SCEMBLIX- asciminib tablet, film coated
SCEMBLIX by
Drug Labeling and Warnings
SCEMBLIX by is a Prescription medication manufactured, distributed, or labeled by Novartis Pharmaceuticals Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SCEMBLIX safely and effectively. See full prescribing information for SCEMBLIX.
SCEMBLIX® (asciminib) tablets, for oral use
Initial U.S. Approval: 2021
INDICATIONS AND USAGE
SCEMBLIX is a kinase inhibitor indicated for the treatment of adult patients with:
- Newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP).
This indication is approved under accelerated approval based on major molecular response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial(s). (1) - Previously treated Ph+ CML in CP. (1)
- Ph+ CML in CP with the T315I mutation. (1)
DOSAGE AND ADMINISTRATION
- Recommended Dosage in Ph+ CML in CP: 80 mg orally once daily or 40 mg orally twice daily. (2.1)
- Recommended Dosage in Ph+ CML in CP with the T315I Mutation: 200 mg orally twice daily. (2.2)
- Avoid food for at least 2 hours before and 1 hour after taking SCEMBLIX. (2.5)
- Swallow tablets whole. Do not break, crush, or chew the tablets. (2.5)
DOSAGE FORMS AND STRENGTHS
- Film-coated tablets: 20 mg, 40 mg, and 100 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Myelosuppression: Severe thrombocytopenia and neutropenia events may occur. Monitor complete blood counts regularly during therapy and manage by treatment interruption or dose reduction. (2.4, 5.1)
- Pancreatic Toxicity: Monitor serum lipase and amylase. Interrupt, then resume at reduced dose or discontinue SCEMBLIX based on severity. Evaluate for pancreatitis when lipase elevation is accompanied by abdominal symptoms. (2.4, 5.2)
- Hypertension: Monitor blood pressure and manage hypertension as clinically indicated. Interrupt, dose reduce, or stop SCEMBLIX if hypertension is not medically controlled. (2.4, 5.3)
- Hypersensitivity: May cause hypersensitivity reactions. Monitor patients for signs and symptoms and initiate appropriate treatment as clinically indicated. (5.4)
- Cardiovascular Toxicity: Cardiovascular toxicity may occur. Monitor patients with history of cardiovascular risk factors for cardiovascular signs and symptoms. Initiate appropriate treatment as clinically indicated. (5.5)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.6, 8.1, 8.3)
ADVERSE REACTIONS
Most common adverse reactions (≥ 20%) are musculoskeletal pain, rash, fatigue, upper respiratory tract infection, headache, abdominal pain, arthralgia, and diarrhea. (6.1)
Most common select laboratory abnormalities (≥ 20%) are lymphocyte count decreased, leukocyte count decreased, platelet count decreased, neutrophil count decreased, calcium corrected decreased, lipase increased, cholesterol increased, uric acid increased, alanine aminotransferase (ALT) increased, alkaline phosphatase (ALP) increased, hemoglobin decreased, triglycerides increased, creatine kinase increased, amylase increased, and aspartate aminotransferase (AST) increased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
- Strong CYP3A4 Inhibitors: Closely monitor for adverse reactions during concomitant use of SCEMBLIX at 200 mg twice daily. (7.1)
- Itraconazole Oral Solution Containing Hydroxypropyl-β-cyclodextrin: Avoid concomitant use of SCEMBLIX at all recommended doses. (7.1)
- Certain Substrates of CYP3A4: Closely monitor for adverse reactions during concomitant use of SCEMBLIX at 80 mg total daily dose. Avoid use of SCEMBLIX at 200 mg twice daily. (7.2)
- Substrates of CYP2C9: Avoid concomitant use of SCEMBLIX at all recommended doses.
- Certain P-gp Substrates: Closely monitor for adverse reactions during concomitant use of SCEMBLIX at all recommended doses. (7.2)
- Substrates of BCRP: Avoid concomitant use with rosuvastatin at all recommended doses. Closely monitor for adverse reactions of other BCRP substrates during concomitant use of SCEMBLIX at all recommended doses. (7.2)
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2025
- Newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP).
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Patients with Newly Diagnosed or Previously Treated Ph+ CML-CP
2.2 Recommended Dosage in Patients with Ph+ CML-CP with the T315I Mutation
2.3 Missed Dose
2.4 Dosage Modifications
2.5 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
5.2 Pancreatic Toxicity
5.3 Hypertension
5.4 Hypersensitivity
5.5 Cardiovascular Toxicity
5.6 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on SCEMBLIX
7.2 Effect of SCEMBLIX on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Newly Diagnosed Ph+ CML-CP
14.2 Ph+ CML-CP, Previously Treated with Two or More TKIs
14.3 Ph+ CML-CP with the T315I Mutation
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
SCEMBLIX is indicated for the treatment of adult patients with:
- Newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP).
This indication is approved under accelerated approval based on major molecular response rate [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial(s). - Previously treated Ph+ CML in CP.
- Ph+ CML in CP with the T315I mutation.
- Newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP).
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Patients with Newly Diagnosed or Previously Treated Ph+ CML-CP
The recommended dose of SCEMBLIX is 80 mg taken orally once daily at approximately the same time each day or 40 mg orally twice daily at approximately 12-hour intervals. The recommended dose of SCEMBLIX is taken orally without food. Avoid food consumption for at least 2 hours before and 1 hour after taking SCEMBLIX [see Clinical Pharmacology (12.2)].
Continue treatment with SCEMBLIX as long as clinical benefit is observed or until unacceptable toxicity occurs.
2.2 Recommended Dosage in Patients with Ph+ CML-CP with the T315I Mutation
The recommended dose of SCEMBLIX is 200 mg taken orally twice daily at approximately 12-hour intervals. The recommended dose of SCEMBLIX is taken orally without food. Avoid food consumption for at least 2 hours before and 1 hour after taking SCEMBLIX [see Clinical Pharmacology (12.2)].
2.3 Missed Dose
Once Daily Dosage Regimen: If a SCEMBLIX dose is missed by more than approximately 12 hours, skip the dose and take the next dose as scheduled.
Twice Daily Dosage Regimens: If a SCEMBLIX dose is missed by more than approximately 6 hours, skip the dose and take the next dose as scheduled.
2.4 Dosage Modifications
Dosage Modifications for Patients with Newly Diagnosed or Previously Treated Ph+ CML-CP
For the management of adverse reactions, reduce the SCEMBLIX dose as described in Table 1.
Dosage Modifications for Patients with Ph+ CML-CP with the T315I Mutation
For the management of adverse reactions, reduce the SCEMBLIX dose as described in Table 1.
Table 1: Recommended Dosage Reductions for SCEMBLIX for Adverse Reactions Dosage reduction Dosage for Patients with newly diagnosed or previously treated Ph+ CML-CP Dosage for patients with Ph+ CML-CP with the T315I mutation First - 40 mg once daily
- 20 mg twice daily
160 mg twice daily Subsequent reduction Permanently discontinue SCEMBLIX in patients unable to tolerate 40 mg once daily OR 20 mg twice daily. Permanently discontinue SCEMBLIX in patients unable to tolerate 160 mg twice daily. The recommended dosage modifications for the management of selected adverse reactions are shown in Table 2.
Table 2: SCEMBLIX Dosage Modification for the Management of Adverse Reactions Abbreviations: ANC, absolute neutrophil count; PLT, platelets; ULN, upper limit of normal.
aBased on Common Terminology Criteria for Adverse Events (CTCAE) version 4.03.Adverse reaction Dosage modification Thrombocytopenia and/or neutropenia [see Warnings and Precautions (5.1)] ANC less than 1 x 109/L and/or PLT less than 50 x 109/L Withhold SCEMBLIX until resolved to ANC greater than or equal to 1 x 109/L and/or PLT greater than or equal to 50 x 109/L.
If resolved:- Within 2 weeks: resume SCEMBLIX at starting dose.
- After more than 2 weeks: resume SCEMBLIX at reduced dose.
Asymptomatic amylase and/or lipase elevation [see Warnings and Precautions (5.2)] Elevation greater than 2 x ULN Withhold SCEMBLIX until resolved to less than 1.5 x ULN.
If resolved:- Resume SCEMBLIX at reduced dose. If events reoccur at reduced dose, permanently discontinue SCEMBLIX.
- Permanently discontinue SCEMBLIX. Perform diagnostic tests to exclude pancreatitis.
Non-hematologic adverse reactions [see Warnings and Precautions (5.3, 5.4, 5.5)] Grade 3a or higher Withhold SCEMBLIX until recovery to Grade 1 or less.
If resolved:- Resume SCEMBLIX at reduced dose.
- Permanently discontinue SCEMBLIX.
-
3 DOSAGE FORMS AND STRENGTHS
- 20 mg asciminib film-coated tablets: pale yellow, unscored, round, biconvex, with beveled edges, film-coated tablet debossed with “20” on one side and the “Novartis” logo on the other side.
- 40 mg asciminib film-coated tablets: violet white, unscored, round, biconvex, with beveled edges, film-coated tablet debossed with “40” on one side and the “Novartis” logo on the other side.
- 100 mg asciminib film-coated tablets: light red, unscored, round, biconvex, with beveled edges, film-coated tablet debossed with “100” on one side and the “Novartis” logo on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
Thrombocytopenia, neutropenia, and anemia have occurred in patients receiving SCEMBLIX. Thrombocytopenia occurred in 156 of 556 (28%) patients receiving SCEMBLIX, with Grade 3 or 4 thrombocytopenia reported in 39 (7%) and 53 (10%) of patients, respectively. Among the patients with Grade 3 or 4 thrombocytopenia, median time to first occurrence of events was 6 weeks (range, 0.1 to 64 weeks). SCEMBLIX was permanently discontinued in 11 (2%) patients, while it was temporarily withheld in 70 (13%) patients due to thrombocytopenia.
