SUNLENCA- lenacapavir sodium tablet, film coated SUNLENCA- lenacapavir sodium kit
Sunlenca by
Drug Labeling and Warnings
Sunlenca by is a Prescription medication manufactured, distributed, or labeled by Gilead Sciences. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SUNLENCA safely and effectively. See full prescribing information for SUNLENCA.
SUNLENCA® (lenacapavir) tablets, for oral use
SUNLENCA® (lenacapavir) injection, for subcutaneous use
Initial U.S. Approval: 2022RECENT MAJOR CHANGES
Dosage and Administration (2.4) 03/2026 INDICATIONS AND USAGE
SUNLENCA, a human immunodeficiency virus type 1 (HIV-1) capsid inhibitor, in combination with other antiretroviral(s), is indicated for the treatment of HIV-1 infection in heavily treatment-experienced adults with multidrug resistant HIV-1 whose current antiretroviral regimen is failing due to resistance, intolerance, or safety considerations. (1)
DOSAGE AND ADMINISTRATION
- Recommended dosage – Initiation with one of two options followed by once every 6 months maintenance injection dosing. Tablets may be taken without regard to food. (2.2)
Initiation Option 1 Day 1 927 mg by subcutaneous injection (2 × 1.5 mL injections)
600 mg orally (2 × 300 mg tablets)Day 2 600 mg orally (2 × 300 mg tablets) Initiation Option 2 Day 1 600 mg orally (2 × 300 mg tablets) Day 2 600 mg orally (2 × 300 mg tablets) Day 8 300 mg orally (1 × 300 mg tablet) Day 15 927 mg by subcutaneous injection (2 × 1.5 mL injections) Maintenance 927 mg by subcutaneous injection (2 × 1.5 mL injections) every 6 months (26 weeks) from the date of the last injection +/-2 weeks. - Planned missed injections: If scheduled injection is to be missed by more than 2 weeks, SUNLENCA tablets may be used for oral bridging for up to 6 months until injections resume. Recommended dosage is 300 mg orally once every 7 days. (2.3)
- Unplanned missed injections: If more than 28 weeks have elapsed since last injection and tablets have not been taken for oral bridging, restart initiation from Day 1 (using Option 1 or Option 2) if clinically appropriate. (2.3)
- SUNLENCA injection is for subcutaneous administration only. Two 1.5 mL injections are required for complete dose. (2.4)
DOSAGE FORMS AND STRENGTHS
Tablets: 300 mg
Injection: 463.5 mg/1.5 mL (309 mg/mL) in single-dose vials. (3)
CONTRAINDICATIONS
Concomitant administration of SUNLENCA is contraindicated with strong CYP3A inducers. (4)
WARNINGS AND PRECAUTIONS
- Immune reconstitution syndrome: May necessitate further evaluation and treatment. (5.1)
- Residual concentrations of lenacapavir may remain in systemic circulation for up to 12 months or longer. Counsel patients regarding the dosing schedule; non-adherence could lead to loss of virologic response and development of resistance. (5.2)
- May increase exposure and risk of adverse reactions to drugs primarily metabolized by CYP3A initiated within 9 months after the last subcutaneous dose of SUNLENCA. (5.2)
- If discontinued, initiate an alternative, fully suppressive antiretroviral regimen where possible no later than 28 weeks after the final injection of SUNLENCA. If virologic failure occurs, switch to an alternative regimen if possible. (5.2)
- Injection site reactions may occur, and nodules and indurations may be persistent. Improper administration (intradermal injection) has been associated with serious injection site reactions. (5.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence greater than or equal to 3%, all grades) are nausea and injection site reactions. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Adherence to Treatment Regimen
2.2 Recommended Dosage
2.3 Recommended Dosing Schedule for Missed Dose
2.4 Preparation and Administration of Subcutaneous Injection
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Immune Reconstitution Syndrome
5.2 Long-Acting Properties and Potential Associated Risks with SUNLENCA
5.3 Injection Site Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on SUNLENCA
7.2 Effect of SUNLENCA on Other Drugs
7.3 Established and Other Potentially Significant Drug Interactions
7.4 Drugs without Clinically Significant Interactions with SUNLENCA
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
SUNLENCA, in combination with other antiretroviral(s), is indicated for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in heavily treatment-experienced adults with multidrug resistant HIV-1 whose current antiretroviral regimen is failing due to resistance, intolerance, or safety considerations.
-
2 DOSAGE AND ADMINISTRATION
2.1 Adherence to Treatment Regimen
Prior to starting SUNLENCA, healthcare providers should carefully select patients who agree to the required every 6 month injection dosing schedule and counsel patients about the importance of adherence to scheduled SUNLENCA dosing visits and concomitant oral antiretroviral therapy to help maintain viral suppression and reduce the risk of viral rebound and potential development of resistance with missed doses [see Warnings and Precautions (5.2), Microbiology (12.4)].
2.2 Recommended Dosage
SUNLENCA can be initiated using one of the two recommended dosage regimens in Table 1 and Table 2 below. Maintenance dosing is administered by subcutaneous injection every 6 months regardless of the initiation regimen. Healthcare providers should determine the appropriate initiation regimen for the patient. SUNLENCA oral tablets may be taken with or without food [see Clinical Pharmacology (12.3)].
Table 1 Recommended Treatment Regimen for SUNLENCA Initiation and Maintenance, Option 1 Treatment Time - * From the date of the last injection.
Dosage of SUNLENCA: Initiation Day 1 927 mg by subcutaneous injection (2 × 1.5 mL injections)
600 mg orally (2 × 300 mg tablets)Day 2 600 mg orally (2 × 300 mg tablets) Dosage of SUNLENCA: Maintenance Every 6 months (26 weeks) * +/-2 weeks 927 mg by subcutaneous injection (2 × 1.5 mL injections) Table 2 Recommended Treatment Regimen for SUNLENCA Initiation and Maintenance, Option 2 Treatment Time - * From the date of the last injection.
Dosage of SUNLENCA: Initiation Day 1 600 mg orally (2 × 300 mg tablets) Day 2 600 mg orally (2 × 300 mg tablets) Day 8 300 mg orally (1 × 300 mg tablet) Day 15 927 mg by subcutaneous injection (2 × 1.5 mL injections) Dosage of SUNLENCA: Maintenance Every 6 months (26 weeks) * +/-2 weeks 927 mg by subcutaneous injection (2 × 1.5 mL injections) 2.3 Recommended Dosing Schedule for Missed Dose
Planned Missed Injections
During the maintenance period, if a patient plans to miss a scheduled 6-month injection visit by more than 2 weeks, SUNLENCA tablets may be taken for up to 6 months until injections resume. Refer to Table 3 below for the recommended dosage after planned missed injections.
Table 3 Recommended Dosage after Planned Missed Injections: Weekly Oral Maintenance Time since Last Injection Recommendation 26 to 28 weeks Maintenance oral dosage of 300 mg taken once every 7 days for up to 6 months.
Resume the maintenance injection dosage within 7 days after the last oral dose.Unplanned Missed Injections
Patients who miss a scheduled injection visit should be clinically reassessed, including consideration of lenacapavir resistance testing, to ensure resumption of therapy remains appropriate. During the maintenance period, if more than 28 weeks have elapsed since the last injection and SUNLENCA tablets have not been taken, see Table 4 below for the recommended dosage after unplanned missed injections. Adherence to the injection dosing schedule is strongly recommended [see Dosage and Administration (2.1) and Microbiology (12.4)].
Table 4 Recommended Dosage after Unplanned Missed Injections Time since Last Injection Recommendation More than 28 weeks Reinitiate with Option 1 (Table 1) or Option 2 (Table 2) and then continue with maintenance injection dosing. 2.4 Preparation and Administration of Subcutaneous Injection
SUNLENCA injection is only for subcutaneous administration into the abdomen by a healthcare provider. Do NOT administer intradermally due to risk of serious injection site reactions [see Warnings and Precautions (5.3)].
Use aseptic technique. Visually inspect the solution in the vials and prepared syringe for particulate matter and discoloration prior to administration. SUNLENCA injection is a yellow solution. Do not use SUNLENCA injection if the solution is discolored or if it contains particulate matter. Once the solution is withdrawn from the vials, the subcutaneous injections should be administered as soon as possible [see How Supplied/Storage and Handling (16)].
There are two available injection kits, which differ only in how SUNLENCA injection is prepared (the components and associated method for withdrawal of the solution from the vials) [see How Supplied/Storage and Handling (16)]. Refer to the figures below for the relevant injection kit.
The injection kit components are for single use only. Two 1.5 mL injections are required for a complete dose.
Vial access device injection kit
Figure 1 identifies the components for use in the administration steps for the vial access device injection kit, and the administration steps are provided in Figure 2. Use of a vial access device is required in this kit.
