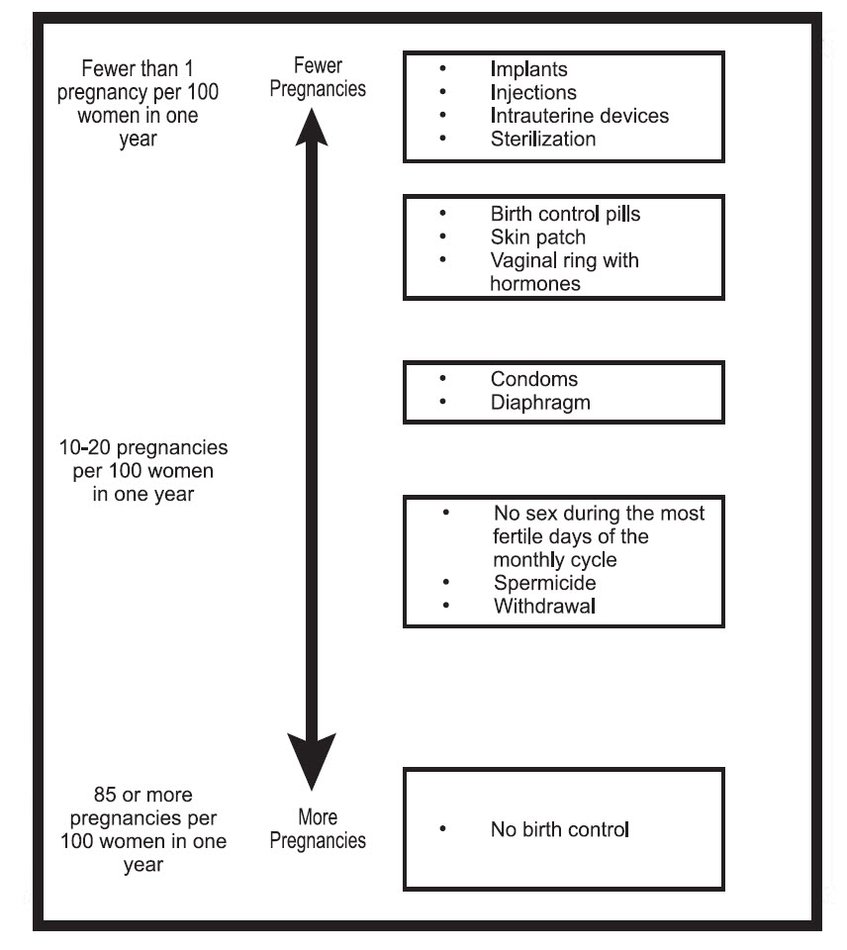
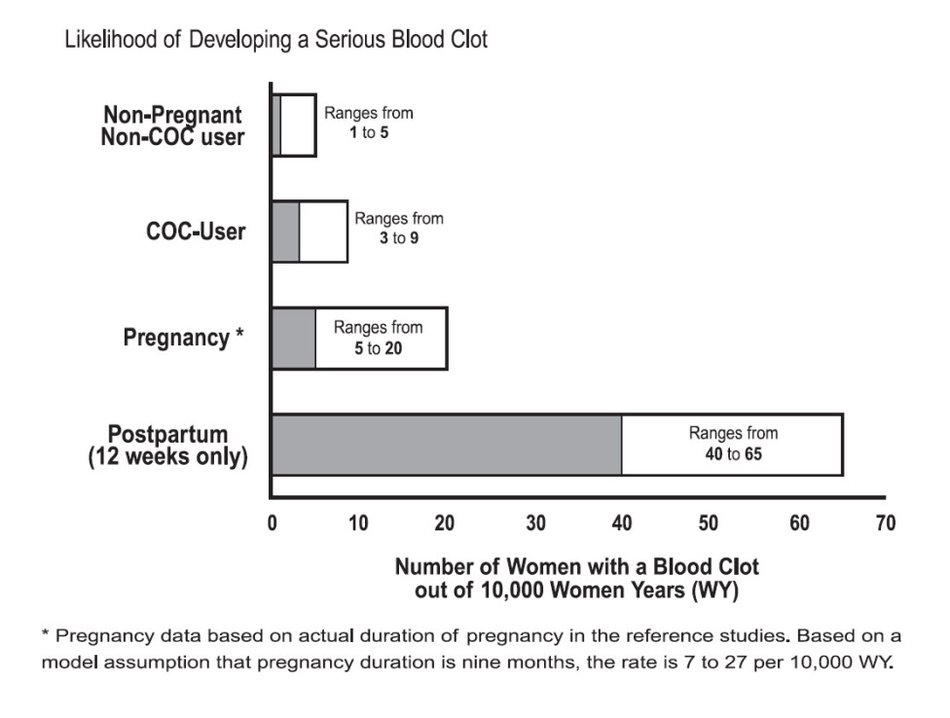
Trademark Results [Syeda]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 SYEDA 77716580 4112175 Live/Registered |
XIROMED PHARMA ESPAÃA S.L. 2009-04-17 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.