VYLOY- zolbetuximab injection, powder, for suspension
VYLOY by
Drug Labeling and Warnings
VYLOY by is a Prescription medication manufactured, distributed, or labeled by Astellas Pharma US, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VYLOY safely and effectively. See full prescribing information for VYLOY.
VYLOY® (zolbetuximab-clzb) for injection, for intravenous use
Initial U.S. Approval: 2024INDICATIONS AND USAGE
VYLOY is a claudin 18.2-directed cytolytic antibody and is indicated in combination with fluoropyrimidine- and platinum-containing chemotherapy for the first-line treatment of adults with locally advanced unresectable or metastatic human epidermal growth factor receptor 2 (HER2)-negative gastric or gastroesophageal junction adenocarcinoma whose tumors are claudin (CLDN) 18.2 positive as determined by an FDA-approved test (1).
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
For injection: 100 mg and 300 mg lyophilized powder in a single-dose vial. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity reactions including serious anaphylaxis reactions and serious and fatal infusion-related reactions have occurred. Monitor patients during and for at least 2 hours after infusion with VYLOY. Interrupt, slow the rate of infusion or permanently discontinue VYLOY based on severity and type of reaction. Premedicate with antihistamines for subsequent infusions after a hypersensitivity reaction. (2.4, 5.1)
- Severe nausea and vomiting: Premedicate patients with antiemetics prior to each infusion. Interrupt or permanently discontinue VYLOY based on the severity of the nausea and/or vomiting. Manage patients during and after infusion with antiemetics or fluid replacement. (2.4, 5.2)
ADVERSE REACTIONS
The most common adverse reactions (≥15%) for VYLOY in combination with mFOLFOX6 or CAPOX were nausea, vomiting, fatigue, decreased appetite, diarrhea, peripheral sensory neuropathy, abdominal pain, constipation, decreased weight, hypersensitivity reactions, and pyrexia.
The most common laboratory abnormalities (≥15%) for VYLOY in combination with mFOLFOX6 or CAPOX were decreased neutrophil count, decreased leucocyte count, decreased albumin, increased creatinine, decreased hemoglobin, increased glucose, decreased lymphocyte count, increased aspartate aminotransferase, decreased platelets, increased alkaline phosphatase, increased alanine aminotransferase, decreased glucose, decreased sodium, decreased phosphate, decreased potassium, and decreased magnesium (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Astellas Pharma US, Inc. at 1-800-727-7003 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Prior to Administration
2.3 Recommended Dosage
2.4 Dosage Modifications for Adverse Reactions
2.5 Preparation
2.6 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity reactions, including anaphylaxis reactions, and infusion related reactions
5.2 Severe Nausea and Vomiting
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
VYLOY, in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of adults with locally advanced unresectable or metastatic human epidermal growth factor receptor 2 (HER2)‑negative gastric or gastroesophageal junction (GEJ) adenocarcinoma whose tumors are claudin (CLDN) 18.2 positive as determined by an FDA-approved test [see Dosage and Administration (2.1) and Clinical Studies (14)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select adult patients with locally advanced unresectable or metastatic HER2-negative gastric or GEJ adenocarcinoma whose tumors are CLDN18.2 positive (defined as ≥75% of tumor cells demonstrating moderate to strong membranous CLDN18 immunohistochemical staining) for treatment with VYLOY in combination with fluoropyrimidine- and platinum-containing chemotherapy using an FDA-approved test [see Clinical Studies (14)].
Information on FDA-approved tests for the detection of CLDN18.2 is available at https://www.fda.gov/CompanionDiagnostics.
2.2 Prior to Administration
If a patient is experiencing nausea and/or vomiting prior to administration of VYLOY, the symptoms should be resolved to Grade ≤1 before administering the first infusion.
Premedication
Prior to each infusion of VYLOY, premedicate patients with a combination of antiemetics (e.g., NK-1 receptor blockers and/or 5-HT3 receptor blockers, as well as other drugs as indicated) for the prevention of nausea and vomiting [see Warnings and Precautions (5.2)].
2.3 Recommended Dosage
Administer VYLOY in combination with fluoropyrimidine- and platinum-containing chemotherapy as follows:
- First dose: 800 mg/m2 intravenously.
-
Subsequent doses:
- o 600 mg/m2 intravenously every 3 weeks, or
- o 400 mg/m2 intravenously every 2 weeks.
- Continue treatment until disease progression or unacceptable toxicity.
2.4 Dosage Modifications for Adverse Reactions
No dose reduction for VYLOY is recommended. Adverse reactions for VYLOY are managed by reducing the infusion rate, interruption of the infusion, withholding the dose, and/or permanently discontinuing VYLOY as described in Table 1.
Table 1. Recommended Dose Modifications for VYLOY for Adverse Reactions - * Toxicity was graded per National Cancer Institute Common Terminology Criteria for Adverse Events Version 5.0 (NCI-CTCAE v5.0).
- † Follow Grade 2 management for Grade 3 infusion-related nausea and vomiting.
Adverse Reaction
Severity*
Dose Modification
Hypersensitivity or Infusion-related reactions [see Warnings and Precautions (5.1)].
Grade 2
- Interrupt the infusion until Grade ≤1, then resume at a reduced infusion rate for the remaining infusion.
- Premedicate and administer the next infusion per the infusion rates in Table 2.
Grade 3† or 4 or anaphylaxis
- Immediately stop the infusion and permanently discontinue.
2.5 Preparation
Reconstitution
- Calculate the recommended dose based on the patient’s body surface area as described in Section 2.3 to determine the total volume and number of vials needed.
-
Reconstitute each vial of VYLOY to achieve a concentration of 20 mg/mL as follows:
- o 100 mg vial add 5 mL of Sterile Water for Injection.
- o 300 mg vial add 15 mL of Sterile Water for Injection.
- Slowly add the Sterile Water for Injection into the VYLOY vial, and direct the stream toward the inside wall of the vial. Do not inject directly onto the lyophilized powder.
- Slowly swirl each vial until the contents are completely dissolved. Allow the reconstituted vial(s) to settle until the bubbles are gone. Do not shake the vial.
- Visually inspect the reconstituted solution for particulate matter and discoloration. The reconstituted solution should be clear to slightly opalescent, colorless to slight yellow and free of visible particles. Discard any vial with visible particles or discoloration.
- Store reconstituted vial(s) at room temperature 15°C to 30°C (59°F to 86°F) for up to 5 hours if not used immediately. This product does not contain a preservative.
Dilution
-
Withdraw the required volume of reconstituted VYLOY vial(s) and transfer into an infusion bag containing 0.9% Sodium Chloride Injection, to a final concentration of 5 mg/mL.
