FINTEPLA- fenfluramine solution
Fintepla by
Drug Labeling and Warnings
Fintepla by is a Prescription medication manufactured, distributed, or labeled by UCB, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FINTEPLA safely and effectively. See full prescribing information for FINTEPLA.
FINTEPLA ®(fenfluramine) oral solution
Initial U.S. Approval: 1973WARNING: VALVULAR HEART DISEASE and PULMONARY ARTERIAL HYPERTENSION
See full prescribing information for complete boxed warning.
RECENT MAJOR CHANGES
Boxed Warning 4/2025 Warnings and Precautions ( 5.1) 4/2025 INDICATIONS AND USAGE
FINTEPLA is indicated for the treatment of seizures associated with Dravet syndrome and Lennox-Gastaut syndrome in patients 2 years of age and older. ( 1)
DOSAGE AND ADMINISTRATION
- FINTEPLA is to be administered orally and may be taken with or without food. ( 2.2)
- Dravet Syndrome
- Lennox-Gastaut Syndrome
- Dravet Syndrome and Lennox-Gastaut Syndrome
- Dosage adjustment is recommended in patients:
DOSAGE FORMS AND STRENGTHS
Oral solution: 2.2 mg/mL fenfluramine ( 3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Decreased Appetite and Decreased Weight: Advise patients that FINTEPLA can cause decreased appetite and decreased weight. ( 5.3)
- Somnolence, Sedation, and Lethargy: Monitor for somnolence and sedation. Advise patients not to drive or operate machinery until they have gained sufficient experience on FINTEPLA. ( 5.4)
- Suicidal Behavior and Ideation: Monitor patients for suicidal behavior and thoughts. ( 5.5)
- Withdrawal of Antiepileptic Drugs: FINTEPLA should be gradually withdrawn to minimize the risk of increased seizure frequency and status epilepticus. ( 5.6)
- Serotonin Syndrome: Advise patients that serotonin syndrome is a potentially life-threatening condition and may occur with FINTEPLA, particularly with concomitant administration of FINTEPLA with other serotonergic drugs. ( 5.7)
- Increase in Blood Pressure: Monitor blood pressure during treatment. ( 5.8)
- Glaucoma: Discontinue therapy in patients with acute decrease in visual acuity or ocular pain. ( 5.9)
ADVERSE REACTIONS
The most common adverse reactions (incidence at least 10% and greater than placebo) in patients with Dravet syndrome were decreased appetite; somnolence, sedation, lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue, malaise, asthenia; ataxia, balance disorder, gait disturbance; blood pressure increased; drooling, salivary hypersecretion; pyrexia; upper respiratory tract infection; vomiting; decreased weight; fall; status epilepticus. ( 6.1)
The most common adverse reactions (incidence at least 10% and greater than placebo) in patients with Lennox-Gastaut syndrome were diarrhea; decreased appetite; fatigue; somnolence; vomiting. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact UCB, Inc. at 1-844-599-2273 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Dose adjustment is required for patients taking stiripentol plus clobazam. ( 2.2, 2.3, 7.1)
- Strong CYP1A2 or CYP2D6 inhibitors: a dose adjustment is recommended ( 2.3, 7.1)
- Strong CYP1A2, CYP2B6, or CYP3A4 inducers: it is recommended to avoid coadministration with FINTEPLA. If coadministration is necessary, consider a FINTEPLA dosage increase. ( 7.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 10/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: VALVULAR HEART DISEASE and PULMONARY ARTERIAL HYPERTENSION
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Assessments Prior to Initiating FINTEPLA
2.2 Dosing Information
2.3 Dosage Modifications for Patients with Concomitant Use of Strong CYP1A2 or CYP2D6 Inhibitors (DS and LGS)
2.4 Dosage Modifications for Patients with Severe Renal Impairment (DS and LGS)
2.5 Dosage Modifications for Patients with Mild, Moderate, and Severe Hepatic Impairment (DS and LGS)
2.6 Assessments During and After Administration of FINTEPLA
2.7 Administration Instructions
2.8 Discontinuation of FINTEPLA
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Valvular Heart Disease and Pulmonary Arterial Hypertension
5.2 FINTEPLA REMS Program
5.3 Decreased Appetite and Decreased Weight
5.4 Somnolence, Sedation, and Lethargy
5.5 Suicidal Behavior and Ideation
5.6 Withdrawal of Antiepileptic Drugs
5.7 Serotonin Syndrome
5.8 Increase in Blood Pressure
5.9 Glaucoma
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on FINTEPLA
7.2 Effects of Serotonin Receptor Antagonists
7.3 Serotonergic Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Dravet Syndrome
14.2 Lennox-Gastaut Syndrome
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: VALVULAR HEART DISEASE and PULMONARY ARTERIAL HYPERTENSION
FINTEPLA can cause valvular heart disease and pulmonary arterial hypertension [see Warnings and Precautions (5.1)].
Echocardiogram assessments are required before, during, and after treatment with FINTEPLA. The benefits versus the risks of initiating or continuing FINTEPLA must be considered, based on echocardiogram findings [see Dosage and Administration (2.1, 2.6) and Warnings and Precautions (5.1)].
Because of the risks of valvular heart disease and pulmonary arterial hypertension, FINTEPLA is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the FINTEPLA REMS [see Warnings and Precautions (5.2)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Assessments Prior to Initiating FINTEPLA
Prior to starting treatment with FINTEPLA, obtain an echocardiogram assessment to evaluate for valvular heart disease and pulmonary arterial hypertension [see Dosage and Administration (2.6)and Warnings and Precautions (5.1)] .
2.2 Dosing Information
FINTEPLA is to be administered orally and may be taken with or without food.
Dravet Syndrome
- The initial starting and maintenance dosage for patients with Dravet syndrome is 0.1 mg/kg twice daily, which can be increased weekly based on efficacy and tolerability. Table 1 provides the recommended titration schedule, if needed.
- Patients with Dravet syndrome not on concomitant stiripentol who are tolerating FINTEPLA at 0.1 mg/kg twice daily and require further reduction of seizures may benefit from a dosage increase up to a maximum recommended maintenance dosage of 0.35 mg/kg twice daily (maximum daily dosage of 26 mg).
Patients with Dravet syndrome taking concomitant stiripentol plus clobazam who are tolerating FINTEPLA at 0.1 mg/kg twice daily and require further reduction of seizures may benefit from a dosage increase up to a maximum recommended maintenance dosage of 0.2 mg/kg twice daily (maximum daily dosage of 17 mg) [see Drug Interactions (7.1)].
Lennox-Gastaut Syndrome
- The initial starting dosage for patients with Lennox-Gastaut syndrome is 0.1 mg/kg twice daily, which should be increased weekly based on tolerability. Table 1 provides the recommended titration schedule.
- Patients with Lennox-Gastaut syndrome not on concomitant stiripentol who are tolerating FINTEPLA should be titrated to the recommended maintenance dosage of 0.35 mg/kg twice daily (maximum daily dosage of 26 mg).
- Patients with Lennox-Gastaut syndrome taking concomitant stiripentol plus clobazam who are tolerating FINTEPLA should be titrated to the recommended maintenance dosage of 0.2 mg/kg twice daily (maximum daily dosage of 17 mg) [see Drug Interactions (7.1)].
Table 1: FINTEPLA Recommended Titration Schedule * Without concomitant stiripentol * With concomitant stiripentol plus clobazam Weight-based Dosage † Maximum Total Daily Dosage ‡ Weight-based Dosage † Maximum Total Daily Dosage ‡ - * For patients not on concomitant stiripentol in whom a more rapid titration is warranted, the dose may be increased every 4 days.
- † To calculate the dose volume: Weight (kg) × Weight-based dosage (mg/kg) ÷ 2.2 mg/mL = mL dose to be taken twice daily
- ‡ For maximum dosage with concomitant use of strong CYP1A2 or CYP2D6 inhibitors, in patients with severe renal impairment, or in patients with hepatic impairment see Dosage and Administration 2.3, 2.4, 2.5.
- § For patients with Dravet syndrome, dosage may be increased based on clinical response to the maximum recommended dosage, as needed.
- ¶ For patients with Lennox-Gastaut syndrome, dosage should be increased as tolerated to the recommended maintenance dosage (i.e., Day 14).
Initial Dosage § 0.1 mg/kg twice daily 26 mg 0.1 mg/kg twice daily 17 mg Day 7 0.2 mg/kg twice daily 26 mg 0.15 mg/kg twice daily 17 mg Day 14 ¶ 0.35 mg/kg twice daily 26 mg 0.2 mg/kg twice daily 17 mg 2.3 Dosage Modifications for Patients with Concomitant Use of Strong CYP1A2 or CYP2D6 Inhibitors (DS and LGS)
For patients with concomitant use of FINTEPLA with a strong CYP1A2 or CYP2D6 inhibitor, a maximum total daily dosage of 20 mg without concomitant stiripentol and 17 mg with concomitant stiripentol plus clobazam is recommended [see Drug Interactions (7.1)] .
2.4 Dosage Modifications for Patients with Severe Renal Impairment (DS and LGS)
For patients with severe renal impairment (estimated glomerular filtration rate (eGFR) 15 to 29 mL/min/1.73m 2), a maximum total daily dosage of 20 mg without concomitant stiripentol and 17 mg with concomitant stiripentol plus clobazam is recommended [see Use in Specific Populations (8.6)] .
2.5 Dosage Modifications for Patients with Mild, Moderate, and Severe Hepatic Impairment (DS and LGS)
See Table 2for dosage adjustments and recommendations for patients with hepatic impairment [see Use in Specific Populations (8.7)] .
Table 2: FINTEPLA Dosage Modifications and Recommendations for Patients with Hepatic Impairment Hepatic Impairment Classification Without concomitant stiripentol * With concomitant stiripentol plus clobazam Maximum total daily dosage Maximum total daily dosage - * titrate as recommended [see Dosage and Administration (2.2)]
Mild
(Child-Pugh A)20 mg 13 mg * Moderate
(Child-Pugh B)20 mg Use not recommended Severe
(Child-Pugh C)17 mg Use not recommended 2.6 Assessments During and After Administration of FINTEPLA
To evaluate for valvular heart disease and pulmonary arterial hypertension, obtain an echocardiogram assessment every 6 months during treatment with FINTEPLA, and 3 to 6 months after the final dose of FINTEPLA [see Warnings and Precautions (5.1)].
2.7 Administration Instructions
A calibrated measuring device (either a 3 mL or 6 mL oral syringe) will be provided by the pharmacy and is recommended to measure and administer the prescribed dose accurately [see How Supplied/Storage and Handling (16.1)] . A household teaspoon or tablespoon is not an adequate measuring device and should not be used.
Discard any unused FINTEPLA oral solution remaining after 3 months of first opening the bottle or the "Discard After" date on the bottle, whichever is sooner.
FINTEPLA is compatible with commercially available gastric and nasogastric feeding tubes.