Figure 1 SUNLENCA Vial Access Device Injection Kit Components

Figure 2 SUNLENCA Injection Steps for Vial Access Device Injection Kit
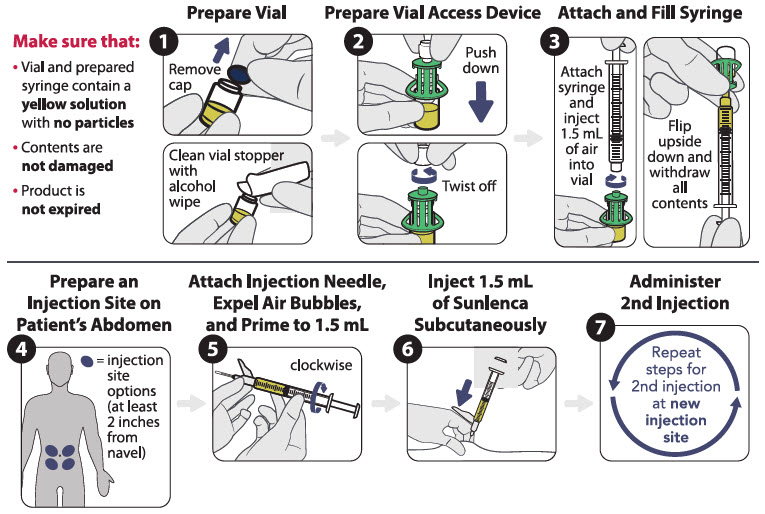
Withdrawal needle injection kit
Figure 3 identifies the components for use in the administration steps for the withdrawal needle injection kit, and the administration steps are provided in Figure 4. The 18-gauge needle is for withdrawal only in this kit.
Figure 3 SUNLENCA Withdrawal Needle Injection Kit Components
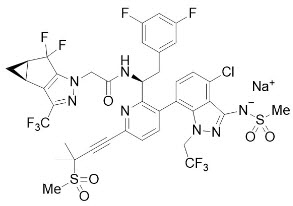
Figure 4 SUNLENCA Injection Steps for Withdrawal Needle Injection Kit

- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Concomitant administration of SUNLENCA with strong CYP3A inducers is contraindicated due to decreased lenacapavir plasma concentrations, which may result in the loss of therapeutic effect and development of resistance to SUNLENCA [see Drug Interactions (7.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections [such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia (PCP), or tuberculosis], which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, Guillain-Barré syndrome, and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.2 Long-Acting Properties and Potential Associated Risks with SUNLENCA
Residual concentrations of lenacapavir may remain in the systemic circulation of patients for prolonged periods (up to 12 months or longer after the last subcutaneous dose). It is important to counsel patients that maintenance dosing by injection is required every 6 months, because missed doses or non-adherence to injections could lead to loss of virologic response and development of resistance [see Dosage and Administration (2.1)].
Lenacapavir, a moderate CYP3A inhibitor, may increase the exposure to, and therefore potential risk of adverse reactions from, drugs primarily metabolized by CYP3A initiated within 9 months after the last subcutaneous dose of SUNLENCA [see Drug Interactions and Clinical Pharmacology (7.2, 12.3)].
If SUNLENCA is discontinued, to minimize the potential risk of developing viral resistance, it is essential to initiate an alternative, fully suppressive antiretroviral regimen where possible no later than 28 weeks after the final injection of SUNLENCA. If virologic failure occurs during treatment, switch the patient to an alternative regimen if possible [see Dosage and Administration (2.1)].
5.3 Injection Site Reactions
Administration of SUNLENCA may result in local injection site reactions (ISRs). If clinically significant ISRs occur, evaluate and institute appropriate therapy and follow-up.
Manifestations of ISRs may include swelling, pain, erythema, nodule, induration, pruritus, extravasation or mass. Nodules and indurations at the injection site may take longer to resolve than other ISRs. In clinical studies, after a median follow-up of 553 days, 30% of nodules and 13% of indurations (in 10% and 1% of participants, respectively) associated with the first injections of SUNLENCA had not fully resolved. Measurements and qualitative assessments of ISRs were not routinely reported. Where described, the majority of the injection site nodules and indurations were palpable but not visible, and had a maximum size of approximately 1 to 4 cm [see Adverse Reactions (6.1)].
The mechanism driving the persistence of injection site nodules and indurations in some patients is not fully understood, but based on available data, they may be related to the presence of the subcutaneous drug depot. In some patients who had a skin biopsy performed of an injection site nodule or induration, dermatopathology revealed foreign body inflammation or granulomatous response.
Improper administration (intradermal injection) has been associated with serious injection site reactions, including necrosis and ulcer [see Adverse Reactions (6)]. Ensure SUNLENCA is only administered subcutaneously in the abdomen [see Dosage and Administration (2.4)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.1)]
- Injection Site Reactions [see Warnings and Precautions (5.3)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The primary safety assessment of SUNLENCA was based on data from heavily treatment-experienced adult participants with HIV who received SUNLENCA in a Phase 2/3 trial (CAPELLA; N=72) through Week 52 (median duration on study of 71 weeks) [see Clinical Studies (14)], as well as supportive data in treatment-naïve adult participants with HIV who received SUNLENCA in a Phase 2 trial (CALIBRATE; N=157) through Week 54 (median duration of exposure of 66 weeks).
The most common adverse reactions (all Grades) reported in at least 3% of participants in CAPELLA were nausea and injection site reactions. The proportion of participants in CAPELLA who discontinued treatment with SUNLENCA due to adverse events, regardless of severity, was 1% (Grade 1 injection site nodule in 1 participant). Table 3 displays the frequency of adverse reactions (all Grades) greater than or equal to 3% in the SUNLENCA group.
Table 3 Adverse Reactions (All Grades) Reported in ≥ 3% * of Heavily Treatment Experienced Adults with HIV-1 Receiving SUNLENCA in CAPELLA (Week 52 Analysis) Adverse Reactions SUNLENCA + Background Regimen
(N=72)- * Frequencies of adverse reactions are based on all adverse events attributed to trial drug by the investigator, based on all participants (cohorts 1 and 2) in CAPELLA.
Injection Site Reactions 65% Nausea 4% The majority (96%) of all adverse reactions associated with SUNLENCA were mild or moderate in severity.
Injection-Associated Adverse Reactions
Local Injection Site Reactions (ISRs):
The most frequent adverse reactions were ISRs. Of the 72 participants in CAPELLA, 65% had experienced an ISR attributed to study drug through at least the Week 52 visit. Most participants had mild (Grade 1, 44%) or moderate (Grade 2, 17%) ISRs. Four percent of participants experienced a severe (Grade 3) ISR (erythema, pain, swelling) that resolved within 15 days. The ISRs reported in more than 1% of participants were swelling (36%), pain (31%), erythema (31%), nodule (25%), induration (15%), pruritus (6%), extravasation (3%) and mass (3%). ISRs reported in 1% of participants included discomfort, hematoma, edema, and ulcer.
Nodules and indurations at the injection site took longer to resolve than other ISRs. The median time to resolution of all ISRs, excluding nodules and indurations, was 5 days (range: 1 to 183). The median time to resolution of nodules and indurations associated with the first injections of SUNLENCA was 148 (range: 41 to 727) and 70 (range: 3 to 252) days, respectively. After a median follow up of 553 days, 30% of nodules and 13% of indurations (in 10% and 1% of participants, respectively) associated with the first injections of SUNLENCA had not fully resolved. Qualitative descriptions of injection site nodules and indurations were not routinely reported, but, where reported, the majority of injection site nodules and indurations were palpable but not visible. Measurements of injection site nodules and indurations were not routinely performed or standardized, but where measurements were reported, the maximum size for the majority of injection site nodules and indurations was approximately 1 to 4 cm [see Warnings and Precautions (5.3)].
Laboratory Abnormalities
The frequency of laboratory abnormalities (Grades 3 to 4) occurring in at least 2% of participants in CAPELLA are presented in Table 4. A causal association between SUNLENCA and these laboratory abnormalities has not been established.
Table 4 Selected Laboratory Abnormalities (Grades 3 to 4) Reported in ≥ 2% of Participants Receiving SUNLENCA in CAPELLA (Week 52 Analysis) Laboratory Parameter Abnormality SUNLENCA + Background Regimen
(N=72) *ALT= alanine aminotransferase; AST= aspartate aminotransferase; ULN = upper limit of normal - * Frequencies are based on treatment-emergent laboratory abnormalities in all participants (cohorts 1 and 2) in CAPELLA. Percentages were calculated based on the number of participants with post-baseline toxicity grades for each laboratory parameter (n=72 for all parameters except hyperglycemia fasting n=57).
- † Grade 3 only (no Grade 4 values reported).
Creatinine ( >1.8 × ULN or ≥1.5 × baseline) 13% Glycosuria (>2+) † 6% Hyperglycemia (fasting) (>250 mg/dL) 5% Proteinuria (>2+) † 4% ALT (≥5 × ULN) † 3% AST (≥5 × ULN) 3% Direct Bilirubin (>ULN) † 3% 6.2 Postmarketing Experience
In addition to adverse reactions reported from clinical trials, the following adverse reactions have been identified during postmarketing use. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
General disorders and administration site conditions
Injection site necrosis [see Warnings and Precautions (5.3)].