- o The diluted solution of VYLOY is compatible with intravenous infusion bags composed of polyethylene (PE), polypropylene (PP), polyvinyl chloride (PVC) [with either Di(2-ethylhexyl) phthalate (DEHP) or, Trioctyl trimellitate (TOTM) plasticizers], ethylene propylene copolymer, ethylene-vinyl acetate (EVA) copolymer, PP and styrene-ethylene-butylene-styrene copolymer.
- o The diluted solution of VYLOY is compatible with infusion tubing composed of PE, PVC [with DEHP, TOTM or Di(2-ethylhexyl) terephthalate plasticizers], polybutadiene (PB), or elastomer modified polypropylene with in-line filter membranes composed of polyethersulfone (PES) or polysulfone.
- Mix diluted solution by gentle inversion. Do not shake the bag.
- Visually inspect the infusion bag for any particulate matter prior to use. The diluted solution should be free of visible particles. Do not use the infusion bag if particulate matter is observed.
- Discard any unused portion left in the single-dose vials.
Storage of diluted infusion
-
Store the prepared infusion bag:
- o At room temperature 15°C to 30°C (59°F to 86°F) for no longer than 6 hours from the end of the preparation of the infusion bag to the completion of the infusion.
- o Under refrigeration at 2°C to 8°C (36°F to 46°F) for no longer than 16 hours from the end of the preparation of the infusion bag to the completion of the infusion. Do not freeze.
2.6 Administration
- Administer VYLOY as an intravenous infusion only. Do NOT administer as an intravenous push or bolus.
- If VYLOY and fluoropyrimidine- and platinum-containing chemotherapy are administered on the same day, VYLOY must be administered first.
-
No incompatibilities have been observed with
- o closed system transfer devices composed of PP, PE, stainless steel, silicone (rubber/oil/resin), polyisoprene, PVC with TOTM plasticizer, acrylonitrile-butadiene-styrene (ABS) copolymer, methyl methacrylate-ABS copolymer, thermoplastic elastomer, polytetrafluoroethylene, polycarbonate, PES, acrylic copolymer, polybutylene terephthalate, PB, or EVA copolymer.
- o central ports composed of silicone rubber, titanium alloy or PVC with TOTM plasticizer.
- In-line filters (pore size of 0.2 μm composed of materials listed above) are recommended to be used during administration.
- Do NOT co-administer other drugs through the same infusion line.
- Immediately administer the infusion as described in Table 2. To minimize the risk of adverse reactions, begin each infusion at a slower rate for 30 to 60 minutes; if tolerated, gradually increase the rate as described in Table 2.
- If the infusion time exceeds the recommended storage time (6 hours from end of preparation of infusion solution at room temperature or 16 hours from end of preparation of infusion solution under refrigeration), the infusion bag must be discarded and a new infusion bag prepared to continue the infusion.
Infusion Rate Recommendations
Table 2. Infusion Rates Recommended for Each VYLOY Infusion - * In the absence of adverse reactions after 30 to 60 minutes, the infusion rate can be increased to the subsequent infusion rate as tolerated.
VYLOY Dose
Initial Infusion Rate
(first 30-60 minutes)*
Subsequent Infusion Rate
First Dose
800 mg/m2
100 mg/m2/hr
200-265 mg/m2/hr
Subsequent Doses
600 mg/m2 every 3 weeks
75 mg/m2/hr
150-265 mg/m2/hr
or
or
or
400 mg/m2 every 2 weeks
50 mg/m2/hr
100-200 mg/m2/hr
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity reactions, including anaphylaxis reactions, and infusion related reactions
Hypersensitivity reactions, including serious anaphylaxis reactions, and serious and fatal infusion-related reactions (IRR) have been reported in clinical studies when VYLOY has been administered.
Any grade hypersensitivity reactions, including anaphylactic reactions, occurring with VYLOY in combination with mFOLFOX6 or CAPOX was 18%. Severe (Grade 3 or 4) hypersensitivity reactions, including anaphylactic reactions, occurred in 2% of patients. Seven patients (1.3%) permanently discontinued VYLOY for hypersensitivity reactions, including two patients (0.4%) who permanently discontinued VYLOY due to anaphylactic reactions. Seventeen (3.2%) patients required dose interruption, and three patients (0.6%) required infusion rate reduction due to hypersensitivity reactions.
All grade IRRs occurred in 3.2% in patients administered VYLOY in combination with mFOLFOX6 or CAPOX. Severe (Grade 3) IRRs occurred in 2 (0.4%) patients who received VYLOY. An IRR led to permanent discontinuation of VYLOY in 2 (0.4%) patients and dose interruption in 7 (1.3%) patients. The infusion rate was reduced for VYLOY for 2 (0.4%) patients due to an IRR.
Monitor patients during infusion with VYLOY and for 2 hours after completion of infusion or longer if clinically indicated, for hypersensitivity reactions with symptoms and signs that are highly suggestive of anaphylaxis (urticaria, repetitive cough, wheeze and throat tightness/change in voice). Monitor patients for signs and symptoms of IRRs including nausea, vomiting, abdominal pain, salivary hypersecretion, pyrexia, chest discomfort, chills, back pain, cough and hypertension.
If a severe or life-threatening hypersensitivity or IRR reaction occurs, discontinue VYLOY permanently, treat symptoms according to standard medical care, and monitor until symptoms resolve. For any Grade 2 hypersensitivity or IRR, interrupt the VYLOY infusion until Grade ≤1, then resume at a reduced infusion rate for the remaining infusion. Premedicate the patient with antihistamines for the subsequent infusions, administer per the infusion rates in Table 2 and closely monitor the patient for symptoms and signs of a hypersensitivity reaction. The infusion rate may be gradually increased as tolerated [see Dosage and Administration (2.4)].
5.2 Severe Nausea and Vomiting
VYLOY is emetogenic. Nausea and vomiting occurred more often during the first cycle of treatment.
All grade nausea and vomiting occurred in 82% and 67%, respectively, of patients treated with VYLOY in combination with mFOLFOX6 and 69% and 66% in combination with CAPOX, respectively. Severe (Grade 3) nausea occurred in 16% and 9% of patients treated with VYLOY in combination with mFOLFOX6 or CAPOX respectively. Severe (Grade 3) vomiting occurred in 16% and 12% of patients treated with VYLOY in combination with mFOLFOX6 or CAPOX.
Nausea led to permanent discontinuation of VYLOY in combination with mFOLFOX6 or CAPOX in 18 (3.4%) patients and dose interruption in 147 (28%) patients. Vomiting led to permanent discontinuation of VYLOY in combination with mFOLFOX6 or CAPOX in 20 (3.8%) patients and dose interruption in 150 (28%) patients.