2.8 Discontinuation of FINTEPLA
When discontinuing FINTEPLA, the dose should be decreased gradually. As with all antiepileptic drugs, abrupt discontinuation should be avoided when possible, to minimize the risk of increased seizure frequency and status epilepticus [see Warnings and Precautions (5.6)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
FINTEPLA is contraindicated in patients with:
- Hypersensitivity to fenfluramine or any of the excipients in FINTEPLA [see Description (11)]
- Concomitant use, or within 14 days of the administration, of monoamine oxidase inhibitors because of an increased risk of serotonin syndrome [see Warnings and Precautions (5.7)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Valvular Heart Disease and Pulmonary Arterial Hypertension
FINTEPLA can cause valvular heart disease (VHD) and pulmonary arterial hypertension (PAH). There is a known association between serotonergic drugs with 5-HT2B receptor agonist activity, including fenfluramine (the active ingredient in FINTEPLA), and valvular heart disease and pulmonary arterial hypertension. Although no patients receiving FINTEPLA developed valvular heart disease or pulmonary arterial hypertension in clinical trials for DS and LGS of up to 3 years in duration, cases of valvular heart disease and pulmonary arterial hypertension have been reported during use of FINTEPLA in the postmarketing setting [see Boxed Warningand Adverse Reactions (6.1)] .
Because of this risk, cardiac monitoring is required prior to starting treatment, during treatment, and after treatment with FINTEPLA concludes .Cardiac monitoring via echocardiogram can identify evidence of valvular heart disease and pulmonary arterial hypertension prior to a patient becoming symptomatic, aiding in early detection of these conditions.
Monitoring
Prior to starting treatment, patients must undergo an echocardiogram to evaluate for valvular heart disease and pulmonary arterial hypertension.
Echocardiograms should be repeated every 6 months, and once 3-6 months post-treatment with FINTEPLA.
The prescriber must consider the benefits versus the risks of initiating or continuing treatment with FINTEPLA if any of the following signs are observed via ECHO:
- Valvular abnormality or new abnormality via echocardiogram.
- VHD as indicated by mild or greater aortic regurgitation or moderate or greater mitral regurgitation, with additional characteristics of VHD (e.g., valve thickening or restrictive valve motion).
- PAH as indicated by elevated right heart/pulmonary artery pressure (PASP > 35 mm Hg).
FINTEPLA is available only through a restricted program under a REMS [see Warnings and Precautions (5.2)].
5.2 FINTEPLA REMS Program
FINTEPLA is available only through a restricted distribution program called the FINTEPLA REMS program because of the risk of valvular heart disease and pulmonary arterial hypertension [see Warnings and Precautions (5.1)].
Notable requirements of the FINTEPLA REMS Program include:
- Prescribers must be certified by enrolling in the FINTEPLA REMS program.
- Prescribers must counsel patients receiving FINTEPLA about the risk of valvular heart disease and pulmonary arterial hypertension, how to recognize signs and symptoms of valvular heart disease and pulmonary arterial hypertension, the need for baseline (pretreatment) and periodic cardiac monitoring via echocardiogram during FINTEPLA treatment, and cardiac monitoring after FINTEPLA treatment.
- Patients must enroll in the REMS program and comply with ongoing monitoring requirements [see Warnings and Precautions (5.1)].
- The pharmacy must be certified by enrolling in the REMS program and must only dispense to patients who are authorized to receive FINTEPLA.
- Wholesalers and distributors must only distribute to certified pharmacies.
Further information is available at www.FinteplaREMS.com or by telephone at 1-877-964-3649.
5.3 Decreased Appetite and Decreased Weight
FINTEPLA can cause decreases in appetite and weight. In placebo-controlled studies for DS (Study 1 and Study 2 combined), approximately 37% of patients treated with FINTEPLA reported, as an adverse reaction, decreased appetite and approximately 9% reported decreased weight, as compared to 8% and 1%, respectively, of patients on placebo .In the placebo- controlled study for LGS (Study 3), approximately 28% of patients treated with FINTEPLA reported, as an adverse reaction, decreased appetite and approximately 5% reported decreased weight, as compared to 15% and 2%, respectively, of patients on placebo [see Adverse Reactions (6.1)] . By the end of the controlled studies, 19% (Studies 1 and 2 combined) of DS patients and 7% (Study 3) of LGS patients treated with FINTEPLA had a measured decrease in weight of 7% or greater from their baseline weight, compared to 2% (Study 1 and 2) and 0% (Study 3) of patients on placebo. This measured decrease in weight appeared to be dose-related. In the controlled studies for DS, 26% of patients on FINTEPLA 0.7 mg/kg/day (Study 1), 19% of patients on FINTEPLA 0.4 mg/kg/day in combination with stiripentol (Study 2), and 13% of patients taking FINTEPLA 0.2 mg/kg/day (Study 1) experienced at least a 7% decrease in weight from baseline. In the controlled study for LGS, 9% of patients on FINTEPLA 0.7 mg/kg/day (Study 3) and 6% of patients on FINTEPLA 0.2 mg/kg/day (Study 3) experienced at least a 7% decrease in weight from baseline. Approximately half of the patients with LGS and most patients with DS resumed the expected measured increases in weight during the open-label extension studies. Given the frequency of these adverse reactions, the growth of pediatric patients treated with FINTEPLA should be carefully monitored. Weight should be monitored regularly during treatment with FINTEPLA, and dose modifications should be considered if a decrease in weight is observed.
5.4 Somnolence, Sedation, and Lethargy
FINTEPLA can cause somnolence, sedation, and lethargy. In controlled studies for DS (Study 1 and Study 2 combined), the incidence of somnolence, sedation, and lethargy was 25% in patients treated with FINTEPLA, compared with 11% of patients on placebo. In the controlled study for LGS (Study 3), the incidence of somnolence, sedation, and lethargy was 19% in patients treated with FINTEPLA, compared with 16% of patients on placebo. In general, these effects may diminish with continued treatment [see Adverse Reactions (6.1)].
Other central nervous system (CNS) depressants, including alcohol, could potentiate these effects of FINTEPLA. Prescribers should monitor patients for somnolence and sedation and should advise patients not to drive or operate machinery until they have gained sufficient experience on FINTEPLA to gauge whether it adversely affects their ability to drive or operate machinery.
5.5 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including FINTEPLA, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with an AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs that did not include FINTEPLA showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as 1 week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5-100 years) in the clinical trials analyzed. Table 3 shows absolute and relative risk by indication for all evaluated AEDs.
Table 3: Risk of Suicidal Thoughts or Behaviors by Indication for Antiepileptic Drugs in the Pooled Analysis Indication Placebo Patients with Events per 1000 Patients Drug Patients with Events per 1000 Patients Relative Risk: Incidence of Events in Drug Patients/ Incidence in Placebo Patients Risk Difference: Additional Drug Patients with Events per 1000 Patients Epilepsy 1.0 3.4 3.5 2.4 Psychiatric 5.7 8.5 1.5 2.9 Other 1.0 1.8 1.9 0.9 Total 2.4 4.3 1.8 1.9 The relative risk for suicidal thoughts or behavior was higher in clinical trials in patients with epilepsy than in clinical trials in patients with psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing FINTEPLA or any other AED must balance the risk of suicidal thoughts or behaviors with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
5.6 Withdrawal of Antiepileptic Drugs
As with most AEDs, FINTEPLA should generally be withdrawn gradually because of the risk of increased seizure frequency and status epilepticus. If withdrawal is needed because of a serious adverse reaction, rapid discontinuation can be considered.
5.7 Serotonin Syndrome
Serotonin syndrome, a potentially life-threatening condition, may occur with FINTEPLA, particularly with concomitant administration of FINTEPLA with other serotonergic drugs, including, but not limited to, selective serotonin-norepinephrine reuptake inhibitors (SNRIs), selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants (TCAs), bupropion, triptans, dietary supplements (e.g., St. John's Wort, tryptophan), drugs that impair metabolism of serotonin (including monoamine oxidase inhibitors [MAOIs], which are contraindicated with FINTEPLA [see Contraindications (4)], dextromethorphan, lithium, tramadol, and antipsychotics with serotonergic agonist activity. Patients should be monitored for the emergence of signs and symptoms of serotonin syndrome, which include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular signs (e.g., hyperreflexia, incoordination), and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). If serotonin syndrome is suspected, treatment with FINTEPLA should be stopped immediately and symptomatic treatment should be started.
5.8 Increase in Blood Pressure
FINTEPLA can cause an increase in blood pressure [see Adverse Reactions (6.1)]. Rare cases of significant elevation in blood pressure, including hypertensive crisis, has been reported in adult patients treated with fenfluramine, including patients without a history of hypertension. In clinical trials of up to 3 years in duration, no pediatric or adult patient receiving FINTEPLA developed a hypertensive crisis. Monitor blood pressure in patients treated with FINTEPLA.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in labeling:
- Valvular Heart Disease and Pulmonary Arterial Hypertension [see Warnings and Precautions (5.1)]
- Decreased Appetite and Decreased Weight [see Warnings and Precautions (5.3)]
- Somnolence, Sedation, and Lethargy [see Warnings and Precautions (5.4)]
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.5)]
- Withdrawal of Antiepileptic Drugs [see Warnings and Precautions (5.6)]
- Serotonin Syndrome [see Warnings and Precautions (5.7)]
- Increase in Blood Pressure [see Warnings and Precautions (5.8)]
- Glaucoma [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In controlled and uncontrolled trials in patients with Dravet syndrome (DS), 341 patients were treated with FINTEPLA, including 312 patients treated for more than 6 months, 284 patients treated for more than 1 year, and 138 patients treated for more than 2 years.
In controlled and uncontrolled trials in patients with Lennox-Gastaut syndrome (LGS), 262 patients were treated with FINTEPLA, including 219 patients treated for more than 6 months, 172 patients treated for more than 1 year, and 127 patients treated for more than 2 years.
Dravet Syndrome
In placebo-controlled trials of patients with DS taking concomitant standard of care AEDs, 122 patients were treated with FINTEPLA and 84 patients received placebo [see Clinical Studies (14.1)] . The duration of treatment in these trials was 16 weeks (Study 1) or 17 weeks (Study 2).
In Study 1 and Study 2, the mean age was 9 years (range 2 to 19 years) and approximately 46% of patients were female and 74% were White. All patients were receiving at least one other AED.
In Study 1 and Study 2, the rates of discontinuation as a result of any adverse reaction were 13%, 0%, and 7% for patients treated with FINTEPLA 0.7 mg/kg/day, 0.2 mg/kg/day, and 0.4 mg/kg/day in combination with stiripentol, respectively, compared to 6% for patients on placebo. The most frequent adverse reaction leading to discontinuation in the patients treated with any dose of FINTEPLA was somnolence (3%).
The most common adverse reactions that occurred in patients treated with FINTEPLA (incidence at least 10% and greater than placebo) were decreased appetite; somnolence, sedation, lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue, malaise, asthenia; ataxia, balance disorder, gait disturbance; blood pressure increased; drooling, salivary hypersecretion; pyrexia; upper respiratory tract infection; vomiting; decreased weight; fall; status epilepticus.
