LEVOCETIRIZINE DIHYDROCHLORIDE tablet, film coated
LEVOCETIRIZINE DIHYDROCHLORIDE by
Drug Labeling and Warnings
LEVOCETIRIZINE DIHYDROCHLORIDE by is a Prescription medication manufactured, distributed, or labeled by Preferred Pharmaceuticals Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LEVOCETIRIZINE DIHYDROCHLORIDE TABLETS safely and effectively. See full prescribing information for LEVOCETIRIZINE DIHYDROCHLORIDE TABLETS.
LEVOCETIRIZINE dihydrochloride tablets, for oral use
Initial U.S. Approval: 1995INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Immediate release breakable (scored) tablets, 5 mg (3)
CONTRAINDICATIONS
Patients with a known hypersensitivity to levocetirizine or any of the ingredients of levocetirizine dihydrochloride tablets or to cetirizine (4.1)
Patients with end-stage renal disease at less than 10 mL/min creatinine clearance or patients undergoing hemodialysis (4.2)
Children 6 months to 11 years of age with renal impairment (4.3)WARNINGS AND PRECAUTIONS
Avoid engaging in hazardous occupations requiring complete mental alertness such as driving or operating machinery when taking levocetirizine dihydrochloride tablets. (5.1)
Avoid concurrent use of alcohol or other central nervous system depressants with levocetirizine dihydrochloride tablets. (5.1)
Use with caution in patients with predisposing factors of urinary retention (e.g. spinal cord lesion, prostatic hyperplasia). Discontinue levocetirizine dihydrochloride tablets if urinary retention occurs. (5.2)ADVERSE REACTIONS
The most common adverse reactions (rate ≥2% and > placebo) were somnolence, nasopharyngitis, fatigue, dry mouth, and pharyngitis in subjects 12 years of age and older, and pyrexia, somnolence, cough, and epistaxis in children 6 to 12 years of age. In subjects 1 to 5 years of age, the most common adverse reactions (rate ≥2% and > placebo) were pyrexia, diarrhea, vomiting, and otitis media. In subjects 6 to 11 months of age, the most common adverse reactions (rate ≥3% and > placebo) were diarrhea and constipation. (6.1). To report SUSPECTED ADVERSE REACTIONS, contact Macleods Pharma USA, Inc., at 1-888-943-3210 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Renal Impairment
Because levocetirizine hydrochloride is substantially excreted by the kidneys, the risk of adverse reactions to this drug may be greater in patients with impaired renal function (8.6, 12.3).
Pediatric Use
Do not exceed the recommended doses of 2.5 mg once daily in children 6 to 11 years. Systemic exposure with these doses in pediatric age group is comparable to that from a 5 mg once daily dose in adults. (12.3)See 17 for PATIENT COUNSELING INFORMATION.
Revised: 10/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS & USAGE
1.2 Chronic Idiopathic Urticaria
2 DOSAGE & ADMINISTRATION
2.2 Chronic Idiopathic Urticaria
3 DOSAGE FORMS & STRENGTHS
4 CONTRAINDICATIONS
4.1 Patients with Known Hypersensitivity
4.2 Patients with End-Stage Renal Disease
4.3 Pediatric Patients with Impaired Renal Function
5 WARNINGS AND PRECAUTIONS
5.1 Somnolence
5.2 Urinary Retention
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Antipyrine, Azithromycin, Cimetidine, Erythromycin, Ketoconazole, Theophylline, and Pseudoephedrine
7.2 Ritonavir
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis & Mutagenesis & Impairment Of Fertility
14 CLINICAL STUDIES
14.1 Perennial Allergic Rhinitis
14.2 Chronic Idiopathic Urticaria
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS & USAGE
-
2 DOSAGE & ADMINISTRATION
Levocetirizine dihydrochloride is available as 5 mg breakable (scored) tablets, allowing for the administration of 2.5 mg, if needed. Levocetirizine dihydrochloride can be taken without regard to food consumption..
2.2 Chronic Idiopathic Urticaria
Adults and Children 12 Years of Age and Older
The recommended dose of levocetirizine dihydrochloride is 5 mg (1 tablet) once daily in the evening. Some patients may be adequately controlled by 2.5 mg (1/2 tablet) once daily in the evening.Children 6 to 11 Years of Age
The recommended dose of levocetirizine dihydrochloride is 2.5 mg (1/2 tablet) once daily in the evening. The 2.5 mg dose should not be exceeded because the systemic exposure with 5 mg is approximately twice that of adults [see Clinical Pharmacology (12.3)].Dose Adjustment for Renal and Hepatic Impairment
In adults and children 12 years of age and older with:
Mild renal impairment (creatinine clearance [CLCR]=50-80 mL/min): a dose of 2.5 mg once daily is recommended;
Moderate renal impairment (CLCR = 30-50 mL/min): a dose of 2.5 mg once every other day is recommended;
Severe renal impairment (CLCR = 10-30 mL/min): a dose of 2.5 mg twice weekly (administered once every 3-4 days) is recommended;
End-stage renal disease patients (CLCR <10 mL/min) and patients undergoing hemodialysis should not receive levocetirizine dihydrochloride.
No dose adjustment is needed in patients with solely hepatic impairment. In patients with both hepatic impairment and renal impairment, adjustment of the dose is recommended. - 3 DOSAGE FORMS & STRENGTHS
-
4 CONTRAINDICATIONS
The use of levocetirizine dihydrochloride is contraindicated in:
4.1 Patients with Known Hypersensitivity
Patients with known hypersensitivity to levocetirizine or any of the ingredients of levocetirizine dihydrochloride tablets, or to cetirizine. Observed reactions range from urticaria to anaphylaxis [see Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Somnolence
In clinical trials the occurrence of somnolence, fatigue, and asthenia has been reported in some patients under therapy with levocetirizine dihydrochloride. Patients should be cautioned against engaging in hazardous occupations requiring complete mental alertness, and motor coordination such as operating machinery or driving a motor vehicle after ingestion of levocetirizine dihydrochloride. Concurrent use of levocetirizine dihydrochloride with alcohol or other central nervous system depressants should be avoided because additional reductions in alertness and additional impairment of central nervous system performance may occur..