Neutropenia occurred in 121 (22%) patients receiving SCEMBLIX, with Grade 3 and 4 neutropenia reported in 42 (8%) and 35 (6%) patients, respectively. Among the patients with Grade 3 or 4 neutropenia, median time to first occurrence of events was 7 weeks (range, 0.1 to 180 weeks). SCEMBLIX was permanently discontinued in 7 (1.3%) patients, while it was temporarily withheld in 52 (9%) patients due to neutropenia.
Anemia occurred in 72 (13%) patients receiving SCEMBLIX, with Grade 3 anemia occurring in 23 (4%) patients. Among the patients with Grade 3 or 4 anemia, median time to first occurrence of events was 24 weeks (range, 0.1 to 207 weeks). SCEMBLIX was temporarily withheld in 3 (0.5%) patients due to anemia [see Adverse Reactions (6.1)].
Perform complete blood counts every two weeks for the first 3 months of treatment and monthly thereafter or as clinically indicated. Monitor patients for signs and symptoms of myelosuppression.
Based on the severity of thrombocytopenia and/or neutropenia, reduce dose, temporarily withhold, or permanently discontinue SCEMBLIX [see Dosage and Administration (2.4)].
5.2 Pancreatic Toxicity
Pancreatitis occurred in 11 of 556 (2%) patients receiving SCEMBLIX, with Grade 3 pancreatitis occurring in 6 (1.1%) patients. SCEMBLIX was permanently discontinued in three (0.5%) patients, while it was temporarily withheld in 6 (1.1%) patients due to pancreatitis. Elevation of serum lipase and amylase occurred in 110 of 556 (20%) patients receiving SCEMBLIX, with Grade 3 and Grade 4 pancreatic enzyme elevations occurring in 41 (7%) and 11 (2%) patients, respectively. SCEMBLIX was permanently discontinued in 11 (2%) patients due to the elevation of serum lipase and amylase [see Adverse Reactions (6.1)].
Assess serum lipase and amylase levels monthly during treatment with SCEMBLIX, or as clinically indicated. Monitor patients for signs and symptoms of pancreatic toxicity. Perform more frequent monitoring in patients with a history of pancreatitis. If lipase and amylase elevation are accompanied by abdominal symptoms, temporarily withhold SCEMBLIX and consider appropriate diagnostic tests to exclude pancreatitis [see Dosage and Administration (2.4)].
Based on the severity of lipase and amylase elevation, reduce dose, temporarily withhold, or permanently discontinue SCEMBLIX [see Dosage and Administration (2.4)].
5.3 Hypertension
Hypertension occurred in 98 of 556 (18%) patients receiving SCEMBLIX, with Grade 3 or 4 hypertension reported in 53 (10%) and 1 (0.2%) patients, respectively. Among the patients with Grade 3 or 4 hypertension, median time to first occurrence was 45 weeks (range, 0.1 to 365 weeks). SCEMBLIX was temporarily withheld in 5 (0.9%) patients due to hypertension [see Adverse Reactions (6.1)].
Monitor and manage hypertension using standard antihypertensive therapy during treatment with SCEMBLIX as clinically indicated; for Grade 3 or higher hypertension, temporarily withhold, reduce dose, or permanently discontinue SCEMBLIX depending on persistence of hypertension [see Dosage and Administration (2.4)].
5.4 Hypersensitivity
Hypersensitivity occurred in 172 of 556 (31%) patients receiving SCEMBLIX, with Grade 3 or 4 hypersensitivity reported in 6 (1.1%) patients [see Adverse Reactions (6.1)]. Reactions included rash, edema, and bronchospasm. Monitor patients for signs and symptoms of hypersensitivity and initiate appropriate treatment as clinically indicated; for Grade 3 or higher hypersensitivity, temporarily withhold, reduce dose, or permanently discontinue SCEMBLIX depending on persistence of hypersensitivity [see Dosage and Administration (2.4)].
5.5 Cardiovascular Toxicity
Cardiovascular toxicity (including ischemic cardiac and CNS conditions, arterial thrombotic and embolic conditions) and cardiac failure occurred in 65 (12%) and in 13 (2.3%) of 556 patients receiving SCEMBLIX, respectively [see Adverse Reactions (6.1)]. Grade 3 cardiovascular toxicity was reported in 14 (2.5%) patients, while Grade 3 cardiac failure was observed in 5 (0.9%) patients. Grade 4 cardiovascular toxicity occurred in 4 (0.7%) patients, with fatalities occurring in 5 (0.9%) patients. Grade 4 cardiac failure was reported in 1 (0.2%) patient with fatality occurring in 1 (0.2%) patient. Permanent discontinuation of SCEMBLIX occurred in 4 (0.7%) patients due to cardiovascular toxicity and in 1 (0.2%) patient due to cardiac failure, respectively. In the majority of patients, cardiovascular toxicity occurred in patients with preexisting cardiovascular conditions or risk factors, and/or prior exposure to multiple TKIs.
Arrhythmia, including QTc prolongation, occurred in 35 of 556 (6%) patients receiving SCEMBLIX, with Grade 3 or 4 arrhythmia reported in 10 (1.8%) and 2 (0.4%) patients, respectively. QTc prolongation occurred in 5 of 556 (0.9%) patients receiving SCEMBLIX, with Grade 3 QTc prolongation reported in 2 (0.4%) patients [see Adverse Reactions (6.1)].
Monitor patients with history of cardiovascular risk factors for cardiovascular signs and symptoms. Initiate appropriate treatment as clinically indicated; for Grade 3 or higher cardiovascular toxicity, temporarily withhold, reduce dose, or permanently discontinue SCEMBLIX depending on persistence of cardiovascular toxicity [see Dosage and Administration (2.4)].
5.6 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, SCEMBLIX can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of asciminib to pregnant rats and rabbits during the period organogenesis caused adverse developmental outcomes, including embryo-fetal mortality and malformations at maternal exposures (AUC) equivalent to or less than those in patients at the recommended doses. Advise pregnant women and females of reproductive potential of the potential risk to a fetus if SCEMBLIX is used during pregnancy or if the patient becomes pregnant while taking SCEMBLIX. Verify the pregnancy status of females of reproductive potential prior to starting treatment with SCEMBLIX. Females of reproductive potential should use effective contraception during treatment with SCEMBLIX and for 1 week after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions can occur with SCEMBLIX and are discussed in greater detail in other sections of the labeling:
- Myelosuppression [see Warnings and Precautions (5.1)]
- Pancreatic Toxicity [see Warnings and Precautions (5.2)]
- Hypertension [see Warnings and Precautions (5.3)]
- Hypersensitivity [see Warnings and Precautions (5.4)]
- Cardiovascular Toxicity [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflect exposure to SCEMBLIX at 10 mg to 200 mg orally twice daily (between 0.25 to 5 times the recommended dosage for the 80 mg daily dosage and between 0.05 times and up to the recommended dosage for the 200 mg twice daily dosage) in 556 patients enrolled in one of three clinical trials, including patients with Ph+ CML in CP receiving SCEMBLIX as monotherapy: study CABL001J12301 (ASC4FIRST), study CABL001A2301 (ASCEMBL) and study CABL001X2101 [see Clinical Studies (14)]. Among the 556 patients receiving SCEMBLIX, the median duration of exposure to SCEMBLIX was 123 weeks (range, 0.1 to 439 weeks), with 79% of patients exposed for at least 48 weeks and 71% of patients exposed for at least 96 weeks, respectively.
The most common (≥ 20%) adverse reactions in patients who received SCEMBLIX were musculoskeletal pain, rash, fatigue, upper respiratory tract infection, headache, abdominal pain, arthralgia, and diarrhea.
Adverse Reactions in Patients with Newly Diagnosed Ph+ CML-CP
The ASC4FIRST clinical trial randomized 405 patients with newly diagnosed Ph+ CML-CP to receive SCEMBLIX 80 mg once daily or investigator selected tyrosine kinase inhibitors (IS-TKIs). IS-TKIs included imatinib (400 mg once daily), nilotinib (300 mg twice daily), dasatinib (100 mg once daily), or bosutinib (400 mg once daily) [see Clinical Studies (14.1)]. The safety population (received at least 1 dose of SCEMBLIX) included 200 patients with newly diagnosed Ph+ CML-CP. Among patients who received SCEMBLIX, 84% were exposed for 96 weeks or longer [see Clinical Studies (14.1)].
Serious adverse reactions occurred in 15% of patients who received SCEMBLIX. Serious adverse reactions in ≥ 1% included pancreatitis (1%), musculoskeletal pain (1%), and peripheral neuropathy (1%).
Permanent discontinuation due to an adverse reaction occurred in 5% of patients receiving SCEMBLIX. Adverse reactions which resulted in permanent discontinuation of SCEMBLIX in ≥ 1% of patients included pancreatic enzymes increased (1.5%), cardiovascular toxicity (1%), and thrombocytopenia (1%).
Dosage interruptions of SCEMBLIX due to an adverse reaction occurred in 33% of patients. Adverse reactions which required dosage interruption in > 5% of patients included thrombocytopenia (13%) and neutropenia (10%).
Dose reductions of SCEMBLIX due to an adverse reaction occurred in 6% of patients. Adverse reactions which required dose reductions in > 1% of patients included thrombocytopenia (2.5%) and neutropenia (1.5%).
The most common (≥ 20%) adverse reactions in patients who received SCEMBLIX were musculoskeletal pain and rash.
The most common select laboratory abnormalities that worsened from baseline in ≥ 20% of patients who received SCEMBLIX were lymphocyte count decreased, leukocyte count decreased, platelet count decreased, neutrophil count decreased, calcium corrected decreased, lipase increased, cholesterol increased, uric acid increased, alanine aminotransferase (ALT) increased, alkaline phosphatase (ALP) increased, hemoglobin decreased, and triglycerides increased.