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on SUNLENCA
Lenacapavir is a substrate of P-gp, UGT1A1, and CYP3A.
Strong or Moderate CYP3A Inducers
Drugs that are strong or moderate inducers of CYP3A may significantly decrease plasma concentrations of lenacapavir [see Clinical Pharmacology (12.3)], which may result in loss of therapeutic effect of SUNLENCA and development of resistance. Concomitant administration of SUNLENCA with strong CYP3A inducers during SUNLENCA treatment is contraindicated [see Contraindications (4)]. Concomitant administration of SUNLENCA with moderate CYP3A inducers during SUNLENCA treatment is not recommended.
7.2 Effect of SUNLENCA on Other Drugs
Lenacapavir is a moderate inhibitor of CYP3A. Due to the long half-life of lenacapavir following subcutaneous administration, SUNLENCA may increase the exposure of drugs primarily metabolized by CYP3A [see Clinical Pharmacology (12.3)] initiated within 9 months after the last subcutaneous dose of SUNLENCA, which may increase the potential risk of adverse reactions. See the prescribing information of the sensitive CYP3A substrate for dosing recommendations with moderate inhibitors of CYP3A.
7.3 Established and Other Potentially Significant Drug Interactions
Table 5 provides a listing of clinically significant drug interactions with recommended prevention or management strategies, but is not all inclusive. The drug interactions described are based on studies conducted with SUNLENCA or are drug interactions that may occur with SUNLENCA [see Contraindications (4) and Clinical Pharmacology (12.3)].
Table 5 Drug Interactions with SUNLENCA Concomitant Drug Class:
Drug NameEffect on Concentration * Clinical Comment - * ↑ = Increase, ↓ = Decrease.
- † Drug-drug interaction study was conducted.
- ‡ The induction potency of St. John's wort may vary widely based on preparation.
Antiarrhythmics:
digoxin↑ digoxin Use with caution and monitor digoxin therapeutic concentration. Anticoagulants:
Direct Oral Anticoagulants (DOACs)
rivaroxaban
dabigatran
edoxaban↑ DOAC Refer to the DOAC prescribing information for concomitant administration with moderate CYP3A inhibitors and/or P-gp inhibitors. Anticonvulsants:
carbamazepine
oxcarbazepine
phenobarbital
phenytoin↓ lenacapavir Concomitant administration of carbamazepine, oxcarbazepine, phenobarbital, or phenytoin may result in loss of therapeutic effect and development of resistance.
Concomitant administration of SUNLENCA with carbamazepine or phenytoin is contraindicated.
Concomitant administration of SUNLENCA with oxcarbazepine or phenobarbital is not recommended. Consider use of alternative anticonvulsants.Antiretroviral Agents:
atazanavir/cobicistat †
atazanavir/ritonavir↑ lenacapavir (atazanavir/cobicistat, atazanavir/ritonavir) Concomitant administration of efavirenz, nevirapine, or tipranavir/ritonavir may result in loss of therapeutic effect and development of resistance. efavirenz †
nevirapine
tipranavir/ritonavir↓ lenacapavir (efavirenz, nevirapine, tipranavir/ritonavir) Concomitant administration with atazanavir/cobicistat, atazanavir/ritonavir, efavirenz, nevirapine, or tipranavir/ritonavir is not recommended. Antimycobacterials:
rifabutin
rifampin †
rifapentine↓ lenacapavir Concomitant administration of rifabutin, rifampin and rifapentine may result in loss of therapeutic effect and development of resistance.
Concomitant administration of SUNLENCA with rifampin is contraindicated [see Contraindications (4)].
Concomitant administration of SUNLENCA with rifabutin or rifapentine is not recommended.Corticosteroids (systemic):
cortisone/hydrocortisone
dexamethasone↑ corticosteroids (systemic) Concomitant administration with systemic corticosteroids whose exposures are significantly increased by CYP3A inhibitors can increase the risk for Cushing's syndrome and adrenal suppression. Initiate with the lowest starting dose and titrate carefully while monitoring for safety. ↓ lenacapavir (dexamethasone) Concomitant administration of systemic dexamethasone may result in loss of therapeutic effect of lenacapavir and development of resistance. Alternative corticosteroids to dexamethasone should be considered, particularly for long-term use. Ergot derivatives:
dihydroergotamine
ergotamine
methylergonovine↑ dihydroergotamine
↑ ergotamine
↑ methylergonovineConcomitant administration of SUNLENCA with dihydroergotamine, ergotamine or methylergonovine is not recommended. Herbal Products:
St. John's wort ‡
(Hypericum perforatum)↓ lenacapavir Concomitant administration of St. John's wort may result in loss of therapeutic effect and development of resistance.
Concomitant administration of SUNLENCA with St. John's wort is contraindicated.HMG-CoA Reductase Inhibitors:
lovastatin
simvastatin↑ lovastatin
↑ simvastatinInitiate lovastatin and simvastatin with the lowest starting dose and titrate carefully while monitoring for safety (e.g., myopathy). Narcotic analgesics metabolized by CYP3A:
e.g., fentanyl, oxycodone↑ fentanyl
↑ oxycodoneCareful monitoring of therapeutic effects and adverse reactions associated with CYP3A-metabolized narcotic analgesics (including potentially fatal respiratory depression) is recommended with co-administration. tramadol ↑ tramadol A decrease in dose may be needed for tramadol with concomitant use. Narcotic analgesic for treatment of opioid dependence:
buprenorphine, methadonebuprenorphine: effects unknown
methadone: effects unknownInitiation of buprenorphine or methadone in patients taking SUNLENCA: Carefully titrate the dose of buprenorphine or methadone to the desired effect; use the lowest feasible initial or maintenance dose.
Initiation of SUNLENCA in patients taking buprenorphine or methadone: A dose adjustment for buprenorphine or methadone may be needed. Monitor clinical signs and symptoms.Opioid Antagonist:
naloxegol↑ naloxegol Avoid use with SUNLENCA; if unavoidable, decrease the dosage of naloxegol and monitor for adverse reactions. Phosphodiesterase-5 (PDE-5) Inhibitors:
sildenafil
tadalafil
vardenafil↑ PDE-5 inhibitors Use of PDE-5 inhibitors for pulmonary arterial hypertension (PAH):
Concomitant administration of SUNLENCA with tadalafil for the treatment of PAH is not recommended.
Use of PDE-5 inhibitors for erectile dysfunction (ED):
Refer to the prescribing information of PDE-5 inhibitors for dose recommendations.Sedatives/Hypnotics:
midazolam (oral) †
triazolam↑ midazolam (oral)
↑ triazolamUse with caution when midazolam or triazolam is concomitantly administered with SUNLENCA 7.4 Drugs without Clinically Significant Interactions with SUNLENCA
Based on drug interaction studies conducted with SUNLENCA, no clinically significant drug interactions have been observed with: darunavir/cobicistat, cobicistat, famotidine, pitavastatin, rosuvastatin, tenofovir alafenamide, and voriconazole.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in individuals exposed to SUNLENCA during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
There are insufficient human data on the use of SUNLENCA during pregnancy to inform a drug-associated risk of birth defects and miscarriage. In animal reproduction studies, no adverse developmental effects were observed when lenacapavir was administered to rats and rabbits at exposures (AUC) ≥16 times the exposure in humans at the recommended human dose (RHD) of SUNLENCA (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. The background rate of major birth defects in a U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) is 2.7%. The rate of miscarriage is not reported in the APR. The estimated background rate of miscarriage in clinically recognized pregnancies in the U.S. general population is 15 to 20%.
Data
Animal Data
Lenacapavir was administered intravenously to pregnant rabbits (up to 20 mg/kg/day on gestation days (GD) 7 to 19), orally to rats (up to 300 mg/kg/day on GD 6 to 17), and subcutaneously to rats (up to 300 mg/kg on GD 6). No significant toxicological effects on embryo-fetal (rats and rabbits) or pre/postnatal (rats) development were observed at exposures (AUC) approximately 16 times (rats) and 39 times (rabbits) the exposure in humans at the RHD of SUNLENCA.
8.2 Lactation
Risk Summary
It is not known whether SUNLENCA is present in human breast milk, affects human milk production, or has effects on the breastfed infant. After administration to pregnant rats, lenacapavir was detected in the plasma of nursing rat pups, without effects on these nursing pups (see Data).
Potential risks of breastfeeding include: (1) HIV-1 transmission (in infants without HIV-1), (2) developing viral resistance (in infants with HIV-1), and (3) adverse reactions in a breastfed infant similar to those seen in adults.
8.4 Pediatric Use
The safety and effectiveness of SUNLENCA have not been established in pediatric patients.
8.5 Geriatric Use
Clinical studies of SUNLENCA did not include sufficient numbers of participants aged 65 and over to determine whether they respond differently from younger patients.