Table 4 lists the adverse reactions that were reported in 5% or more of patients treated with FINTEPLA and at a rate greater than those on placebo during the titration and maintenance phases of Study 1 and Study 2.
Table 4: Adverse Reactions in 5% or More of Patients Treated with FINTEPLA and Greater Than Placebo in Placebo-Controlled Trials for Dravet Syndrome (Study 1 and 2) Adverse Reaction FINTEPLA Dose Group Combined Placebo Group * Study 1 Study 2 0.2 mg/kg/day 0.7 mg/kg/day 0.4 mg/kg/day † N=39
%N=40
%N=43
%N=84
%- * Patients in placebo groups from Studies 1 and 2 were pooled.
- † 0.4 mg/kg/day was not an intermediate dose. Patients on the 0.4 mg/kg/day dose were also taking concomitant stiripentol plus clobazam, which increases exposure of FINTEPLA.
- ‡ Consisted of trace and mild mitral regurgitation, and trace aortic regurgitation, which are considered physiologic.
Decreased appetite 23 38 49 8 Somnolence, sedation, lethargy 26 25 23 11 Abnormal echocardiogram ‡ 18 23 9 6 Diarrhea 31 15 23 6 Constipation 3 10 7 0 Fatigue, malaise, asthenia 15 10 30 5 Ataxia, balance disorder, gait disturbance 10 10 7 1 Abnormal behavior 0 8 9 0 Blood pressure increased 13 8 0 5 Drooling, salivary hypersecretion 13 8 2 0 Hypotonia 0 8 0 0 Rash 8 8 5 4 Blood prolactin increased 0 5 0 0 Chills 0 5 2 0 Decreased activity 0 5 0 1 Dehydration 0 5 0 0 Insomnia 0 5 5 2 Pyrexia 15 5 21 14 Stereotypy 0 5 0 0 Upper respiratory tract infection 21 5 7 10 Vomiting 10 5 5 8 Weight decreased 13 5 7 1 Croup 5 3 0 1 Ear infection 8 3 9 5 Gastroenteritis 8 3 2 0 Increased heart rate 5 3 0 2 Irritability 0 3 9 2 Rhinitis 8 3 7 2 Tremor 3 3 9 0 Urinary incontinence 5 3 0 0 Decreased blood glucose 0 0 9 1 Bronchitis 3 0 9 1 Contusion 5 0 0 0 Eczema 0 0 5 0 Enuresis 5 0 0 0 Fall 10 0 0 4 Headache 8 0 0 2 Laryngitis 0 0 5 0 Negativism 5 0 0 0 Status epilepticus 3 0 12 2 Urinary tract infection 5 0 5 0 Viral infection 0 0 5 1 Lennox-Gastaut Syndrome
In the placebo-controlled trial of patients with LGS taking concomitant standard of care AEDs (Study 3), 176 patients were treated with FINTEPLA and 87 patients received placebo [see Clinical Studies (14.2)] . The duration of treatment in this trial was 16 weeks. The mean age was 13.7 years (range 2 to 35 years) and 29% of patients were at least 18 years of age, 45% of patients were female, and 79% were White. All patients were receiving at least one other AED.
The rates of discontinuation as a result of any adverse reaction were 6% and 5% for patients treated with FINTEPLA 0.7 mg/kg/day and 0.2 mg/kg/day, respectively, compared to 1% for patients on placebo. The most frequent adverse reactions leading to discontinuation in the patients treated with any dose of FINTEPLA were seizure (2%) and somnolence (2%).
The common adverse reactions that occurred in patients treated with FINTEPLA (incidence at least 10% and greater than placebo) were diarrhea; decreased appetite; fatigue; somnolence; vomiting.
Table 5 lists the adverse reactions that were reported in 5% or more of patients treated with FINTEPLA and at a rate greater than those on placebo during the titration and maintenance phases of Study 3.
Table 5: Adverse Reactions in 5% or More of Patients Treated with FINTEPLA and Greater Than Placebo in the Placebo-Controlled Trial for Lennox Gastaut Syndrome (Study 3) Adverse Reaction FINTEPLA Dose Group Study 3 Placebo Group 0.2 mg/kg/day 0.7mg/kg/day N=89
%N=87
%N=87
%Decreased appetite 20 36 12 Fatigue, malaise, asthenia 14 24 16 Somnolence, sedation, lethargy 12 22 16 Diarrhea 11 13 5 Constipation 6 9 6 Vomiting 14 8 6 Weight decreased 2 8 2 Upper respiratory tract infection 8 7 3 Seizure 9 5 7 Irritability 8 3 6 Echocardiographic Safety Assessments of Valvular Heart Disease and Pulmonary Arterial Hypertension
Valvular heart disease and pulmonary arterial hypertension were evaluated in the placebo- controlled and open-label extension studies via echocardiography for up to 3 years in duration for 341 DS patients and 263 LGS patients [see Warnings and Precautions (5.1)]. Screening for valvular heart disease assessed for mild or greater aortic regurgitation or moderate or greater mitral regurgitation, and assessed for additional characteristics of VHD (e.g., valve thickening or restrictive valve motion).
In these clinical studies, two patients with LGS exhibited mild aortic regurgitation (AR) but neither patient had any cardiac signs or symptoms or evidence of valvular structural changes. Neither patient had VHD. The rates of mild AR are consistent with those seen in the screening period prior to treatment (3 patients in LGS and 1 patient in DS clinical trials).
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of FINTEPLA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Psychiatric disorders: aggression
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on FINTEPLA
Stiripentol Plus Clobazam
Coadministration of FINTEPLA with stiripentol plus clobazam, with or without valproate, increases fenfluramine plasma concentrations [see Clinical Pharmacology (12.3)]. If FINTEPLA is coadministered with stiripentol plus clobazam, the maximum daily dosage of FINTEPLA is 0.2 mg/kg twice daily (maximum daily dosage of 17 mg) [see Dosage and Administration (2.2)] .
Strong CYP1A2, CYP2B6, or CYP3A Inducers
Coadministration of FINTEPLA with strong CYP1A2, CYP2B6, or CYP3A inducers will decrease fenfluramine plasma concentrations, which may lower the efficacy of FINTEPLA [see Clinical Pharmacology (12.3)] .
It is recommended to avoid coadministration of strong CYP1A2, CYP2B6 or CYP3A inducers. If coadministration of a strong CYP1A2, CYP2B6, or CYP3A inducer with FINTEPLA is necessary, monitor the patient for reduced efficacy and consider increasing the dosage of FINTEPLA as needed; however, do not exceed the maximum daily dosage of FINTEPLA [see Dosage and Administration (2.2)].
If a strong CYP1A2, CYP2B6, or CYP3A inducer is discontinued during maintenance treatment with FINTEPLA, consider gradual reduction in the FINTEPLA dosage to the dose administered prior to initiating the inducer [see Warnings and Precautions (5.6)].
Strong CYP1A2 or CYP2D6 Inhibitors
Coadministration of FINTEPLA with strong CYP1A2 or CYP2D6 inhibitors will increase fenfluramine plasma concentrations [see Clinical Pharmacology (12.3)] . If FINTEPLA is coadministered with strong CYP1A2 or CYP2D6 inhibitors, the maximum daily dosage of FINTEPLA is 20 mg [see Dosage and Administration (2.3)].
If a strong CYP1A2 or CYP2D6 inhibitor is discontinued during maintenance treatment with FINTEPLA, consider gradual increase in the FINTEPLA dosage to the dose recommended without CYP1A2 or CYP2D6 inhibitors; however, do not exceed the maximum daily dosage of FINTEPLA [see Dosage and Administration (2.2)].
If FINTEPLA is coadministered with stiripentol and a strong CYP1A2 or CYP2D6 inhibitor, do not exceed the maximum daily dosage of FINTEPLA of 17 mg [see Dosage and Administration (2.3)].
7.2 Effects of Serotonin Receptor Antagonists
Cyproheptadine and potent 5-HT1A, 5-HT1D, 5-HT2A, and 5-HT2C serotonin receptor antagonists may decrease the efficacy of FINTEPLA. If cyproheptadine or potent 5--HT1A, 5--HT1D, 5-HT2A, or 5-HT2C serotonin receptor antagonists are coadministered with FINTEPLA, patients should be monitored appropriately.
7.3 Serotonergic Drugs
Concomitant administration of FINTEPLA and drugs (e.g., SSRIs, SNRIs, TCAs, MAO inhibitors, trazodone, etc.), over-the-counter medications (e.g., dextromethorphan), or herbal supplements (e.g., St. John's Wort) that increase serotonin may increase the risk of serotonin syndrome [see Warnings and Precautions (5.7)]. Concomitant use of FINTEPLA is contraindicated within 14 days of taking MAOIs. Use FINTEPLA with caution in patients taking other medications that increase serotonin.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antiepileptic drugs (AEDs), such as FINTEPLA, during pregnancy. Encourage women who are taking FINTEPLA during pregnancy to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry by calling the toll-free number 1-888-233-2334 or visiting http://www.aedpregnancyregistry.org.
Risk Summary
There are no data on FINTEPLA use in pregnant women. Available data from epidemiologic studies with fenfluramine or dexfenfluramine are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. FINTEPLA can cause decreased appetite and decreased weight [see Warnings and Precautions (5.3)]; monitor for adequate weight gain during pregnancy. In animal studies, administration of fenfluramine throughout organogenesis (rat and rabbit) or throughout gestation and lactation (rat) resulted in adverse effects on development (fetal malformations, embryofetal and offspring mortality and growth impairment) in the presence of maternal toxicity at clinically relevant maternal plasma levels of fenfluramine and its major active metabolite (see Data) .
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-associated Maternal and/or Embryofetal Risk
Epilepsy, with or without exposure to antiepileptic drugs, has been associated with several adverse outcomes during pregnancy, including preeclampsia, preterm labor, antepartum and postpartum hemorrhage, placental abruption, poor fetal growth, prematurity, fetal death, and maternal mortality. The risk of maternal or fetal injury may be greatest for patients with untreated or poorly controlled convulsive seizures. Women with epilepsy who become pregnant should not abruptly discontinue antiepileptic drugs, including FINTEPLA, due to the risk of status epilepticus or severe seizures, which may be life-threatening [see Warnings and Precautions (5.6)].
Data
Animal Data
Oral administration of fenfluramine (0, 4.5, 8.6, or 34.6 mg/kg/day) to pregnant rats during organogenesis resulted in decreased fetal body weights and marked increases in fetal malformations (external, visceral, and skeletal) at the highest dose tested, which was associated with maternal toxicity. At the no-effect dose (8.6 mg/kg/day) for adverse effects on embryofetal development in rats, maternal plasma exposures (AUC) of fenfluramine and norfenfluramine (the major metabolite) were approximately 2 and 5 times, respectively, those in humans at the maximum recommended human dose (MRHD) of 26 mg/day.
