MRESVIA- respiratory syncytial virus vaccine suspension
mRESVIA by
Drug Labeling and Warnings
mRESVIA by is a Other medication manufactured, distributed, or labeled by Moderna US, INC., ModernaTX, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use MRESVIA safely and effectively. See full prescribing information for MRESVIA.
MRESVIATM (Respiratory Syncytial Virus Vaccine) injectable suspension, for intramuscular use
Initial U.S. Approval: 2024INDICATIONS AND USAGE
MRESVIATM is a vaccine indicated for active immunization for the prevention of lower respiratory tract disease (LRTD) caused by respiratory syncytial virus (RSV) in
individuals 60 years of age and older. (1)
individuals 18 through 59 years of age who are at increased risk for LRTD caused by RSV. (1)
DOSAGE AND ADMINISTRATION
For intramuscular use.
Administer a single dose (0.5 mL). (2.1)
DOSAGE FORMS AND STRENGTHS
Injectable suspension. A single dose is 0.5 mL. (3)
CONTRAINDICATIONS
History of severe allergic reaction (e.g., anaphylaxis) to any component of MRESVIA. (4)
ADVERSE REACTIONS
The most commonly reported (≥10%) adverse reactions in individuals 60 years and older were injection-site pain (55.9%), fatigue (30.8%), headache (26.7%), myalgia (25.6%), arthralgia (21.7%), axillary (underarm) swelling or tenderness (15.2%), and chills (11.6%). (6.1)
The most commonly reported (≥10%) adverse reactions in individuals 18 through 59 years who are at increased risk for LRTD caused by RSV were injection site pain (73.9%), fatigue (36.9%), headache (33.3%), myalgia (28.9%), arthralgia (22.7%), chills (19.9%), axillary (underarm) swelling or tenderness (17.1%), and nausea/vomiting (10.8%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact ModernaTX, Inc. at 1-866-663-3762 or VAERS at 1-800-822-7967 or www.vaers.hhs.gov.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dose and Schedule
2.2 Preparation for Administration
2.3 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Management of Acute Allergic Reactions
5.2 Syncope
5.3 Altered Immunocompetence
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Efficacy in Participants 60 Years of Age and Older
14.2 Immunogenicity in Participants 18 through 59 Years of Age at Increased Risk for LRTD Caused by RSV
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.2 Preparation for Administration
MRESVIA is supplied as a pre-filled syringe that contains a frozen suspension that must be thawed prior to administration.
Thaw each syringe before use, either in the refrigerator or at room temperature, following the instructions in Table 1.
Table 1: Thawing Conditions and Times Configuration Thaw in Refrigerator Thaw at Room Temperature Carton of 1 pre-filled syringe
Thaw between 2°C to 8°C (36°F to 46°F) for 100 minutes.
Thaw between 15°C to 25°C (59°F to 77°F) for 40 minutes.
Carton of 2 pre‑filled syringes
Thaw between 2°C to 8°C (36°F to 46°F) for 100 minutes.
Thaw between 15°C to 25°C (59°F to 77°F) for 40 minutes.
Carton of 10 pre-filled syringes
Thaw between 2°C to 8°C (36°F to 46°F) for 160 minutes.
Thaw between 15°C to 25°C (59°F to 77°F) for 80 minutes.
One syringe (removed from carton)
Thaw between 2°C to 8°C (36°F to 46°F) for 100 minutes.
Thaw between 15°C to 25°C (59°F to 77°F) for 40 minutes.
- After thawing, do not refreeze.
- Syringes should not be returned to the refrigerator after standing at room temperature.
- Pre-filled syringes may be stored at 8°C to 25°C (46°F to 77°F) for a total of 24 hours after removal from refrigerated conditions. Discard the thawed pre-filled syringe if not used within this time.
- Do not shake.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- MRESVIA is a white to off-white suspension that may contain visible white or translucent product-related particulates. Do not administer if the vaccine is discolored or contains other particulate matter.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Do not administer MRESVIA to individuals with a history of a severe allergic reaction (e.g., anaphylaxis) to any component of MRESVIA [see Description (11)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Management of Acute Allergic Reactions
Appropriate medical treatment must be immediately available to manage potential anaphylactic reactions following administration of MRESVIA.
-
6 ADVERSE REACTIONS
In a clinical trial conducted in participants 60 years of age and older, the most commonly reported (≥10%) adverse reactions were injection‑site pain (55.9%), fatigue (30.8%), headache (26.7%), myalgia (25.6%), arthralgia (21.7%), axillary (underarm) swelling or tenderness (15.2%), and chills (11.6%).
In a clinical trial conducted in participants 18 through 59 years of age at increased risk for LRTD caused by RSV, the most commonly reported (≥10%) adverse reactions were injection site pain (73.9%), fatigue (36.9%), headache (33.3%), myalgia (28.9%), arthralgia (22.7%), chills (19.9%), axillary (underarm) swelling or tenderness (17.1%), and nausea/vomiting (10.8%).
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice.
Individuals 60 Years of Age and Older
The safety of MRESVIA was evaluated in Study 1 (NCT05127434), a placebo-controlled, observer blinded clinical study conducted in 22 countries that includes participants from North America/ Europe, Central/Latin America, Africa and Asian/Pacific regions. A total of 18,231 participants received MRESVIA and 18,181 received saline placebo (0.5 mL).
In Study 1, the median age of the participants was 67 years (range 60‑108 years). Overall, 51.0% of the participants were male, 49.0% were female, 33.6% were Hispanic or Latino, 61.8% were White, 12.0% were Black or African American, 11.0% were Asian, 4.9% were American Indian or Alaska Native, 0.1% were Native Hawaiian or Pacific Islander, 5.5% were other races, and 4.1% were Multiracial.
Demographic characteristics were comparable between participants who received MRESVIA and those who received placebo.
Solicited Adverse Reactions: Local and systemic adverse reactions (ARs) were solicited in an electronic diary (eDiary) for 7 days following injection (i.e., the day of injection and 6 subsequent days) among participants receiving MRESVIA (n=18,160) and participants receiving placebo (n=18,098). Events that persisted for more than 7 days were followed until resolution, but not to exceed 28 days after the study injection.
The percentage of participants who reported solicited local and systemic adverse reactions are presented in Table 2 and Table 3. Solicited local and systemic adverse reactions had a median duration of 1 to 2 days.
Table 2: Percentage of Participants with Solicited Local Adverse Reactions Any Grade and ≥Grade 3 Starting Within 7 Days* of Vaccination Local Adverse Reactions‡ MRESVIA
(N=18,154 – 18,156)
%Placebo†
(N=18,093 – 18,094)
%Abbreviations: Any = Grade 1 or above; Percentages were based on the number of exposed participants who submitted any data for the event.
N = number of vaccinated participants with available data for the events listed.