8.6 Renal Impairment
No dosage adjustment of SUNLENCA is recommended in patients with mild, moderate or severe renal impairment (estimated creatinine clearance greater than or equal to 15 mL per minute). SUNLENCA has not been studied in patients with ESRD (estimated creatinine clearance less than 15 mL per minute) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosage adjustment of SUNLENCA is recommended in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment. SUNLENCA has not been studied in patients with severe hepatic impairment (Child-Pugh Class C) [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
No data are available on overdose of SUNLENCA in patients. If overdose occurs, monitor the patient for evidence of toxicity. Treatment of overdose with SUNLENCA consists of general supportive measures including monitoring of vital signs as well as observation of the clinical status of the patient. As lenacapavir is highly bound to plasma proteins, it is unlikely to be significantly removed by dialysis.
-
11 DESCRIPTION
SUNLENCA tablets and SUNLENCA injection contain lenacapavir sodium, a capsid inhibitor.
The chemical name of lenacapavir sodium is: Sodium (4-chloro-7-(2-((S)-1-(2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamido)-2-(3,5-difluorophenyl)ethyl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-3-yl)-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)(methylsulfonyl)amide.
Lenacapavir sodium has a molecular formula of C39H31ClF10N7NaO5S2, a molecular weight of 990.3, and the following structural formula:

Lenacapavir sodium is a light yellow to yellow solid and is practically insoluble in water.
SUNLENCA tablets are for oral administration. Each film-coated tablet contains 300 mg of lenacapavir (present as 306.8 mg lenacapavir sodium) and the following inactive ingredients: copovidone, croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, and poloxamer 407. The tablets are film-coated with a coating material containing iron oxide black, iron oxide red, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
SUNLENCA injection is for subcutaneous administration. Each single-dose vial contains 463.5 mg/1.5 mL (309 mg/mL) of lenacapavir (present as 473.1 mg/1.5 mL of lenacapavir sodium) as a sterile, preservative-free, clear, yellow solution and the following inactive ingredients: 896.3 mg of polyethylene glycol 300 (as solvent) and water for injection. The apparent pH range of the injection is 9.0–10.2.
The vial stoppers are not made with natural rubber latex.
-
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
Exposure-Response
In CAPELLA, oral loading doses (600 mg on Day 1 and Day 2, 300 mg on Day 8) followed by subcutaneous doses (927 mg every 6 months starting on Day 15) of SUNLENCA in heavily treatment-experienced participants with multiclass resistant HIV-1, efficacy outcomes (change in plasma HIV-1 RNA from Day 1 to Day 14, and percentage of participants with HIV-1 RNA less than 50 copies/mL at Week 26) were similar across the range of observed lenacapavir exposures.
12.3 Pharmacokinetics
The pharmacokinetic (PK) properties of lenacapavir are provided in Table 6 and Table 7. The estimated lenacapavir exposures are comparable between the two recommended dosing regimens.
Table 6 Pharmacokinetic Properties of Lenacapavir Oral Subcutaneous - * Values reflect absolute bioavailability following subcutaneous administration of the 927 mg dose.
- † Values reflect administration of lenacapavir with or without food.
- ‡ Due to slow release from the site of subcutaneous administration, the absorption profile of subcutaneously administered lenacapavir is complex.
- § Values refer to geometric mean ratio [low-fat meal/fasting] in PK parameters and (90% confidence interval). Low fat meal is approximately 400 kcal, 25% fat.
- ¶ Values refer to geometric mean ratio [high-fat meal/fasting] in PK parameters and (90% confidence interval). High fat meal is approximately 1000 kcal, 50% fat.
- # Values reflect the blood-to-plasma ratio of lenacapavir following a single dose intravenous administration of [14C] lenacapavir through 336 hours postdose.
- Þ Dosing in mass balance studies: single dose intravenous administration of [14C] lenacapair to participants without HIV-1.
- ß Metabolized via oxidation, N-dealkylation, hydrogenation, amide hydrolysis, glucuronidation, hexose conjugation, pentose conjugation, and glutathione conjugation; primarily via CYP3A and UGT1A1 and no single circulating metabolite accounted for >10% of plasma drug-related exposure.
Absorption % Absolute bioavailability 6 to 10 100 * Tmax † 4 hours 77 to 84 days ‡ Effect of Food Effect of low-fat meal (relative to fasting) § AUCinf ratio 98.6 (58.2,167.2) - Cmax ratio 115.8 (55.4, 242.1) - Effect of high-fat meal (relative to fasting) ¶ AUCinf ratio 115.2 (72.0, 184.5) - Cmax ratio 145.2 (77.9, 270.5) - Distribution Apparent volume of distribution (Vd/F, L) 19240 9500 to 11700 % bound to human plasma proteins >98.5 Blood-to-plasma ratio 0.5 to 0.7 # Elimination t1/2 10 to 12 days 8 to 12 weeks Clearance (mean apparent clearance, L/h) 55 4.2 % of dose of unchanged drug in plasma Þ 69 Metabolism Metabolic pathway(s) CYP3A (minor)
UGT1A1 (minor)Excretion Major routes of elimination Excretion of unchanged drug into feces ß % of dose excreted in urine Þ <1 % of dose excreted in feces (% unchanged) ß 76 (33) Table 7 Lenacapavir Exposures Following Oral and Subcutaneous Administration of SUNLENCA in Heavily Treatment Experienced Participants with HIV Parameter Mean (%CV) Recommended Dosing Regimen, Option 1 * Recommended Dosing Regimen, Option 2 * Day 1: 600 mg (oral) + 927 mg (SC)
Day 2: 600 mg (oral)Days 1 and 2: 600 mg (oral),
Day 8: 300 mg (oral),
Day 15: 927 mg (SC)Day 1 to end of Month 6 Days 1 to 15 Day 15 to end of Month 6 CV = coefficient of variation; NA = not applicable; SC = subcutaneous - * Predicted exposures utilizing population PK analysis.
Cmax
(ng/mL)101.4 (53.1) 88.0 (72.4) 86.5 (51.7) AUCtau
(h∙ng/mL)242266 (46.0) 19496 (72.6) 239163 (47.2) Ctrough
(ng/mL)32.5 (57.2) 46.9 (72.3) 32.5 (57.5) Lenacapavir exposures after subcutaneous administration were similar between heavily treatment experienced participants with HIV-1 and participants without HIV-1 based on population pharmacokinetics analysis. Lenacapavir exposures (AUCtau, Cmax and Ctrough) after oral administration were 28% to 43% higher in participants with HIV-1 who were heavily treatment experienced, compared to participants without HIV-1 based on population PK analysis. These differences were not considered clinically relevant.
Specific Populations
There were no clinically significant differences in the pharmacokinetics of lenacapavir based on age (18 to 78 years), sex, ethnicity (hispanic or non-hispanic), race (white, black, asian or other), body weight (41.4 to 164 kg), severe renal impairment (creatinine clearance of 15 to less than 30 mL per minute, estimated by Cockroft-Gault method), or moderate hepatic impairment (Child-Pugh Class B). The effect of end-stage renal disease (including dialysis), or severe hepatic impairment (Child-Pugh Class C), on the pharmacokinetics of lenacapavir is unknown. As lenacapavir is greater than 98.5% protein bound, dialysis is not expected to alter exposures of lenacapavir [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical Studies
Clinical drug-drug interaction study indicated that lenacapavir is a substrate of CYP3A, P-gp, and UGT1A1. Table 8 summarizes the pharmacokinetic effects of other drugs on lenacapavir.
Lenacapavir is a moderate inhibitor of CYP3A. Lenacapavir is an inhibitor of P-gp and BCRP but does not inhibit OATP. Table 9 summarizes the pharmacokinetic effects of lenacapavir on other drugs.
Table 8 Effect of Other Drugs on Lenacapavir *,† Coadministered Drug Dose of Coadministered Drug (mg) Mean Ratio of Lenacapavir Pharmacokinetic Parameters (90% CI); No effect = 1.00 Cmax AUC - * Single dose of lenacapavir 300 mg administered orally.
- † All interaction studies conducted in participants without HIV-1.
- ‡ 400 mg loading dose twice daily for a day, followed by 200 mg maintenance dose twice daily.
Cobicistat (fed)
(Inhibitor of CYP3A [strong] and P-gp)150 once daily 2.10
(1.62, 2.72)2.28
(1.75, 2.96)Darunavir / cobicistat (fed)
(Inhibitor of CYP3A [strong] and inhibitor and inducer of P-gp)800/150 once daily 2.30
(1.79, 2.95)1.94
(1.50, 2.52)Voriconazole (fasted)
(Inhibitor of CYP3A [strong])400 twice daily, 200 twice daily ‡ 1.09
(0.81, 1.47)1.41
(1.10, 1.81)Atazanavir / cobicistat (fed)
(Inhibitor of CYP3A [strong] and UGT1A1 and P-gp)300/150 once daily 6.60
(4.99, 8.73)4.21
(3.19, 5.57)Rifampin (fasted)
(Inducer of CYP3A [strong] and P-gp and UGT)600 once daily 0.45
(0.34, 0.60)0.16
(0.12, 0.20)Efavirenz (fasted)
(Inducer of CYP3A [moderate] and P-gp)600 once daily 0.64
(0.45, 0.92)0.44
(0.32, 0.59)Famotidine (2 hours before, fasted) 40 once daily 1.01
(0.75, 1.34)1.28
(1.00, 1.63)Table 9 Effect of Lenacapavir on Other Drugs *,† Coadministered Drug Dose of Coadministered Drug (mg) Mean Ratio of Coadministered Drug Pharmacokinetic Parameters (90% CI) ‡; No effect = 1.00 Cmax AUC - * All interaction studies conducted in participants without HIV-1.