Pretreat with antiemetics prior to each infusion of VYLOY [see Dosage and Administration (2.2)]. Manage patients during and after infusion with antiemetics or fluid replacement.
Interrupt the infusion, or permanently discontinue VYLOY based on severity [see Dosage and Administration (2.4)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
- Hypersensitivity Reactions, including anaphylaxis, and infusion related reactions [see Warnings and Precautions (5.1)].
- Severe Nausea and Vomiting [see Warnings and Precautions (5.2)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflect exposure to VYLOY in 533 patients at an 800 mg/m2 initial dose followed by subsequent doses of 600 mg/m2 every 3 weeks in combination with fluoropyrimidine- and platinum-containing chemotherapy in the SPOTLIGHT (279) and GLOW (254) studies. Among 533 patients who received VYLOY in these studies, 47% were exposed for ≥6 months and 20% were exposed for ≥12 months.
In this pooled population, the most common (≥15%) adverse reactions, were nausea, vomiting, fatigue, decreased appetite, diarrhea, peripheral sensory neuropathy, abdominal pain, constipation, decreased weight, hypersensitivity reactions, and pyrexia. The most common (≥15%) laboratory abnormalities in the pooled population were decreased neutrophil count, decreased leucocyte count, decreased albumin, increased creatinine, decreased hemoglobin, increased glucose, decreased lymphocyte count, increased aspartate aminotransferase, decreased platelets, increased alkaline phosphatase, increased alanine aminotransferase, decreased glucose, decreased sodium, decreased phosphate, decreased potassium, and decreased magnesium.
SPOTLIGHT
The safety of VYLOY was evaluated in SPOTLIGHT in patients with locally advanced unresectable or metastatic gastric or GEJ cancer who received at least one dose of VYLOY at an 800 mg/m2 initial dose followed by 600 mg/m2 subsequent doses every 3 weeks in combination with mFOLFOX6 [see Clinical Studies (14)]. The median duration of exposure to VYLOY in combination with mFOLFOX6 was 6.2 months (range: 1 day to 40.9 months).
Serious adverse reactions occurred in 45% of patients treated with VYLOY in combination with mFOLFOX6; the most common serious adverse reactions (≥2%) were vomiting (8%), nausea (7%), neutropenia (2.9%), febrile neutropenia (2.9%), diarrhea (2.9%), intestinal obstruction (3.2%), pyrexia (2.5%), pneumonia (2.5%), respiratory failure (2.2%), pulmonary embolism (2.2%), decreased appetite (2.1%) and sepsis (2.0%). Fatal adverse reactions occurred in 5% of patients who received VYLOY in combination with mFOLFOX6 including sepsis (1.4%), pneumonia (1.1%), respiratory failure (1.1%), intestinal obstruction (0.7%), acute hepatic failure (0.4%), acute myocardial infarction (0.4%), death (0.4%), disseminated intravascular coagulation (0.4%), encephalopathy (0.4%), and upper gastrointestinal hemorrhage (0.4%).
Permanent discontinuation of VYLOY due to an adverse reaction occurred in 20% of patients; the most common adverse reactions leading to discontinuation (≥2%) were nausea and vomiting.
Dosage interruptions of VYLOY due to an adverse reaction occurred in 75% of patients; the most common adverse reactions leading to dose interruption (≥5%) were nausea, vomiting, neutropenia, abdominal pain, fatigue, and hypertension.
Tables 3 and 4 summarize the most common (≥15%) adverse reactions and laboratory abnormalities with a difference between arms of ≥5%, respectively, compared to placebo in SPOTLIGHT.
Table 3. Adverse Reactions (≥15%) in Patients Treated with VYLOY in SPOTLIGHT with a Difference Between Arms of ≥5% Compared to Placebo
Adverse ReactionVYLOY
with mFOLFOX6
n=279Placebo
with mFOLFOX6
n=278All Grades
%Grade 3 or 4
%All Grades
%Grade 3 or 4
%Gastrointestinal disorders
Nausea
82
16
61
7
Vomiting
67
16
36
6
Metabolism and nutrition disorders
Decreased appetite
47
6
34
3.2
General disorders and administration site conditions
Peripheral edema
18
0.7
9
0
Table 4. Laboratory Abnormalities (≥ 15%) in SPOTLIGHT with a Difference Between Arms of ≥ 5% Compared to Placebo Laboratory Abnormality VYLOY with mFOLFOX6* Placebo with mFOLFOX6* All Grades
%Grade 3 or 4
%All Grades
%Grade 3 or 4
%- * The denominator used to calculate the rate varied from 271 to 272 based on the number of patients with a baseline value and at least one post-treatment value.
Albumin decreased
78
4.4
47
1.1
Potassium decreased
28
11
21
6
Glucose decreased
45
0.4
35
0.4
Sodium decreased
29
5
21
2.9
GLOW
The safety of VYLOY was evaluated in GLOW in patients with locally advanced unresectable or metastatic gastric/GEJ cancer who received at least one dose of VYLOY at an 800 mg/m2 initial dose followed by 600 mg/m2 subsequent doses every 3 weeks in combination with CAPOX [see Clinical Studies (14)]. The median duration of exposure to VYLOY in combination with CAPOX was 4.4 months (range: 0.03 to 30.7 months).
Serious adverse reactions occurred in 47% of patients treated with VYLOY in combination with CAPOX; the most common serious adverse reactions (≥2%) were vomiting (6%), nausea (4.3%), decreased appetite (3.9%), decreased platelet count (3.1%), upper gastrointestinal hemorrhage (2.8%), diarrhea (2.8%), pneumonia (2.4%), pulmonary embolism (2.3%), and pyrexia (2.0%). Fatal adverse reactions occurred in 8% of patients who received VYLOY in combination with CAPOX including sepsis (1.2%), pneumonia (0.4%), death (0.8%), upper gastrointestinal hemorrhage (0.8%), cerebral hemorrhage (0.8%), abdominal infection (0.4%), acute respiratory distress syndrome (0.4%), cardio‑respiratory arrest (0.4%), decreased platelet count (0.4%), disseminated intravascular coagulation (0.4%), dyspnea (0.4%), gastric perforation (0.4%), hemorrhagic ascites (0.4%), procedural complication (0.4%), sudden death (0.4%), and syncope (0.4%).
Permanent discontinuation of VYLOY due to an adverse reaction occurred in 19% of patients; the most common adverse reaction leading to discontinuation (≥2%) was vomiting.
Dosage interruption of VYLOY due to an adverse reaction occurred in 55% of patients; the most common adverse reactions leading to dose interruption (≥2%) were nausea, vomiting, neutropenia, thrombocytopenia, anemia, fatigue, infusion-related reaction, and abdominal pain.