* 7 days included day of vaccination and the subsequent 6 days. Adverse reactions and use of pain medication were collected in the electronic diary (e-diary).
† Placebo is 0.9% sodium chloride (normal saline) injection.
‡ No Grade 4 solicited local adverse reactions were reported.
§ Injection site pain grading scale: Does not interfere with activity (Grade 1); repeated use of over-the-counter pain reliever >24 hours or interferes with activity (Grade 2); any use of prescription pain reliever or prevents daily activity (Grade 3).
¶ Axillary (underarm) swelling or tenderness grading scale: No interference with activity (Grade 1); repeated use of over-the-counter pain reliever >24 hours or some interference with activity (Grade 2); any use of prescription pain reliever or prevents daily activity (Grade 3).Injection Site Pain, Any Grade§
55.9
13.8
Injection Site Pain, Grade 3§
1.7
1.1
Erythema (Redness), ≥ 2.5 cm
2.0
0.6
Erythema (Redness), Grade 3, >10 cm
0.6
0.3
Swelling (Hardness), ≥ 2.5 cm
3.7
0.3
Swelling (Hardness), Grade 3, >10 cm
0.9
<0.1
Axillary (underarm) swelling or tenderness, Any Grade¶
15.2
6.1
Axillary (underarm) swelling or tenderness, Grade 3¶
0.8
0.6
Table 3: Percentage of Participants with Solicited Systemic Adverse Reactions Any Grade and ≥Grade 3 Starting Within 7 Days* of Vaccination Systemic Adverse Reactions‡ MRESVIA
(N=18,146 – 18,153)
%Placebo†
(N=18,092 – 18,093)
%Abbreviations: Any = Grade 1 or above; Percentages were based on the number of exposed participants who submitted any data for the event. N = number of vaccinated participants with available data for the events listed. * 7 days included day of vaccination and the subsequent 6 days. Adverse reactions and use of pain medication were collected in the electronic diary (e-diary). † Placebo is 0.9% sodium chloride (normal saline) injection. ‡ With the exception of fever, no Grade 4 solicited systemic adverse reactions were reported. § Headache grading scale: No interference with activity (Grade 1); repeated use of over-the-counter pain reliever >24 hours or some interference with activity (Grade 2); significant, any use of prescription pain reliever or prevents daily activity (Grade 3). ¶ Fatigue grading scale: No interference with activity (Grade 1); some interference with activity (Grade 2); significant, prevents daily activity (Grade 3). # Myalgia and arthralgia grading scales: No interference with activity (Grade 1); some interference with activity (Grade 2); significant, prevents daily activity (Grade 3). ♠ Nausea/vomiting grading scale: No interference with activity or 1-2 episodes per 24 hours (Grade 1); some interference with activity or >2 episodes per 24 hours (Grade 2); prevents daily activity, requires outpatient intravenous hydration (Grade 3). ♥ Chills grading scale: No interference with activity (Grade 1); some interference with activity not requiring medical intervention (Grade 2); prevents daily activity and requires medical intervention (Grade 3). Fever, Any Grade (≥38°C / ≥100.4°F)
2.7
1.3
Fever, Grade 3 (39.0°C – 40.0°C /
102.1°F – 104.0°F)
0.4
0.2
Fever, Grade 4 (>40.0°C / >104.0°F)
0.2
0.2
Headache, Any Grade§
26.7
18.8
Headache, Grade 3§
1.5
1.1
Fatigue, Any Grade¶
30.8
20.0
Fatigue, Grade 3¶
1.7
1.2
Myalgia, Any Grade#
25.6
14.4
Myalgia, Grade 3#
1.4
0.8
Arthralgia, Any Grade#
21.7
14.0
Arthralgia, Grade 3#
1.1
0.7
Nausea/vomiting, Any Grade♠
7.0
5.2
Nausea/vomiting, Grade 3♠
0.4
0.4
Chills, Any Grade♥
11.6
6.8
Chills, Grade 3♥
0.6
0.4
Unsolicited Adverse Events: Incidence of unsolicited adverse events, serious adverse events, and medically attended adverse events within 28 days of vaccination were similar in the groups that received MRESVIA or placebo. Unsolicited adverse events within 28 days considered related to the study vaccination were numerically higher in the recipients of MRESVIA (5.7%) than in the placebo recipients (4.4%), primarily attributed to events that were consistent with solicited adverse reactions.
There was a numerically higher incidence of urticaria in the MRESVIA group than the placebo group within 7 days post injection (8 and 2 participants, respectively) and within 28 days post injection (15 and 5 participants, respectively).
Serious Adverse Events: The median duration of safety follow-up was 311 days (range 1 to 585 days), and 96.6% of participants had at least a 6-month follow-up duration after vaccination. SAEs throughout the study were reported by 7.8% and 7.9% of participants in the MRESVIA group and the placebo group, respectively. One participant in the MRESVIA group had an SAE of facial paralysis with onset four days after vaccination assessed as related to MRESVIA. Within 28 days and 42 days post vaccination, there was no imbalance in reports of facial paralysis (including Bell’s palsy) between treatment groups. There were no other notable patterns or numerical imbalances between treatment groups for specific categories of serious adverse events that would suggest a causal relationship to MRESVIA.
Individuals 18 through 59 Years of Age at Increased Risk for LRTD Caused by RSV
The safety of MRESVIA was evaluated in Study 2 (NCT06067230) in which 502 participants aged 18 through 59 years at increased risk for LRTD caused by RSV received MRESVIA. Study 2 was conducted in the US, Canada and the United Kingdom.
From the day of vaccination (Day 1) through data cutoff for the safety analysis, the median duration of follow-up was 253 days (range: 8 to 349 days) in the MRESVIA group. As of the data cutoff date, 492/502 (98.0%) participants had completed at least 180 days of post-injection follow-up.
In Study 2, the median age of the participants in the MRESVIA group was 53.0 years (range 19‑59 years). Overall, 46.4% of the participants were male, 53.6% were female, 27.9% were Hispanic or Latino, 79.9% were White, 16.9% were Black or African American, 0.8% were Asian, 0.4% were American Indian or Alaska Native, 0.6% were Native Hawaiian or Pacific Islander, and 0.8% were Multiracial.
Conditions increasing the risk for RSV-LRTD were reported by the following percentages of participants in the MRESVIA group (individual participants may have reported more than 1 condition): diabetes mellitus (DM) in 59.6%, asthma in 38.8%, coronary artery disease (CAD) in 20.7%, chronic obstructive pulmonary disease (COPD) in 10.4%, congestive heart failure (CHF) in 9.0%, and chronic respiratory disease other than COPD or asthma in 2.4% of participants.