5.2 Urinary Retention
Urinary retention has been reported postmarketing with levocetirizine dihydrochloride. Levocetirizine dihydrochloride should be used with caution in patients with predisposing factors of urinary retention (e.g. spinal cord lesion, prostatic hyperplasia) as levocetirizine dihydrochloride may increase the risk of urinary retention. Discontinue levocetirizine dihydrochloride if urinary retention occurs.
-
6 ADVERSE REACTIONS
Use of levocetirizine dihydrochloride has been associated with somnolence, fatigue, asthenia, and urinary retention [see Warnings and Precautions (5)].
6.1 Clinical Trials Experience
The safety data described below reflect exposure to levocetirizine dihydrochloride in 2708 patients with allergic rhinitis or chronic idiopathic urticaria in 14 controlled clinical trials of 1 week to 6 months duration.
The short-term (exposure up to 6 weeks) safety data for adults and adolescents are based upon eight clinical trials in which 1896 patients (825 males and 1071 females aged 12 years and older) were treated with levocetirizine dihydrochloride 2.5, 5, or 10 mg once daily in the evening.
The short-term safety data from pediatric patients are based upon two clinical trials in which 243 children with allergic rhinitis (162 males and 81 females 6 to 12 years of age) were treated with levocetirizine dihydrochloride 5 mg once daily for 4 to 6 weeks, one clinical trial in which 114 children (65 males and 49 females 1 to 5 years of age) with allergic rhinitis or chronic idiopathic urticaria were treated with levocetirizine dihydrochloride 1.25 mg twice daily for 2 weeks, and one clinical trial in which 45 children (28 males and 17 females 6 to 11 months of age) with symptoms of allergic rhinitis or chronic urticaria were treated with levocetirizine dihydrochloride 1.25 mg once daily for 2 weeks.
The long-term (exposure of 4 or 6 months) safety data in adults and adolescents are based upon two clinical trials in which 428 patients (190 males and 238 females) with allergic rhinitis were exposed to treatment with levocetirizine dihydrochloride 5 mg once daily. Long term safety data are also available from an 18-month trial in 255 levocetirizine dihydrochloride -treated subjects 12-24 months of age.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trial of another drug and may not reflect the rates observed in practice.Adults and Adolescents 12 years of Age and Older
In studies up to 6 weeks in duration, the mean age of the adult and adolescent patients was 32 years, 44% of the patients were men and 56% were women, and the large majority (more than 90%) was Caucasian.In these trials 43% and 42% of the subjects in the levocetirizine dihydrochloride 2.5 mg and 5 mg groups, respectively, had at least one adverse event compared to 43% in the placebo group.
In placebo-controlled trials of 1-6 weeks in duration, the most common adverse reactions were somnolence, nasopharyngitis, fatigue, dry mouth, and pharyngitis, and most were mild to moderate in intensity. Somnolence with levocetirizine dihydrochloride showed dose ordering between tested doses of 2.5, 5 and 10 mg and was the most common adverse reaction leading to discontinuation (0.5%).
Table 1 lists adverse reactions that were reported in greater than or equal to 2% of subjects aged 12 years and older exposed to levocetirizine dihydrochloride 2.5 mg or 5 mg in eight placebo-controlled clinical trials and that were more common with levocetirizine dihydrochloride than placebo.
Table 1: Adverse Reactions Reported in ≥ 2%* of Subjects Aged 12 Years and Older Exposed to Levocetirizine Dihydrochloride 2.5 mg or 5 mg Once Daily in Placebo-Controlled Clinical Trials 1-6 Weeks in Duration
Adverse Reactions
Levocetirizine Dihydrochloride 2.5 mg
(n = 421)
Levocetirizine Dihydrochloride 5 mg (n = 1070)
Placebo (n = 912)
Somnolence22 (5%)
61 (6%)
16 (2%)
Nasopharyngitis25 (6%)
40 (4%)
28 (3%)
Fatigue5 (1%)
46 (4%)
20 (2%)
Dry Mouth12 (3%)
26 (2%)
11 (1%)
Pharyngitis10 (2%)
12 (1%)
9 (1%)
*Rounded to the closest unit percentage
Additional adverse reactions of medical significance observed at a higher incidence than in placebo in adults and adolescents aged 12 years and older exposed to levocetirizine dihydrochloride are syncope (0.2%) and weight increased (0.5%).
Pediatric Patients 6 to 12 Years of Age
A total of 243 pediatric patients 6 to 12 years of age received levocetirizine dihydrochloride 5 mg once daily in two short-term placebo controlled double-blind trials. The mean age of the patients was 9.8 years, 79 (32%) were 6 to 8 years of age, and 50% were Caucasian. Table 2 lists adverse reactions that were reported in greater than or equal to 2% of subjects aged 6 to 12 years exposed to levocetirizine dihydrochloride 5 mg in placebo-controlled clinical trials and that were more common with levocetirizine dihydrochloride than placebo.