Table 3 summarizes the adverse reactions in ASC4FIRST.
Table 3: Adverse Reactions (≥ 10%) in Patients with Newly Diagnosed Ph+ CML in CP, Who Received SCEMBLIX in ASC4FIRST Abbreviations: Ph+ CML in CP, Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP); IS-TKIs, investigator selected tyrosine kinase inhibitors.
aMusculoskeletal pain includes: musculoskeletal pain, myalgia, pain in extremity, back pain, non-cardiac chest pain, bone pain, neck pain, musculoskeletal stiffness, musculoskeletal discomfort, musculoskeletal chest pain, arthritis, and spinal pain.
bRash includes: rash, rash maculo-papular, rash pustular, rash macular, dermatitis exfoliative, drug eruption, dermatitis acneiform, eczema, rash pruritic and rash vesicular.
cFatigue includes: fatigue and asthenia.
dDiarrhea includes: diarrhea, colitis and enteritis.
eAbdominal pain includes: abdominal pain, abdominal pain upper, abdominal discomfort, abdominal pain lower and gastrointestinal pain.
fUpper respiratory tract infection includes: upper respiratory tract infection, nasopharyngitis, pharyngitis, rhinitis and respiratory tract infection.
gDyslipidemia includes: dyslipidemia, hypertriglyceridemia, blood cholesterol increased, hypercholesterolemia, hyperlipidemia and blood triglycerides increased.
hHeadache contains: headache and migraine.
iHypertension contains: blood pressure increased, blood pressure systolic increased, diastolic hypertension, and hypertension.SCEMBLIX
N = 200IS-TKIs
N = 201Adverse Reaction All Grades
%Grade 3 or 4
%All Grades
%Grade 3 or 4
%Musculoskeletal and connective tissue disorders Musculoskeletal paina 29 1.5 34 0.5 Arthralgia 13 2 10 0.5 Skin and subcutaneous tissue disorders Rashb 20 0 27 2 General disorders and administration site conditions Fatiguec 19 1 22 1 Gastrointestinal disorders Diarrhead 18 0 28 1.5 Abdominal paine 17 0.5 12 0.5 Constipation 10 0 9 0.5 Infections and infestations Upper respiratory tract infectionf 18 0 20 0.5 Metabolism and nutrition disorders Dyslipidemiag 19 1 11 0.5 Nervous system disorders Headacheh 17 0.5 17 0 Vascular disorders Hypertensioni 11 6 6 4 Clinically relevant adverse reactions in < 10% of patients treated with SCEMBLIX in ASC4FIRST included: nausea, cough, pruritus, dry eye, pyrexia, vomiting, dizziness, edema, lower respiratory tract infection, decreased appetite, hypothyroidism, urticaria, arrhythmia, influenza, neuropathy peripheral, hemorrhage, urinary tract infection, pneumonia, dyspnea, pancreatitis, vision blurred, febrile neutropenia, and palpitations.
Table 4 summarizes the laboratory abnormalities in ASC4FIRST.
Table 4: Select Laboratory Abnormalities (≥ 20%) That Worsened From Baseline in Patients with Newly Diagnosed Ph+ CML in CP, Who Received SCEMBLIX in ASC4FIRST aThe denominator used to calculate the rate for SCEMBLIX and IS TKIs varied from 198 to 200 and 201, respectively, based on the number of patients with a baseline value and at least one post-treatment value.
bWorst post-baseline laboratory abnormalities based on normal ranges.
CTCAE version 5.0SCEMBLIXa IS-TKIsa Laboratory Abnormality All Grades
%Grade 3 or 4
%All Grades
%Grade 3 or 4
%Hematologic parameters Lymphocyte count decreased 72 3.5 84 11 Leukocyte count decreased 55 5 68 13 Platelet count decreased 48 13 56 11 Neutrophil count decreased 47 12 63 21 Hemoglobin decreased 25 3 51 5 Biochemical parameters Calcium corrected decreased 46 0.5 76 2 Lipase increased 39 12 50 12 Cholesterol increased 40 0 34 0 Uric acid increasedb 37 - 22 - Alanine aminotransferase (ALT) increased 33 2.5 45 6 Alkaline phosphatase (ALP) increased 26 0.5 36 0 Triglycerides increased 25 1 20 1.5 Adverse Reactions in Patients with Ph+ CML-CP, Previously Treated with Two or More TKIs
The clinical trial randomized and treated 232 patients with Ph+ CML-CP, previously treated with two or more TKIs to receive SCEMBLIX 40 mg twice daily or bosutinib 500 mg once daily (ASCEMBL) [see Clinical Studies (14.2)]. The safety population (received at least 1 dose of SCEMBLIX) included 156 patients with Ph+ CML-CP, previously treated with two or more TKIs. Among patients who received SCEMBLIX, 83% were exposed for 24 weeks or longer and 56% were exposed for 96 weeks or longer.
Serious adverse reactions occurred in 18% of patients who received SCEMBLIX. Serious adverse reactions in ≥ 1% included cardiac failure congestive (1.9%), pyrexia (1.9%), urinary tract infection (1.9%), headache (1.3%), and thrombocytopenia (1.3%). Two patients (1.3%) had a fatal adverse reaction, one each for mesenteric artery thrombosis and ischemic stroke.
Permanent discontinuation of SCEMBLIX due to an adverse reaction occurred in 8% of patients. Adverse reactions which resulted in permanent discontinuation of SCEMBLIX in > 2% of patients included thrombocytopenia (3.2%) and neutropenia (2.6%).
Dosage interruptions of SCEMBLIX due to an adverse reaction occurred in 41% of patients. Adverse reactions which required dosage interruption in > 5% of patients included thrombocytopenia (19%) and neutropenia (18%).
Dose reductions of SCEMBLIX due to an adverse reaction occurred in 6% of patients. Adverse reactions which required dose reductions in > 1% of patients included thrombocytopenia (4.5%) and neutropenia (1.3%).
The most common (≥ 20%) adverse reactions in patients who received SCEMBLIX were upper respiratory tract infections, musculoskeletal pain, headache, and fatigue.
The most common select laboratory abnormalities that worsened from baseline in ≥ 20% of patients who received SCEMBLIX were platelet count decreased, triglycerides increased, neutrophil count decreased, hemoglobin decreased, creatine kinase increased, alanine aminotransferase (ALT) increased, aspartate aminotransferase (AST) increased, uric acid increased, and lymphocyte count decreased.
Table 5 summarizes the adverse reactions in ASCEMBL.
Table 5: Adverse Reactions (≥ 10%) in Patients with Ph+ CML in CP, Previously Treated with Two or More TKIs Who Received SCEMBLIX in ASCEMBL Abbreviations: Ph+ CML in CP, Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP); TKIs, tyrosine kinase inhibitors.
aUpper respiratory tract infection includes: nasopharyngitis, upper respiratory tract infection, rhinitis, pharyngitis, respiratory tract infection, and pharyngotonsillitis.
bMusculoskeletal pain includes: pain in extremity, back pain, myalgia, non-cardiac chest pain, neck pain, bone pain, spinal pain, arthritis, musculoskeletal pain, and musculoskeletal chest pain.
cHeadache includes: headache and post-traumatic headache.
dFatigue includes: fatigue and asthenia.
eRash includes: rash, rash maculopapular, dermatitis acneiform, rash pustular, eczema, dermatitis, skin exfoliation, dermatitis exfoliative generalized, rash morbilliform, drug eruption, erythema multiforme, and rash erythematous.
fHypertension includes: hypertension and hypertensive crisis.
gDiarrhea includes: diarrhea and colitis.
hAbdominal pain includes: abdominal pain, abdominal pain upper, abdominal discomfort, abdominal pain lower, abdominal tenderness, and epigastric discomfort.
SCEMBLIX
N = 156Bosutinib
N = 76Adverse Reaction All Grades
%Grade 3 or 4
%All Grades
%Grade 3 or 4
%Infections and infestations Upper respiratory tract infectiona 26 0.6 12 1.3 Musculoskeletal and connective tissue disorders Musculoskeletal painb 24 2.6 17 1.3 Arthralgia 13 0.6 3.9 0 Nervous system disorders Headachec 21 1.9 16 0 General disorders and administration-site conditions Fatigued 20 0.6 11 1.3 Skin and subcutaneous tissue disorders Rashe 18 0.6 30 8 Vascular disorders Hypertensionf 14 7 5 3.9 Gastrointestinal disorders Diarrheag 13 0 72 11 Nausea 12 0.6 46 0 Abdominal painh 14 0 24 2.6 Clinically relevant adverse reactions in < 10% of patients treated with SCEMBLIX in ASCEMBL included: cough, dyspnea, pleural effusion, dizziness, neuropathy peripheral, edema, pyrexia, vomiting, constipation, dyslipidemia, decreased appetite, pruritus, urticaria, lower respiratory tract infection, influenza, urinary tract infection, pneumonia, hemorrhage, arrhythmia (including electrocardiogram QT prolonged), palpitations, cardiac failure congestive, vision blurred, dry eye, hypothyroidism, and febrile neutropenia.
Table 6 summarizes the laboratory abnormalities in ASCEMBL.
Table 6: Select Laboratory Abnormalities (≥ 10%) That Worsened From Baseline in Patients with Ph+ CML in CP, Previously Treated with Two or More TKIs Who Received SCEMBLIX in ASCEMBL aThe denominator used to calculate the rate for SCEMBLIX and bosutinib varied from 152 to 156 and 75 to 76, respectively, based on the number of patients with a baseline value and at least one post-treatment value.