Tables 5 and 6 summarize the most common (≥15%) adverse reactions and laboratory abnormalities with a difference between arms of ≥5%, respectively compared to placebo in GLOW.
Table 5. Adverse Reactions (≥15%) in Patients Treated with VYLOY in GLOW with a Difference Between Arms of ≥5% Compared to Placebo Adverse Reaction
VYLOY
with CAPOX
n=254
Placebo
with CAPOX
n=249
All Grades
%
Grade 3 or 4
%
All Grades
%
Grade 3 or 4
%
Gastrointestinal disorders
Nausea
69
9
50
2.4
Vomiting
66
12
31
3.6
Metabolism and nutrition disorders
Decreased appetite
41
7
34
1.6
Blood and lymphatic system disorders
Neutropenia
20
7
14
2.8
Investigations
Weight decreased
20
0.4
10
0.4
Other clinically relevant adverse reactions (<15%) in GLOW with a difference between arms of 5% compared to placebo included peripheral edema.
Table 6. Laboratory Abnormalities (≥15%) in Patients Treated with VYLOY in GLOW with a Difference Between Arms of ≥5% Compared to Placebo - * The denominator used to calculate the rate varied from 237 to 238 based on the number of patients with a baseline value and at least one post-treatment value.
Laboratory Abnormality
VYLOY with CAPOX*
Placebo with CAPOX*
All Grades
%
Grade 3 or 4
%
All Grades
%
Grade 3 or 4
%
Albumin decreased
66
3.8
47
1.7
Leukocytes decreased
66
6
60
8
Neutrophils decreased
76
21
70
14
Glucose decreased
24
0
18
0
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no data with VYLOY use in pregnant women to inform any drug-associated risks. Embryo-fetal toxicity was not observed in pregnant mice intravenously administered zolbetuximab-clzb [see Data]. VYLOY should only be given to a pregnant woman if the benefit outweighs the potential risk.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Data
Animal Data
In an embryo-fetal development toxicity study, zolbetuximab-clzb was intravenously administered to pregnant mice during the period of organogenesis and did not result in embryo-fetal toxicity at doses up to 300 mg/kg (approximately 1.9 times the recommended clinical dose based on AUC). Zolbetuximab-clzb crossed the placental barrier resulting in higher fetal serum concentrations on Day 18 of gestation than maternal serum concentrations on Day 16 of gestation.
8.2 Lactation
Risk Summary
There are no data on the presence of zolbetuximab-clzb in human milk, the effects on the breastfed child, or the effects on milk production. Because antibodies may be excreted in human milk and because of the potential for adverse reactions in a breastfed child, advise a lactating woman not to breastfeed during treatment with VYLOY and for 8 months after the last dose.
8.3 Females and Males of Reproductive Potential
VYLOY is used in combination with fluoropyrimidine- or platinum-containing chemotherapy. Refer to the Full Prescribing Information of fluoropyrimidine- and platinum-containing chemotherapy products for pregnancy testing, contraception, and infertility information.
8.4 Pediatric Use