Table 2: Adverse Reactions Reported in ≥ 2%* of Subjects Aged 6-12 Years Exposed to Levocetirizine Dihydrochloride 5 mg Once Daily in Placebo-Controlled Clinical Trials 4 and 6 Weeks in DurationAdverse
Reactions
Levocetirizine Dihydrochloride
5 mg (n = 243)
Placebo
(n = 240)
Pyrexia10 (4%)
5 (2%)
Cough8 (3%)
2 (<1%)
Somnolence7 (3%)
1 (<1%)
Epistaxis6 (2%)
1 (<1%)
*Rounded to the closest unit percentage
Pediatric Patients 1 to 5 Years of Age
A total of 114 pediatric patients 1 to 5 years of age received levocetirizine dihydrochloride 1.25 mg twice daily in a two week placebo-controlled double-blind safety trial. The mean age of the patients was 3.8 years, 32% were 1 to 2 years of age, 71% were Caucasian and 18% were Black. Table 3 lists adverse reactions that were reported in greater than or equal to 2% of subjects aged 1 to 5 years exposed to levocetirizine dihydrochloride 1.25 mg twice daily in the placebo-controlled safety trial and that were more common with levocetirizine dihydrochloride than placebo.Table 3: Adverse Reactions Reported in ≥2%* of Subjects Aged 1-5 Years Exposed to Levocetirizine Dihydrochloride 1.25 mg Twice Daily in a 2-Week Placebo-Controlled Clinical Trial
Adverse
Reactions
Levocetirizine Dihydrochloride
1.25 mg Twice Daily
(n = 114)
Placebo
(n = 59)
Pyrexia5 (4%)
1 (2%)
Diarrhea4 (4%)
2 (3%)
Vomiting4 (4%)
2 (3%)
Otitis Media3 (3%)
0 (0%)
*Rounded to the closest unit percentage
Pediatric Patients 6 to 11 Months of Age
A total of 45 pediatric patients 6 to 11 months of age received levocetirizine dihydrochloride 1.25 mg once daily in a two week placebo-controlled double-blind safety trial. The mean age of the patients was 9 months, 51% were Caucasian and 31% were Black. Adverse reactions that were reported in more than 1 subject (i.e. greater than or equal to 3% of subjects) aged 6 to 11 months exposed to levocetirizine dihydrochloride 1.25 mg once daily in the placebo-controlled safety trial and that were more common with levocetirizine dihydrochloride than placebo included diarrhea and constipation which were reported in 6 (13%) and 1 (4%) and 3 (7%) and 1 (4%) children in the levocetirizine dihydrochloride and placebo-treated groups, respectively.Long-Term Clinical Trials Experience
In two controlled clinical trials, 428 patients (190 males and 238 females) aged 12 years and older were treated with levocetirizine dihydrochloride 5 mg once daily for 4 or 6 months. The patient characteristics and the safety profile were similar to that seen in the short-term studies. Ten (2.3%) patients treated with Levocetirizine dihydrochloride discontinued because of somnolence, fatigue or asthenia compared to 2 (<1%) in the placebo group.
There are no long term clinical trials in children below 12 years of age with allergic rhinitis or chronic idiopathic urticaria.Laboratory Test Abnormalities
Elevations of blood bilirubin and transaminases were reported in <1% of patients in the clinical trials. The elevations were transient and did not lead to discontinuation in any patient.6.2 Postmarketing Experience
In addition to the adverse reactions reported during clinical trials and listed above, the following adverse reactions have also been identified during postapproval use of levocetirizine dihydrochloride. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiac disorders: palpitations, tachycardia
Ear and labyrinth disorders: vertigo
Eye disorders: blurred vision, visual disturbances
Gastrointestinal disorders: nausea, vomiting
General disorders and administration site conditions: edema
Hepatobiliary disorders: hepatitis
Immune system disorders: anaphylaxis and hypersensitivity
Metabolism and nutrition disorders: increased appetite
Musculoskeletal, connective tissues, and bone disorders: arthralgia, myalgia
Nervous system disorders: dizziness, dysgeusia, febrile seizure, movement disorders (including dystonia and oculogyric crisis), paresthesia, seizure (reported in subjects with and without a known seizure disorder),
tremor
Psychiatric disorders: aggression and agitation, depression, hallucinations, insomnia, nightmare, suicidal ideation
Renal and urinary disorders: dysuria, urinary retention
Respiratory, thoracic, and mediastinal disorders: dyspnea
Skin and subcutaneous tissue disorders: angioedema, fixed drug eruption, pruritus, rash and urticaria
Besides these reactions reported under treatment with levocetirizine dihydrochloride, other potentially severe adverse events have been reported from the postmarketing experience with cetirizine. Since levocetirizine is the principal pharmacologically active component of cetirizine, one should take into account the fact that the following adverse events could also potentially occur under treatment with levocetirizine dihydrochloride.
Cardiac disorders: severe hypotension
Gastrointestinal disorders: cholestasis
Nervous system disorders: extrapyramidal symptoms, myoclonus, orofacial dyskinesia, tic
Pregnancy, puerperium and perinatal conditions: stillbirth
Renal and urinary disorders: glomerulonephritis
Skin and subcutaneous tissue disorders: acute generalized exanthematous pustulosis (AGEP); rebound pruritus - pruritus within a few days after discontinuation of cetirizine, usually after long-term use (e.g. months to years) of cetirizine. -
7 DRUG INTERACTIONS
In vitro data indicate that levocetirizine is unlikely to produce pharmacokinetic interactions through inhibition or induction of liver drug-metabolizing enzymes. No in vivo drug-drug interaction studies have been performed with levocetirizine. Drug interaction studies have been performed with racemic cetirizine.