Solicited Adverse Reactions: Reactogenicity was assessed using a prespecified list of local and systemic adverse reaction terms that were actively solicited daily via eDiaries during the 7 days following vaccination (i.e., the day of vaccination and 6 subsequent days). The reported frequencies of specific solicited local and systemic adverse reactions among participants 18 through 59 years of age at increased risk for LRTD caused by RSV who received MRESVIA are presented in Table 4 and Table 5. Solicited local and systemic adverse reactions had a median onset of 2.0 days after vaccination and a median duration of 3 days.
Table 4: Percentage of Participants with Solicited Local Adverse Reactions Any Grade and ≥Grade 3 Starting Within 7 Days* of Vaccination with MRESVIA, Study 2
Local Adverse Reactions(N=502)
%Abbreviations: Any = Grade 1 or above; Percentages were based on the number of exposed participants who submitted any data for the event. N = number of vaccinated participants with available data for the events listed. * 7 days included day of vaccination and the subsequent 6 days. Adverse reactions and use of pain medication were collected in the electronic diary (e‑diary). † Injection site pain grading scale: Does not interfere with activity (Grade 1); repeated use of over-the-counter pain reliever >24 hours or interferes with activity (Grade 2); any use of prescription pain reliever or prevents daily activity (Grade 3); requires emergency room visit or hospitalization (Grade 4). ‡ A single solicited local AR of injection site pain in the left arm was classified as Grade 4 on Day 3 due to an emergency room visit to rule out a cardiac event. The injection site pain resolved without treatment on Day 4. § Erythema grading scale: 25 to 50 mm / 2.5 to 5 cm (Grade 1); 51 to 100 mm / 5.1 to 10 cm (Grade 2); >100 mm / >10 cm (Grade 3); necrosis or exfoliative dermatitis (Grade 4). There were no Grade 4 solicited local ARs for erythema. ¶ There were no Grade 4 solicited local ARs for swelling. # Axillary (underarm) swelling or tenderness grading scale: No interference with activity (Grade 1); repeated use of over-the-counter pain reliever >24 hours or some interference with activity (Grade 2); any use of prescription pain reliever or prevents daily activity (Grade 3). There were no Grade 4 solicited local ARs for axillary swelling or tenderness. Injection Site Pain, Any Grade†
73.9
Injection Site Pain, Grade 3 or Grade 4†, ‡
1.6
Erythema (Redness), ≥ 2.5 cm§
2.4
Erythema (Redness), Grade 3, > 10 cm§
0.0
Swelling (Hardness), ≥ 2.5 cm¶
4.6
Swelling (Hardness), Grade 3, >10 cm¶
0.2
Axillary (underarm) swelling or tenderness, Any Grade#
17.1
Axillary (underarm) swelling or tenderness, Grade 3#
0.6
Table 5: Percentage of Participants with Solicited Systemic Adverse Reactions Any Grade and ≥Grade 3 Starting Within 7 Days* of Vaccination with MRESVIA, Study 2
Systemic Adverse Reactions(N=502)
%Abbreviations: Any = Grade 1 or above; Percentages were based on the number of exposed participants who submitted any data for the event. N = number of vaccinated participants with available data for the events listed. * 7 days included day of vaccination and the subsequent 6 days. Adverse reactions and use of pain medication were collected in the electronic diary (e‑diary). † Fever grading scale: 38.0 to 38.4°C / 100.4 to 101.1°F (Grade 1); 38.5 to 38.9°C / 101.2 to 102.0°F (Grade 2); 39.0 to 40.0°C / 102.1 to 104.0°F (Grade 3); >40.0°C / >104.0°F (Grade 4) ‡ Headache grading scale: No interference with activity (Grade 1); repeated use of over-the-counter pain reliever >24 hours or some interference with activity (Grade 2); significant, any use of prescription pain reliever or prevents daily activity (Grade 3). No Grade 4 solicited systemic ARs were reported. § Fatigue grading scale: No interference with activity (Grade 1); some interference with activity (Grade 2); significant, prevents daily activity (Grade 3). ¶ Myalgia and arthralgia grading scales: No interference with activity (Grade 1); some interference with activity (Grade 2); significant, prevents daily activity (Grade 3). # Nausea/vomiting grading scale: No interference with activity or 1-2 episodes per 24 hours (Grade 1); some interference with activity or >2 episodes per 24 hours (Grade 2); prevents daily activity, requires outpatient intravenous hydration (Grade 3). ♠ Chills grading scale: No interference with activity (Grade 1); some interference with activity not requiring medical intervention (Grade 2); prevents daily activity and requires medical intervention (Grade 3). Fever, Any Grade (≥38°C / ≥100.4°F)†
3.6
Fever, Grade 3 (39.0°C – 40.0°C / 102.1°F – 104.0°F)†
0.6
Headache, Any Grade‡
33.3
Headache, Grade 3‡
1.6
Fatigue, Any Grade§
36.9
Fatigue, Grade 3§
2.8
Myalgia, Any Grade¶
28.9
Myalgia, Grade 3¶
2.2
Arthralgia, Any Grade¶
22.7
Arthralgia, Grade 3¶
2.0
Nausea/vomiting, Any Grade#
10.8
Chills, Any Grade♠
19.9
Chills, Grade 3♠
0.8
Serious Adverse Events: Overall, by the data cutoff, serious adverse events were reported for 19 participants (3.8%) in Study 2. No SAEs were assessed as related to MRESVIA.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
All pregnancies have a risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
There are no human data to establish whether there is a vaccine-associated risk with use of MRESVIA in pregnancy.
A developmental toxicity study was performed in female rats administered a vaccine formulation that included approximately twice the amount of nucleoside-modified messenger ribonucleic acid (mRNA), encoding the same RSV fusion (F) glycoprotein stabilized in the prefusion conformation, as in MRESVIA. The vaccine formulation was administered twice prior to mating and twice during gestation. The study revealed no evidence of harm to the fetus due to the vaccine (see Data).
Animal Data
In a developmental toxicity study, 0.2 mL of a vaccine formulation containing 96 mcg of nucleoside‑modified mRNA per dose (a full human dose of MRESVIA contains 50 mcg of nucleoside‑modified mRNA) was administered to female rats by the intramuscular route on four occasions: 28 and 14 days prior to mating, and on gestation days 1 and 13. No vaccine‑related fetal malformations or variations and no adverse effects on postnatal development were observed in the study. The developmental toxicity study revealed no evidence of impaired female fertility.
8.2 Lactation
Risk Summary
It is not known whether MRESVIA is excreted in human milk. No human or animal data are available to assess the effects of MRESVIA on the breastfed infant or on milk production/excretion.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for MRESVIA and any potential adverse effects on the breastfed child from MRESVIA or from the underlying maternal condition. For preventive vaccines, the underlying condition is susceptibility to disease prevented by the vaccine.
8.4 Pediatric Use
Safety and effectiveness of MRESVIA in individuals younger than 18 years of age have not been established.