The safety and effectiveness of VYLOY in pediatric patients have not been established.
8.5 Geriatric Use
Of the 533 patients in clinical studies of VYLOY in combination with mFOLFOX6 or CAPOX, 34% (n=179) were over 65 years, and 5% were over 75 years (n=28) [see Clinical Studies (14)]. No overall differences in safety or effectiveness were observed between patients aged 65 years or older and younger patients.
-
11 DESCRIPTION
Zolbetuximab-clzb is a chimeric (mouse/human) antibody composed of variable regions derived from mouse anti-human claudin-18 isoform 2 monoclonal antibody and constant regions derived from human IgG1. The molecular weight is approximately 147 kDa.
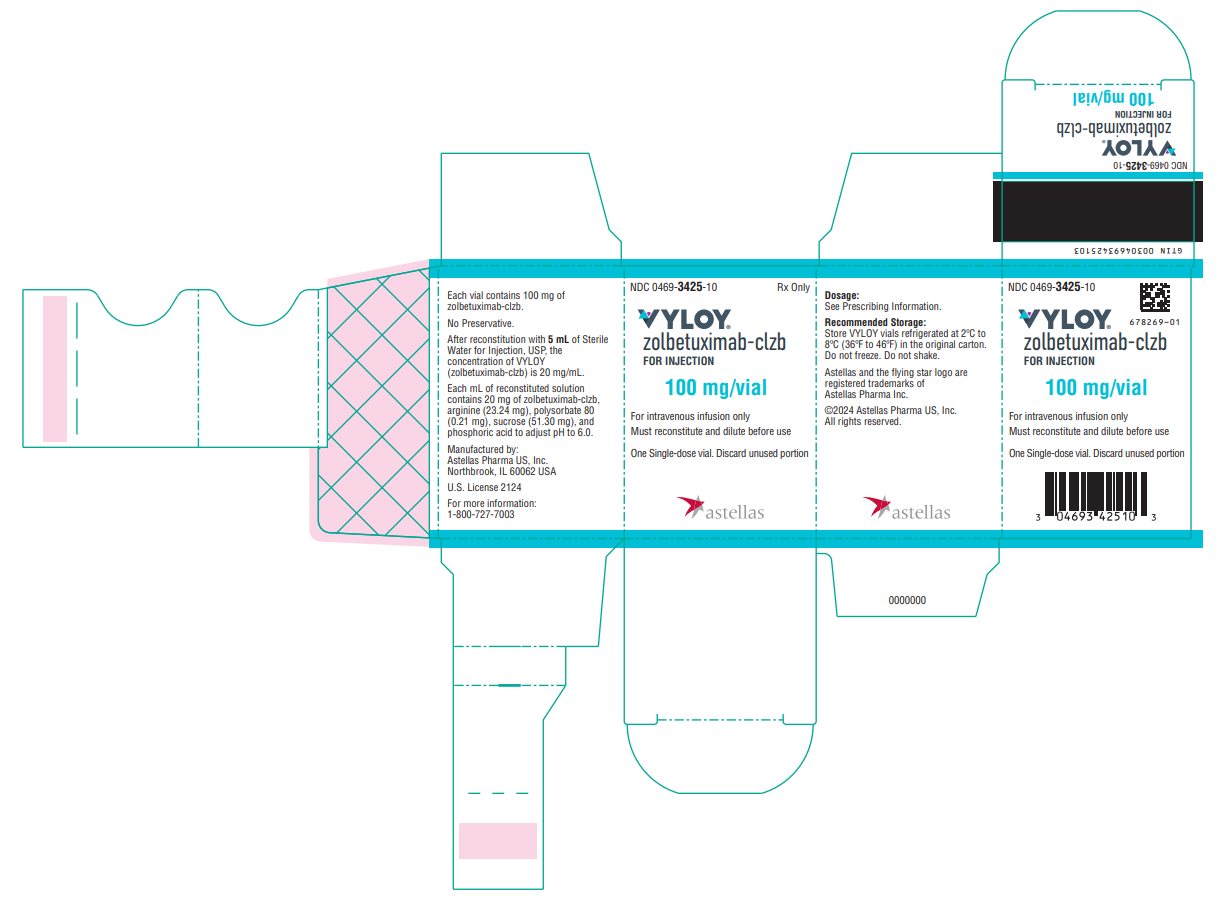
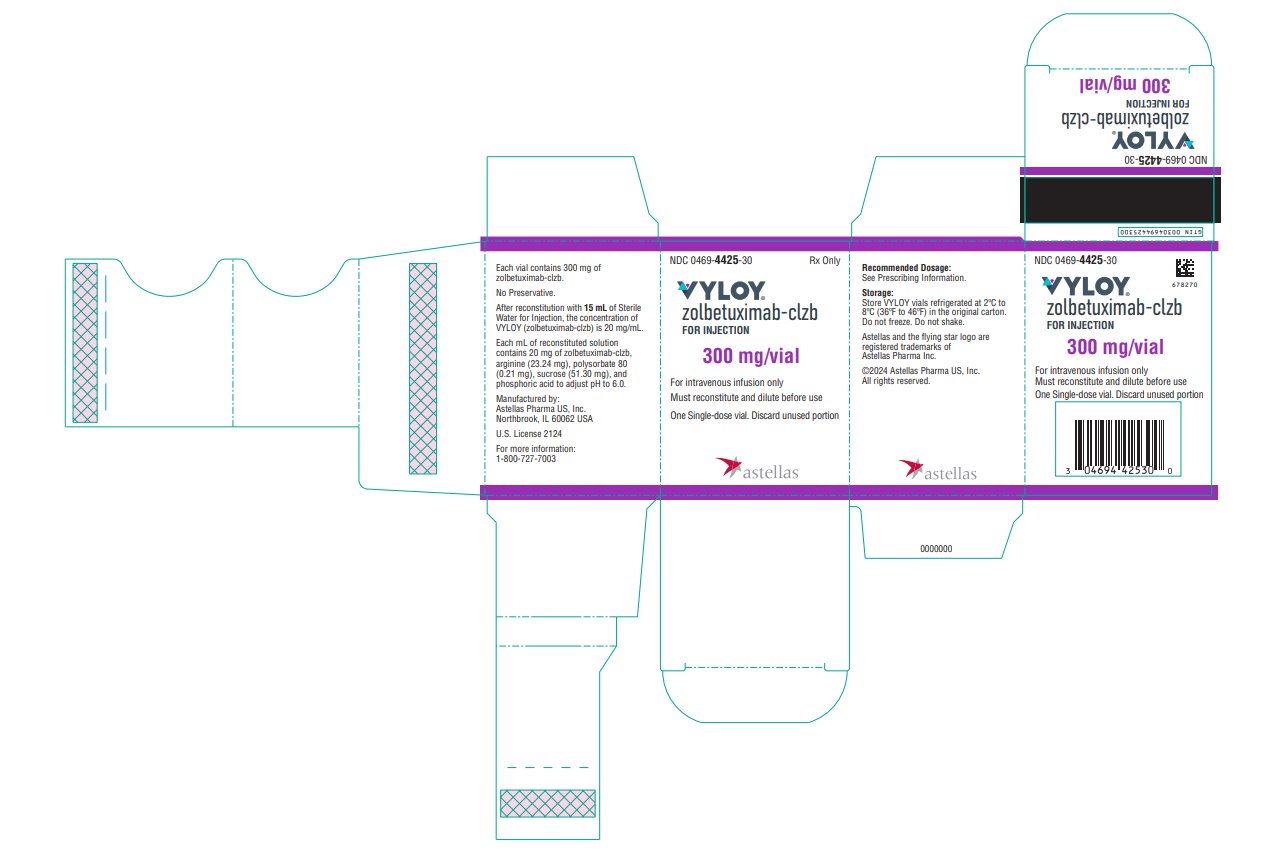
VYLOY (zolbetuximab-clzb) for injection is provided as a sterile, preservative-free, white to off-white lyophilized powder in single-dose vials for intravenous use. VYLOY is supplied as 100 mg or 300 mg per vial and requires reconstitution with Sterile Water for Injection, USP, (5 mL or 15 mL) resulting in a clear to slightly opalescent, colorless to slightly yellow solution with a final concentration of 20 mg/mL. Each mL of reconstituted solution contains 20 mg of zolbetuximab-clzb, arginine (23.24 mg), polysorbate 80 (0.21 mg), sucrose (51.30 mg), and phosphoric acid to adjust pH to 6.0.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Zolbetuximab-clzb is a claudin 18.2 (CLDN18.2)-directed cytolytic antibody that depletes CLDN18.2-positive cells via antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). Zolbetuximab-clzb in combination with chemotherapy had increased antitumor activity in CLDN18.2-expressing mouse tumor models compared to zolbetuximab-clzb or chemotherapy alone.
12.2 Pharmacodynamics
The exposure-response relationships for efficacy and safety at the recommended dosages of zolbetuximab-clzb in patients with locally advanced unresectable or metastatic HER2-negative gastric or gastroesophageal junction adenocarcinoma whose tumors are CLDN18.2 positive have not been fully characterized.
12.3 Pharmacokinetics
Following a 2-hour intravenous infusion, zolbetuximab-clzb exhibited dose-proportional pharmacokinetics at doses ranging from 33 mg/m2 to 1000 mg/m2 (0.04 times to 1.25 times the recommended first dose). When administered at a first dose of 800 mg/m2 followed by subsequent doses of 600 mg/m2 every 3 weeks, steady state was achieved by 18 weeks with a geometric mean (coefficient of variation [CV]%) Cmax of 415 (22%) mcg/mL and AUCtau of 3149 (37%) daymcg/mL.
Distribution
The estimated geometric mean (CV%) of the steady state volume of distribution of zolbetuximab-clzb was 14.0 (59%) L.
Metabolism
Zolbetuximab-clzb is expected to be catabolized into small peptides and amino acids.
Elimination
The estimated geometric mean (CV%) of the clearance (CL) and t1/2 of zolbetuximab-clzb was 0.013 (44%) L/h and 41 (62%) days, respectively.