CTCAE version 4.03.SCEMBLIXa Bosutiniba Laboratory Abnormality All Grades
%Grade 3 or 4
%All Grades
%Grade 3 or 4
%Hematologic parameters Platelet count decreased 46 24 36 12 Neutrophil count decreased 43 22 33 15 Hemoglobin decreased 37 2 54 5 Lymphocyte count decreased 20 3.3 34 2.6 Biochemical parameters Triglycerides increased 44 5 30 2.6 Creatine kinase increased 30 2.6 24 5 Alanine aminotransferase (ALT) increased 26 0.6 50 16 Aspartate aminotransferase (AST) increased 21 1.9 46 7 Uric acid increased 21 6 18 2.6 Phosphate decreased 18 6 20 7 Corrected calcium decreased 16 0.6 22 0 Lipase increased 15 4.5 18 7 Creatinine increased 15 0 26 0 Amylase increased 13 1.3 13 0 Alkaline phosphatase (ALP) increased 13 0 12 0 Bilirubin increased 12 0 3.9 0 Cholesterol increased 12 0 8 0 Potassium decreased 11 0 9 0 Adverse Reactions in Patients with Ph+ CML-CP with the T315I Mutation
The single-arm clinical trial enrolled patients with Ph+ CML-CP with the T315I mutation [see Clinical Studies (14.3)]. The safety population (received at least 1 dose of SCEMBLIX) included 48 patients with Ph+ CML-CP with the T315I mutation who received 200 mg of SCEMBLIX twice daily. Among these patients, 83% were exposed for 24 weeks or longer, and 75% were exposed for 48 weeks or longer.
Serious adverse reactions occurred in 23% of patients who received SCEMBLIX. Serious adverse reactions in > 1% included abdominal pain (4.2%), vomiting (4.2%), pneumonia (4.2%), musculoskeletal pain (2.1%), headache (2.1%), hemorrhage (2.1%), constipation (2.1%), arrhythmia (2.1%), and pleural effusion (2.1%).
Permanent discontinuation of SCEMBLIX due to an adverse reaction occurred in 10% of patients. Adverse reactions which resulted in permanent discontinuation of SCEMBLIX in > 2% of patients included pancreatic enzymes increased (2.1%).
Dosage interruptions of SCEMBLIX due to an adverse reaction occurred in 31% of patients. Adverse reactions which required dosage interruption in > 5% of patients included pancreatic enzymes increased (17%) and thrombocytopenia (8%).
Dose reductions of SCEMBLIX due to an adverse reaction occurred in 23% of patients. Adverse reactions which required dose reductions in > 1% of patients included pancreatic enzymes increased (10%), abdominal pain (4.2%), anemia (2.1%), blood bilirubin increased (2.1%), dizziness (2.1%), fatigue (2.1%), hepatic enzymes increased (2.1%), musculoskeletal pain (2.1%), nausea (2.1%), neutropenia (2.1%), pruritus (2.1%), and thrombocytopenia (2.1%).
The most common (≥ 20%) adverse reactions in patients who received SCEMBLIX were musculoskeletal pain, fatigue, nausea, rash, and diarrhea.
The most common select laboratory abnormalities that worsened from baseline in ≥ 20% of patients who received SCEMBLIX were alanine aminotransferase (ALT) increased, lipase increased, triglycerides increased, hemoglobin decreased, neutrophil count decreased, lymphocyte count decreased, phosphate decreased, aspartate aminotransferase (AST) increased, amylase increased, platelet count decreased, and bilirubin increased.
Table 7 summarizes adverse reactions in study X2101.
Table 7: Adverse Reactions (≥ 10%) in Patients with Ph+ CML in CP with the T315I Mutation Who Received SCEMBLIX in X2101 aMusculoskeletal pain includes: pain in extremity, back pain, myalgia, musculoskeletal pain, non-cardiac chest pain, bone pain, arthritis, and musculoskeletal chest pain.
bFatigue includes: fatigue and asthenia.
cAbdominal pain includes: abdominal pain and hepatic pain.
dRash includes: rash, rash maculopapular, dermatitis acneiform, eczema, rash papular, skin exfoliation, and dyshidrotic eczema.
eHeadache includes: headache and migraine.
fCough includes: cough and productive cough.
gHemorrhage includes: epistaxis, ear hemorrhage, mouth hemorrhage, post procedural hemorrhage, skin hemorrhage, and vaginal hemorrhage.
hHypertension includes: hypertension and hypertensive crisis.
iUpper respiratory tract infection includes: upper respiratory tract infection, nasopharyngitis, rhinitis, and pharyngitis.SCEMBLIX
200 mg twice daily
N = 48Adverse Reaction All Grades
%Grade 3 or 4
%Musculoskeletal and connective tissue disorders Musculoskeletal paina 42 4.2 Arthralgia 17 0 General disorders and administration-site conditions Fatigueb 31 2.1 Edema 10 4.2 Gastrointestinal disorders Nausea 27 0 Diarrhea 21 2.1 Vomiting 19 6 Abdominal painc 17 8 Skin and subcutaneous tissue disorders Rashd 27 0 Pruritus 13 0 Nervous system disorders Headachee 19 2.1 Respiratory, thoracic, and mediastinal disorders Coughf 15 0 Vascular disorders Hemorrhageg 15 2.1 Hypertensionh 13 8 Infections and infestations Upper respiratory tract infectioni 13 0 Clinically relevant adverse reactions in < 10% of patients treated with SCEMBLIX in X2101 included: constipation, pancreatitis, pyrexia, dizziness, neuropathy peripheral, pneumonia, lower respiratory tract infection, dyspnea, pleural effusion, dry eye, vision blurred, arrhythmia, palpitations, cardiac failure congestive, decreased appetite, dyslipidemia, hypersensitivity, and urticaria.
Table 8 summarizes laboratory abnormalities in X2101.
Table 8: Select Laboratory Abnormalities (≥ 10%) That Worsened From Baseline in Patients with Ph+ CML in CP with the T315I Mutation in X2101 aThe denominator used to calculate the rate was 48 based on the number of patients with a baseline value and at least one post-treatment value.
CTCAE version 4.03.SCEMBLIXa
200 mg twice dailyLaboratory Abnormality All Grades
%Grade 3-4
%Hematologic parameters Hemoglobin decreased 44 4.2 Neutrophil count decreased 44 15 Lymphocyte count decreased 42 4.2 Platelet count decreased 25 15 Biochemical parameters Alanine aminotransferase (ALT) increased 48 6 Potassium increased 48 2.1 Triglycerides increased 46 2.1 Lipase increased 46 21 Phosphate decreased 40 6 Uric acid increased 40 4.2 Aspartate aminotransferase (AST) increased 35 2.1 Calcium corrected decreased 33 0 Creatinine increased 31 0 Amylase increased 29 10 Bilirubin increased 23 0 Cholesterol increased 15 0 Alkaline phosphatase (ALP) increased 13 0 -
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on SCEMBLIX
Strong CYP3A4 Inhibitors
Asciminib is a CYP3A4 substrate. Concomitant use of SCEMBLIX with a strong CYP3A4 inhibitor increases both the asciminib Cmax and AUC, which may increase the risk of adverse reactions [see Clinical Pharmacology (12.3)]. Closely monitor for adverse reactions in patients treated with SCEMBLIX at 200 mg twice daily with concomitant use of strong CYP3A4 inhibitors.
Itraconazole Oral Solution Containing Hydroxypropyl-β-cyclodextrin
Concomitant use of SCEMBLIX with itraconazole oral solution containing hydroxypropyl-β-cyclodextrin decreases asciminib Cmax and AUC, which may reduce SCEMBLIX efficacy [see Clinical Pharmacology (12.3)]. Avoid coadministration of SCEMBLIX at all recommended doses with itraconazole oral solution containing hydroxypropyl-β-cyclodextrin.
Strong CYP3A4 Inducers
Concomitant use of SCEMBLIX with strong CYP3A4 inducers decreases both the asciminib Cmax and AUC [see Clinical Pharmacology (12.3)].
7.2 Effect of SCEMBLIX on Other Drugs
Certain CYP3A4 Substrates
Asciminib is a CYP3A4 inhibitor. Concomitant use of SCEMBLIX increases the Cmax and AUC of CYP3A4 substrates, which may increase the risk of adverse reactions of these substrates [see Clinical Pharmacology (12.3)].
Closely monitor for adverse reactions in patients treated with SCEMBLIX at 80 mg total daily dose with concomitant use of certain CYP3A4 substrates, where minimal concentration changes may lead to serious adverse reactions. Avoid coadministration of SCEMBLIX at 200 mg twice daily with certain CYP3A4 substrates, where minimal concentration changes may lead to serious adverse reactions.
CYP2C9 Substrates
Asciminib is a CYP2C9 inhibitor. Concomitant use of SCEMBLIX increases the Cmax and AUC of CYP2C9 substrates, which may increase the risk of adverse reactions of these substrates [see Clinical Pharmacology (12.3)].
Avoid coadministration of SCEMBLIX at 80 mg total daily dose with certain CYP2C9 substrates, where minimal concentration changes may lead to serious adverse reactions. If coadministration is unavoidable, reduce the CYP2C9 substrate dosage as recommended in its prescribing information.
Avoid coadministration of SCEMBLIX at 200 mg twice daily with sensitive CYP2C9 substrates and certain CYP2C9 substrates, where minimal concentration changes may lead to serious adverse reactions. If coadministration is unavoidable, consider alternative therapy with a non-CYP2C9 substrate.
Certain P-gp Substrates
Asciminib is a P-gp inhibitor. Concomitant use of SCEMBLIX increases the plasma concentrations of P-gp substrates, which may increase the risk of adverse reactions of these substrates [see Clinical Pharmacology (12.3)].
Closely monitor for adverse reactions in patients treated with SCEMBLIX at all recommended doses with concomitant use of P-gp substrates, where minimal concentration changes may lead to serious toxicities.
Substrates of BCRP
Asciminib is a BCRP inhibitor. Concomitant use of SCEMBLIX may increase the plasma concentration of BCRP substrates [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions associated with these substrates.