Oral administration of fenfluramine (0, 4.3, 8.6, 13.0 mg/kg/day) to pregnant rabbits throughout organogenesis resulted in increased embryofetal mortality at all doses and increases in fetal malformations (external and skeletal) at the highest dose tested, which was associated with maternal toxicity. A no-adverse-effect dose for adverse effects on embryofetal development in rabbits was not identified. At the lowest dose tested in rabbits (4.3 mg/kg/day), maternal plasma exposures of fenfluramine and norfenfluramine were lower than those in humans at the MRHD.
Oral administration of fenfluramine (0, 4.3, 8.6, or 34.6 mg/kg/day) to female rats throughout gestation and lactation resulted in marked increases in stillborn pups and neonatal offspring deaths at the highest dose tested and delayed growth and reflex development during the pre- weaning period at all doses. Maternal body weight gain was decreased at all doses during pregnancy and at the two highest doses during lactation. A no-effect dose for adverse effects on pre- and postnatal development in rats was not determined. At the lowest dose tested in rats (4.3 mg/kg/day), maternal plasma exposures of fenfluramine and norfenfluramine were approximately 0.5 and 3 times, respectively, those in humans at the MRHD.
8.2 Lactation
Risk Summary
There are no data on the presence of fenfluramine or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for FINTEPLA and any potential adverse effects on the breastfed infant from FINTEPLA or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Infertility
In animal studies, oral administration of fenfluramine resulted in adverse reproductive effects in males and females at clinically relevant doses in the presence of parental toxicity [see Nonclinical Toxicology (13.1)] .
8.4 Pediatric Use
The safety and effectiveness of FINTEPLA for the treatment of seizures associated with DS and LGS have been established in patients 2 years of age and older.
Use of FINTEPLA for the treatment of seizures associated with DS in patients 2 years of age and older is supported by two randomized, double-blind, placebo-controlled trials in 202 patients 2 to 18 years of age. Use of FINTEPLA for the treatment of seizures associated with LGS is supported by a randomized, double-blind, placebo-controlled study in 263 patients aged 2 to 35 years, including 187 patients less than 18 years [see Boxed Warning, Warnings and Precautions (5), Adverse Reaction (6.1), and Clinical Studies (14)].
FINTEPLA can cause decreases in appetite and weight. The growth of pediatric patients treated with FINTEPLA should be carefully monitored.
Safety and effectiveness in patients less than 2 years of age have not been established.
Juvenile Animal Data
Oral administration of fenfluramine (0, 3.0, 7.8, or 17.3 mg/kg/day) to young rats for 10 weeks starting on postnatal day 7 resulted in reduced body weight and neurobehavioral changes (decreased locomotor activity and learning and memory deficits) at all doses tested. Neurobehavioral effects persisted after dosing was discontinued. Bone size was decreased at the mid and high doses; brain size was decreased at the highest dose. Partial or complete recovery was seen for these endpoints. A no-effect dose for postnatal developmental toxicity was not identified. The lowest dose tested (3.0 mg/kg/day) was associated with plasma fenfluramine exposures (AUC) less than that in humans at the maximum recommended human dose (MRHD) of 26 mg/day and norfenfluramine (metabolite) exposures (AUC) approximately 2 times that in humans at the MRHD.
8.5 Geriatric Use
Clinical studies of FINTEPLA for the treatment of DS or LGS did not include patients 65 years of age and over to determine whether they respond differently from younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
In patients with estimated glomerular filtration rate (eGFR) 15 to 29 mL/min/1.73m 2, do not exceed the maximum daily dosage of FINTEPLA of 20 mg .In patients with eGFR 15 to 29 ml/min/1.73m 2and concomitant stiripentol use, do not exceed the maximum daily dosage of FINTEPLA of 17 mg [see Dosage and Administration (2.4)and Clinical Pharmacology (12.3)]. FINTEPLA has not been studied in patients with eGFR < 15 mL/min/1.73m 2.
8.7 Hepatic Impairment
Combined molar exposures of fenfluramine and norfenfluramine were increased in subjects with various degrees of hepatic impairment (Child-Pugh Class A, B, and C), necessitating a dosage adjustment in these patients [see Dosage and Administration (2.5)and Clinical Pharmacology (12.3)] .
-
10 OVERDOSAGE
Overdose has not been observed in the FINTEPLA clinical trial program. However, overdose of fenfluramine, the active ingredient in FINTEPLA, has been reported at higher doses than those included in the clinical trial program. Some of the cases were fatal. Events reported after overdose include mydriasis, tachycardia, flushing, tremors/twitching/muscle spasms, agitation/restlessness/anxiety, increased muscle tone/rigor/opisthotonos, respiratory distress or failure, and seizure. Seizure, coma, and cardiorespiratory arrest were reported in most of the fatal overdoses.
There is no available specific antidote to the overdose reactions of FINTEPLA. In the event of overdose, standard medical practice for the management of drug overdosage should be used. An adequate airway, oxygenation, and ventilation should be ensured; monitoring of cardiac rhythm and vital sign measurement is recommended. A certified poison control center should be contacted for updated information on the management of overdose with FINTEPLA.
-
11 DESCRIPTION
FINTEPLA oral solution contains 2.2 mg/mL fenfluramine, equivalent to 2.5 mg/mL of the hydrochloride salt.
The active ingredient, fenfluramine hydrochloride, is designated chemically as N-ethyl-α- methyl-3-(trifluoromethyl)phenethylamine hydrochloride.
The structural formula is:
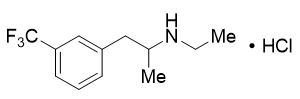
Fenfluramine hydrochloride is a white to off-white crystalline solid. The pKa of fenfluramine is 10.2.
FINTEPLA is a clear, colorless solution, pH 5.
FINTEPLA contains the following inactive ingredients: cherry flavor, citric acid, ethylparaben hydroxyethylcellulose, methylparaben, potassium citrate, sucralose, and water.
FINTEPLA contains no ingredient made from gluten-containing grain (wheat, barley, or rye), and contains not more than 0.1% of carbohydrates, which is solely derived from the cherry flavor.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The precise mechanism by which fenfluramine exerts its therapeutic effects in the treatment of seizures associated with Dravet syndrome and Lennox-Gastaut syndrome is unknown. Fenfluramine and the metabolite, norfenfluramine, exhibit agonist activity at serotonin 5-HT2 receptors. There is an association between serotonergic drugs with 5-HT2B receptor agonist activity, including fenfluramine and norfenfluramine, and valvular heart disease and pulmonary arterial hypertension.
12.3 Pharmacokinetics
The pharmacokinetics of fenfluramine and norfenfluramine were studied in healthy subjects, in pediatric patients with DS, and in pediatric and adult patients with LGS. The steady-state systemic exposure (C maxand AUC) of fenfluramine was slightly greater than dose proportional over the dose range of 13 to 51.8 mg twice-daily fenfluramine (i.e., 1 to 4 times the maximum recommended dose). In pediatric patients with DS who received FINTEPLA 0.7 mg/kg/day, up to a total daily dose of 26 mg fenfluramine, the geometric mean steady-state fenfluramine (coefficient of variation) C maxwas 68.0 (41%) ng/mL and AUC 0-24hwas 1390 (44%) ng*h/mL.
Absorption
Fenfluramine has a time to maximum plasma concentration (T max) of 3 to 5 hours at steady state. The absolute bioavailability of fenfluramine is approximately 68-74%. There was no effect of food on the pharmacokinetics of fenfluramine or norfenfluramine.
Distribution
The geometric mean (CV%) apparent volume of distribution (Vz/F) of fenfluramine is 11.9 (16.5%) L/kg following oral administration of FINTEPLA in healthy subjects. Fenfluramine is 50% bound to human plasma proteins in vitro and binding is independent of drug concentrations.
Elimination
The elimination half-life of fenfluramine was 20 hours and the geometric mean (CV%) clearance (CL/F) was 24.8 (29%) L/h, following oral administration of FINTEPLA in healthy subjects.
Metabolism
Over 75% of fenfluramine is metabolized to norfenfluramine prior to elimination, primarily by CYP1A2, CYP2B6, and CYP2D6. Other CYP enzymes involved to a minor extent are CYP2C9, CYP2C19, and CYP3A4/5. Both fenfluramine and norfenfluramine are pharmacologically active. Norfenfluramine is further deaminated and oxidized to form inactive metabolites.
Excretion
Most of an orally administered dose of fenfluramine (greater than 90%) is excreted in the urine as fenfluramine, norfenfluramine, or other metabolites with fenfluramine and norfenfluramine accounting for less than 25% of the total; less than 5% is found in feces.
Specific Populations
The effect of age (range: 2 to 50 years), sex, and race had no clinically meaningful effect on the pharmacokinetics of fenfluramine.
Patients with Renal Impairment
In a dedicated clinical study comparing the pharmacokinetics of a single dose of 0.35 mg/kg FINTEPLA in subjects with severe renal impairment (eGFR < 30 mL/min/1.73m 2determined by MDRD) and matched healthy volunteers, C maxand AUC 0-infof fenfluramine increased by 20% and 88%, respectively, and C maxand AUC 0-infof norfenfluramine increased by 13% and 21%, respectively in subjects with severe renal impairment [see Use in Specific Populations (8.6)] . FINTEPLA has not been studied in patients with eGFR < 15 mL/min/1.73m 2(determined by MDRD). It is not known if fenfluramine or norfenfluramine is dialyzable.
Patients with Hepatic Impairment
In a study comparing the pharmacokinetics of a single dose of 0.35 mg/kg FINTEPLA in subjects with mild, moderate, or severe hepatic impairment (Child-Pugh Class A, B, or C) and subjects with normal liver function, AUC 0-tof fenfluramine increased by 95%, 113%, and 185% in subjects with mild, moderate, and severe hepatic impairment, respectively. C maxof fenfluramine increased by 19%, 16%, and 29% in subjects with mild, moderate, and severe hepatic impairment, respectively. AUC 0-tof norfenfluramine increased by 18% in mild hepatic impairment, 4% in moderate hepatic impairment, and decreased by 11% in severe hepatic impairment. C maxof norfenfluramine decreased by 21%, 36%, and 45% in subjects with mild, moderate, and severe hepatic impairment, respectively. Combined molar AUC 0-tof fenfluramine and norfenfluramine increased by 55%, 56%, and 82% in subjects with mild, moderate, and severe hepatic impairment, respectively. Combined molar C maxof fenfluramine and norfenfluramine increased by 7.5%, 1.3%, and 8% in subjects with mild, moderate, and severe hepatic impairment, respectively. The maximum daily dosage of FINTEPLA should be reduced in patients with mild hepatic impairment with/without stiripentol plus clobazam. The maximum daily dosage of FINTEPLA should be reduced in patients with moderate or severe hepatic impairment without stiripentol plus clobazam [see Dosage and Administration (2.5)and Use in Specific Populations (8.7)].