7.1 Antipyrine, Azithromycin, Cimetidine, Erythromycin, Ketoconazole, Theophylline, and Pseudoephedrine
Pharmacokinetic interaction studies performed with racemic cetirizine demonstrated that cetirizine did not interact with antipyrine, pseudoephedrine, erythromycin, azithromycin, ketoconazole, and cimetidine. There was a small decrease (~16%) in the clearance of cetirizine caused by a 400 mg dose of theophylline. It is possible that higher theophylline doses could have a greater effect.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from published literature and postmarketing experience with levocetirizine use in pregnant women are insufficient to identify any drug-associated risks of miscarriage, birth defects, or adverse maternal or fetal outcomes. In animal reproduction studies, there was no evidence of fetal harm with administration of levocetirizine by the oral route to pregnant rats and rabbits, during the period of organogenesis, at doses up to 390 times and 470 times, respectively, the maximum recommended human dose (MRHD) in adults. In rats treated during late gestation and the lactation period, cetirizine had no effects on pup development at oral doses up to approximately 60 times the MRHD in adults. In mice treated during late gestation and the lactation period, cetirizine administered by the oral route to the dams had no effects on pup development at a dose that was approximately 25 times the MRHD in adults; however, lower pup weight gain during lactation was observed at a dose that was 95 times the MRHD in adults [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal data
In embryo-fetal development studies, pregnant rats received daily doses of levocetirizine up to 200 mg/kg/day from gestation days 6 to 15 and pregnant rabbits received daily doses of levocetirizine up to 120 mg/kg/day from gestation days 6 to 18. Levocetirizine produced no evidence of fetal harm in rats and rabbits at doses up to 390 and 470 times the MRHD, respectively (on a mg/m2 basis with maternal oral doses of 200 and 120 mg/kg/day in rats and rabbits, respectively).
No prenatal and postnatal development (PPND) studies in animals have been conducted with levocetirizine. In a PPND study conducted in mice, cetirizine was administered at oral doses up to 96 mg/kg/day from gestation day 15 through lactation day 21. Cetirizine lowered pup body weight gain during lactation at an oral dose in dams that was approximately 95 times the MRHD (on a mg/m2 basis with a maternal oral dose of 96 mg/kg/day); however, there were no effects on pup weight gain at an oral dose in dams that was approximately 25 times the MRHD (on a mg/m2 basis with a maternal oral dose of 24 mg/kg/day). In a PPND study conducted in rats, cetirizine was administered at oral doses up to 180 mg/kg/day from gestation day 17 to lactation day 22. Cetirizine did not have any adverse effects on rat dams or offspring development at doses up to approximately 60 times the MRHD (on a mg/m2 basis with a maternal oral dose of 30 mg/kg/day). Cetirizine caused excessive maternal toxicity at an oral dose in dams that was approximately 350 times the MRHD (on a mg/m2 basis with a maternal oral dose of 180 mg/kg/day).8.2 Lactation
Risk Summary
There are no data on the presence of levocetirizine in human milk, the effects on the breastfed infant, or the effects on milk production. However, cetirizine has been reported to be present in human breast milk. In mice and beagle dogs, studies indicated that cetirizine was excreted in milk [see Data]. When a drug is present in animal milk, it is likely the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for levocetirizine and any potential adverse effects on the breastfed child from levocetirizine or from the underlying maternal condition.
Data
Animal data
Cetirizine was detected in the milk of mice. No adverse developmental effects on pups were seen when cetirizine was administered orally to dams during lactation at a dose that was approximately 25 times the MRHD in adults [see Use in Specific Populations (8.1)]. Studies in beagle dogs indicated that approximately 3% of the dose of cetirizine was excreted in milk. The concentration of drug in animal milk does not necessarily predict the concentration of drug in human milk.
8.4 Pediatric Use
The recommended dose of levocetirizine dihydrochloride for the treatment of the uncomplicated skin manifestations of chronic idiopathic urticaria in patients 6 months to 17 years of age is based on extrapolation of efficacy from adults 18 years of age and older [see Clinical Studies (14)].
The recommended dose of levocetirizine dihydrochloride in patients 6 years to 11 years of age with chronic idiopathic urticaria is based on cross-study comparisons of the systemic exposure of levocetirizine dihydrochloride in adults and pediatric patients and on the safety profile of levocetirizine dihydrochloride in both adult and pediatric patients at doses equal to or higher than the recommended dose for patients 6 months to 11 years of age.
The safety of levocetirizine dihydrochloride 5 mg once daily was evaluated in 243 pediatric patients 6 to 12 years of age in two placebo-controlled clinical trials lasting 4 and 6 weeks. The safety of levocetirizine dihydrochloride 1.25 mg twice daily was evaluated in one 2-week clinical trial in 114 pediatric patients 1 to 5 years of age and the safety of levocetirizine dihydrochloride 1.25 mg once daily was evaluated in one 2-week clinical trial in 45 pediatric patients 6 to 11 months of age [see Adverse Reactions (6.1)].
The effectiveness of levocetirizine dihydrochloride 2.5 mg once daily (6 to 11 years of age) for the treatment of the symptoms of chronic idiopathic urticaria is supported by the extrapolation of demonstrated efficacy of levocetirizine dihydrochloride 5 mg once daily in patients 12 years of age and older based on the pharmacokinetic comparison between adults and children.
Cross-study comparisons indicate that administration of a 5 mg dose of levocetirizine dihydrochloride to 6 to 12 year old pediatric patients resulted in about 2-fold the systemic exposure (AUC) observed when 5 mg of levocetirizine dihydrochloride was administered to healthy adults. Therefore, in children 6 to 11 years of age the recommended dose of 2.5 mg once daily should not be exceeded. In a population pharmacokinetics study the administration of 1.25 mg once daily in children 6 months to 5 years of age resulted in systemic exposure comparable to 5 mg once daily in adults [see Dosage and Administration (2.2), Clinical Studies (14), and Clinical Pharmacology (12.3)].
8.5 Geriatric Use
Clinical studies of levocetirizine dihydrochloride for each approved indication did not include sufficient numbers of patients aged 65 years and older to determine whether they respond differently than younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy.
8.6 Renal Impairment
Levocetirizine dihydrochloride is known to be substantially excreted by the kidneys and the risk of adverse reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection and it may be useful to monitor renal function [see Dosage and Administration (2) and Clinical Pharmacology (12.3) ].