8.5 Geriatric Use
Of the total number of participants (N = 36,412) who received MRESVIA or placebo in Study 1 (NCT05127434), 22,554 (61.9%) were 60 to 69 years of age, 10,972 (30.1%) were 70 to 79 years of age, and 2,886 (7.9%) were 80 years of age and older [see Adverse Reactions (6.1) and Clinical Studies (14)].
-
11 DESCRIPTION
MRESVIA is a sterile white to off-‑white injectable suspension for intramuscular use.
Each 0.5 mL dose of MRESVIA contains 50 mcg of nucleoside modified mRNA encoding the F glycoprotein from RSV subtype A stabilized in the prefusion conformation (pre-F protein).
Each 0.5 mL dose of MRESVIA also contains the following ingredients: a total lipid content of 1.02 mg (SM-102 (heptadecan-9-yl 8-((2-hydroxyethyl) (6-oxo-6-(undecyloxy) hexyl) amino) octanoate), polyethylene glycol 2000 dimyristoyl glycerol [PEG2000-DMG], cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphocholine [DSPC]), 0.25 mg tromethamine, 1.2 mg tromethamine hydrochloride, 0.021 mg acetic acid, 0.10 mg sodium acetate trihydrate, 44 mg sucrose, and water for injection.
MRESVIA does not contain a preservative. The rubber tip cap and plunger used for the pre-filled syringe are not made with natural rubber latex.
- 12 CLINICAL PHARMACOLOGY
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Efficacy in Participants 60 Years of Age and Older
Study 1 (NCT05127434) is a randomized, placebo‑controlled, observer‑blind, case‑driven clinical study to evaluate the safety and efficacy of MRESVIA to prevent RSV‑LRTD in individuals 60 years of age and older with or without underlying medical conditions after receipt of a single dose of MRESVIA. Study 1 is being conducted in 22 countries and includes participants from North America/ Europe, Central/Latin America, Africa, and Asian/Pacific regions and is designed to follow participants for up to 24 months after vaccination.
Participants were randomized to a single dose of MRESVIA or placebo (in a 1:1 ratio). Randomization was stratified by age (60 to 74 years; ≥75 years) and risk factors for LRTD, which were defined as congestive heart failure (CHF) and/or chronic obstructive pulmonary disease (COPD) at screening.
The primary efficacy analysis population [Per‑Protocol (PP) Efficacy Set] included 35,064 participants who received either MRESVIA (n=17,561) or placebo (n=17,503), with a data cutoff of 30 Nov 2022. This study population included 49.1% female, 50.9% male, 63.4% White, 12.2% Black or African American, 8.7% Asian, 5.1% American Indian or Alaska Native, and 10.6% other. Among participants, 34.7% identified as Hispanic or Latino. The median age of participants was 67 years (range 60‑96 years), with 30.9% of participants between 70 and 79 years and 5.6% of participants ≥80 years. There were no notable differences in demographics or pre‑existing medical conditions between participants who received MRESVIA and those who received placebo. A total of 7.0% had protocol-defined LRTD risk factors (CHF and/or COPD) and 29.5% had one or more comorbidity of interest (COPD, asthma, chronic respiratory disease, diabetes, CHF, advanced liver disease, or advanced renal disease).
Study exclusion criteria included history of myocarditis, pericarditis, or myopericarditis within 2 months prior to screening; autoimmune conditions requiring systemic immunosuppressants (stable HIV‑positive participants were permitted); history of serious reaction to any prior vaccination. Individuals were not eligible for inclusion in the Per-Protocol Efficacy Set if they received any other vaccine within 28 days before or after administration of the study injection.
The primary efficacy endpoints were the prevention of a first episode of RSV‑LRTD with either ≥2 signs/symptoms or ≥3 signs/symptoms starting 14 days after vaccination. RSV-LRTD was defined based on the following criteria: The participant must have had RT-PCR-confirmed RSV infection and experienced new or worsening of ≥2 (or ≥3) of the following signs/symptoms for at least 24 hours: shortness of breath, cough and/or fever (≥37.8°C [100.0°F]), wheezing and/or rales and/or rhonchi, sputum production, tachypnea (≥20 breaths per minute or increase of ≥2 breaths per minute from baseline measurement in those who have baseline tachypnea), hypoxemia (new oxygen saturation ≤93% or new or increasing use of supplemental oxygen), or pleuritic chest pain. If signs/symptoms could not be captured, radiologic evidence of pneumonia with RT-PCR-confirmed RSV infection was also counted as RSV-LRTD.
The primary efficacy analyses were performed when at least 50% of targeted RSV-LRTD cases had accrued [which occurred after a median of 3.7 months of follow-up (range 15 to 379 days) when 20.2% of participants had reached 6 months of follow-up]. Both primary efficacy analyses met the predefined success criterion (lower bound of the alpha-adjusted CI of the VE was >20%). Additional analyses of efficacy were performed after a median of 8.6 months of follow-up (range 15 to 530 days) when 94.2% of participants had reached 6 months of follow-up after vaccination and met the same success criterion (lower bound of the 95% CI of the VE was >20%). Analyses of efficacy for both timepoints are presented in Table 6.
Table 6: Efficacy of MRESVIA to Prevent First Episode of Protocol‑Defined RSV-LRTD (Per‑Protocol Efficacy Set) Abbreviations: RSV-LRTD = Respiratory Syncytial Virus-Lower Respiratory Tract Disease; N = number of participants in Per-Protocol Efficacy set; n = number of participants with protocol defined RSV-LRTD; CI = Confidence Interval. * Vaccine efficacy (VE) is defined as 100% x (1 - hazard ratio (MRESVIA vs. placebo)). The CI for VE is based on a stratified Cox proportional hazard model with Efron's method of tie handling and with the treatment group as a fixed effect, adjusting for stratification factors at randomization. Stratification factors at randomization are Age Group (60 to 74 years or 75 years and older) and LRTD Risk (Present or Absent). † For primary analysis for RSV-LRTD with 2 or more symptoms, 95.04% CI where the alpha value of 4.96% was derived from the Lan-DeMets approximation to the Pocock stopping boundary with an information fraction of 0.99 (85 out of total of 86 cases). For primary analysis for RSV-LRTD with 3 or more symptoms, 95.10% CI where the alpha value of 4.90% was derived from the Lan-DeMets approximation to the Pocock stopping boundary with an information fraction of 0.97 (31 out of total of 32 cases). ‡ For additional analyses for RSV-LRTD with 2 or more and 3 or more symptoms, 95% CI. Primary Analyses
3.7 months median follow-up
MRESVIA
(N=17,561)
n (%)
Placebo
(N=17,503)
n (%)
Vaccine Efficacy* Based on Hazard Ratio (%)
(% CI†)
RSV-LRTD With 2 or More Signs/Symptoms
15 (0.09)
70 (0.40)
78.7
(62.8, 87.9)
RSV-LRTD With 3 or More Signs/Symptoms
5 (0.03)
26 (0.15)
80.9
(50.1, 92.7)
Additional Analyses
8.6 months median follow-up
MRESVIA
(N=18,074)
n (%)
Placebo
(N=18,010)
n (%)
Vaccine Efficacy* Based on Hazard Ratio (%)
(% CI‡)
RSV-LRTD With 2 or More Signs/Symptoms
48 (0.27)
127 (0.71)
62.5
(47.7, 73.1)
RSV-LRTD With 3 or More Signs/Symptoms
20 (0.11)
51 (0.28)
61.1
(34.7, 76.8)Descriptive vaccine efficacy analyses by age subgroup and for participants with at least one comorbidity are presented in Table 7.