- † Following 600 mg twice daily for 2 days, single 600 mg doses of lenacapavir were administered with each coadministered drug, resulting in lenacapavir exposures similar to or higher than those at the recommended dosage regimen.
- ‡ All No Effect Boundaries are 70% to 143%.
- § Tenofovir alafenamide is converted to tenofovir in vivo.
- ¶ Major active metabolite of midazolam.
Tenofovir alafenamide (fed)
(substrate of P-gp)25 single dose 1.24
(0.98, 1.58)1.32
(1.09, 1.59)Tenofovir §
(substrate of P-gp)1.23
(1.05, 1.44)1.47
(1.27, 1.71)Pitavastatin (simultaneous administration, fed)
(substrate of OATP)2 single dose 1.00
(0.84, 1.19)1.11
(1.00, 1.25)Pitavastatin (3 days after lenacapavir, fed)
(substrate of OATP)2 single dose 0.85
(0.69, 1.05)0.96
(0.87, 1.07)Rosuvastatin (fed)
(substrate of BCRP and OATP)5 single dose 1.57
(1.38, 1.80)1.31
(1.19, 1.43)Midazolam (simultaneous administration, fed)
(substrate of CYP3A)2.5 single dose 1.94
(1.81, 2.08)3.59
(3.30, 3.91)1-hydroxymidazolam ¶
(substrate of CYP3A)0.54
(0.50, 0.59)0.76
(0.72, 0.80)Midazolam (1 day after lenacapavir, fed)
(substrate of CYP3A)2.5 single dose 2.16
(2.02, 2.30)4.08
(3.77, 4.41)1-hydroxymidazolam ¶
(substrate of CYP3A)0.52
(0.48, 0.57)0.84
(0.80, 0.88)In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Lenacapavir is not a substrate, inducer, or inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2D6. Lenacapavir is not an inducer of CYP3A4.
12.4 Microbiology
Mechanism of Action
Lenacapavir is a multistage, selective inhibitor of HIV-1 capsid function that directly binds to the interface between capsid protein (p24) subunits in hexamers. Surface plasmon resonance sensorgrams showed dose-dependent and saturable binding of lenacapavir to cross-linked wild-type capsid hexamer with an equilibrium binding constant (KD) of 1.4 nM. Lenacapavir inhibits HIV-1 replication by interfering with multiple essential steps of the viral lifecycle, including capsid-mediated nuclear uptake of HIV-1 proviral DNA (by blocking nuclear import proteins binding to capsid), virus assembly and release (by interfering with Gag/Gag-Pol functioning, reducing production of capsid protein subunits), and capsid core formation (by disrupting the rate of capsid subunit association, leading to malformed capsids).
Antiviral Activity in Cell Culture
Lenacapavir has antiviral activity that is specific to human immunodeficiency virus (HIV-1 and HIV-2). The antiviral activity of lenacapavir against laboratory and clinical isolates of HIV-1 was assessed in T-lymphoblastoid cell lines, PBMCs, primary monocyte/macrophage cells, and CD4+ T-lymphocytes with EC50 values ranging from 30 to 190 pM. Lenacapavir displayed antiviral activity in cell culture against all HIV-1 groups (M, N, O), including subtypes A, A1, AE, AG, B, BF, C, D, E, F, G with EC50 values ranging from 20 and 160 pM. The median EC50 value for subtype B isolates (n=8) was 40 pM. Lenacapavir was 15- to 25-fold less active against HIV-2 isolates relative to HIV-1.
In a study of lenacapavir in combination with representatives from the major classes of anti-retroviral agents (INSTIs, NNRTIs, NRTIs, and PIs), no antagonism of antiviral activity was observed.
Resistance
In Cell Culture
HIV-1 variants with reduced susceptibility to lenacapavir have been selected in cell culture. Resistance selections with lenacapavir identified 7 substitutions in capsid: L56I, M66I, Q67H, K70N, N74D/S, and T107N singly or in dual combination that conferred 4- to >3,226-fold reduced phenotypic susceptibility to lenacapavir relative to wild-type (WT) virus. The M66I substitution alone or in combination conferred >3,226-fold decreased susceptibility to lenacapavir in a single-cycle infectivity assay; substitutions Q67H and T107N, conferred 4- to 6.3-fold decreased susceptibility; K70N, N74D and Q67H/N74S conferred 22- to 32-fold decreased susceptibility; and L56I conferred 239-fold decreased susceptibility.
In Clinical Trials
In CAPELLA, 39% (28/72) of heavily treatment-experienced participants met the criteria for resistance analyses through Week 156 (HIV-1 RNA ≥ 50 copies/mL at confirmed virologic failure [suboptimal virologic response at Week 4, virologic rebound, or viremia at last visit]) and were analyzed for lenacapavir resistance-associated substitution emergence. Lenacapavir resistance-associated capsid substitutions were found in 57% (n=16) of participants with confirmed virologic failure who had post-baseline capsid genotypic resistance data (n=28).
In the 16 participants with LEN resistance emergence through Week 156, 6 participants had emergence of the M66I substitution along with other substitutions (Q67H/K/N, K70N/R/S, N74D, A105T, T107A/C/S), 4 participants had emergence of Q67H + K70R with or without substitutions A105T and/or T107N or S, 2 participants had emergence of T107A or S alone, 1 participant had emergence of Q67K + K70H, 1 participant had emergence of K70N + N74K + T107T/N, 1 participant had emergence of Q67H alone, and 1 participant had emergence of N74D alone.
Phenotypic analyses of the confirmed virologic failure isolates with emergent lenacapavir resistance-associated substitutions showed 4.5- to >1428-fold decreases in lenacapavir susceptibility when compared to WT.
Among the 16 participants with virologic failure who developed lenacapavir resistance-associated substitutions in capsid, 6 received SUNLENCA in combination with a background regimen with no fully active antiretrovirals based on the baseline genotypic and/or phenotypic resistance. Therefore, given the risk of developing resistance in situations of functional monotherapy, careful consideration should be given to having fully active drugs in addition to SUNLENCA in the treatment regimen.
Nine participants with virologic failure had emergent resistance substitutions to components of the optimized background regimen (OBR) including NNRTI substitutions K103N/S and V106M, NRTI substitutions L74I, M184I/V, K65K/R, and K65R and S68N from mixtures at baseline, and emergent protease resistance substitutions M46I, I47I/V, I54I/L, V82V/A, I84I/V, L90L/M.
Oral Maintenance for Missed Injections
Of the 57 participants who received oral maintenance, 1 participant in Cohort 2 met the criteria for resistance analysis. The participant had emergence of lenacapavir resistance substitution N74D in capsid with no phenotypic data due to assay failure.
Lenacapavir Resistance with Suboptimal Adherence to Other Antiretroviral Drugs in the Regimen
The development of lenacapavir-associated capsid resistance substitutions can occur with suboptimal adherence to the other oral antivirals in the regimen. Seven of the 16 participants who had virologic rebound and developed lenacapavir resistance substitutions achieved resuppresion to HIV-1 RNA < 50 copies/mL on background oral regimens that included darunavir and/or dolutegravir. With lenacapavir resistance substitutions and decreased susceptibility to lenacapavir, it is unclear how much antiviral activity lenacapavir is contributing to the resuppression of HIV-1 RNA in these participants. Three of the 16 participants had a subsequent viral load rebound associated with additional emergent lenacapavir resistance substitutions and higher fold changes to lenacapavir. This supports that there was reduced lenacapavir antiviral activity from the previous emergent lenacapavir resistance substitutions, which contributed to further lenacapavir resistance emergence. Additionally, in these 3 participants, there was no evidence of emergent resistance to the oral OBR, signifying suboptimal adherence to the oral OBR and functional lenacapavir monotherapy.
Thus, adherence to other antiretroviral drugs in the regimen should be optimized while the patient is receiving SUNLENCA. During SUNLENCA treatment, if suboptimal adherence to the oral antiretroviral regimen occurs with concurrent HIV-1 RNA viral load increases (i.e., virologic failure), lenacapavir resistance should be assessed, and if detected, consideration should be given to discontinuing SUNLENCA treatment [see Dosage and Administration (2.1), Warnings and Precautions (5.2)].