8.7 Hepatic Impairment
As levocetirizine is mainly excreted unchanged by the kidneys, it is unlikely that the clearance of levocetirizine is significantly decreased in patients with solely hepatic impairment [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Overdosage has been reported with levocetirizine dihydrochloride.
Symptoms of overdose may include drowsiness in adults. In children agitation and restlessness may initially occur, followed by drowsiness. There is no known specific antidote to levocetirizine dihydrochloride. Should overdose occur, symptomatic or supportive treatment is recommended. Levocetirizine dihydrochloride is not effectively removed by dialysis, and dialysis will be ineffective unless a dialyzable agent has been concomitantly ingested.
The acute maximal non-lethal oral dose of levocetirizine was 240 mg/kg in mice (approximately 190 times the maximum recommended daily oral dose in adults, approximately 230 times the maximum recommended daily oral dose in children 6 to 11 years of age, and approximately 180 times the maximum recommended daily oral dose in children 6 months to 5 years of age on a mg/m2 basis). In rats the maximal non-lethal oral dose was 240 mg/kg (approximately 390 times the maximum recommended daily oral dose in adults, approximately 460 times the maximum recommended daily oral dose in children 6 to 11 years of age, and approximately 370 times the maximum recommended daily oral dose in children 6 months to 5 years of age on a mg/m2 basis).
-
11 DESCRIPTION
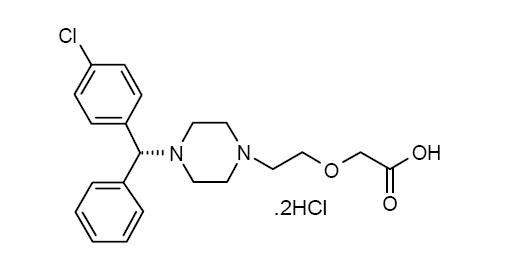
Levocetirizine dihydrochloride, USP the active component of levocetirizine dihydrochloride tablets USP is an orally active H1-receptor antagonist. The chemical name is (R)-[2-[4-[(4-chlorophenyl) phenylmethyl]-1-piperazinyl] ethoxy] acetic acid dihydrochloride. Levocetirizine dihydrochloride USP is the R enantiomer of cetirizine hydrochloride, a racemic compound with antihistaminic properties. The molecularl formula of levocetirizine dihydrochloride USP is C21H25ClN2O32HCl. The molecular weight is 461.82 and the chemical structure is shown below:
Levocetirizine dihydrochloride USP is a white, crystalline powder and is water soluble.
Levocetirizine dihydrochloride 5 mg tablets are formulated as immediate release, white to off-white, biconvex, film-coated, oval-shaped scored tablets for oral administration. The tablets are debossed on one side with ‘M 17’. Inactive ingredients are: microcrystalline cellulose, croscarmellose sodium, lactose monohydrate, colloidal anhydrous silica, and magnesium stearate. The film coating contains hypromellose, titanium dioxide, and macrogol 400.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Levocetirizine, the active enantiomer of cetirizine, is an antihistamine; its principal effects are mediated via selective inhibition of H1 receptors. The antihistaminic activity of levocetirizine has been documented in a variety of animal and human models. In vitro binding studies revealed that levocetirizine has an affinity for the human H1-receptor 2-fold higher than that of cetirizine (Ki = 3 nmol/L vs. 6 nmol/L, respectively). The clinical relevance of this finding is unknown.
12.2 Pharmacodynamics
Studies in adult healthy subjects showed that levocetirizine at doses of 2.5 mg and 5 mg inhibited the skin wheal and flare caused by the intradermal injection of histamine. In contrast, dextrocetirizine exhibited no clear change in the inhibition of the wheal and flare reaction. Levocetirizine at a dose of 5 mg inhibited the wheal and flare caused by intradermal injection of histamine in 14 pediatric subjects (aged 6 to 11 years) and the activity persisted for at least 24 hours. The clinical relevance of histamine wheal skin testing is unknown.
A QT/QTc study using a single dose of 30 mg of levocetirizine did not demonstrate an effect on the QTc interval. While a single dose of levocetirizine had no effect, the effects of levocetirizine may not be at steady state following single dose. The effect of levocetirizine on the QTc interval following multiple dose administration is unknown. Levocetirizine is not expected to have QT/QTc effects because of the results of QTc studies with cetirizine and the long post-marketing history of cetirizine without reports of QT prolongation.
12.3 Pharmacokinetics
Levocetirizine exhibited linear pharmacokinetics over the therapeutic dose range in adult healthy subjects.