Avoid coadministration of SCEMBLIX at all recommended doses with rosuvastatin. Closely monitor for adverse reactions in patients treated with SCEMBLIX at all recommended doses with concomitant use of other BCRP substrates. Reduce the dosage of the other BCRP substrates as recommended in their Prescribing Information when used concomitantly with SCEMBLIX at all recommended doses.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and the mechanism of action, SCEMBLIX can cause embryo-fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on SCEMBLIX use in pregnant women to evaluate a drug-associated risk.
Animal reproduction studies in pregnant rats and rabbits demonstrated that oral administration of asciminib during organogenesis induced structural abnormalities, embryo-fetal mortality, and alterations to growth (see Data).
Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In embryo-fetal development studies, pregnant animals received oral doses of asciminib at 25, 150, and 600 mg/kg/day in rats and at 15, 50, and 300 mg/kg/day in rabbits during the period of organogenesis.
In rats, maternal toxicity at the asciminib dose of 600 mg/kg/day resulted in the early termination of the dose group; a complete embryo-fetal examination was not conducted for this group. Adverse embryo-fetal findings were observed at 25 and 150 mg/kg; these doses did not cause maternal toxicities. Increases in fetal weights at 25 and 150 mg/kg/day were observed, which may be related to increased ossification (i.e., increased rate of development). Malformations were evident at 150 mg/kg and included cleft palate, anasarca (edema), and cardiac abnormalities. Additional fetal findings included urinary tract and skeletal variations, observed primarily at 150 mg/kg/day. At the dose of 25 mg/kg/day, the area under the curve (AUC) exposures were equivalent to or below those achieved in patients at the 40 mg twice daily or 80 mg once daily doses, respectively. At the dose of 25 mg/kg/day, the AUC exposures were below those achieved in patients at the 200 mg twice daily dose.
In rabbits, maternal toxicities at the asciminib dose of 300 mg/kg/day resulted in the early termination of the dose group; a complete embryo-fetal examination was not conducted for this group. Adverse embryo-fetal findings were observed at 50 mg/kg; this dose did not cause maternal toxicities. Findings at the 50 mg/kg dose included increases in early resorptions and post-implantation loss, decreases in the number of live fetuses, and cardiac malformations. At the dose of 50 mg/kg/day, the AUC exposures were 4-fold those achieved in patients at the 40 mg twice daily or 80 mg once daily doses. At the dose of 50 mg/kg/day, the AUC exposures were below those achieved in patients at the 200 mg twice daily dose.
8.2 Lactation
Risk Summary
There are no data on the presence of asciminib or its metabolites in human milk, the effects on the breastfed child, or milk production.
Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with SCEMBLIX and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
Based on findings from animal studies, SCEMBLIX can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to starting treatment with SCEMBLIX.
Contraception
Females
Females of reproductive potential should use effective contraception during treatment with SCEMBLIX and for 1 week after the last dose.
Infertility
Based on findings in animals, SCEMBLIX may impair fertility in females of reproductive potential [see Nonclinical Toxicology (13.1)]. The reversibility of the effect on fertility is unknown.
8.4 Pediatric Use
The safety and efficacy of SCEMBLIX in pediatric patients have not been established.
8.5 Geriatric Use
Among the 556 patients receiving SCEMBLIX in the ASC4FIRST, ASCEMBL and X2101 studies, 130 (23%) were 65 years of age and older and 31 (6%) were 75 years of age and older.
Overall, no differences in safety or efficacy of SCEMBLIX were observed between patients 65 years of age and older compared to younger patients.
8.6 Renal Impairment
No dose adjustment is required for patients with mild to severe renal impairment (estimated glomerular filtration rate [eGFR] 15 to 89 mL/min/1.73 m2) and not requiring dialysis receiving SCEMBLIX [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment is required for patients with mild [total bilirubin ≤ upper limit of normal (ULN) and aspartate aminotransferase (AST) > ULN or total bilirubin > 1 to 1.5 times ULN and any AST] to severe hepatic impairment (total bilirubin > 3 times ULN and any AST) receiving SCEMBLIX [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
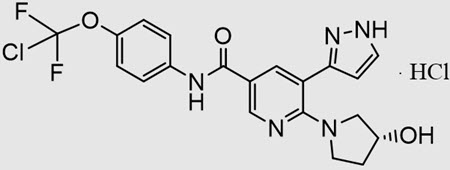
SCEMBLIX (asciminib) is a kinase inhibitor. The chemical name of the drug substance is N-[4-(Chlorodifluoromethoxy)phenyl]-6-[(3R)-3-hydroxypyrrolidin-1-yl]-5-(1H-pyrazol-3-yl)pyridine-3-carboxamide-hydrogen chloride (1/1). Asciminib hydrochloride is a white to slightly yellow powder. The molecular formula of asciminib hydrochloride is C20H18ClF2N5O3.HCl, and the relative molecular mass is 486.30 g/mol for the hydrochloride salt and 449.84 g/mol for the free base. The chemical structure of asciminib hydrochloride is shown below:
SCEMBLIX film-coated tablets are supplied for oral use with three strengths that contain 20 mg, 40 mg and 100 mg of asciminib (equivalent to 21.62 mg, 43.24 mg and 108.10 mg, respectively, of asciminib hydrochloride). The tablets contain colloidal silicon dioxide, croscarmellose sodium, ferric oxide, hydroxypropyl cellulose, lactose monohydrate, lecithin, magnesium stearate, microcrystalline cellulose, polyvinyl alcohol, talc, titanium dioxide, and xanthan gum. The 20 mg tablets contain ferric oxide, yellow and ferric oxide, red. The 40 mg and 100 mg tablets contain ferrosoferric oxide and ferric oxide, red.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Asciminib is an ABL/BCR-ABL1 tyrosine kinase inhibitor. Asciminib inhibits the ABL1 kinase activity of the BCR::ABL1 fusion protein, by binding to the ABL myristoyl pocket. In studies conducted in vitro or in animal models of CML, asciminib showed activity against wild-type BCR::ABL1 and several mutant forms of the kinase, including the T315I mutation.
12.2 Pharmacodynamics
Exposure-Response Relationships
Over asciminib dosages of 10 mg to 200 mg twice daily (0.25 to 5 times the recommended 80 mg daily dosage), a lower exposure was associated with a smaller decrease in BCR::ABL1 level and a lower MMR rate at Week 24.
Over asciminib dosages of 10 mg to 280 mg twice daily (0.25 to 7 times the recommended 80 mg daily dosage), a higher exposure was associated with slightly higher incidence of some adverse reactions (e.g., Grade ≥ 3 lipase increase, Grade ≥ 3 hemoglobin decrease, Grade ≥ 2 ALT increase, Grade ≥ 2 AST increase, Grade ≥ 2 bilirubin increase, and any grade lipase increase).
Cardiac Electrophysiology
Asciminib does not cause a large mean increase in QTc interval (i.e., > 20 msec) at the maximum recommended clinical dosage (200 mg twice daily). Based on available clinical data, small mean QTc increase (< 10 msec) cannot be excluded.
12.3 Pharmacokinetics
Asciminib steady-state exposure (AUC and Cmax) increase slightly more than dose proportional across the dose range of 10 to 200 mg (0.25 to 5 times the recommended 80 mg daily dosage) administered once or twice daily.
Pharmacokinetic parameters are presented as geometric mean (CV%) unless otherwise stated. The steady state Cmax and AUCtau of asciminib at recommended dosages are listed in Table 9.
Table 9: Steady Statea Asciminib Exposure at Recommended Dosages aSteady state is achieved within 3 days.
bAUCtau represents AUC0-12h for twice daily dosing and AUC0-24h for once daily dosing.Asciminib dosage Cmax (ng/mL) AUCtaub (ng*h/mL) Accumulation ratio 80 mg once daily 1781 (23%) 15112 (28%) 1.30 40 mg twice daily 793 (49%) 5262 (48%) 1.65 200 mg twice daily 5642 (40%) 37547 (41%) 1.92 Absorption
The median (range) Tmax of asciminib is 2.5 hours (2 to 3 hours).
Effect of Food
The AUC and Cmax of asciminib decreased by 62% and 68%, respectively, with a high-fat meal (1000 calories, 50% fat) and by 30% and 35%, respectively, with a low-fat meal (400 calories, 25% fat) compared to the fasted state following administration of SCEMBLIX.
Distribution
The apparent volume of distribution of asciminib at steady state is 151 L (135%). Asciminib is the main circulating component in plasma (93% of the administered dose).
Asciminib is 97% bound to human plasma proteins in vitro.
Elimination
The total apparent clearance of asciminib is 6.7 L/hour (48%) at 40 mg twice daily and 80 mg once daily, and 4.1 L/hour (38%) at 200 mg twice daily. The terminal elimination half-life of asciminib is 5.5 hours (38%) at 40 mg twice daily and 80 mg once daily, and 9.0 hours (33%) at 200 mg twice daily.
Metabolism
Asciminib is metabolized by CYP3A4-mediated oxidation, UGT2B7- and UGT2B17-mediated glucuronidation.
Excretion
Eighty percent (57% as unchanged) and 11% (2.5% as unchanged) of the asciminib dose were recovered in the feces and in the urine of healthy subjects, respectively, following oral administration of a single 80 mg dose of radio-labeled asciminib.
Asciminib is eliminated by biliary secretion via breast cancer-resistant protein (BCRP).
Specific Populations
No clinically significant differences in the pharmacokinetics of asciminib were observed based on sex, age (20 to 88 years), race (Asian 20%, White 70%, Black/African American 4%), or body weight (42-184 kg), mild to moderate renal impairment (eGFR 30 to 89 mL/min/1.73 m2), or mild (total bilirubin ≤ ULN and AST > ULN or total bilirubin > 1 to 1.5 times ULN and any AST) to moderate (total bilirubin > 1.5 to 3 times ULN and any AST) hepatic impairment.