Cross-Resistance
The antiviral activity in cell culture of lenacapavir was determined against a broad spectrum of HIV-1 site-directed mutants and patient-derived HIV-1 isolates with resistance to the four main classes of anti-retroviral agents (INSTI, NNRTI, NRTI, and PI; n=58), as well as to viruses resistant to the gp120-directed attachment inhibitor fostemsavir, the CD4+-directed post-attachment inhibitor ibalizumab, the CCR5 co-receptor antagonist maraviroc, and the gp41 fusion inhibitor enfuvirtide (n=42). These data indicated that lenacapavir remained fully active against all variants tested, thereby demonstrating a non-overlapping resistance profile. In addition, the antiviral activity of lenacapavir in patient isolates was unaffected by the presence of naturally occurring Gag polymorphisms and substitutions at protease cleavage sites.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Lenacapavir was not carcinogenic in a 6-month rasH2 transgenic mouse study in males or females at doses of up to 300 mg/kg/dose once every 13 weeks.
A 104-week carcinogenicity study was conducted in male and female rats at lenacapavir doses of 0, 102, 309, or 927 mg/kg by subcutaneous injection once every 13-weeks. A treatment-related increase in the incidence of malignant sarcoma at the injection site was observed in males and a treatment-related increase in combined benign fibroma and malignant fibrosarcoma at the injection site was observed in females, at the highest dose (927 mg/kg). This dose in rats resulted in an exposure approximately 34-times the human exposure at the RHD, based on AUC. These tumors are considered to be a secondary response to chronic tissue irritation and granulomatous inflammation, due to the depot effect of lenacapavir following subcutaneous injection. The clinical relevance of these findings are unknown.
-
14 CLINICAL STUDIES
The efficacy and safety of SUNLENCA in heavily treatment-experienced participants with multidrug resistant HIV-1 is based on 52-week data from CAPELLA, a randomized, placebo-controlled, double-blind, multicenter trial (NCT 04150068).
CAPELLA was conducted in 72 heavily treatment-experienced participants with multiclass resistant HIV-1. Participants were required to have a viral load ≥ 400 copies/mL, documented resistance to at least two antiretroviral medications from each of at least 3 of the 4 classes of antiretroviral medications (NRTI, NNRTI, PI and INSTI), and ≤ 2 fully active antiretroviral medications from the 4 classes of antiretroviral medications remaining at baseline due to resistance, intolerability, drug access, contraindication, or other safety concerns.
The trial was composed of two cohorts. Participants were enrolled into the randomized cohort (cohort 1, N=36) if they had a < 0.5 log10 HIV-1 RNA decline compared to the screening visit. Participants were enrolled into the non-randomized cohort (cohort 2, N=36) if they had a ≥ 0.5 log10 HIV-1 RNA decline compared to the screening visit or after cohort 1 reached its planned sample size.
In the 14-day functional monotherapy period, participants in cohort 1 were randomized in a 2:1 ratio in a blinded fashion to receive either SUNLENCA or placebo, while continuing their failing regimen. This period was to establish the virologic activity of SUNLENCA. After the functional monotherapy period, participants who had received SUNLENCA continued on SUNLENCA along with an OBR; participants who had received placebo during this period initiated SUNLENCA along with an OBR.
Participants in cohort 1 had a mean age of 52 years (range: 24 to 71), 72% were male, 46% were White, 46% were Black, and 9% were Asian. 29% percent of participants identified as Hispanic/Latino. The mean baseline plasma HIV-1 RNA was 4.3 log10 copies/mL (range: 2.3 to 5.4). 19% of participants had baseline viral loads greater than 100,000 copies/mL. The mean baseline CD4+ cell count was 161 cells/mm3 (range: 6 to 827). 75% of participants had CD4+ cell counts below 200 cells/mm3. The mean number of years since participants first started HIV treatment was 24 years (range: 7 to 33); the mean number of antiretroviral agents in failing regimens at baseline was 4 (range: 1 to 7). The percentage of participants in the randomized cohort with known resistance to at least 2 agents from the NRTI, NNRTI, PI and INSTI classes was 97%, 94%, 78% and 75%, respectively. In cohort 1, 53% of participants had no fully active agents, 31% had 1 fully active agent, and 17% had 2 or more fully active agents within their initial failing regimen, including 6% of participants were who were receiving fostemsavir, which was an investigational agent at the start of the CAPELLA trial.
Participants in cohort 2 initiated SUNLENCA and an OBR on Day 1.
Participants in cohort 2 had a mean age of 48 years (range: 23 to 78), 78% were male, 36% were White, 31% were Black, 33% were Asian, and 14% of participants identified as Hispanic/Latino. The mean baseline plasma HIV-1 RNA was 4.1 log10 copies/mL (range: 1.3 to 5.7). 19% of participants had baseline viral loads greater than 100,000 copies/mL. The mean baseline CD4+ cell count was 258 cells/mm3 (range: 3 to 1296). 53% of participants had CD4+ cell counts below 200 cells/mm3. The mean number of years since participants first started HIV treatment was 19 years (range: 3 to 35); the mean number of antiretroviral agents in failing regimens at baseline was 4 (range: 2 to 7). The percentage of participants in the non-randomized cohort with known resistance to at least 2 agents from the NRTI, NNRTI, PI and INSTI classes was 100%, 100%, 83% and 64%, respectively. In cohort 2, 31% of participants had no fully active agents, 42% had 1 fully active agent, and 28% had 2 or more fully active agents within their initial failing regimen, including 6% of participants who were receiving fostemsavir, which was an investigational agent at the start of the CAPELLA trial.
The primary efficacy endpoint was the proportion of participants in cohort 1 achieving ≥ 0.5 log10 copies/mL reduction from baseline in HIV-1 RNA at the end of the functional monotherapy period. The results of the primary endpoint analysis are shown in Table 10.
Table 10 Proportion of Participants Achieving a ≥ 0.5 log10 Decrease in Viral Load at the End of the Functional Monotherapy Period in the CAPELLA Trial (Cohort 1) SUNLENCA
(N=24)Placebo
(N=12)- * p < 0.0001
Proportion of Participants Achieving a ≥ 0.5 log10 Decrease in Viral Load 87.5% 16.7% Treatment Difference (95% CI) 70.8% (34.9% to 90.0%) * The results at Weeks 26 and 52 are provided in Table 11 and Table 12.
Table 11 Virologic Outcomes (HIV-1 RNA < 50 copies/mL) at Weeks 26 * and 52 † with SUNLENCA plus OBR in the CAPELLA Trial (Cohort 1) SUNLENCA plus OBR
(N=36)Week 26 Week 52 OBR = optimized background regimen - * Week 26 window was between Days 184 and 232 (inclusive).
- † Week 52 window was between Days 324 and 414 (inclusive).
- ‡ Includes participants who had ≥ 50 copies/mL in the Week 26 or 52 window; participants who discontinued early due to lack or loss of efficacy; participants who discontinued for reasons other than an adverse event (AE), death or lack or loss of efficacy and at the time of discontinuation had a viral value of ≥ 50 copies/mL.
- § Includes participants who discontinued due to AE or death at any time point from Day 1 through the time window if this resulted in no virologic data on treatment during the specified window.
- ¶ Includes participants who discontinued for reasons other than an AE, death or lack or loss of efficacy, e.g., withdrew consent, loss to follow-up, etc.
HIV-1 RNA < 50 copies/mL 81% 83% HIV-1 RNA ≥ 50 copies/mL‡ 19% 14% No virologic data in Week 26 or 52 Window 0 3% Discontinued Study Drug Due to AE or Death § 0 0 Discontinued Study Drug Due to Other Reasons ¶ and Last Available HIV-1 RNA < 50 copies/mL 0 3% Missing Data During Window but on Study Drug 0 0 Table 12 Virologic Outcomes (HIV-1 RNA < 50 copies/mL) by Baseline Covariates at Weeks 26 * and 52 † with SUNLENCA plus OBR in the CAPELLA trial (Cohort 1) SUNLENCA plus OBR
(N=36)Week 26 Week 52 ARV = antiretroviral; DRV=darunavir; DTG=dolutegravir; INSTI = integrase strand-transfer inhibitor; OBR = optimized background regimen; - * Week 26 window was between Days 184 and 232 (inclusive).
- † Week 52 window was between Days 324 and 414 (inclusive).