Absorption
Levocetirizine is rapidly and extensively absorbed following oral administration. In adults, peak plasma concentrations are achieved 0.9 hour after administration of the oral tablet. The accumulation ratio following daily oral administration is 1.12 with steady state achieved after 2 days. Peak concentrations are typically 270 ng/mL and 308 ng/mL following a single and a repeated 5 mg once daily dose, respectively. Food had no effect on the extent of exposure (AUC) of the levocetirizine tablet, but Tmax was delayed by about 1.25 hours and Cmax was decreased by about 36% after administration with a high fat meal; therefore, levocetirizine can be administered with or without food.Distribution
The mean plasma protein binding of levocetirizine in vitro ranged from 91 to 92%, independent of concentration in the range of 90-5000 ng/mL, which includes the therapeutic plasma levels observed. Following oral dosing, the average apparent volume of distribution is approximately 0.4 L/kg, representative of distribution in total body water.Metabolism
The extent of metabolism of levocetirizine in humans is less than 14% of the dose and therefore differences resulting from genetic polymorphism or concomitant intake of hepatic drug metabolizing enzyme inhibitors are expected to be negligible. Metabolic pathways include aromatic oxidation, N- and O-dealkylation, and taurine conjugation. Dealkylation pathways are primarily mediated by CYP 3A4 while aromatic oxidation involves multiple and/or unidentified CYP isoforms.Elimination
The plasma half-life in adult healthy subjects was about 8 to 9 hours after administration of oral tablets and oral solution, and the mean oral total body clearance for levocetirizine was approximately 0.63 mL/kg/min. The major route of excretion of levocetirizine and its metabolites is via urine, accounting for a mean of 85.4% of the dose. Excretion via feces accounts for only 12.9% of the dose. Levocetirizine is excreted both by glomerular filtration and active tubular secretion. Renal clearance of levocetirizine correlates with that of creatinine clearance. In patients with renal impairment the clearance of levocetirizine is reduced [see Dosage and Administration (2.2)].Drug Interaction Studies
In vitro data on metabolite interaction indicate that levocetirizine is unlikely to produce, or be subject to metabolic interactions. Levocetirizine at concentrations well above Cmax level achieved within the therapeutic dose ranges is not an inhibitor of CYP isoenzymes 1A2, 2C9, 2C19, 2A1, 2D6, 2E1, and 3A4, and is not an inducer of UGT1A or CYP isoenzymes 1A2, 2C9 and 3A4.No formal in vivo drug interaction studies have been performed with levocetirizine. Studies have been performed with the racemic cetirizine [see Drug Interactions (7)].
Pediatric patients
Data from a pediatric pharmacokinetic study with oral administration of a single dose of 5 mg levocetirizine in 14 children age 6 to 11 years with body weight ranging between 20 and 40 kg show that Cmax and AUC values are about 2-fold greater than that reported in healthy adult subjects in a cross-study comparison. The mean Cmax was 450 ng/mL, occurring at a mean time of 1.2 hours, weight-normalized, total body clearance was 30% greater, and the elimination half-life 24% shorter in this pediatric population than in adults.Dedicated pharmacokinetic studies have not been conducted in pediatric patients younger than 6 years of age. A retrospective population pharmacokinetic analysis was conducted in 323 subjects (181 children 1 to 5 years of age, 18 children 6 to 11 years of age, and 124 adults 18 to 55 years of age) who received single or multiple doses of levocetirizine ranging from 1.25 mg to 30 mg. Data generated from this analysis indicated that administration of 1.25 mg once daily to children 6 months to 5 years of age results in plasma concentrations similar to those of adults receiving 5 mg once daily.
Geriatric patients
Limited pharmacokinetic data are available in elderly subjects. Following once daily repeat oral administration of 30 mg levocetirizine for 6 days in 9 elderly subjects (65–74 years of age), the total body clearance was approximately 33% lower compared to that in younger adults. The disposition of racemic cetirizine has been shown to be dependent on renal function rather than on age. This finding would also be applicable for levocetirizine, as levocetirizine and cetirizine are both predominantly excreted in urine. Therefore, the levocetirizine dihydrochloride dose should be adjusted in accordance with renal function in elderly patients [seeDosage and Administration (2)].Gender
Pharmacokinetic results for 77 patients (40 men, 37 women) were evaluated for potential effect of gender. The half-life was slightly shorter in women (7.08 ± 1.72 hr) than in men (8.62 ± 1.84 hr); however, the body weight-adjusted oral clearance in women (0.67 ± 0.16 mL/min/kg) appears to be comparable to that in men (0.59 ± 0.12 mL/min/kg). The same daily doses and dosing intervals are applicable for men and women with normal renal function.Race
The effect of race on levocetirizine has not been studied. As levocetirizine is primarily renally excreted, and there are no important racial differences in creatinine clearance, pharmacokinetic characteristics of levocetirizine are not expected to be different across races. No race-related differences in the kinetics of racemic cetirizine have been observed.Renal impairment
Levocetirizine exposure (AUC) exhibited 1.8-, 3.2-, 4.3-, and 5.7-fold increase in mild, moderate, severe, renal impaired, and end-stage renal disease patients, respectively, compared to healthy subjects. The corresponding increases of half-life estimates were 1.4-, 2.0-, 2.9-, and 4-fold, respectively.The total body clearance of levocetirizine after oral dosing was correlated to the creatinine clearance and was progressively reduced based on severity of renal impairment. Therefore, it is recommended to adjust the dose and dosing intervals of levocetirizine based on creatinine clearance in patients with mild, moderate, or severe renal impairment. In end-stage renal disease patients (CLCR < 10 mL/min) levocetirizine is contraindicated. The amount of levocetirizine removed during a standard 4-hour hemodialysis procedure was <10%.
The dosage of levocetirizine dihydrochloride should be reduced in patients with mild renal impairment. Both the dosage and frequency of administration should be reduced in patients with moderate or severe renal impairment [see Dosage and Administration (2.2)].
Hepatic impairment
Levocetirizine dihydrochloride has not been studied in patients with hepatic impairment. The non-renal clearance (indicative of hepatic contribution) was found to constitute about 28% of the total body clearance in healthy adult subjects after oral administration.As levocetirizine is mainly excreted unchanged by the kidney, it is unlikely that the clearance of levocetirizine is significantly decreased in patients with solely hepatic impairment [see Dosage and Administration (2)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis & Mutagenesis & Impairment Of Fertility
No carcinogenicity studies have been performed with levocetirizine. However, evaluation of cetirizine carcinogenicity studies is relevant for determination of the carcinogenic potential of levocetirizine. In a 2-year carcinogenicity study, in rats, cetirizine was not carcinogenic at dietary doses up to 20 mg/kg (approximately 40, 40, 25, and 10 times the MRHDs in adults, children 6 to 11 years of age, children 2-5 years, and children 6 months to 2 years of age, respectively, on a mg/m2 basis). In a 2-year carcinogenicity study in mice, cetirizine caused an increased incidence of benign hepatic tumors in males at a dietary dose of 16 mg/kg (approximately 15, 15, 9, and 5 times the MRHDs in adults, children 6 to 11 years of age, children 2-5 years, and children 6 months to 2 years of age, respectively, on a mg/m2 basis). No increased incidence of benign tumors was observed at a dietary dose of 4 mg/kg (approximately 4, 4, 2, and 1 times the MRHDs in adults, children 6 to 11 years of age, children 2-5 years, and children 6 months to 2 years of age, respectively on a mg/m2 basis). The clinical significance of these findings during long-term use of levocetirizine is not known.