Specific Populations
The following factors had no clinically important effect on the clearance of zolbetuximab-clzb: age (range: 22 to 83 years), sex, race including White (50%), Asian (42%), Black (0.8%), mild to moderate (CLcr ≥30 to <90 mL/min) renal impairment and mild hepatic impairment (total bilirubin (TB) ≤upper limit of normal (ULN) and AST >ULN, or TB >1 to 1.5 x ULN and any AST). The effect of severe renal impairment, and moderate to severe hepatic impairment is unknown.
12.6 Immunogenicity
The observed incidence of anti-drug antibody (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of VYLOY or of other zolbetuximab products.
During the approximately 30-month period of treatment with VYLOY 800/600 mg/m2 every 3 weeks in combination with mFOLFOX6 or CAPOX in the clinical studies SPOTLIGHT and GLOW, the incidence of anti-zolbetuximab-clzb antibody formation was 9.5% (46 of 485 ADA-evaluable patients).
Because of the low occurrence of ADAs, the effect of these antibodies on the pharmacokinetics, safety and/or effectiveness of VYLOY is unknown.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
SPOTLIGHT
The efficacy of VYLOY in combination with mFOLFOX6 was evaluated in SPOTLIGHT (NCT03504397), a double‑blind, randomized, multicenter study that enrolled 565 patients with locally advanced unresectable or metastatic HER2-negative gastric or GEJ adenocarcinoma whose tumors were CLDN18.2 positive. CLDN18.2 positivity (defined as ≥75% of tumor cells demonstrating moderate to strong membranous CLDN18 staining) was determined by immunohistochemistry on gastric or GEJ tumor tissue specimens from all patients with the VENTANA CLDN18 (43‑14A) RxDx Assay performed in a central laboratory. Patients were excluded from the study if they had a complete or partial gastric outlet syndrome, or history of central nervous system metastases.
Patients were randomized 1:1 to receive VYLOY in combination with mFOLFOX6 (n=283) or placebo in combination with mFOLFOX6 (n=282). VYLOY was administered intravenously at an initial dose of 800 mg/m2 (Day 1 of cycle 1) followed by subsequent doses of 600 mg/m2 every 3 weeks in combination with up to 12 treatments (4 cycles) of mFOLFOX6 (oxaliplatin 85 mg/m2, folinic acid (leucovorin or local equivalent) 400 mg/m2, fluorouracil 400 mg/m2 given as a bolus and fluorouracil 2400 mg/m2 given as a continuous infusion) administered on Days 1, 15 and 29 of a 42-day cycle. After 12 treatments, patients were allowed to continue treatment with VYLOY, 5-fluorouracil and folinic acid (leucovorin or local equivalent) at the discretion of the investigator, until progression of disease or unacceptable toxicity.
Treatment with VYLOY continued until RECIST v1.1-defined progression of disease as determined by an independent review committee (IRC) or a subsequent anticancer treatment was initiated. Tumor assessments were performed every 9 weeks up to and including Week 54, then every 12 weeks thereafter.
The major efficacy outcome measure was progression free survival (PFS) as assessed per RECIST v1.1 by IRC. Additional efficacy outcome measures were overall survival (OS), objective response rate (ORR) and duration of response (DOR) as assessed per RECIST v1.1 by IRC.
The study population characteristics were median age of 61 (range: 20-86); 62% were male; 48% were White, 34% Asian, 3.0% American Indian or Alaska, 1.2% Black or African American, 4.1% other racial groups, and race in 9% was unknown or missing; 78% non-Hispanic or Latino, 13% Hispanic or Latino, and ethnicity in 10% was missing; 98% had ECOG performance status (PS) of 0 or 1; 76% had gastric cancer, 24% had GEJ cancer; 84% were metastatic, 16% were locally advanced; and 29% had undergone prior gastrectomy. Subsequent anticancer therapy was received by 135 (48%) patients in the VYLOY in combination with mFOLFOX6 arm and 148 (53%) patients in the placebo in combination with mFOLFOX6 arm.
VYLOY in combination with mFOLFOX6 demonstrated a statistically significant improvement in PFS and OS compared with placebo in combination with mFOLFOX6.
Table 7, Figures 1 and 2 summarize the efficacy results for the SPOTLIGHT study.
Table 7. Efficacy Results in SPOTLIGHT Endpoint VYLOY
with mFOLFOX6
n=283Placebo
with mFOLFOX6
n=282- * Based on Kaplan-Meier estimate.
- † Stratification factors were region, number of metastatic sites and prior gastrectomy from IRT.
- ‡ Based on a stratified Cox proportional hazards model.
- § Based on a 1-sided stratified log-rank test.
- ¶ Based on confirmed response.
- # Based on binomial distribution (Clopper-Pearson).
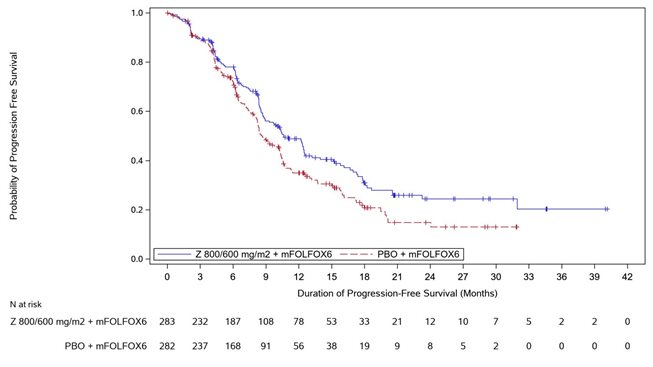
Progression Free Survival
Number (%) of patients with events
146 (51.6)
167 (59.2)
Median in months (95% CI)*
10.6 (8.9, 12.5)
8.7 (8.2, 10.3)
0.751 (0.598, 0.942)
0.0066
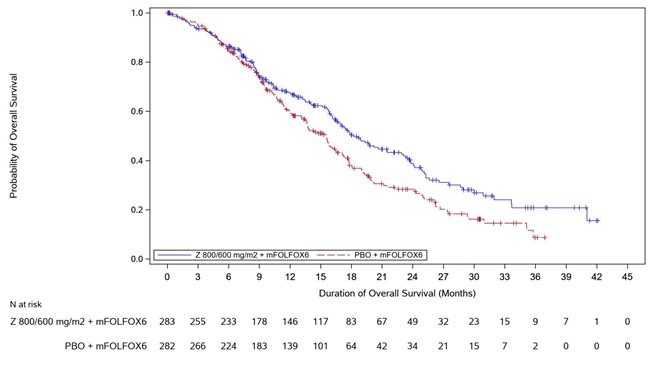
Overall survival
Number (%) of patients with events
149 (52.7)
177 (62.8)
Median in months (95% CI)*
18.2 (16.4, 22.9)
15.5 (13.5, 16.5)
0.750 (0.601, 0.936)
0.0053
Objective Response Rate (CR + PR)¶
ORR (%) (95% CI)#
40.3 (34.5, 46.3)
39.7 (34.0, 45.7)
Complete response rate (%)
14 (4.9)
8 (2.8)
Partial response rate (%)
100 (35.3)
104 (36.9)
Duration of Response
N=114
N=112
Median in months (95% CI)
10.3 (8.3, 10.9)
10.5 (7.7, 13.3)
Figure 1. Kaplan Meier Plot of Progression Free Survival, SPOTLIGHT Study
Figure 2. Kaplan Meier Plot of Overall Survival, SPOTLIGHT Study
GLOW
The efficacy of VYLOY in combination with CAPOX was evaluated in GLOW (NCT03653507), a double-blind, randomized, multicenter study that enrolled 507 patients with locally advanced unresectable or metastatic HER2-negative gastric or GEJ adenocarcinoma whose tumors were CLDN18.2 positive. CLDN18.2 positivity (defined as ≥75% of tumor cells demonstrating moderate to strong membranous CLDN18 staining) was determined by immunohistochemistry on gastric or GEJ tumor tissue specimens from all patients with the VENTANA CLDN18 (43-14A) RxDx Assay performed in a central laboratory. Patients were excluded from the study if they had a complete or partial gastric outlet syndrome, or history of central nervous system metastases.