Patients with Renal Impairment
Asciminib AUCinf and Cmax are increased by 57% and 6%, respectively, in subjects with eGFR between 13 to < 30 mL/min/1.73 m2 and not requiring dialysis compared to subjects with normal renal function (eGFR ≥ 90 mL/min/1.73 m2) following oral administration of a single 40 mg dose of SCEMBLIX. The exposure changes in patients with severe renal impairment are not considered clinically meaningful.
Patients with Hepatic Impairment
Asciminib AUCinf and Cmax are increased by 33% and 4%, respectively, in subjects with severe hepatic impairment (total bilirubin > 3 times ULN and any AST), compared to subjects with normal hepatic function (total bilirubin ≤ ULN and AST ≤ ULN) following oral administration of a single 40 mg dose of SCEMBLIX. The exposure changes are not considered clinically meaningful.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Drugs That Affect Asciminib Plasma Concentration
Strong CYP3A Inhibitors: The asciminib AUCinf and Cmax increased by 36% and 19%, respectively, following coadministration of a single SCEMBLIX dose of 40 mg with a strong CYP3A4 inhibitor (clarithromycin). No clinically significant differences in the pharmacokinetics of asciminib were observed when coadministered with itraconazole, which is also a strong CYP3A4 inhibitor.
Strong CYP3A Inducers: The asciminib AUCinf decreased by 34% and Cmax decreased by 22% following coadministration of a single SCEMBLIX dose of 200 mg with a strong CYP3A4 inducer (phenytoin) 100 mg three times daily. The asciminib AUCinf decreased by 15% while Cmax increased by 9%, following coadministration of a single SCEMBLIX dose of 40 mg with a strong CYP3A4 inducer (rifampicin).
Itraconazole Oral Solution: Coadministration of multiple doses of itraconazole oral solution containing hydroxypropyl-β-cyclodextrin with a single SCEMBLIX dose of 40 mg decreased asciminib AUCinf and Cmax by 40% and 50%, respectively. Concomitant use of oral products containing hydroxypropyl-β-cyclodextrin with SCEMBLIX other than itraconazole oral solution has not been fully characterized.
Imatinib: The asciminib AUCinf and Cmax increase by 108% and 59%, respectively following coadministration of a single SCEMBLIX dose of 40 mg with imatinib (an inhibitor of BCRP, CYP3A4, UGT2B17, and UGT1A3/4). The exposure changes are not considered clinically meaningful. Concomitant use of imatinib with SCEMBLIX at 200 mg twice daily has not been fully characterized.
Other Drugs: No clinically significant differences in the pharmacokinetics of asciminib were observed when coadministered with rabeprazole (acid-reducing agent) and quinidine (P-gp inhibitor).
Drugs That are Affected by Asciminib
CYP3A4 Substrates: The midazolam AUCinf and Cmax increased by 28% and 11%, respectively, following coadministration of a CYP3A4 substrate (midazolam) with SCEMBLIX 40 mg twice daily. The midazolam AUCinf and Cmax increased by 24% and 17%, respectively, following coadministration with SCEMBLIX at 80 mg once daily and 88% and 58%, respectively, at 200 mg twice daily.
CYP2C9 Substrates: The S-warfarin AUCinf and Cmax increased by 41% and 8%, respectively, following coadministration of CYP2C9 substrate (warfarin) with SCEMBLIX at 40 mg twice daily. The S-warfarin AUCinf and Cmax increased by 52% and 4%, respectively, following coadministration with SCEMBLIX at 80 mg once daily and 314% and 7%, respectively, at 200 mg twice daily.
CYP2C8 Substrates: The repaglinide (substrate of CYP2C8, CYP3A4, and OATP1B) AUCinf and Cmax increased by 8% and 14%, respectively, following coadministration of repaglinide with SCEMBLIX 40 mg twice daily. The repaglinide AUCinf and Cmax increased by 12% and 8%, respectively, following coadministration with SCEMBLIX at 80 mg once daily and 42% and 25%, respectively, at 200 mg twice daily. The rosiglitazone (substrate of CYP2C8 and CYP2C9) AUCinf and Cmax increased by 20% and 3%, respectively, following coadministration of rosiglitazone with SCEMBLIX 40 mg twice daily. The rosiglitazone AUCinf and Cmax increased by 24% and 2%, respectively, following coadministration with SCEMBLIX at 80 mg once daily and 66% and 8%, respectively, at 200 mg twice daily.
P-gp Substrates: Coadministration of SCEMBLIX with a drug that is a substrate of P-gp may result in a clinically relevant increase in the plasma concentrations of P-gp substrates, where minimal concentration changes may lead to serious toxicities.
OATP1B Substrates: The atorvastatin (substrate of OATP1B, CYP3A4, and P-gp) AUCinf and Cmax increased by 14% and 24%, respectively, following coadministration of atorvastatin with SCEMBLIX 80 mg once daily.
In Vitro Studies
CYP450 and UGT Enzymes
Asciminib may reversibly inhibit UGT1A1 at plasma concentrations reached at a total daily dose of 80 mg and 200 mg twice daily. In addition, asciminib may reversibly inhibit CYP2C19 at concentrations reached at 200 mg twice daily dose.
Transporter Systems
Asciminib is a substrate of BCRP and P-gp. Asciminib inhibits BCRP, P-gp, OATP1B1, OATP1B3, and OCT1. Asciminib may increase the exposure of BCRP substrates in a dose dependent manner.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2-year carcinogenicity study, rats were administered oral doses of asciminib at 20, 66, and 200 mg/kg/day in males and 10, 30, and 66 mg/kg/day in females. Asciminib induced a statistically significant increase in the incidence of benign Sertoli cell tumors in the ovaries of high-dose females. The exposures to asciminib (AUC) in female rats at 66 mg/kg/day were approximately 8-fold or 6-fold higher than the exposure (AUC) achieved in patients at the dose of 40 mg twice daily or 80 mg once daily, respectively, and equivalent to those achieved in patients at the dose of 200 mg twice daily. The clinical relevance of these findings is currently unknown.
Asciminib was not genotoxic in an in vitro bacterial mutagenicity (Ames) assay, an in vitro micronucleus assay in human peripheral blood lymphocytes (HPBL), or an in vivo rat peripheral blood reticulocyte micronucleus assay.
In a combined male and female fertility and early embryonic development study in rats, animals were administered asciminib doses of 10, 50, or 200 mg/kg/day orally. Male animals were dosed once daily for at least 28 days prior to mating, during the 2-week mating period, and until terminal necropsy (Days 63-67). Female animals were dosed once daily for the 2-week premating period, during the 2-week mating period, and through gestation day (GD) 6. Decreased sperm count and motility were observed at 200 mg/kg/day. While there were no effects on fertility indices or conception rates, a decreased mean number of live embryos was observed at 200 mg/kg/day and was attributed to a lower number of implantations and an increased number of early resorptions. Increased early resorptions were also observed in the embryo-fetal development study in rabbits [see Use in Specific Populations (8.1)].
At the dose of 200 mg/kg, the AUC exposures were approximately 19-fold, 13-fold, or 2-fold higher than those achieved in patients at the 40 mg twice daily, 80 mg once daily, or 200 mg twice daily doses, respectively.
-
14 CLINICAL STUDIES
14.1 Newly Diagnosed Ph+ CML-CP
The efficacy of SCEMBLIX in the treatment of patients with newly diagnosed Ph+ CML in chronic phase (Ph+ CML-CP) was evaluated in the multi-center, randomized, active-controlled, and open-label study ASC4FIRST (NCT04971226).
In this study, a total of 405 patients were randomized in a 1:1 ratio to receive either SCEMBLIX or investigator selected tyrosine kinase inhibitors (IS-TKIs). Prior to randomization, the investigator selected the TKI (imatinib, nilotinib, dasatinib, or bosutinib) to be used in the event of randomization to the comparator arm, based on patient characteristics and comorbidities. Patients were stratified according to EUTOS long-term survival (ELTS) risk group (low, intermediate, high), and pre-randomization selection of TKI (imatinib or other TKIs stratum composed of nilotinib, dasatinib, and bosutinib). Patients received either SCEMBLIX or IS-TKIs, and continued treatment until unacceptable toxicity or treatment failure occurred.
Patients were 37% female and 63% male with median age 51 years (range, 18 to 86 years). Of the 405 patients, 24% were 65 years or older, while 6% were 75 years or older. Patients were White (54%), Asian (44%), Black or African American (1%), and 1% unknown.
Of the 405 patients, 200 received SCEMBLIX, while 201 received IS-TKIs. Of the 201 patients receiving IS-TKIs, 99 received imatinib, 49 received nilotinib, 42 received dasatinib, and 11 received bosutinib. Four patients did not receive any treatment.
The median duration of treatment was 27 months (range, 0.2 to 36 months) for patients receiving SCEMBLIX, and 25 months (range, 0.3 to 35 months) for patients receiving IS-TKIs. With a minimum of 96 weeks of follow-up, 82% of patients on SCEMBLIX, and 60% of patients on IS-TKIs were still receiving treatment.
The main efficacy outcome was major molecular response rate (MMR) at 48 weeks. Efficacy was established based on SCEMBLIX compared to IS-TKIs and SCEMBLIX compared to IS-TKIs within the imatinib stratum. The key secondary objective evaluated MMR at 96 weeks for SCEMBLIX compared both to IS-TKIs and to IS-TKIs within the imatinib stratum. Secondary objectives evaluated MMR at 48 and 96 weeks for SCEMBLIX compared to IS-TKIs within the other TKIs stratum. The main efficacy outcomes from ASC4FIRST are summarized in Table 10.