Table 7: Efficacy of MRESVIA to Prevent First Episode of RSV-LRTD With 2 or More Signs/Symptoms by Subgroup (8.6 Months Median Follow-up, Per‑Protocol Efficacy Set) Abbreviations: RSV-LRTD = Respiratory Syncytial Virus-associated Lower Respiratory Tract Disease * Vaccine efficacy (VE) is defined as 100% x (1 - hazard ratio (MRESVIA vs. placebo)). The CI for VE is based on a stratified Cox proportional hazard model with Efron's method of tie handling and with the treatment group as a fixed effect, adjusting for stratification factors at randomization. All the VE analyses presented are descriptive. † Based on the number of participants in each subgroup. ‡ VE cannot be reliably estimated due to the low number of cases accrued in this age group. § Comorbidities included in this analysis were chronic cardiopulmonary conditions, including CHF, COPD, asthma and chronic respiratory conditions as well as diabetes, advanced liver, and advanced kidney disease. Subgroup
MRESVIA
Cases, n/N†
Placebo
Cases, n/N†VE*, %
(95% CI)Overall (≥60 years)
48/18,074
127/18,010
62.5 (47.7, 73.1)
60 to 69 years
32/11,193
77/11,146
58.8 (37.8, 72.7)
70 to 79 years
10/5,455
45/5,431
78.0 (56.3, 88.9)
≥80 years
6/1,426
5/1,433
-20.0 (-293.3, 63.4)‡
≥60 years with ≥1 comorbidity§
17/5,365
51/5,244
67.4 (43.6, 81.2)
14.2 Immunogenicity in Participants 18 through 59 Years of Age at Increased Risk for LRTD Caused by RSV
Study 2 evaluated the immunogenicity of MRESVIA in adults aged 18 through 59 years at increased risk for LRTD caused by RSV. Participants had documented confirmation of at least one of the following conditions: CAD and/or CHF, chronic lung disease (including but not limited to COPD or persistent asthma), and Type 1 or Type 2 DM. The demographic characteristics of participants in the Per-Protocol (PP) set in Study 2 were similar to those of the Safety Set. Effectiveness of MRESVIA in this population was assessed by comparison of neutralizing antibody (nAb) levels against the two major RSV subtypes, RSV-A and RSV-B, at Day 29 post-vaccination to those in a subset of Study 1 participants ≥60 years of age. The percentage of Study 1 subset participants who had at least one chronic medical condition (COPD, asthma, chronic respiratory disease, diabetes, CHF, advanced liver disease or advanced renal disease) was 57.2%.
In analyses of the primary endpoints, non-inferiority was demonstrated for the nAb Geometric Mean Titers (GMTs) for RSV-A and RSV-B [Study 2 PP/Study 1 Per-Protocol Immunogenicity (PPI) subset; lower bound of the 2‑sided 95% CIs of the Geometric Mean Ratio (GMR) > 0.667] (Table 8).
Table 8: Comparison of Model-Adjusted RSV Neutralizing GMTs at Day 29 after Vaccination with MRESVIA, 18 through 59 Years of Age at Increased Risk of LRTD Caused by RSV (Study 2) versus 60 Years of Age and Older (Study 1) Study 2
18 through 59 years of age at increased risk of RSV-LRTD
(PP Set)
(N=489-492)Study 1
≥ 60 years of age
(PPI Set)
(N=1511-1513)Study 2
versus
Study 1RSV subgroups GMTa
(95% CI)GMTa
(95% CI)GMR
95% CIPP=Per-Protocol PPI=Per-Protocol Immunogenicity GMT=Geometric Mean Titer GMR=Geometric Mean Ratio N = Number of participants with available antibody data at baseline (Day 1) and Day 29. Antibody values reported as below the lower limit of quantification (LLOQ) are replaced by 0.5 x LLOQ. Values greater than the upper limit of quantification (ULOQ) are replaced by the ULOQ. a The model-adjusted GMT is estimated on Analysis of covariance (ANCOVA) model. In the ANCOVA model, the log-transformed antibody levels at Day 29 post baseline are treated as a dependent variable, with the treatment group as an explanatory variable and the log-transformed baseline antibody level as a covariate. The resulted least square (LS) means, difference of LS means, and 95% CI are back transformed to the original scale for presentation. RSV-A
23245
(21326, 25336)
19988
(19038, 20985)
1.2
(1.1, 1.3)
RSV-B
7831
(7242, 8467)
6901
(6603, 7213)
1.1
(1.0, 1.2)
Seroresponse Rate Difference between Study 2 and Study 1 for Day 29 nAb (RSV-A and RSV-B)
Seroresponse for RSV-A and RSV-B nAb is defined as a post-primary dose titer ≥ 4 × LLOQ if baseline is < LLOQ or a ≥ 4-fold increase from baseline if baseline is ≥ LLOQ. The difference in seroresponse rate (SRR) for RSV‑A was 11.8% (95% CI: 7.8, 15.5) and for RSV-B was 10.8% (95% CI: 5.9, 15.6). These differences met prespecified non-inferiority criteria (lower bound of the 95% CI > -10%). Study 2 SRR (95% CI) at Day 29 for RSV-A was 85.8% (82.4, 88.7) and for RSV-B was 67.3% (62.9, 71.4). Study 1 SRR (95% CI) at Day 29 for RSV‑A was 74.0% (71.7, 76.2) and for RSV-B was 56.5% (53.9, 59.0).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
MRESVIA is supplied as follows:
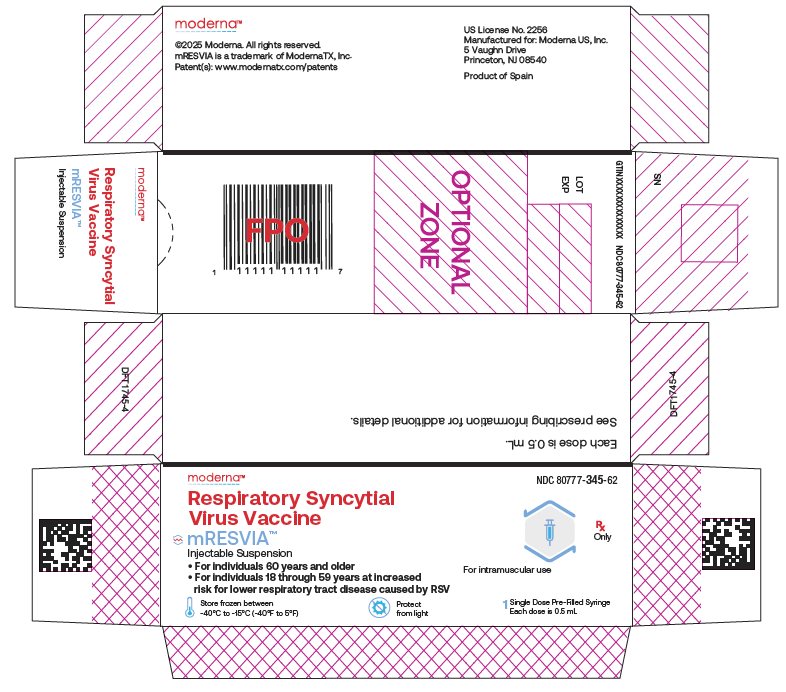
NDC 80777‑345‑62 Carton of 1 single‑dose pre-filled syringe in a paperboard tray. The syringe contains 1 dose of 0.5 mL (NDC: 80777-345-01).