Patients were randomized 1:1 to receive VYLOY in combination with CAPOX (n=254) or placebo in combination with CAPOX (n=253). VYLOY was administered intravenously at an initial dose of 800 mg/m2 (Day 1 of cycle 1) followed by a subsequent dose of 600 mg/m2 every 3 weeks in combination with up to 8 treatments (8 cycles) of CAPOX administered on Day 1 (oxaliplatin 130 mg/m2) and on Days 1 to 14 (capecitabine 1000 mg/m2) of a 21-day cycle. After 8 treatments of oxaliplatin, patients were allowed to continue treatment of VYLOY and capecitabine at the discretion of the investigator, until progression of disease or unacceptable toxicity.
Treatment with VYLOY continued until RECIST v1.1-defined progression of disease as determined by IRC or subsequent anticancer treatment was initiated. Tumor assessments were performed every 9 weeks up to and including Week 54, then every 12 weeks thereafter.
The major efficacy outcome measure was PFS as assessed per RECIST v1.1 by IRC. Additional efficacy outcome measures were OS, ORR, and DOR as assessed per RECIST v1.1 by IRC.
The study population characteristics were median age of 60 years (range: 21-83); 62% were male; 62% were Asian, 36% were White and race in 1.4% was missing; 95% non-Hispanic or Latino, 3.4% were Hispanic or Latino and ethnicity in 1.4% was missing; 99% had ECOG performance status (PS) of 0 or 1; 84% had primary gastric cancer, 16% had primary gastroesophageal adenocarcinoma; 88% were metastatic, 12% were locally advanced; and 27% had undergone prior gastrectomy.
VYLOY in combination with CAPOX demonstrated a statistically significant improvement in PFS and OS compared with placebo in combination with CAPOX.
Table 8, Figures 3 and 4 summarize the efficacy results for the GLOW study.
Table 8. Efficacy Results in GLOW Endpoint VYLOY
with CAPOX
n=254Placebo
with CAPOX
n=253- * Based on Kaplan-Meier estimate.
- † Stratification factors were region, number of metastatic sites and prior gastrectomy from IRT.
- ‡ Based on a stratified Cox proportional hazards model.
- § Based on a 1-sided stratified log-rank test.
- ¶ Based on confirmed response.
- # Based on binomial distribution (Clopper-Pearson).
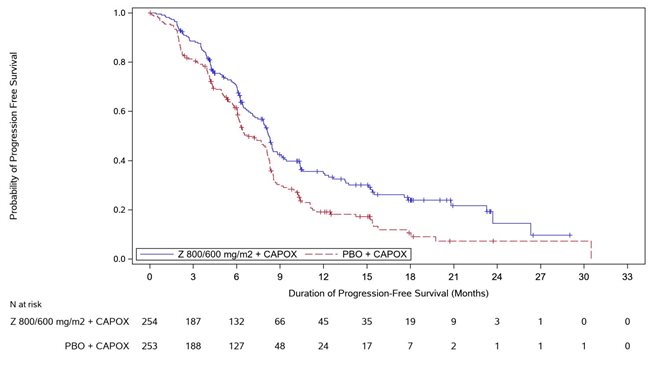
Progression Free Survival
Number (%) of patients with events
137 (53.9)
172 (68.0)
Median in months (95% CI)*
8.2 (7.5, 8.8)
6.8 (6.1, 8.1)
0.687 (0.544, 0.866)
0.0007
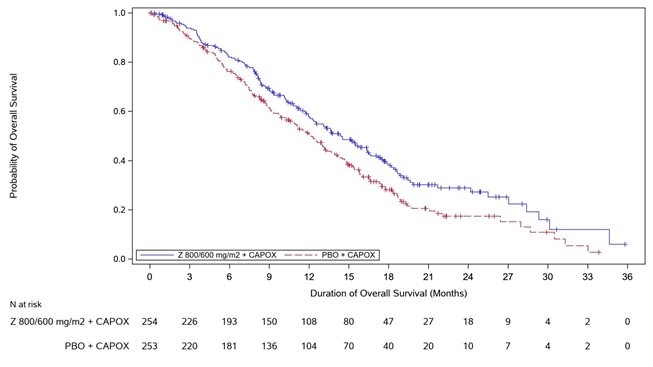
Overall survival
Number (%) of patients with events
144 (56.7)
174 (68.8)
Median in months (95% CI)*
14.4 (12.3, 16.5)
12.2 (10.3, 13.7)
0.771 (0.615, 0.965)
0.0118
Objective Response Rate (CR + PR)¶
ORR (%) (95% CI)#
32.3 (26.6, 38.4)
31.2 (25.6, 37.3)
Complete response rate (%)
6 (2.4)
2 (0.8)
Partial response rate (%)
76 (29.9)
77 (30.4)
Duration of Response
N=82
N=79
Median in months (95% CI)
8.3 (6.3, 11.4)
6.2 (6.0, 7.6)
Figure 3. Kaplan Meier Plot of Progression Free Survival, GLOW Study
-
16 HOW SUPPLIED/STORAGE AND HANDLING
VYLOY (zolbetuximab-clzb) for injection 100 mg and 300 mg are supplied as a sterile, preservative-free, white to off-white lyophilized powder in single-dose vials. VYLOY vials are available in the following packages:
- Carton of one 100 mg single-dose vial (NDC: 0469-3425-10)
- Carton of one 300 mg single-dose vial (NDC: 0469-4425-30)
Store VYLOY vials refrigerated at 2ºC to 8ºC (36ºF to 46ºF) in the original carton. Do not freeze. Do not shake.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Hypersensitivity reactions, including anaphylaxis and infusion-related reactions
Advise patients of the risk of hypersensitivity reactions including anaphylaxis and infusion-related reactions and to contact their healthcare provider right away if they experience symptoms of a hypersensitivity or infusion-related reaction during or after the administration of VYLOY. [see Warnings and Precautions (5.1)].Severe nausea and vomiting
Advise patients of the risk of severe nausea and vomiting and to immediately contact their healthcare provider if they experience persistent or worsening nausea or vomiting [see Warnings and Precautions (5.2)].Lactation
Advise women not to breastfeed during treatment with VYLOY and for 8 months after the last dose of VYLOY [see Use in Specific Populations (8.2)].Manufactured by:
Astellas Pharma US, Inc.