Table 10: Efficacy Results in Patients with Newly Diagnosed Ph+ CML-CP (ASC4FIRST) Abbreviations: MMR, major molecular response (BCR::ABL1IS ≤ 0.1%); IS-TKIs, investigator selected tyrosine kinase inhibitors.
aIS-TKIs include imatinib (400 mg once daily) and other TKIs of nilotinib (300 mg twice daily), dasatinib (100 mg once daily) or bosutinib (400 mg once daily).
bEstimated using a common risk difference stratified by PRS-TKI and baseline ELTS risk groups.
cAdjusted p-value using a Cochran-Mantel-Haenszel 1-sided test stratified by PRS-TKI and baseline ELTS risk groups.
dAdjusted p-value using a Cochran-Mantel-Haenszel 1-sided test stratified by baseline ELTS risk groups.Response SCEMBLIX
80 mg once daily
All patients
(N = 201)IS-TKIsa
All patients
(N = 204)Difference
(95% CI)bp-value MMR at 48 weeks
MMR (%)
(95% CI)68
(61, 74)49
(42, 56)19
(10, 28)< 0.001c MMR at 96 weeks
MMR (%)
(95% CI)74
(68, 80)52
(45, 59)22
(14, 31)< 0.001c Response SCEMBLIX
80 mg once daily
Imatinib stratum
(N = 101)IS-TKIsa
Imatinib stratum
(N = 102)Difference
(95% CI)bp-value MMR at 48 weeks
MMR (%)
(95% CI)69
(59, 78)40
(31, 50)30
(17, 42)< 0.001d MMR at 96 weeks
MMR (%)
(95% CI)76
(67, 84)47
(37, 57)30
(18, 42)< 0.001d MMR rates at 96 weeks in patients receiving SCEMBLIX and IS-TKIs within the other TKIs stratum were 72% (95% CI: 62%, 81%) and 57% (95% CI: 47%, 67%), respectively.
Median time to MMR in patients receiving SCEMBLIX, IS-TKIs, and IS-TKIs within the imatinib stratum were: 24 weeks (95% CI: 24 to 25 weeks), 36 weeks (95% CI: 36 to 49 weeks), and 49 weeks (95% CI: 36 to 60 weeks), respectively.
14.2 Ph+ CML-CP, Previously Treated with Two or More TKIs
The efficacy of SCEMBLIX in the treatment of patients with Ph+ CML-CP, previously treated with two or more TKIs was evaluated in the multi-center, randomized, active-controlled, and open-label study ASCEMBL (NCT03106779).
In this study, a total of 233 patients were randomized in a 2:1 ratio and stratified according to major cytogenetic response (MCyR) status to receive either SCEMBLIX 40 mg twice daily (N = 157) or bosutinib 500 mg once daily (N = 76). Patients continued treatment until unacceptable toxicity or treatment failure occurred.
Patients were 52% female and 48% male with a median age of 52 years (range, 19 to 83 years). Of the 233 patients, 19% were 65 years or older, while 2.6% were 75 years or older. Patients were White (75%), Asian (14%), and Black or African American (4.3%). Of the 233 patients, 81% and 18% had Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1, respectively. Patients who had previously received 2, 3, 4, or 5 or more prior lines of TKIs were 48%, 31%, 15%, and 6%, respectively. The median duration of treatment was 103 weeks (range, 0.1 to 201 weeks) for patients receiving SCEMBLIX and 31 weeks (range, 1 to 188 weeks) for patients receiving bosutinib.
The main efficacy outcomes from ASCEMBL are summarized in Table 11.
Table 11: Efficacy Results in Patients with Ph+ CML-CP, Previously Treated with Two or More TKIs (ASCEMBL) Abbreviations: MMR, major molecular response (BCR::ABL1IS ≤ 0.1%); CCyR, complete cytogenetic response (0% of Philadelphia-positive metaphases in bone marrow aspirate with at least 20 examined).
aEstimated using a common risk difference stratified by baseline major cytogenetic response status.
bEstimated using a Cochran-Mantel-Haenszel two-sided test stratified by baseline major cytogenetic response status.
cCCyR analysis based on patients who were not in CCyR at baseline.SCEMBLIX
40 mg
twice dailyBosutinib
500 mg
once daily
Difference
(95% CI)
p-value
MMR rate,
% (95% CI)
at 24 weeksN = 157
25
(19, 33)N = 76
13
(6.5, 23)
12a
(2.2, 22)
0.029b
MMR rate,
% (95% CI)
at 96 weeksN = 157
38
(30, 46)N = 76
16
(8, 26)
22a
(11, 33)
0.001b
CCyR rate,
% (95% CI)
at 24 weeksN = 103c
41
(31, 51)N = 62c
24
(14, 37)
17
(3.6, 31)CCyR rate,
% (95% CI)
at 96 weeksN = 103c
40
(30, 50)N = 62c
16
(8, 28)
24a
(10, 37)With a median duration of follow-up of 28 months (range, 1 day to 45 months), the median duration of response for patients treated with SCEMBLIX had not yet been reached.
14.3 Ph+ CML-CP with the T315I Mutation
The efficacy of SCEMBLIX in the treatment of patients with Ph+ CML-CP with the T315I mutation was evaluated in a multi-center open-label study CABL001X2101 (NCT02081378). Testing for T315I mutation utilized a qualitative p210 BCR::ABL1 mutation test on peripheral blood using Sanger Sequencing.
Efficacy was based on 45 patients with Ph+ CML-CP with the T315I mutation who received SCEMBLIX at a dose of 200 mg twice daily. Patients continued treatment until unacceptable toxicity or treatment failure occurred.
Of the 45 patients, 80% were male and 20% female; 31% were 65 years or older, while 9% were 75 years or older with a median age of 54 years (range, 26 to 86 years). The patients were White (47%), Asian (27%), and Black or African American (2.2%), and 24% were unreported or unknown. Seventy-three percent and 27% of patients had ECOG performance status 0 and 1, respectively. Patients who had previously received 1, 2, 3, 4, and 5 or more TKIs were 18%, 31%, 36%, 13%, and 2.2%, respectively. MMR was achieved by 24 weeks in 42% (19/45, 95% CI: 28% to 58%) of the 45 patients treated with SCEMBLIX. MMR was achieved by 96 weeks in 49% (22/45, 95% CI: 34% to 64%) of the 45 patients treated with SCEMBLIX. The median duration of treatment was 108 weeks (range, 2 to 215 weeks).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
SCEMBLIX tablets are available as:
Table 12: SCEMBLIX Package Configurations and NDC Numbers Package configuration Tablet strength NDC number Bottle of 60 tablets 20 mg NDC: 0078-1091-20 Bottle of 60 tablets 40 mg NDC: 0078-1098-20 Bottle of 60 tablets 100 mg NDC: 0078-1196-20 - SCEMBLIX (asciminib) 20 mg film-coated tablets are supplied as pale yellow, unscored, round, biconvex, with beveled edges, film-coated tablets containing 20 mg of asciminib. Each tablet is debossed with “20” on one side and the “Novartis” logo on the other side.
- SCEMBLIX (asciminib) 40 mg film-coated tablets are supplied as violet white, unscored, round, biconvex, with beveled edges, film-coated tablets containing 40 mg of asciminib. Each tablet is debossed with “40” on one side and the “Novartis” logo on the other side.
- SCEMBLIX (asciminib) 100 mg film-coated tablets are supplied as light red, unscored, round, biconvex, with beveled edges, film-coated tablets containing 100 mg of asciminib. Each tablet is debossed with “100” on one side and the “Novartis” logo on the other side.
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature]. Dispense and store in the original container in order to protect from moisture.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Myelosuppression
Inform patients of the possibility of developing low blood cell counts. Advise patients to immediately report fever, any suggestion of infection, or signs or symptoms suggestive of bleeding or easy bruising [see Warnings and Precautions (5.1)].
Pancreatic Toxicity
Inform patients of the possibility of developing pancreatitis that may be accompanied by nausea, vomiting, severe abdominal pain, or abdominal discomfort, and to promptly report these symptoms [see Warnings and Precautions (5.2)].
Hypertension
Inform patients of the possibility of developing hypertension. Advise patients to contact their healthcare provider for elevated blood pressure or if symptoms of hypertension occur, including confusion, headache, dizziness, chest pain, or shortness of breath [see Warnings and Precautions (5.3)].
Hypersensitivity
Advise the patient to discontinue SCEMBLIX and seek immediate medical attention if any signs or symptoms of a hypersensitivity reaction, such as rash, edema, or bronchospasm occur [see Warnings and Precautions (5.4)].
Cardiovascular Toxicity
Inform patients of the possibility of the occurrence of cardiovascular toxicity, especially those with a history of cardiovascular risk factors. Advise patients to immediately contact their healthcare provider or get medical help if they develop cardiovascular signs and symptoms [see Warnings and Precautions (5.5)].
Embryo-Fetal Toxicity
Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the potential risk to a fetus [see Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment and for 1 week after receiving the last dose of SCEMBLIX [see Warnings and Precautions (5.6), Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during treatment with SCEMBLIX and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise females of reproductive potential of the potential for impaired fertility from SCEMBLIX [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Drug Interactions
Advise patients that SCEMBLIX and certain other medicines, including over-the-counter medications or herbal supplements, can interact with each other and may alter the effects of SCEMBLIX [see Drug Interactions (7)].
Instructions for Taking SCEMBLIX
Advise patients to take SCEMBLIX exactly as prescribed and not to change their dose or schedule or to stop taking SCEMBLIX unless they are told to do so by their healthcare provider.
Advise patients to take SCEMBLIX orally without food. Advise patients to avoid food for at least 2 hours before and 1 hour after taking SCEMBLIX. SCEMBLIX tablets should be swallowed whole. Patients should not break, crush, or chew the tablets [see Dosage and Administration (2.5)].
Advise patients that if they take SCEMBLIX once daily and miss a dose by more than 12 hours to skip the missed dose. Advise patients that if they take SCEMBLIX twice daily and miss a dose by more than 6 hours to skip the missed dose. Advise patients to take the next dose as scheduled [see Dosage and Administration (2.3)].