Effect of a single dose of stiripentol, clobazam, and valproic acid combination:
Coadministration of a single 0.7 mg/kg dose of FINTEPLA, with a single dose of a stiripentol, clobazam, and valproic acid combination in healthy volunteers, increased the AUC 0-infof fenfluramine by 69% and the C maxby 18%, and decreased the AUC 0-72hours of norfenfluramine by 41% and the C maxby 42%, as compared to FINTEPLA administered alone.
Effect of steady state stiripentol plus clobazam, with or without valproate:
Fenfluramine pharmacokinetic data were collected from patients after receiving multiple fenfluramine administrations in Study 1 as well as Study 2. Population pharmacokinetic modeling and simulation were used to assess the effect of stiripentol plus clobazam with or without valproate on fenfluramine pharmacokinetics. The effect of stiripentol plus clobazam, with or without valproate, on fenfluramine pharmacokinetics is greater when FINTEPLA is at steady-state than for the first dose of FINTEPLA. At steady state in the patient population, the coadministration of 0.1 mg/kg twice daily (0.2 mg/kg/day), maximum 17 mg/day, of FINTEPLA with stiripentol plus clobazam with or without valproate, is expected to result in a 166% increase in fenfluramine AUC 0-24and a 38% decrease in norfenfluramine AUC 0-24, as compared to 0.2 mg/kg/day, maximum 26 mg/day, FINTEPLA dose administered alone [see Dosage and Administration (2.1, 2.2)and Drug Interactions (7.1)] .
Effect of steady state cannabidiol:
Coadministration of a single 0.35 mg/kg dose of FINTEPLA with repeated doses of cannabidiol increased the AUC 0-infof fenfluramine by 59% and the C maxby 10%, and decreased the AUC 0-infof norfenfluramine by 22% and the C maxby 33%, as compared to FINTEPLA administered alone. This interaction is not expected to be clinically significant.
Effect of strong CYP1A2 or CYP2D6 inhibitors:
Coadministration of a single 0.35 mg/kg dose of FINTEPLA with fluvoxamine (a strong CYP1A2 inhibitor) at steady state (50 mg once daily) in healthy volunteers increased the AUC 0- infof fenfluramine by 102% and the C maxby 22%, and decreased the AUC 0-infof norfenfluramine by 22% and the C maxby 44%, as compared to FINTEPLA administered alone [see Drug Interactions (7.1)].
Coadministration of a single 0.35 mg/kg dose of FINTEPLA with paroxetine (a strong CYP2D6 inhibitor) at steady state (30 mg once daily) in healthy volunteers increased the AUC 0-infof fenfluramine by 81% and the C maxby 13%, and decreased the AUC 0-infof norfenfluramine by 13% and the C maxby 29%, as compared to FINTEPLA administered alone [see Drug Interactions (7.1)].
Effect of strong CYP1A2, CYP2B6 or CYP3A inducers:
Coadministration of a single 0.35 mg/kg dose of FINTEPLA with rifampin (a CYP1A2, CYP2B6, and CYP3A inducer) at steady state (600 mg once daily) in healthy volunteers decreased the AUC 0-infof fenfluramine by 58% and the C maxby 40%, and decreased the AUC 0-infof norfenfluramine by 50%, and increased the C maxof norfenfluramine by 13%, as compared to FINTEPLA administered alone [see Drug Interactions (7.1)].
Effect of FINTEPLA on other drugs:
Coadministration of a single 0.7 mg/kg dose of FINTEPLA, with a single dose of a stiripentol, clobazam, and valproic acid combination, did not affect the pharmacokinetics of stiripentol, nor the pharmacokinetics of clobazam or its N‑desmethyl-metabolite norclobazam, nor the pharmacokinetics of valproic acid, as compared to the stiripentol, clobazam, and valproic acid combination alone. Coadministration of a single 0.35 mg/kg dose of FINTEPLA, with repeated doses of cannabidiol, did not affect the pharmacokinetics of cannabidiol, as compared to cannabidiol alone.
In Vitro Studies
Fenfluramine is primarily metabolized by CYP1A2, CYP2B6, and CYP2D6 in vitro. Other CYP enzymes involved to a minor extent are CYP2C9, CYP2C19, and CYP3A4/5.
Effect of fenfluramine and norfenfluramine on CYP Substrates: fenfluramine and norfenfluramine are not inhibitors or inducers of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4 at clinically relevant concentrations.
Effect of transporters on fenfluramine and norfenfluramine: fenfluramine and norfenfluramine are not substrates of the P-g, BCRP, OAT1, OAT3, OCT2, MATE1, or MATE2-K transporters.
Effect of FINTEPLA on Transporters: fenfluramine and norfenfluramine are not inhibitors of P-gp, BCRP, OAT1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, or MATE2-K transporters.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Oral administration of fenfluramine to Tg.rasH2 mice (0, 4.3, 13.0, 34.6, or 51.8 mg/kg/day) for 26 weeks and to male and female rats (0, 0.9, 2.2, or 6.9 mg/kg/day) for 89 and 97 weeks, respectively, resulted in no evidence of drug-induced tumors in either species. In rats, plasma exposures (AUC) of fenfluramine and norfenfluramine (the major metabolite) at the highest dose tested were approximately 5 and 11 times, respectively, those in humans at the maximum recommended human dose (MRHD) of 26 mg/day.
Mutagenesis
Fenfluramine was negative in an in vitro bacterial mutation (Ames) assay and an in vivo micronucleus and comet assay in rats.
Impairment of Fertility
Oral administration of fenfluramine (0, 3.0, 6.9, or 17.3 mg/kg/day) to male and female rats prior to and throughout mating and continuing in females to day 7 of gestation resulted in a decrease in fertility and increases in abnormal sperm and epithelial vacuolation of the epididymis at the highest dose tested and altered estrous cyclicity, decreased corpora lutea and implantations, and increased embryolethality at the mid and high dose. These doses were associated with parental toxicity. The no-effect doses for adverse effects on fertility and reproductive performance in rats (6.9 and 3.0 mg/kg/day in males and females, respectively) were associated with plasma fenfluramine exposures (AUC) approximately 3 and 0.6 times, respectively, and norfenfluramine exposures approximately 5 and 3 times, respectively, those in humans at the MRHD.
-
14 CLINICAL STUDIES
14.1 Dravet Syndrome
The effectiveness of FINTEPLA for the treatment of seizures associated with DS in patients 2 years of age and older was established in two randomized, double-blind, placebo-controlled trials in patients 2 to 18 years of age.
Study 1 (N=117) compared a 0.7 mg/kg/day and a 0.2 mg/kg/day dose of FINTEPLA with placebo in patients who were not receiving stiripentol (NCT02682927 and NCT02826863). Study 2 (N=85) compared a 0.4 mg/kg/day dose of FINTEPLA with placebo in patients who were receiving stiripentol and either clobazam, valproate, or both (NCT02926898). In both studies, patients had a clinical diagnosis of DS and were inadequately controlled on at least one AED or other antiseizure treatment including vagal nerve stimulation or a ketogenic diet. Both trials had a 6-week baseline period, during which patients were required to have a minimum of 6 convulsive seizures while on stable AED therapy. Convulsive seizures included tonic, clonic, generalized tonic-clonic, tonic-atonic, secondarily generalized tonic-clonic, hemiclonic, and focal with observable motor signs. The baseline period was followed by randomization into a 2- week (Study 1) or 3-week (Study 2) titration period and a subsequent 12-week maintenance period, where the dose of FINTEPLA remained stable.
In Study 1, 98% of patients were taking between 1 and 4 concomitant AEDs. The most frequently used concomitant AEDs (in at least 25% of patients), were valproate (61%), clobazam (59%), and topiramate (25%). In Study 2, 100% of patients were taking between 2 and 4 concomitant AEDs. The most frequently used concomitant AEDs (in at least 25% of patients), were stiripentol (100%), clobazam (94%), and valproate (89%).
The primary efficacy endpoint in both studies was the change from baseline in the frequency of convulsive seizures per 28 days during the combined 14-week (Study 1) or 15-week (Study 2) titration and maintenance periods (i.e., treatment period). The median longest interval between convulsive seizures was also assessed.
In Study 1 and Study 2, the reduction in convulsive seizure frequency per 28 days was statistically significantly greater for all dose groups of FINTEPLA compared to placebo (Table 6). A reduction in convulsive seizures was observed within 3 to 4 weeks of starting FINTEPLA, and the effect remained generally consistent over the 14- or 15-week treatment period.
Table 6: Change in Convulsive Seizure Frequency During the Treatment Period in Patients with Dravet Syndrome (Study 1 and Study 2) Convulsive Seizure Frequency
(per 28 days)Placebo FINTEPLA
0.2 mg/kg/dayFINTEPLA
0.7 mg/kg/dayFINTEPLA
0.4 mg/kg/day±All 0.4 mg/kg/day patients were also taking concomitant stiripentol, which increases the exposure of FINTEPLA. - * Derived from the primary analysis model
Study 1 N=39 N=38 N=40 NA Baseline Period Median 29.4 18.1 18.7 NA % Difference Relative to Placebo * -31.7% -70.0% NA p-value compared to placebo 0.043 <0.001 Study 2 N=42 NA NA N=43 Baseline Period Median 11.5 NA NA 15.0 % Difference Relative to Placebo * NA NA -59.5% p-value compared to placebo <0.001 Figure 1 and Figure 2 display the percentage of patients by category of seizure response from baseline in convulsive seizure frequency (per 28 days) during the treatment period in Study 1 and Study 2, respectively.
Figure 1: Proportion of Patients by Category of Seizure Response for FINTEPLA and Placebo in Patients with Dravet Syndrome (Study 1)
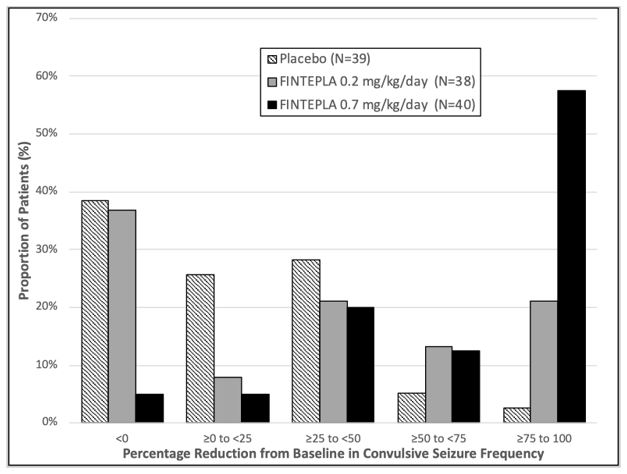
Figure 2: Proportion of Patients by Category of Seizure Response for FINTEPLA and Placebo in Patients with Dravet Syndrome (Study 2)
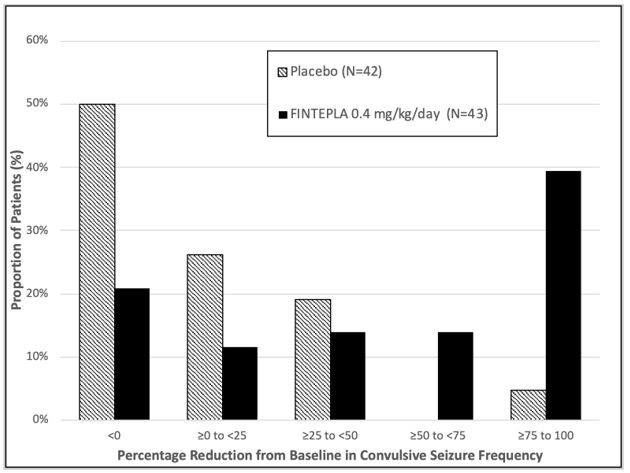
In Study 1, 3 of 40 (8%) patients in the FINTEPLA 0.7 mg/kg/day group and 3 of 38 (8%) patients in the FINTEPLA 0.2 mg/kg/day group reported no convulsive seizures during the 14-week treatment period, compared to 0 patients in the placebo group. In Study 2, 1 of 43 (2%) patients in the FINTEPLA 0.4 mg/kg/day group reported no convulsive seizures during the 15-week treatment period, compared to 0 patients in the placebo group.