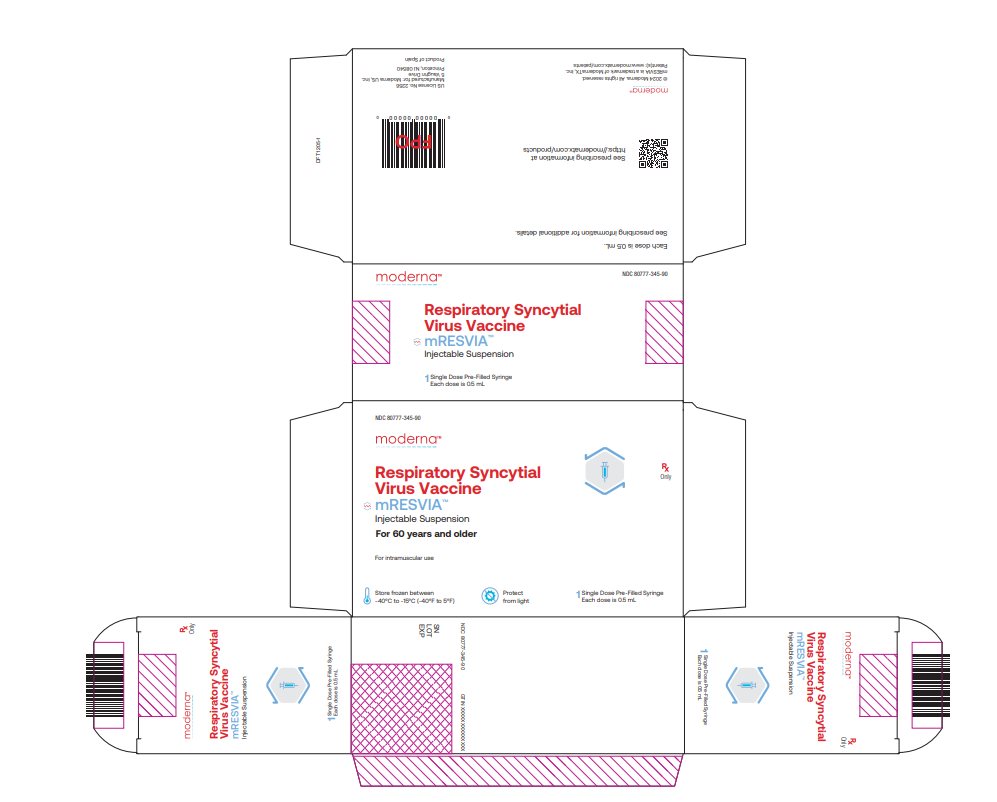
NDC 80777‑345‑90 Carton of 1 single‑dose pre-filled syringe in a blister pack. The syringe contains 1 dose of 0.5 mL (NDC: 80777-345-01).
NDC: 80777-345-63 Carton of 2 single-dose pre-filled syringes in a paperboard tray; each syringe contains 1 dose of 0.5 mL (NDC: 80777-345-01).
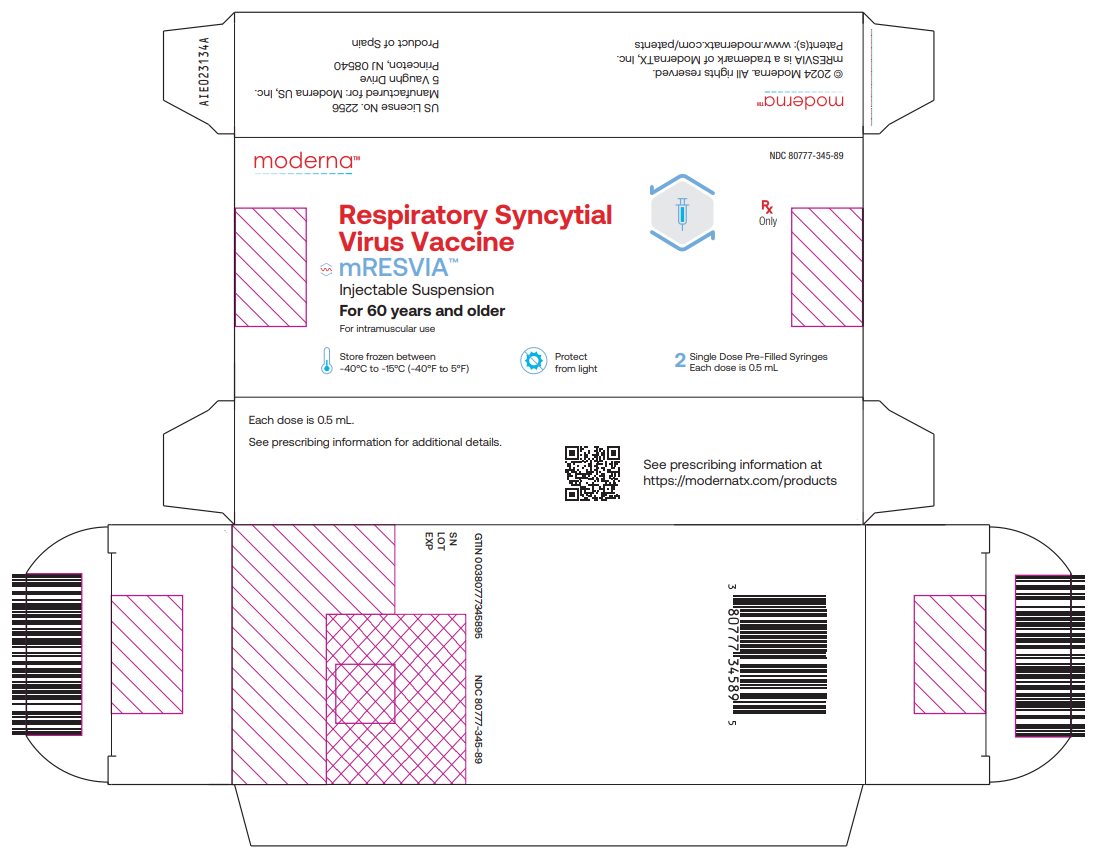
NDC: 80777-345-89 Carton of 2 single-dose pre-filled syringes in a blister pack; each syringe contains 1 dose of 0.5 mL (NDC: 80777-345-01). Each carton contains 1 blister, and each blister contains two syringes. Use one syringe per dose.