Levocetirizine was not mutagenic in the Ames test, and not clastogenic in the human lymphocyte assay, the mouse lymphoma assay, and in vivo micronucleus test in mice.
Fertility and reproductive performance were unaffected in male and female mice and rats that received cetirizine at oral doses up to 64 and 200 mg/kg/day, respectively (approximately 60 and 390 times the MRHD in adults on a mg/m2 basis).
-
14 CLINICAL STUDIES
14.1 Perennial Allergic Rhinitis
Adults and Adolescents 12 Years of Age and Older
The efficacy of levocetirizine dihydrochloride tablet was evaluated in four randomized, placebo-controlled, double-blind clinical trials in adult and adolescent patients 12 years and older with symptoms of perennial allergic rhinitis. The four clinical trials include two dose-ranging trials of 4 weeks duration and two efficacy trials (one 6-week and one 6-month) in patients with perennial allergic rhinitis.These trials included a total of 1729 patients (752 males and 977 females) of whom 227 were adolescents 12 to 17 years of age. Efficacy was assessed using a total symptom score from patient recording of 4 symptoms (sneezing, rhinorrhea, nasal pruritus, and ocular pruritus) in three studies and 5 symptoms (sneezing, rhinorrhea, nasal pruritus, ocular pruritus, and nasal congestion) in one study. Patients recorded symptoms using a 0-3 categorical severity scale (0 = absent, 1 = mild, 2 = moderate, 3 = severe) once daily in the evening reflective of the 24 hour treatment period. The primary endpoint was the mean total symptom score averaged over the first week and over 4 weeks for perennial allergic rhinitis trials.
The two dose-ranging trials were conducted to evaluate the efficacy of levocetirizine dihydrochloride tablet 2.5, 5, and 10 mg once daily in the evening. These trials were 4 weeks in duration and included patients with perennial allergic rhinitis. In these trials, each of the three doses of levocetirizine dihydrochloride tablet demonstrated greater decrease in the reflective total symptom score than placebo and the difference was statistically significant for all three doses in the two studies. Results for one of these trials are shown in Table 4.
Table 4: Mean Reflective Total Symptom Score* in Allergic Rhinitis Dose-Ranging TrialsTreatment
N
Base-
line
On Treatment Adjusted Mean
Difference from Placebo
Esti-
mate
95% CI
p-value
Perennial Allergic Rhinitis Trial – Reflective total symptom score
Levocetirizine Dihydrochloride
2.5 mg
133
7.14
4.12
1.17
(0.71, 1.63)
<0.001
Levocetirizine Dihydrochloride
5 mg
127
7.18
4.07
1.22
(0.76, 1.69)
<0.001
Levocetirizine Dihydrochloride
10 mg
129
7.58
4.19
1.10
(0.64, 1.57)
<0.001
Placebo
128
7.22
5.29
*Total symptom score is the sum of individual symptoms of sneezing, rhinorrhea, nasal pruritus, and ocular pruritus as assessed by patients on a 0-3 categorical severity scale.
One clinical trial evaluated the efficacy of levocetirizine dihydrochloride 5 mg once daily in the evening compared to placebo in patients with perennial allergic rhinitis over a 6-week treatment period. Another trial conducted over a 6-month treatment period assessed efficacy at 4 weeks. Levocetirizine dihydrochloride 5 mg demonstrated a greater decrease from baseline in the reflective total symptom score than placebo and the difference from placebo was statistically significant. Results of the former are shown in Table 5.
Table 5: Mean Reflective Total Symptom Score* in Allergic Rhinitis TrialTreatment
N
Baseline
On Treatment Adjusted Mean
Difference from Placebo
Estimate
95% CI
p-value
Perennial Allergic Rhinitis Trial – Reflective total symptom score
Levocetirizine Dihydrochloride
5 mg
150
7.69
3.93
1.17
(0.70, 1.64)
<0.001
Placebo
142
7.44
5.10
*Total symptom score is the sum of individual symptoms of sneezing, rhinorrhea, nasal pruritus, and ocular pruritus as assessed by patients on a 0-3 categorical severity scale.
Onset of action was evaluated in two environmental exposure unit studies in allergic rhinitis patients with a single dose of levocetirizine dihydrochloride 2.5 or 5 mg. Levocetirizine dihydrochloride 5 mg was found to have an onset of action 1 hour after oral intake. Onset of action was also assessed from the daily recording of symptoms in the evening before dosing in the allergic rhinitis trials. In these trials, onset of effect was seen after 1 day of dosing.
Pediatric Patients Less than 12 Years of Age
There are no clinical efficacy trials with levocetirizine dihydrochloride 2.5 mg once daily in pediatric patients under 12 years of age, and no clinical efficacy trials with levocetirizine dihydrochloride 1.25 mg once daily in pediatric patients 6 months to 5 years of age. The clinical efficacy of levocetirizine dihydrochloride in pediatric patients under 12 years of age has been extrapolated from adult clinical efficacy trials based on pharmacokinetic comparisons [see Use in Specific Populations (8.4)].