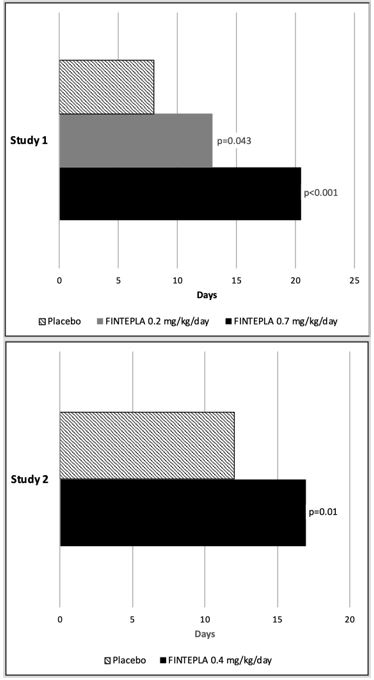
In Study 1 and Study 2, FINTEPLA was associated with a statistically significant longer interval between convulsive seizures compared to placebo (Figure 3).
Figure 3: Median Longest Interval Between Convulsive Seizures in Patients with Dravet Syndrome (Study 1 and Study 2)
14.2 Lennox-Gastaut Syndrome
The effectiveness of FINTEPLA for the treatment of seizures associated with LGS in patients 2 years of age and older was established in a randomized, double-blind, placebo-controlled study in 263 patients 2 to 35 years of age (Study 3; NCT03355209).
Study 3 compared a 0.7 mg/kg/day and a 0.2 mg/kg/day dose of FINTEPLA with placebo. Patients had a diagnosis of LGS and were inadequately controlled on at least one AED, with or without vagal nerve stimulation and/or ketogenic diet. The study had a 4-week baseline period, during which patients were required to have a minimum of 8 drop seizures while on stable AED therapy. Drop seizures were generalized tonic-clonic, secondarily generalized tonic-clonic, tonic, atonic, or tonic-atonic seizures that were confirmed to result in drops. The baseline period was followed by randomization into a 2-week titration period and a subsequent 12-week maintenance period, where the dose of FINTEPLA remained stable.
In Study 3, 99% of patients were taking between 1 and 4 concomitant AEDs. The most frequently used concomitant AEDs (in at least 25% of patients) were clobazam (45%), lamotrigine (34%), and valproate (56%).
The primary efficacy endpoint in Study 3 was the median percent change from baseline in the frequency of drop seizures per 28 days during the combined 14-week titration and maintenance periods (i.e., treatment period). The proportion of patients who achieve improvement (minimally, much, or very much improved) in the Clinical Global Impression of Change (CGI-I) as assessed by Principal Investigator was a secondary endpoint.
In Study 3, the median percent change from baseline (reduction) in the frequency of drop seizures per 28 days was significantly greater for the 0.7 mg/kg/day dose group of FINTEPLA compared with placebo (Table 7). A reduction in drop seizures was observed within 2 weeks of initiating treatment with FINTEPLA, and the effect remained generally consistent over the 14-week treatment period.
The median percent reduction from baseline in drop seizure frequency per 28 days for the lower dose of FINTEPLA (0.2 mg/kg/day) did not reach statistical significance compared to placebo (Table 7).
Table 7: Change in Drop Seizure Frequency during the Treatment Period in Patients with Lennox-Gastaut Syndrome (Study 3) Drop Seizure Frequency
(per 28 days)Placebo FINTEPLA
0.2 mg/kg/dayFINTEPLA
0.7 mg/kg/day- * The total number of patients upon which the efficacy analysis was based is less than the total number randomized in the double-blind, placebo-controlled study because patients with missing data were excluded from the efficacy analysis.
- † Not statistically significant
Study 3 N=85 * N=86 * N=83 * Baseline Period Median Seizure Frequency 55.0 77.8 80.0 Median Percentage Change from Baseline During Treatment -8.7% -13.2% -23.7% p-value compared to placebo 0.1917 † 0.0037 Figure 4 displays the percentage of patients by category of reduction from baseline in drop seizure frequency per 28 days during the treatment period in Study 3.
Figure 4: Proportion of Patients by Category of Seizure Response for FINTEPLA and Placebo in Patients with Lennox–Gastaut Syndrome (Study 3)
Numerically greater improvements on the CGI-I by Investigator were observed in patients treated with FINTEPLA compared with placebo.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
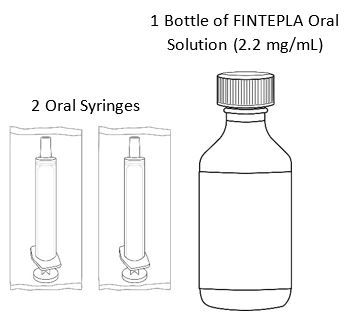
FINTEPLA oral solution is a clear, colorless, cherry flavored liquid containing 2.2 mg/mL fenfluramine and is supplied in a white plastic bottle with a child resistant closure as follows:
- Carton containing one 360 mL bottle (NDC: 43376-322-36)
- Carton containing one 30 mL bottle (NDC: 43376-322-30)
Before dispensing, the pharmacist will insert a press-in bottle adapter into the dispensing bottle. The pharmacy will provide 3 mL or 6 mL calibrated oral dosing syringes.
16.2 Storage and Handling
Store FINTEPLA at room temperature between 20°C to 25°C (68°F to 77°F); excursions are permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Do not refrigerate or freeze. Store the bottle and syringe together.
Discard any unused portion 3 months after first opening the bottle or the "Discard After" date on the bottle, whichever is sooner.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Administration Information
Advise patients who are prescribed FINTEPLA to use the oral dosing syringes provided by the pharmacy [see Dosage and Administration (2.7)and Instructions for Use] . Instruct patients to discard any unused FINTEPLA 3 months after first opening the bottle or if the "discard after" date on the dispensing bottle has passed, whichever is sooner [see How Supplied/Storage and Handling (16.1), (16.2)].
Valvular Heart Disease and Pulmonary Arterial Hypertension
Advise patients that cardiac monitoring must be performed using echocardiography to monitor for serious heart valve changes or high blood pressure in the arteries of the lungs [see Warnings and Precautions (5.1)].
FINTEPLA REMS Program
FINTEPLA is available only through a restricted program called the FINTEPLA REMS program [see Warnings and Precautions (5.2)] . Inform the patient of the following notable requirements:
- Patients must enroll in the program and comply with ongoing echocardiogram monitoring requirements [see Warnings and Precautions (5.1)].
FINTEPLA is only prescribed by certified health care providers and only dispensed from certified pharmacies participating in the program. Therefore, provide patients with the telephone number and website for information on how to obtain the product [see Warnings and Precautions (5.2)].
Decreased Appetite and Decreased Weight
Advise patients that decreased appetite is frequent during treatment with FINTEPLA, which can cause decrease in weight [see Warnings and Precautions (5.3)] .
Somnolence, Sedation, and Lethargy
Inform patients that FINTEPLA can cause somnolence, sedation, and lethargy. Caution patients about operating hazardous machinery, including motor vehicles, until they are reasonably certain that FINTEPLA does not affect them adversely (e.g., impair judgment, thinking, or motor skills) [see Warnings and Precautions (5.4)] .
Suicidal Thinking and Behavior
Counsel patients, their caregivers, and their families that antiepileptic drugs may increase the risk of suicidal thoughts and behavior and advise them to be alert for the emergence or worsening of symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts of self-harm. Instruct patients, caregivers, and families to report behaviors of concern immediately to healthcare providers [see Warnings and Precautions (5.5)] .
Withdrawal of Antiepileptic Drugs (AEDs)
Advise patients not to discontinue use of FINTEPLA without consulting with their healthcare provider. FINTEPLA should normally be gradually withdrawn to reduce the potential for increased seizure frequency and status epilepticus [see Dosage and Administration (2.8), Warnings and Precautions (5.6)] .
Serotonin Syndrome
Inform patients about the risk of serotonin syndrome, which can be life-threatening. Advise patients on the signs and symptoms of serotonin syndrome and that certain over-the-counter medications and herbal supplements can increase this risk [see Warnings and Precautions (5.7)].
Increase in Blood Pressure
Inform patients that FINTEPLA can cause an increase in blood pressure [see Warnings and Precautions (5.8)].
Glaucoma
Inform patients that FINTEPLA can cause mydriasis and can precipitate angle closure glaucoma. Instruct patients to contact their healthcare provider if they have any acute decreases in visual acuity or ocular pain [see Warnings and Precautions (5.9)].
Pregnancy Registry
Advise patients to notify their healthcare provider if they become pregnant or intend to become pregnant during FINTEPLA therapy. Encourage women who are taking FINTEPLA to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry if they become pregnant. This registry is collecting information about the safety of antiepileptic drugs during pregnancy [see Use in Specific Populations (8.1)] .
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
MEDICATION GUIDE
FINTEPLA ®(fin-TEP-la)
(fenfluramine)
oral solutionThis Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 4/2025 Read this Medication Guide before you start taking FINTEPLA and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. What is the most important information I should know about FINTEPLA?
FINTEPLA can cause serious side effects, including:-
Problems with the valves in the heart (valvular heart disease) and high blood pressure in the arteries of the lungs (pulmonary arterial hypertension)have been associated with FINTEPLA. Your healthcare provider will do a test called an echocardiogram to check your heart and for high blood pressure in the arteries of the lungs before you start taking FINTEPLA, again every 6 months during treatment, and one time 3 to 6 months after you take your last dose of FINTEPLA.