Northbrook, Illinois 60062U.S. License 2124
All trademarks are the property of their respective owners.
©2025 Astellas Pharma US, Inc.
00119-ZOL-USA
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
VYLOY® (vye-LOY)
(zolbetuximab-clzb)
for injection
What is VYLOY?
VYLOY is a prescription medicine used to treat adults with cancer of the stomach (gastric cancer) or cancer located where the esophagus joins the stomach (gastroesophageal junction cancer). VYLOY is used in combination with chemotherapy that contains fluoropyrimidine and platinum as the first treatment when your gastric or gastroesophageal junction cancer:
- cannot be removed with surgery or has spread to other parts of the body,
- is HER2-negative, and
- your tumor tests positive for “claudin (CLDN) 18.2.”
It is not known if VYLOY is safe and effective in children.
Before receiving VYLOY, tell your healthcare provider about all of your medical conditions, including if you:
- have nausea or vomiting.
- are pregnant or plan to become pregnant. It is not known if VYLOY will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if VYLOY passes into your breast milk. Do not breastfeed during treatment with VYLOY and for 8 months after the last dose.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How will I receive VYLOY?
- VYLOY will be given to you by intravenous (IV) infusion into your vein.
- Your healthcare provider will decide how much VYLOY you will receive.
- You will usually receive VYLOY every 2 or 3 weeks based on the chemotherapy chosen by your healthcare provider.
- Your healthcare provider will decide how many treatments you need.
What are the possible side effects of VYLOY?
VYLOY may cause serious side effects, including:
- Allergic reactions, including anaphylaxis and infusion related reactions. Allergic reactions are common during treatment with VYLOY and can sometimes be serious. Serious allergic reactions can happen during or after your VYLOY infusion, including life-threatening allergic reactions and serious infusion-related reactions that may lead to death. Your healthcare provider will monitor you during your infusion and for 2 hours after or longer if needed. Tell your healthcare provider or get emergency medical help right away if you get any of the following symptoms of a serious allergic reaction during or after your infusion of VYLOY:
- o itchy, raised bumps on the skin (hives)
- o coughing that does not go away
- o nausea or vomiting
- o stomach (abdominal) pain
- o increased saliva
- o breathing problems such as wheezing
- o throat tightness or change in voice
- o fever
- o chest discomfort
- o chills or shaking
- o back pain
- Severe nausea and vomiting. Nausea and vomiting are common during treatment with VYLOY and can sometimes be severe. Nausea and vomiting happened more often during the first treatment cycle. Before you receive each VYLOY infusion, your healthcare provider will give you medicines to help prevent nausea and vomiting. Tell your healthcare provider right away if nausea or vomiting does not go away or gets worse.
The most common side effects of VYLOY include:
- tiredness
- decreased appetite
- diarrhea
- tingling or numbness of the arms or legs
- stomach (abdominal) pain
- constipation
- decreased weight
- fever
- decreased white blood cells, red blood cells and platelets
- decreased protein (albumin) in the blood
- changes in kidney function tests
- changes in blood sugar (glucose)
- changes in liver function tests
- changes in body salts (electrolytes) in your blood
Your healthcare provider may slow the rate of your infusion, temporarily stop, or completely stop treatment with VYLOY if you have certain side effects.
These are not all of the possible side effects of VYLOY.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of VYLOY.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about VYLOY that is written for health professionals.
What are the ingredients in VYLOY?
Active ingredient: zolbetuximab-clzb
Inactive ingredients: arginine, polysorbate 80, sucrose, and phosphoric acid to adjust pH.
Manufactured by:
Astellas Pharma US, Inc.
Northbrook, Illinois 60062U.S. License 2124
©2025 Astellas Pharma US, Inc.
00119-ZOL-USA
For more information, go to www.VYLOY.com or call 1-888-727-7003.
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 6/2025
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL- 100 mg/vial
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL- 300 mg/vial
-
INGREDIENTS AND APPEARANCE
VYLOY
zolbetuximab injection, powder, for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0469-3425 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ZOLBETUXIMAB (UNII: TF5MPQ8WGY) (ZOLBETUXIMAB - UNII:TF5MPQ8WGY) ZOLBETUXIMAB 20 mg in 1 mL Inactive Ingredients Ingredient Name Strength ARGININE (UNII: 94ZLA3W45F) 23.24 mg in 1 mL SUCROSE (UNII: C151H8M554) 51.30 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.21 mg in 1 mL PHOSPHORIC ACID (UNII: E4GA8884NN) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0469-3425-10 1 in 1 BOX 10/24/2024 1 5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761365 10/24/2024 VYLOY
zolbetuximab injection, powder, for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0469-4425 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ZOLBETUXIMAB (UNII: TF5MPQ8WGY) (ZOLBETUXIMAB - UNII:TF5MPQ8WGY) ZOLBETUXIMAB 20 mg in 1 mL Inactive Ingredients Ingredient Name Strength ARGININE (UNII: 94ZLA3W45F) 23.24 mg in 1 mL SUCROSE (UNII: C151H8M554) 51.30 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.21 mg in 1 mL PHOSPHORIC ACID (UNII: E4GA8884NN) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0469-4425-30 1 in 1 BOX 04/24/2025 1 15 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761365 04/24/2025 Labeler - Astellas Pharma US, Inc. (605764828)
Trademark Results [VYLOY]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VYLOY 79356468 not registered Live/Pending |
Astellas Pharma Inc. 2022-10-13 |
 VYLOY 79199846 5211444 Live/Registered |
Astellas Pharma Inc. 2016-10-14 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.