Age (Years) < 50 100% (9/9) 89% (8/9) ≥ 50 74% (20/27) 81% (22/27) Gender Male 77% (20/26) 77% (20/26) Female 90% (9/10) 100% (10/10) Race Black 81% (13/16) 75% (12/16) Non-Black 84% (16/19) 89% (17/19) Baseline plasma viral load (copies/mL) ≤ 100,000 86% (25/29) 86% (25/29) > 100,000 57% (4/7) 71% (5/7) Baseline CD4+ (cells/mm3) < 200 78% (21/27) 78% (21/27) ≥ 200 89% (8/9) 100% (9/9) Baseline INSTI resistance profile With INSTI resistance 85% (23/27) 81% (22/27) Without INSTI resistance 63% (5/8) 88% (7/8) Number of fully active ARV agents in the OBR 0 67% (4/6) 67% (4/6) 1 86% (12/14) 79% (11/14) ≥ 2 81% (13/16) 94% (15/16) Use of DTG and/or DRV in the OBR With DTG and DRV 83% (10/12) 83% (10/12) With DTG, without DRV 83% (5/6) 83% (5/6) Without DTG, with DRV 78% (7/9) 89% (8/9) Without DTG or DRV 78% (7/9) 78% (7/9) In cohort 1, at Weeks 26 and 52, the mean change from baseline in CD4+ cell count was 81 cells/mm3 (range: -101 to 522) and 82 cells/mm3 (range: -194 to 467), respectively.
In cohort 2, at Weeks 26 and 52, 81% (29/36) and 72% (26/36) of participants achieved HIV-1 RNA < 50 copies/mL, respectively, and the mean change from baseline in CD4+ cell count was 97 cells/mm3 (range: -103 to 459) and 113 cells/mm3 (range: -124 to 405), respectively.
Oral bridging
In CAPELLA across cohorts 1 and 2, 79% of participants (57/72) received SUNLENCA 300 mg once every 7 days as oral bridging. A total of 13, 29, and 15 participants started oral bridging following Weeks 26, 52, and 78 injections, respectively. The median (Q1, Q3) duration of oral bridging was 19 weeks (11, 22), and 12% (7/57) received oral bridging for at least 28 weeks.
In a post-hoc analysis, rates of virologic suppression and change from baseline in CD4+ cell counts in the subset of patients who received oral bridging were consistent before and during the oral bridging period.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
SUNLENCA tablets, 300 mg are beige, capsule-shaped, and film-coated with "GSI" debossed on one side and "62L" on the other side. SUNLENCA tablets are available in a bottle and blister packs, packaged as follows:
Bottle
- SUNLENCA bottle contains 4 tablets (NDC: 61958-3001-3). The bottle also contains a silica gel desiccant and polyester coil, and is closed with a child-resistant closure. Do not remove the desiccant packet.
Keep bottle tightly closed.
Blister Packs
- SUNLENCA 4-Tablets™ blister pack contains 4 tablets (NDC: 61958-3001-1)
- SUNLENCA 5-Tablets™ blister pack contains 5 tablets (NDC: 61958-3001-2)
Within the blister packs, tablets are packaged in a clear blister film sealed to a foil lidding material. The blister card is fitted between two paperboard cards, and packaged with silica gel desiccant in a sealed child-resistant flexible laminated pouch.
SUNLENCA injection is packaged in one of two different injection kits containing the following:
Vial access device injection kit (NDC: 61958-3002-1):
- 2 single-dose clear glass vials, each containing sufficient volume to allow withdrawal of 463.5 mg/1.5 mL (309 mg/mL) of lenacapavir. The injection solution is sterile, preservative-free, clear, and yellow with no visible particles. Vials are sealed with a stopper and aluminium overseal with flip-off cap.
- 2 vial access devices, 2 disposable syringes, and 2 injection safety needles for subcutaneous injection (22-gauge, ½ inch).
Withdrawal needle injection kit (NDC: 61958-3005-1):
- 2 single-dose clear glass vials, each containing sufficient volume to allow withdrawal of 463.5 mg/1.5 mL (309 mg/mL) of lenacapavir. The injection solution is sterile, preservative-free, clear, and yellow with no visible particles. Vials are sealed with a stopper and aluminium overseal with flip-off cap.
- 2 disposable syringes, 2 withdrawal needles (18-gauge, 1.5 inch), and 2 injection safety needles for subcutaneous injection (22-gauge, ½ inch).
The vial stoppers are not made with natural rubber latex.
Store at 20 °C – 25 °C (68 °F – 77 °F), excursions permitted to 15 °C – 30 °C (59 °F – 86 °F).
Keep the vials in the original carton until just prior to preparation of the injections in order to protect from light.
Once the solution has been drawn into the syringes, the injections should be administered as soon as possible.
Discard any unused portion of the solution.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Drug Interactions
SUNLENCA may interact with certain drugs; therefore, advise patients to report to their healthcare provider the use of any other prescription or non-prescription medication or herbal products, including St. John's wort, during treatment with SUNLENCA [see Contraindications (4) and Drug Interactions (7)].
If SUNLENCA is discontinued, advise patients that SUNLENCA may remain in the body and affect certain other drugs for up to 9 months after receiving their last injection [see Drug Interactions (7.2, 7.3)].
Immune Reconstitution Syndrome
Advise patients to inform their healthcare provider immediately of any symptoms of infection, as in some patients with advanced HIV (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started [see Warnings and Precautions (5.1)].
Adherence to SUNLENCA
Counsel patients about the importance of continued medication adherence and scheduled visits to maintain viral suppression and to reduce risk of loss of virologic response and development of resistance. Advise patients to contact their healthcare provider immediately if they stop taking SUNLENCA or any other drug in their antiretroviral regimen [see Dosage and Administration (2.1) and Warnings and Precautions (5.2)].
Missed Dose
Inform patients that SUNLENCA can remain in the body for up to 12 months or longer after receiving their last injection. Advise patients to contact their healthcare provider if they miss or plan to miss a scheduled injection visit and that oral SUNLENCA therapy may be used for up to 6 months to replace missed injections. Advise patients that oral dosing should be used on an interim basis only and that the maintenance injection dosage should be resumed at the earliest possible opportunity [see Dosage and Administration (2.3) and Warnings and Precautions (5.2)].
Injection Site Reactions
Inform patients that injection site reactions (ISRs), such as swelling, pain, erythema, nodule, induration, pruritus, extravasation or mass, may occur. Nodules and indurations at the injection site may take longer to resolve than other ISRs and may be persistent. Instruct patients when to contact their healthcare provider about these reactions [see Warnings and Precautions (5.3)].
Pregnancy Registry
Inform patients that there is an antiretroviral pregnancy registry to monitor fetal outcomes of pregnant individuals exposed to SUNLENCA [see Use in Specific Populations (8.1)].
Lactation
Inform individuals with HIV-1 that the potential risks of breastfeeding include: (1) HIV-1 transmission (in infants without HIV-1), (2) developing viral resistance (in infants with HIV), and (3) adverse reactions in a breastfed infant similar to those seen in adults [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION SUNLENCA® (sun-LEN-kuh)
(lenacapavir)
tabletsSUNLENCA® (sun-LEN-kuh)
(lenacapavir)
injectionThis Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 03/2026 What is SUNLENCA?
SUNLENCA is a prescription medicine that is used with other human immunodeficiency virus-1 (HIV-1) medicines to treat HIV-1 in adults:- who have received HIV-1 medicines in the past, and
- who have HIV-1 virus that is resistant to many HIV-1 medicines, and
- whose current HIV-1 medicines are failing. Your HIV-1 medicines may be failing because the HIV-1 medicines are not working or no longer work, you are not able to tolerate the side effects, or there are safety reasons why you cannot take them.
It is not known if SUNLENCA is safe and effective in children.Do not receive or take SUNLENCA if you also take certain other medicines called strong CYP3A inducers. Ask your healthcare provider if you are not sure. Before receiving or taking SUNLENCA, tell your healthcare provider about all your medical conditions, including if you: - are pregnant or plan to become pregnant. It is not known if SUNLENCA can harm your unborn baby. Tell your healthcare provider if you become pregnant during treatment with SUNLENCA.
Pregnancy Registry: There is a pregnancy registry for women who take SUNLENCA during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk with your healthcare provider about how you can take part in this registry. - are breastfeeding or plan to breastfeed. It is not known whether SUNLENCA will pass to your baby in your breast milk. Talk with your healthcare provider about the following risks to your baby from breastfeeding during treatment with SUNLENCA:
- The HIV-1 virus may pass to your baby if your baby does not have HIV-1.
- The HIV-1 virus may become harder to treat if your baby has HIV-1.
- Your baby may get side effects from SUNLENCA.
Some medicines may interact with SUNLENCA. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.- You can ask your healthcare provider or pharmacist for a list of medicines that interact with SUNLENCA.
- Do not start a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take SUNLENCA with other medicines.
- SUNLENCA may affect certain other medicines for up to 9 months after your last injection.
How should I receive and take SUNLENCA? - Your SUNLENCA treatment will consist of injections and tablets.
- SUNLENCA injections will be given to you by your healthcare provider under the skin (subcutaneous injection) in your stomach-area (abdomen).
- Take SUNLENCA tablets by mouth, with or without food.
- There are two options (Option 1 and Option 2) to start treatment with SUNLENCA. Your healthcare provider will decide which starting option is for you.
- If Option 1 is chosen:
- On Day 1, you will receive 2 SUNLENCA injections and take 2 SUNLENCA tablets.
- On Day 2, you will take 2 SUNLENCA tablets.
- If Option 2 is chosen:
- On Day 1 and Day 2, you will take 2 SUNLENCA tablets each day.