NDC 80777‑345‑61 Carton of 10 single‑dose pre-filled syringes in a paperboard tray; each syringe contains 1 dose of 0.5 mL (NDC: 80777-345-01).
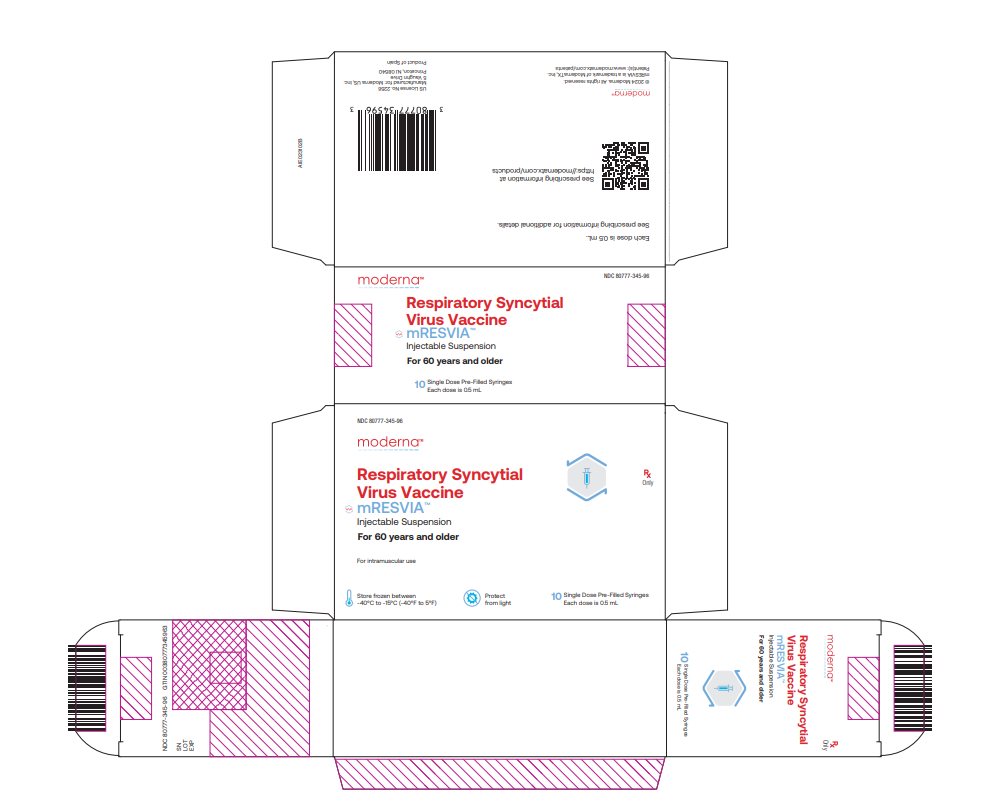
NDC 80777‑345‑96 Carton of 10 single‑dose pre-filled syringes in a blister pack; each syringe contains 1 dose of 0.5 mL (NDC: 80777-345-01). Each carton contains 5 blister packs, and each blister pack contains two syringes. Use one syringe per dose..
16.2 Storage and Handling
During storage, minimize exposure to room light, and avoid exposure to direct sunlight and ultraviolet light.
Frozen Storage
Store frozen between -40°C to ‑15°C (-40°F to 5°F).
Storage after Thawing
Storage at 2°C to 8°C (36°F to 46°F):
- Pre-filled syringes may be stored refrigerated between 2°C to 8°C (36°F to 46°F) for up to 90 days prior to use.
Storage at 8°C to 25°C (46°F to 77°F):
- Pre-filled syringes may be stored between 8°C to 25°C (46°F to 77°F) for a total of 24 hours after removal from refrigerated conditions. Discard the pre-filled syringe if not used within this time. Syringes should not be returned to the refrigerator after being thawed at room temperature.
- Total storage at 8°C to 25°C (46°F to 77°F) must not exceed 24 hours.
- Do not refreeze once thawed. Do not shake.
Transportation of Thawed Pre-filled Syringes
Thawed pre-filled syringes can be transported at 2°C to 8°C (36°F to 46°F) using shipping containers which have been qualified to maintain 2°C to 8°C (36°F to 46°F). Once thawed and transported at 2°C to 8°C (36°F to 46°F), pre-filled syringes should not be refrozen and should be stored at 2°C to 8°C (36°F to 46°F) until use.
-
17 PATIENT COUNSELING INFORMATION
Advise the vaccine recipient or caregiver to read the FDA-approved patient labeling (INFORMATION FOR RECIPIENTS AND CAREGIVERS).
Prior to administration of MRESVIA:
- Inform vaccine recipient or caregiver of the potential benefits and risks of vaccination with MRESVIA.
- Instruct vaccine recipient or caregiver to report any adverse events to their healthcare provider or to the Vaccine Adverse Event Reporting System at 1-800-822-7967 and www.vaers.hhs.gov.
This product’s labeling may have been updated. For the most recent prescribing information, please visit modernatx.com/products/mresvia or mRESVIApro.com.
Manufactured for:
Moderna US, Inc.
5 Vaughn Drive
Princeton, NJ 08540©2025 ModernaTX, Inc. All rights reserved.
MRESVIA is a trademark of ModernaTX, Inc.
Patent(s): www.modernatx.com/patents
Revised: 06/2025
-
INFORMATION FOR RECIPIENTS AND CAREGIVERS
MRESVIA (pronounced em res’ vee ah)
(Respiratory Syncytial Virus Vaccine)
Please read this information sheet before getting MRESVIA. This summary is not intended to take the place of talking with your healthcare provider. If you have questions or would like more information, please talk with your healthcare provider.
What is MRESVIA?
MRESVIA is a vaccine to protect you against lower respiratory tract disease caused by Respiratory Syncytial Virus (RSV).