Call your healthcare provider right away if you develop any of these signs and symptoms of heart or lung problems during treatment with FINTEPLA:
- shortness of breath
- tiredness or weakness, especially with increased activity
- lightheadedness or fainting
- swollen ankles or feet
- chest pain
- sensations of a rapid, fluttering heartbeat (palpitations)
- irregular pulse
- bluish color to your lips and skin (cyanosis)
Because of the risk of heart valve problems (valvular heart disease) and high blood pressure in arteries of lungs (pulmonary arterial hypertension) FINTEPLA is only available through a restricted program called the FINTEPLA Risk Evaluation and Mitigation Strategy (REMS) Program. Before you or your child receives FINTEPLA, your healthcare provider or pharmacist will make sure you understand how to take FINTEPLA safely. If you have any questions about FINTEPLA, ask your healthcare provider, visit www.FinteplaREMS.com, or call 1-877-964-3649. -
Decreased appetite and decreased weight.Decreased appetite and decreased weight are both serious and common side effects of FINTEPLA in people with Dravet syndrome (DS) or Lennox-Gastaut syndrome (LGS).
- Your weight should be checked regularly during your treatment with FINTEPLA.
- Your healthcare provider may need to make changes to your FINTEPLA dose if your weight decreases. In some cases, FINTEPLA may need to be stopped.
- Sleepiness, sedation, and lack of energy (lethargy).These are both serious and common side effects of FINTEPLA in people with Dravet syndrome (DS) or Lennox-Gastaut syndrome (LGS). Taking FINTEPLA with central nervous system (CNS) depressants including alcohol may increase sleepiness. Do notdrive, operate heavy machinery, or do other dangerous activities until you know how FINTEPLA affects you.
-
Like all other antiepileptic drugs, FINTEPLA may cause suicidal thoughts or actionsin a very small number of people (about 1 in 500).
Call your healthcare provider right away if you have any of these symptoms, especially if they are new, worse, or worry you:
- thoughts about suicide or dying
- trouble sleeping (insomnia)
- attempts to commit suicide
- new or worse irritability
- new or worse depression
- acting aggressive, being angry, or violent
- new or worse anxiety
- acting on dangerous impulses
- feeling agitated or restless
- an extreme increase in activity and talking (mania)
- panic attacks
- other unusual changes in behavior or mood
How can I watch for early symptoms of suicidal thoughts and actions? - Pay attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings.
- Keep all follow-up visits with your healthcare provider as scheduled.
- Do not stop taking FINTEPLA without first talking to your healthcare provider. Stopping a seizure medicine such as FINTEPLA suddenly can cause you to have seizures more often or seizures that do not stop (status epilepticus).
What is FINTEPLA? - FINTEPLA is a prescription medicine used to treat the seizures associated with Dravet syndrome (DS) and Lennox- Gastaut syndrome (LGS) in patients 2 years of age and older.
- It is not known if FINTEPLA is safe and effective in children less than 2 years of age.
Do not take FINTEPLA if you: - are allergic to fenfluramine or any of the ingredients in FINTEPLA. See the end of this Medication Guide for a complete list of ingredients in FINTEPLA.
- are taking or have stopped taking medicines called monoamine oxidase inhibitors (MAOI) in the last 14 days. This may cause a serious or life-threatening problem called serotonin syndrome.If you are not sure whether or not you are taking one of these medicines, contact your healthcare provider.
Before taking FINTEPLA, tell your healthcare provider about all of your medical conditions, including if you: - have heart problems
- have or have had weight loss
- have or have had depression, mood problems, or suicidal thoughts or behavior
- have kidney problems
- have liver problems
- are pregnant or plan to become pregnant. Tell your healthcare provider right away if you become pregnant while taking FINTEPLA. You and your healthcare provider will decide if you should take FINTEPLA while you are pregnant.
- If you become pregnant while taking FINTEPLA, talk to your healthcare provider about registering with the North American Antiepileptic Drug Pregnancy Registry. You can enroll in this registry by calling 1-888-233-2334 or go to www.aedpregnancyregistry.org. The purpose of this registry is to collect information about the safety of antiepileptic drugs during pregnancy.
- are breastfeeding or plan to breastfeed. It is not known if FINTEPLA passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby while taking FINTEPLA.
Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine.How should I take FINTEPLA? - Read the Instructions for Useat the end of this Medication Guide for information on the right way to use FINTEPLA.
- Take FINTEPLA exactly as your healthcare provider tells you to take it.
- Your healthcare provider will tell you how much FINTEPLA to take and when to take it.
- FINTEPLA may be taken with or without food.
- Measure your dose of FINTEPLA using the dosing syringe that is provided by the pharmacy. Do not use a household teaspoon or tablespoon.
- FINTEPLA can be given through gastric and nasogastric feeding tubes
What should I avoid while taking FINTEPLA? - Do notdrive, operate heavy machinery, or do other dangerous activities until you know how FINTEPLA affects you. FINTEPLA may cause you to feel sleepy.
What are the possible side effects of FINTEPLA?
FINTEPLA may cause serious side effects, including:- See " What is the most important information I should know about FINTEPLA?"
- serotonin syndrome.Serotonin syndrome is a life-threatening problem that can happen in people taking FINTEPLA, especially if FINTEPLA is taken with certain other medicines to include:
- anti-depressant medicines called SSRIs, SNRIs, TCAs, and MAOIs
- tryptophan
- lithium
- antipsychotics
- St. John's Wort
- dextromethorphan
- tramadol
Call your healthcare provider right away if you have any of the following symptoms of serotonin syndrome. - mental status changes such as seeing things that are not there (hallucinations), agitation, or coma
- changes in blood pressure
- tight muscles
- fast heartbeat
- nausea, vomiting, diarrhea
- high body temperature
- trouble walking
- high blood pressure (hypertension).Hypertension is both a serious and common side effect. FINTEPLA can cause your blood pressure to increase even if you have never had high blood pressure before. Your healthcare provider will check your blood pressure while you are taking FINTEPLA.
- increased pressure in your eye (glaucoma).Symptoms of glaucoma may include:
- red eyes
- seeing halos or bright colors around lights
- nausea or vomiting
- decreased vision
- eye pain or discomfort
- blurred vision
If you have any of these symptoms, call your healthcare provider right away.
The most common side effects of FINTEPLA when used to treat Dravet syndrome (DS) include:- decreased appetite
- diarrhea
- low energy
- respiratory infection
- decreased weight
- fever
- constipation
- abnormal echocardiogram
- sleepiness
- problems with movement, balance, and walking
- increased drooling
- increased blood pressure
- vomiting
- falls
- seizures that do not stop
- weakness
The most common side effects of FINTEPLA when used to treat Lennox-Gastaut syndrome (LGS) include: - diarrhea
- tiredness
- vomiting
- sleepiness
- decreased appetite
These are not all the possible side effects of FINTEPLA. For more information, ask your healthcare provider or pharmacist.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store FINTEPLA? - Store FINTEPLA at room temperature between 68°F to 77°F (20°C and 25°C).
- Do notrefrigerate or freeze.
- Store the FINTEPLA bottle and syringe together in a clean area.
- Throw away (discard) any unused FINTEPLA 3 months after first opening the bottle or if the Discard After date on the package or bottle has passed. Whichever one comes first.
General information about the safe and effective use of FINTEPLA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use FINTEPLA for a condition for which it was not prescribed. Do not give FINTEPLA to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your pharmacist or healthcare provider for information about FINTEPLA that is written for health professionals.What are the ingredients in FINTEPLA?
Active ingredient:fenfluramine hydrochloride
Inactive ingredients: cherry flavor, citric acid, ethylparaben, hydroxyethylcellulose, methylparaben, potassium citrate, sucralose, and water.
FINTEPLA contains no ingredient made from gluten-containing grain (wheat, barley, or rye) and contains not more than 0.1% of carbohydrates, which is from the cherry flavoring.
Manufactured for: UCB, Inc., Smyrna, GA 30080
FINTEPLA ®is a registered trademark of the UCB Group of Companies. ©2025. All rights reserved.
For more information about FINTEPLA, go to www.fintepla.com or call 1-844-599-2273. -
Problems with the valves in the heart (valvular heart disease) and high blood pressure in the arteries of the lungs (pulmonary arterial hypertension)have been associated with FINTEPLA. Your healthcare provider will do a test called an echocardiogram to check your heart and for high blood pressure in the arteries of the lungs before you start taking FINTEPLA, again every 6 months during treatment, and one time 3 to 6 months after you take your last dose of FINTEPLA.
-
INSTRUCTIONS FOR USE
FINTEPLA
®(fin-TEP-la)
(fenfluramine)
oral solution
2.2 mg/mL
Be sure that you read, understand, and follow these instructions before you start using FINTEPLA oral solution and each time you get a refill. There may be new information.
This Instructions for Use contains information on how to take FINTEPLA. This information does not take the place of talking to your healthcare provider about your medical condition or treatment.
What is included with FINTEPLA?
The following items are included to prepare and give an oral dose of FINTEPLA:
- 1 bottle of FINTEPLA oral solution (2.2 mg/mL)
- 2 reusable oral syringes
1 Bottle of FINTEPLA Oral Solution (2.2 mg/mL) 2 Oral Syringes 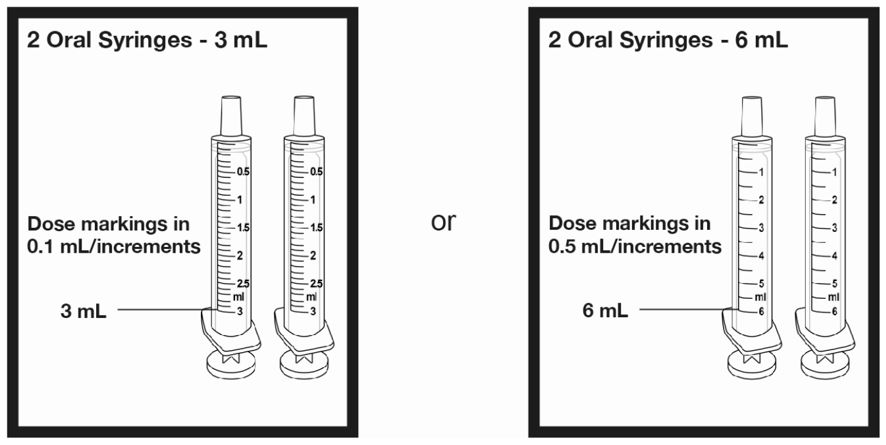
Call the pharmacist at 1-844-288-5007 if you did not receive the items listed above, or if you need help using them.
Important information about FINTEPLA
- FINTEPLA is an oralmedicine (taken by mouth) and is given 2 times each day. Follow your healthcare provider's instructions for taking or giving doses of FINTEPLA.
- If you have questions about how to prepare or give FINTEPLA, contact your healthcare provider or call your pharmacist.
- Always use the oral syringes provided with FINTEPLA to make sure the right dose is given. If you need a new syringe contact your pharmacist. Do notuse a household teaspoon or tablespoon.
Oral syringes provided with FINTEPLA by the pharmacy.
With FINTEPLA you will receive 2 reusable oral syringes.