- On Day 8, you will take 1 SUNLENCA tablet.
- On Day 15, you will receive 2 SUNLENCA injections.
- If Option 1 is chosen:
- After completing Option 1 or Option 2, you will receive 2 SUNLENCA injections every 6 months (26 weeks) from the date of your last injection.
- Stay under the care of a healthcare provider during treatment with SUNLENCA. It is important that you attend your planned appointments to receive your injections of SUNLENCA.
- If you miss or plan to miss your scheduled every 6 months injection of SUNLENCA, call your healthcare provider right away to discuss your treatment options.
- If you plan to miss a scheduled SUNLENCA injection, there is the option to temporarily take SUNLENCA tablets. You will take 1 SUNLENCA tablet by mouth 1 time every 7 days, until your injections resume.
- It is important to continue SUNLENCA treatment as your healthcare provider tells you. Missing SUNLENCA treatment may cause the HIV-1 virus to change (mutate) and become harder to treat (resistant).
- Tell your healthcare provider right away if you stop treatment with SUNLENCA or stop treatment with any other HIV-1 medicines. If you stop treatment with SUNLENCA you will need other medicines to treat your HIV-1. If you do not take other HIV-1 medicines, the amount of virus in your blood may increase and the virus may become harder to treat. Call your healthcare provider right away to discuss your treatment options.
- If you take too many SUNLENCA tablets, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of SUNLENCA?
SUNLENCA may cause serious side effects, including:- Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider right away if you start having any new symptoms after starting your HIV-1 medicine.
- Injection site reactions may happen when you receive SUNLENCA injections and may include swelling, pain, redness, skin hardening, small mass or lump, and itching. Hardened skin or lumps at the injection site usually can be felt but not seen. If you develop hardened skin or a lump, it may take longer than other reactions at the injection site to go away, and the injection site may not completely heal on its own. Tell your healthcare provider if you have any injection site reactions.
These are not all of the possible side effects of SUNLENCA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store SUNLENCA tablets? - Store SUNLENCA tablets at room temperature between 68 °F to 77 °F (20 °C to 25 °C).
- SUNLENCA bottle contains a desiccant packet to help keep your medicine dry (protect it from moisture). Keep the desiccant packet in the bottle. Do not eat the desiccant packet.
- Keep SUNLENCA tablets in their original bottle or blister pack.
- Keep the bottle tightly closed.
General information about the safe and effective use of SUNLENCA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use SUNLENCA for a condition for which it was not prescribed. Do not give SUNLENCA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about SUNLENCA that is written for health professionals.What are the ingredients in SUNLENCA?
Active ingredient: lenacapavir
Inactive ingredients:
SUNLENCA tablets: copovidone, croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, and poloxamer 407. The tablets are film-coated with a coating material containing iron oxide black, iron oxide red, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
SUNLENCA injection: polyethylene glycol 300 and water for injection.
Manufactured and distributed by: Gilead Sciences, Inc. Foster City, CA 94404
SUNLENCA is a trademark of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners.
© 2026 Gilead Sciences, Inc. All rights reserved. 215973-GS-005/IFU-002/IFU-WD-001
For more information, call 1-800-445-3235 or go to www.SUNLENCA.com. -
PRINCIPAL DISPLAY PANEL - 300 mg Tablet Blister Pack Pouch Carton
NDC: 61958-3001-1
Sunlenca®
(lenacapavir) tablets
300 mg per tablet1 pouch containing 4 tablets
Each tablet contains: 300 mg of lenacapavir
(present as 306.8 mg lenacapavir sodium).Store at 20 °C - 25 °C (68 °F - 77 °F).
Dispense only in original container.
Recommended Dosage: See prescribing information.
KEEP OUT OF THE REACH OF CHILDREN
GILEAD
Manufactured for:
Gilead Sciences, Inc.
Foster City, CA 94404
Made in Canada© 2022 Gilead Sciences, Inc.

-
PRINCIPAL DISPLAY PANEL - 5 Tablet Blister Pack Pouch Carton
NDC: 61958-3001-2
Sunlenca®
(lenacapavir) tablets
300 mg per tablet1 pouch containing 5 tablets
Each tablet contains: 300 mg of lenacapavir
(present as 306.8 mg lenacapavir sodium).Store at 20 °C - 25 °C (68 °F - 77 °F).
Dispense only in original container.
Recommended Dosage: See prescribing information.
KEEP OUT OF THE REACH OF CHILDREN
Talk to your healthcare provider before taking
Sunlenca tablets.Your healthcare provider will tell you when to take
Sunlenca tablets.GILEAD
Manufactured for:
Gilead Sciences, Inc.
Foster City, CA 94404
Made in Canada© 2022 Gilead Sciences, Inc.

-
PRINCIPAL DISPLAY PANEL - 4 Tablet Bottle Label
NDC: 61958-3001-3
4 tabletsSunlenca®
(lenacapavir) tablets
300 mg per tabletTalk to your healthcare provider
before taking Sunlenca tablets.Your healthcare provider will tell you
when to take Sunlenca tablets.
-
PRINCIPAL DISPLAY PANEL - Kit Carton
Rx only
NDC: 61958-3002-1Sunlenca®
(lenacapavir) injection463.5 mg/1.5 mL (309 mg/mL)
For Subcutaneous Injection
Contents
- 2 x 1.5 mL lenacapavir single-dose vials
- 2 vial access devices
- 2 syringes
- 2 injection needles (22 gauge, 1/2 inch)
- Prescribing Information
- Instructions for Use
- Patient Information
Both 463.5 mg/1.5 mL (2 single-dose vials) must be
administered to receive the 927 mg dose.For Healthcare Professional administration only.
GILEAD

-
PRINCIPAL DISPLAY PANEL - Kit Carton - 3005
Rx only
NDC: 61958-3005-1Sunlenca®
(lenacapavir) injection463.5 mg/1.5 mL (309 mg/mL)
For Subcutaneous Injection
Contents
- 2 x 1.5 mL lenacapavir single-dose vials
- 2 withdrawal needles (18 gauge, 1½ inch)
- 2 syringes
- 2 injection needles (22 gauge, ½ inch)
- Prescribing Information
- Instructions for Use
- Patient Information
Both 463.5 mg/1.5 mL (2 single-dose vials) must be
administered to receive the 927 mg dose.For Healthcare Professional administration only.
GILEAD

-
INGREDIENTS AND APPEARANCE
SUNLENCA
lenacapavir sodium tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-3001 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LENACAPAVIR SODIUM (UNII: BDT58WJ9WE) (LENACAPAVIR - UNII:A9A0O6FB4H) LENACAPAVIR 300 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) POLOXAMER 407 (UNII: TUF2IVW3M2) METHYL ALCOHOL (UNII: Y4S76JWI15) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color BROWN (Beige) Score no score Shape OVAL Size 21mm Flavor Imprint Code GSI;62L Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-3001-1 1 in 1 CARTON 12/22/2022 1 1 in 1 POUCH 1 4 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 61958-3001-2 1 in 1 CARTON 12/22/2022 2 1 in 1 POUCH 2 5 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 61958-3001-3 4 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 11/13/2024 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215974 12/22/2022 SUNLENCA
lenacapavir sodium kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-3002 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-3002-1 1 in 1 CARTON 12/22/2022 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 2 VIAL 3 mL Part 1 of 1 SUNLENCA
lenacapavir sodium injectionProduct Information Item Code (Source) NDC: 61958-3004 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LENACAPAVIR SODIUM (UNII: BDT58WJ9WE) (LENACAPAVIR - UNII:A9A0O6FB4H) LENACAPAVIR 463.5 mg in 1.5 mL Inactive Ingredients Ingredient Name Strength POLYETHYLENE GLYCOL 300 (UNII: 5655G9Y8AQ) WATER (UNII: 059QF0KO0R) Product Characteristics Color YELLOW Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-3004-1 1.5 mL in 1 VIAL; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215973 03/18/2025 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215973 12/22/2022 SUNLENCA
lenacapavir sodium kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-3005 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-3005-1 1 in 1 CARTON 03/18/2025 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 2 VIAL 3 mL Part 1 of 1 SUNLENCA
lenacapavir sodium injectionProduct Information Item Code (Source) NDC: 61958-3004 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LENACAPAVIR SODIUM (UNII: BDT58WJ9WE) (LENACAPAVIR - UNII:A9A0O6FB4H) LENACAPAVIR 463.5 mg in 1.5 mL Inactive Ingredients Ingredient Name Strength POLYETHYLENE GLYCOL 300 (UNII: 5655G9Y8AQ) WATER (UNII: 059QF0KO0R) Product Characteristics Color YELLOW Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-3004-1 1.5 mL in 1 VIAL; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215973 03/18/2025 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215973 03/18/2025 Labeler - Gilead Sciences, Inc. (185049848)
Trademark Results [Sunlenca]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 SUNLENCA 88823358 not registered Live/Pending |
Gilead Sciences Ireland UC 2020-03-05 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.