MRESVIA is for people 60 years of age and older and also for people 18 through 59 years of age who are at increased risk for RSV (people with medical conditions such as diabetes or with diseases affecting the lungs and heart). Vaccination with MRESVIA may not protect all people who receive the vaccine.
MRESVIA does not contain RSV. MRESVIA cannot give you lower respiratory tract disease caused by RSV.
Who should not get MRESVIA?
You should not get MRESVIA if you had
- a severe allergic reaction to any ingredient in MRESVIA (see What are the ingredients in MRESVIA?)
What should I tell my healthcare provider?
Tell your healthcare provider about all of your medical conditions, including if you:
- have any allergies
- had a severe allergic reaction after receiving a previous dose of any other vaccine
- have a fever
- have a bleeding disorder or are on a blood thinner
- are immunocompromised or are on a medicine that affects your immune system
- have received any other RSV vaccine
- have ever fainted in association with an injection
How is MRESVIA given?
MRESVIA is given as an injection into the muscle.
What are the risks of MRESVIA?
There is a very small chance that MRESVIA could cause a severe allergic reaction. A severe allergic reaction would usually occur within a few minutes to one hour after getting a dose of MRESVIA. For this reason, your healthcare provider may ask you to stay for a short time at the place where you received your vaccine. Signs of a severe allergic reaction may include:
- Trouble breathing
- Swelling of your face and throat
- A fast heartbeat
- A rash all over your body
- Dizziness and weakness
Side effects that have been reported in clinical trials with MRESVIA include:
- Injection‑site reactions: pain, underarm swelling or tenderness in the same arm of the injection, swelling (hardness), and redness
- Fatigue, headache, muscle pain, joint pain, chills, nausea or vomiting, fever, hives, and facial paralysis
These may not be all of the possible side effects of MRESVIA. Ask your healthcare provider about any side effects that concern you. You may report side effects to the Vaccine Adverse Event Reporting System (VAERS) at 1-800-822-7967 or https://vaers.hhs.gov.
What are the ingredients in MRESVIA?
MRESVIA contains the following ingredients:
- messenger ribonucleic acid (mRNA)
- lipids (SM-102, polyethylene glycol dimyristoyl glycerol [PEG2000-DMG], cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphocholine [DSPC])
- tromethamine
- tromethamine hydrochloride
- acetic acid
- sodium acetate trihydrate
- sucrose
- water
MRESVIA does not contain preservative.
What if I have additional questions?
If you would like more information, talk to your healthcare provider, or visit MRESVIA.com or call 1‑866‑MODERNA (1‑866‑663‑3762).
Manufactured for:
Moderna US, Inc.
5 Vaughn Drive
Princeton, NJ 08540©2025 ModernaTX, Inc. All rights reserved.
MRESVIA is a trademark of ModernaTX, Inc.
Patent(s): www.modernatx.com/patents
Revised: 06/2025
-
Package/Label Display Panel
Respiratory Syncytial Virus Vaccine NDC: 80777-345-01 0.5 mL Single Dose Rx Only For IM Use Mfd. for: Moderna US, Inc.
-
Package/Label Display Panel
NDC: 80777-345-90 Respiratory Syncytial Virus Vaccine mRESVIATM Injectable Suspension For 60 years and older For intramuscular use
-
Package/Label Display Panel
NDC: 80777-345-96 Respiratory Syncytial Virus Vaccine mRESVIATM Injectable Suspension For 60 years and older For intramuscular use
-
Package/Label Display Panel
NDC: 80777-345-89 Respiratory Syncytial Virus Vaccine mRESVIATM Injectable Suspension For 60 years and older For intramuscular use
-
Package/Label Display Panel
NDC: 80777-345-62 Respiratory Syncytial Virus Vaccine mRESVIATM Injectable Suspension For individuals 60 years and older For individuals 18 through 59 years at increased
risk for lower respiratory tract disease caused by RSV For intramuscular use
-
Package/Label Display Panel
NDC: 80777-345-63 Respiratory Syncytial Virus Vaccine mRESVIATM Injectable Suspension For individuals 60 years and older For individuals 18 through 59 years at increased
risk for lower respiratory tract disease caused by RSV For intramuscular use
-
Package/Label Display Panel
NDC: 80777-345-61 Respiratory Syncytial Virus Vaccine mRESVIATM Injectable Suspension For individuals 60 years and older For individuals 18 through 59 years at increased
risk for lower respiratory tract disease caused by RSV For intramuscular use
-
INGREDIENTS AND APPEARANCE
MRESVIA
respiratory syncytial virus vaccine suspensionProduct Information Product Type VACCINE Item Code (Source) NDC: 80777-345 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RNA-100-AR02 (UNII: 2ZKG2M978D) (RNA-100-AR02 - UNII:2ZKG2M978D) RNA-100-AR02 50 ug in 0.5 mL Inactive Ingredients Ingredient Name Strength 1,2-DIMYRISTOYL-RAC-GLYCERO-3-METHOXYPOLYETHYLENE GLYCOL 2000 (UNII: 9X2596CIE0) 1,2-DISTEAROYL-SN-GLYCERO-3-PHOSPHOCHOLINE (UNII: 043IPI2M0K) ACETIC ACID (UNII: Q40Q9N063P) CHOLESTEROL (UNII: 97C5T2UQ7J) SM-102 (UNII: T7OBQ65G2I) SODIUM ACETATE (UNII: 4550K0SC9B) SUCROSE (UNII: C151H8M554) TROMETHAMINE (UNII: 023C2WHX2V) TROMETHAMINE HYDROCHLORIDE (UNII: 383V75M34E) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 80777-345-90 1 in 1 CARTON 1 NDC: 80777-345-01 0.5 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC: 80777-345-96 10 in 1 CARTON 2 NDC: 80777-345-01 0.5 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 3 NDC: 80777-345-89 2 in 1 CARTON 3 NDC: 80777-345-01 0.5 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 4 NDC: 80777-345-62 1 in 1 CARTON 4 NDC: 80777-345-01 0.5 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 5 NDC: 80777-345-63 2 in 1 CARTON 5 NDC: 80777-345-01 0.5 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 6 NDC: 80777-345-61 10 in 1 CARTON 6 NDC: 80777-345-01 0.5 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125796 05/31/2024 Labeler - Moderna US, INC. (117626450) Registrant - ModernaTX, Inc. (116912313) Establishment Name Address ID/FEI Business Operations ModernaTX, Inc. 116912313 API MANUFACTURE(80777-345) , ANALYSIS(80777-345)





Trademark Results [mRESVIA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 MRESVIA 98401817 not registered Live/Pending |
ModernaTX, Inc. 2024-02-12 |
 MRESVIA 97757619 not registered Live/Pending |
ModernaTx, Inc. 2023-01-17 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.