14.2 Chronic Idiopathic Urticaria
Adult Patients 18 Years of Age and Older
The efficacy of levocetirizine dihydrochloride for the treatment of the uncomplicated skin manifestations of chronic idiopathic urticaria was evaluated in two multi-center, randomized, placebo-controlled, double-blind clinical trials of 4 weeks duration in adult patients 18 to 85 years of age with chronic idiopathic urticaria. The two trials included one 4-week dose-ranging trial and one 4-week single-dose level efficacy trial. These trials included 423 patients (139 males and 284 females). Most patients (>90%) were Caucasian and the mean age was 41. Of these patients, 146 received levocetirizine dihydrochloride 5 mg once daily in the evening. Efficacy was assessed based on patient recording of pruritus severity on a severity score of 0–3 (0 = none to 3 = severe). The primary efficacy endpoint was the mean reflective pruritus severity score over the first week and over the entire treatment period. Additional efficacy variables were the instantaneous pruritus severity score, the number and size of wheals, and duration of pruritus.The dose-ranging trial was conducted to evaluate the efficacy of levocetirizine dihydrochloride 2.5, 5, and 10 mg once daily in the evening. In this trial, each of the three doses of levocetirizine dihydrochloride demonstrated greater decrease in the reflective pruritus severity score than placebo and the difference was statistically significant for all three doses (see Table 6).
The single dose level trial evaluated the efficacy of levocetirizine dihydrochloride 5 mg once daily in the evening compared to placebo in patients with chronic idiopathic urticaria over a 4-week treatment period. Levocetirizine dihydrochloride 5 mg demonstrated a greater decrease from baseline in the reflective pruritus severity score than placebo and the difference from placebo was statistically significant.
Duration of pruritus, number and size of wheals, and instantaneous pruritus severity score also showed significant improvement over placebo. The significant improvement in the instantaneous pruritus severity score over placebo confirmed end of dosing interval efficacy (see Table 6).
Table 6: Mean Reflective Pruritus Severity Score in Chronic Idiopathic Urticaria TrialsTreatment
N
Baseline
On
Treatment
Adjusted
Mean
Difference
from
Placebo
Estimate
95%
CI
p-
value
Dose-Ranging Trial – Reflective pruritus severity score
Levocetirizine
Dihydrochloride
2.5 mg69
2.08
1.02
0.82
(0.58,
1.06)
<0.001
Levocetirizine
dihydrochloride
5 mg62
2.07
0.92
0.91
(0.66,
1.16)
<0.001
Levocetirizine
Dihydrochloride
10 mg55
2.04
0.73
1.11
(0.85,
1.37)
<0.001
Placebo60
2.25
1.84
Chronic Idiopathic Urticaria Trial – Reflective pruritus severity score
Levocetirizine
dihydrochloride
5 mg80
2.07
0.94
0.62
(0.38,
0.86)
<0.001
Placebo82
2.06
1.56
Pediatric Patients
There are no clinical efficacy trials in pediatric patients with chronic idiopathic urticaria [see Use in Specific Populations (8.4)]. -
16 HOW SUPPLIED/STORAGE AND HANDLING
Levocetirizine dihydrochloride tablets, USP are white to off white, film-coated, biconvex, oval-shaped, scored, debossed with 'M 17' on one side and contain 5 mg levocetirizine dihydrochloride USP.
This tablet is having functional scoring.
They are supplied in unit of use HDPE bottles.
30 Tablets (NDC: 68788-7189-3)
60 Tablets (NDC: 68788-7189-6)
90 Tablets (NDC: 68788-7189-9)
100 Tablets (NDC: 68788-7189-1)
Storage:
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Somnolence
Caution patients against engaging in hazardous occupations requiring complete mental alertness, and motor coordination such as operating machinery or driving a motor vehicle after ingestion of levocetirizine dihydrochloride.Concomitant Use of Alcohol and other Central Nervous System Depressants
Instruct patients to avoid concurrent use of levocetirizine dihydrochloride with alcohol or other central nervous system depressants because additional reduction in mental alertness may occur.Dosing of Levocetirizine Dihydrochloride
Do not exceed the recommended daily dose in adults and adolescents 12 years of age and older of 5 mg once daily in the evening. In children 6 to 11 years of age the recommended dose is 2.5 mg once daily in the evening. Advise patients to not ingest more than the recommended dose of levocetirizine dihydrochloride because of the increased risk of somnolence at higher doses.
Manufactured for :
Macleods Pharma USA, Inc.
Plainsboro, NJ 08536Manufactured by:
Macleods Pharmaceuticals Ltd.
Baddi, Himachal Pradesh, INDIARevised: 04/2019
PM02114009Repackaged By: Preferred Pharmaceuticals Inc.
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
LEVOCETIRIZINE DIHYDROCHLORIDE
levocetirizine dihydrochloride tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68788-7189(NDC:33342-200) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LEVOCETIRIZINE DIHYDROCHLORIDE (UNII: SOD6A38AGA) (LEVOCETIRIZINE - UNII:6U5EA9RT2O) LEVOCETIRIZINE DIHYDROCHLORIDE 5 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) Product Characteristics Color WHITE (white to off white) Score 2 pieces Shape OVAL (biconvex) Size 8mm Flavor Imprint Code ML17 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68788-7189-3 30 in 1 BOTTLE; Type 0: Not a Combination Product 07/17/2018 2 NDC: 68788-7189-6 60 in 1 BOTTLE; Type 0: Not a Combination Product 07/17/2018 3 NDC: 68788-7189-9 90 in 1 BOTTLE; Type 0: Not a Combination Product 07/17/2018 4 NDC: 68788-7189-1 100 in 1 BOTTLE; Type 0: Not a Combination Product 07/17/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA205564 07/17/2018 Labeler - Preferred Pharmaceuticals Inc. (791119022) Registrant - Preferred Pharmaceuticals Inc. (791119022) Establishment Name Address ID/FEI Business Operations Preferred Pharmaceuticals Inc. 791119022 REPACK(68788-7189)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.