Distributed by:
Novartis Pharmaceuticals Corporation
One Health Plaza
East Hanover, New Jersey 07936© Novartis
T2025-68
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 10/2024 PATIENT INFORMATION
SCEMBLIX® (sem blix)
(asciminib) tabletsWhat is SCEMBLIX?
SCEMBLIX is a prescription medicine used to treat adults with:- newly diagnosed or previously treated Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP).
- Ph+ CML in CP with the T315I mutation.
Before taking SCEMBLIX, tell your healthcare provider about all of your medical conditions, including if you: - have a history of inflammation of your pancreas (pancreatitis)
- have a history of heart problems or blood clots in your arteries and veins (types of blood vessels)
- are pregnant or plan to become pregnant. SCEMBLIX can harm your unborn baby.
Females who are able to become pregnant:- Your healthcare provider will do a pregnancy test before you start treatment with SCEMBLIX.
- You should use effective birth control (contraception) during treatment and for 1 week after your last dose of SCEMBLIX. Talk to your healthcare provider about birth control methods that may be right for you.
- Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with SCEMBLIX.
- are breastfeeding or plan to breastfeed. It is not known if SCEMBLIX passes into your breast milk. Do not breastfeed during treatment and for 1 week after your last dose of SCEMBLIX.
Taking SCEMBLIX with certain other medicines may affect each other causing side effects and may affect the way that SCEMBLIX works.How should I take SCEMBLIX? - Take SCEMBLIX exactly as your healthcare provider tells you to take it.
- Do not change your dose or schedule or stop taking SCEMBLIX unless your healthcare provider tells you to.
- Take SCEMBLIX without food. You should avoid eating for at least 2 hours before and 1 hour after taking SCEMBLIX.
- Swallow SCEMBLIX tablets whole. Do not break, crush, or chew SCEMBLIX tablets.
- If you take SCEMBLIX 1 time a day and miss a dose by more than 12 hours, skip the missed dose and take your next dose at your regular time.
- If you take SCEMBLIX 2 times a day and miss a dose by more than 6 hours, skip the missed dose and take your next dose at your regular time.
What are the possible side effects of SCEMBLIX?
SCEMBLIX may cause serious side effects, including:- Low blood cell counts. SCEMBLIX may cause low platelet counts (thrombocytopenia), low white blood cell counts (neutropenia), and low red blood cell counts (anemia). Your healthcare provider will do blood tests to check your blood cell counts every 2 weeks for the first 3 months of treatment and then monthly or as needed during treatment with SCEMBLIX. Tell your healthcare provider right away if you have unexpected bleeding or easy bruising, blood in your urine or stools, fever, or any signs of an infection.
- Pancreas problems. SCEMBLIX may increase enzymes in your blood called amylase and lipase, which may be a sign of inflammation of the pancreas (pancreatitis). Your healthcare provider may do blood tests monthly or as needed during treatment with SCEMBLIX to check for problems with your pancreas. Tell your healthcare provider right away if you have severe stomach-area (abdominal) pain or discomfort, nausea, or vomiting.
- High blood pressure. Your healthcare provider may check your blood pressure and treat any high blood pressure during treatment with SCEMBLIX as needed. Tell your healthcare provider if you develop high blood pressure or symptoms of high blood pressure, including confusion, headaches, dizziness, chest pain, or shortness of breath.
- Allergic reaction. Stop taking SCEMBLIX and get medical help right away if you get any signs or symptoms of an allergic reaction, including:
○ trouble breathing or swallowing
○ swelling of the face, lips, or tongue
○ skin rash or flushing of your skin○ feeling dizzy or faint
○ fever
○ fast heartbeat- Heart and blood vessel (cardiovascular) problems. SCEMBLIX may cause heart and blood vessel problems, including heart attack, stroke, blood clots or blockage of your arteries, heart failure, and abnormal heartbeat, which can be serious and may sometimes lead to death. In the majority of cases, these heart and blood vessel problems happened in people with risk factors or a history of these problems, and previously treated with other tyrosine kinase inhibitor (TKI) medicines. Your healthcare provider may monitor you for heart and blood vessel problems and treat you as needed during treatment with SCEMBLIX. Tell your healthcare provider or get medical help right away if you get:
○ shortness of breath
○ chest pain or pressure
○ feeling like your heart is beating too fast or you feel abnormal heartbeats
○ swelling in your ankles or feet
○ dizziness○ weight gain
○ numbness or weakness on one side of your body
○ decreased vision or loss of vision
○ trouble talking
○ pain in your arms, legs, back, neck or jaw
○ headache
○ severe stomach-area (abdominal) painThe most common side effects of SCEMBLIX include: muscle, bone, or joint pain
rash
tiredness
nose, throat, or sinus (upper respiratory tract) infections
headache
stomach-area (abdominal) pain
diarrheadecreased white blood cell counts, platelet counts, and red blood cell counts
decreased blood calcium corrected levels
increased blood pancreas enzyme (lipase and amylase) levels
increased blood fat (cholesterol and triglycerides) levels
increased blood uric acid levels
increased blood liver enzyme levels
increased blood alkaline phosphatase levels
increased blood creatine kinase levelsYour healthcare provider may change your dose or temporarily or permanently stop treatment with SCEMBLIX if you have certain side effects.
SCEMBLIX may cause fertility problems in females. This may affect your ability to have a child. Talk to your healthcare provider if this is a concern for you.
These are not all of the possible side effects of SCEMBLIX.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store SCEMBLIX? - Store SCEMBLIX at room temperature between 68°F to 77°F (20°C to 25°C).
- Store SCEMBLIX in the original container to protect it from moisture.
General information about the safe and effective use of SCEMBLIX.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use SCEMBLIX for a condition for which it was not prescribed. Do not give SCEMBLIX to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for more information about SCEMBLIX that is written for health professionals.What are the ingredients in SCEMBLIX?
Active ingredient: asciminib
Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, ferric oxide, hydroxypropyl cellulose, lactose monohydrate, lecithin, magnesium stearate, microcrystalline cellulose, polyvinyl alcohol, talc, titanium dioxide, and xanthan gum. The 20 mg tablets contain ferric oxide, yellow and ferric oxide, red. The 40 mg and 100 mg tablets contain ferrosoferric oxide and ferric oxide, red.
Distributed by: Novartis Pharmaceuticals Corporation, One Health Plaza, East Hanover, New Jersey 07936
For more information, go to www.SCEMBLIX.com or call 1-888-669-6682.© Novartis
T2024-79
-
PRINCIPAL DISPLAY PANEL
NDC: 0078-1091-20
SCEMBLIX®
(asciminib)
Tablets20 mg*
Rx only
60 Tablets
Swallow tablets whole. Do not break,
crush, or chew the tablets.NOVARTIS

-
PRINCIPAL DISPLAY PANEL
NDC: 0078-1098-20
SCEMBLIX®
(asciminib)
Tablets40 mg*
Rx only
60 Tablets
Swallow tablets whole. Do not break,
crush, or chew the tablets.NOVARTIS

-
PRINCIPAL DISPLAY PANEL
NDC: 0078-1196-20
SCEMBLIX®
(asciminib)
Tablets100 mg*
Rx only
60 Tablets
Swallow tablets whole. Do not break,
crush, or chew the tablets.NOVARTIS

-
INGREDIENTS AND APPEARANCE
SCEMBLIX
asciminib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-1091 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ASCIMINIB HYDROCHLORIDE (UNII: C5U34S9XFV) (ASCIMINIB - UNII:L1F3R18W77) ASCIMINIB 20 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) LECITHIN, SOYBEAN (UNII: 1DI56QDM62) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) XANTHAN GUM (UNII: TTV12P4NEE) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color YELLOW (pale yellow) Score no score Shape ROUND (biconvex, with beveled edges) Size 6mm Flavor Imprint Code 20;IMPRINT Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-1091-20 60 in 1 BOTTLE; Type 0: Not a Combination Product 10/29/2021 2 NDC: 0078-1091-94 14 in 1 BOTTLE; Type 0: Not a Combination Product 10/29/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215358 10/29/2021 SCEMBLIX
asciminib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-1098 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ASCIMINIB HYDROCHLORIDE (UNII: C5U34S9XFV) (ASCIMINIB - UNII:L1F3R18W77) ASCIMINIB 40 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) LECITHIN, SOYBEAN (UNII: 1DI56QDM62) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) XANTHAN GUM (UNII: TTV12P4NEE) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color WHITE (violet white) Score no score Shape ROUND (biconvex, with beveled edges) Size 8mm Flavor Imprint Code 40;IMPRINT Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-1098-20 60 in 1 BOTTLE; Type 0: Not a Combination Product 10/29/2021 2 NDC: 0078-1098-30 5 in 1 CARTON 10/29/2021 08/31/2023 2 NDC: 0078-1098-05 60 in 1 BOTTLE; Type 0: Not a Combination Product 3 NDC: 0078-1098-94 14 in 1 BOTTLE; Type 0: Not a Combination Product 10/29/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215358 10/29/2021 SCEMBLIX
asciminib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-1196 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ASCIMINIB HYDROCHLORIDE (UNII: C5U34S9XFV) (ASCIMINIB - UNII:L1F3R18W77) ASCIMINIB 100 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) LECITHIN, SOYBEAN (UNII: 1DI56QDM62) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) XANTHAN GUM (UNII: TTV12P4NEE) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color RED (light red) Score no score Shape ROUND (biconvex, with beveled edges) Size 11mm Flavor Imprint Code 100;IMPRINT Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-1196-20 60 in 1 BOTTLE; Type 0: Not a Combination Product 04/18/2024 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215358 10/29/2021 Labeler - Novartis Pharmaceuticals Corporation (002147023)
Trademark Results [SCEMBLIX]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 SCEMBLIX 79281911 not registered Live/Pending |
Novartis AG 2020-02-13 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.