2 oral syringes that can measure up to 3 mL OR 2 oral syringes that can measure up to 6 mL 
or 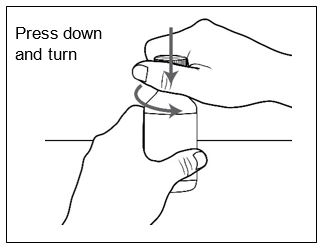
Call the pharmacist at 1-844-288-5007 if you have any questions about the syringes provided with FINTEPLA.
Parts of the Oral Syringe 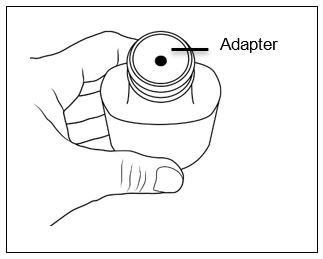
Preparing a Dose 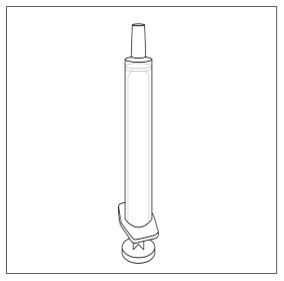
Step 1.Make sure you have: - The bottle of FINTEPLA oral solution, and
- A clean, dry reusable oral syringe that was provided with FINTEPLA.
- Do notuse the medicine if the "Throw Away" (Discard) date has passed.
- If the date is near, contact your pharmacy or healthcare provider to get a refill or new prescription.
- If the date has passed, dispose of any unused FINTEPLA.
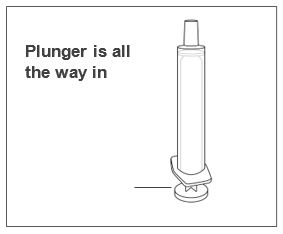
Step 3.Press down and turn the childproof cap to the left (counterclockwise) and remove it from the bottle. - Set the cap aside (do not throw away).
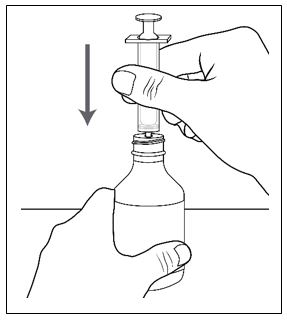
Step 4.Make sure the adapter is on the bottle. - If the bottle does not have an adapter, contact the pharmacist.
- Always leave the adapter in placein the bottle of medicine.
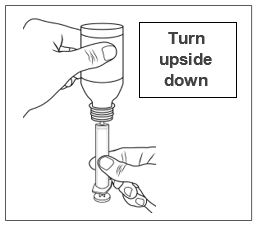
Step 5.Remove an oral syringe from its packaging, if needed.
Only use the oral syringes provided with FINTEPLA.
If an oral syringe is damaged, or you cannot read the dose markings:- Use the other oral syringe provided, or
- Contact the pharmacist to get a new one.
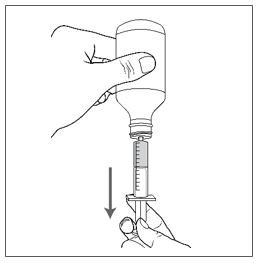
Step 6.Make sure the plunger is pushed all the way into the oral syringe. 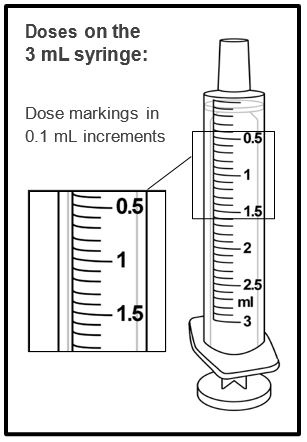
Step 7.Hold the bottle of medicine firmly on a hard, flat surface.
Step 8.Push the tip of the oral syringe into the opening of the adapter until it cannot be pushed further.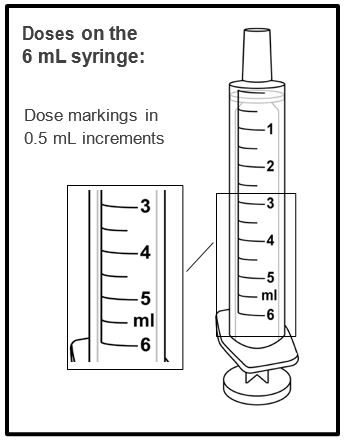
Step 9.Hold the oral syringe and bottle together and turn upside down. 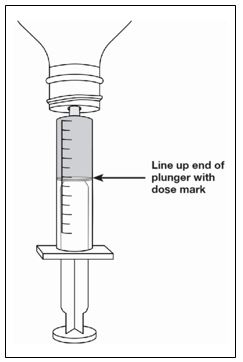
Step 10.Slowly pull the plunger of the oral syringe to withdraw the prescribed dose. 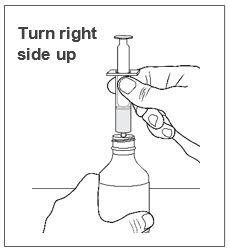
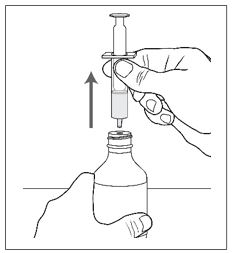
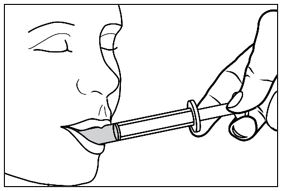
Step 11.Line up the end of the plunger with the mark for the prescribed dose on the oral syringe.
Tips to Getting the Correct Dose- If you draw out too much medicine:
- Leave the oral syringe in the adapter.
- Push the plunger slowly back into the syringe until you reach the prescribed dose.
- If you see air bubbles in the medicine:
- Leave the oral syringe in the adapter.
- Pull the plunger further down.
- Allow the bubbles to rise to the tip of the syringe.
- Push the plunger in all the way.
-
Slowlypull the plunger out to the prescribed dose.
Note:Very small bubbles in the liquid are normal.
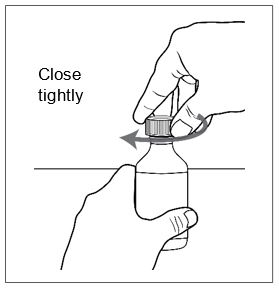
Step 12.Hold the oral syringe and bottle together and then turn the bottle right side up. 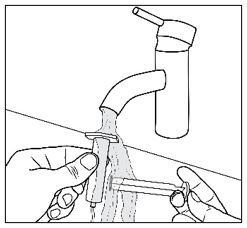
Step 13.Holding the bottle firmly, gently pull the oral syringe out of the bottle adapter. Step 14.Make sure the dose in the oral syringe still matches the prescribed dose.
If the dose does not match:- Put the syringe back into adapter.
- See steps 9 to 11 to adjust the dose, as needed.
Giving FINTEPLA 
Step 15.Place the tip of the oral syringe against the inside of the cheek.
Step 16.Gently push the plunger in until all the medicine in the oral syringe is given.- Do notsquirt or forcefully push the medicine into the back of the throat. This may cause choking.
Step 17.Place the cap back on the bottle tightly by turning the cap right (clockwise) until it stops. - Always leave the adapter in place in the bottle
- The cap will fit over it.
Cleaning the syringe 
- Rinse the oral syringe with cold water and allow it to air dry after each use.
- Make sure you rinse the inside of the syringe and the plunger.
- Pull cold water into the syringe with the plunger and push it out several times to clean the syringe.
- Remove the plunger from the barrel of the oral syringe.
- Rinse both parts under cold water.
- Make sure the syringe and plunger are completely dry before the next use.
- Do not use detergent to clean the syringe and plunger.
- Do not clean in the dishwasher.
How should I store FINTEPLA?
- Store FINTEPLA at room temperature between 68°F to 77°F (20°C to 25°C).
- Do notrefrigerate or freeze.
- Keep the cap tightly closed and the bottle upright.
- Store the FINTEPLA bottle and syringe together in a clean area.
- Throw away (discard) any unused FINTEPLA 3 months after first opening the bottle orif the Discard After date on the package or bottle has passed. Whichever one comes first.
- Keep FINTEPLA and all medicines out of the reach of children.
Manufactured for: UCB, Inc., Smyrna, GA 30080
FINTEPLA ®is a registered trademark of the UCB Group of Companies. ©2025. All rights reserved.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 10/2025 -
PRINCIPAL DISPLAY PANEL - 30 mL Bottle Label - NDC: 3376-322-30
NDC: 43376-322-30
Fintepla ®
(fenfluramine)
oral solution
2.2 mg/mLRx only
For oral use onlyContents 30 mL

-
PRINCIPAL DISPLAY PANEL - 30 mL Bottle Carton - NDC: 3376-322-30
NDC: 43376-322-30
Fintepla ®
(fenfluramine)
oral solution
2.2 mg/mLFor oral use only
Attention Pharmacist:
Dispense the enclosed
Medication Guide to each patientContents:
One 30 mL bottleRx only
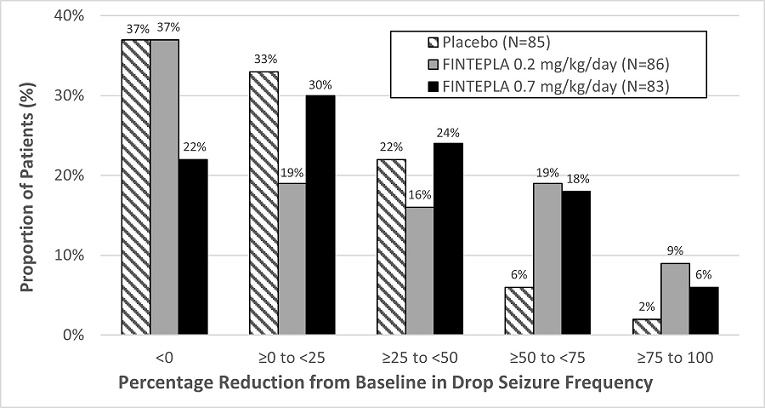
-
PRINCIPAL DISPLAY PANEL - 360 mL Bottle Label - NDC: 43376-322-36
NDC: 43376-322-36
Fintepla ®
(fenfluramine)
oral solution
2.2 mg/mLFor oral use only
Rx only
Contents 360 mL

- PRINCIPAL DISPLAY PANEL - 360 mL Bottle Carton - NDC: 43376-322-36
-
INGREDIENTS AND APPEARANCE
FINTEPLA
fenfluramine solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 43376-322 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FENFLURAMINE (UNII: 2DS058H2CF) (FENFLURAMINE - UNII:2DS058H2CF) FENFLURAMINE 2.2 mg in 1 mL Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) POTASSIUM CITRATE (UNII: EE90ONI6FF) ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) HYDROXYETHYL CELLULOSE (2000 MPA.S AT 1%) (UNII: S38J6RZN16) METHYLPARABEN (UNII: A2I8C7HI9T) SUCRALOSE (UNII: 96K6UQ3ZD4) ETHYLPARABEN (UNII: 14255EXE39) Product Characteristics Color Score Shape Size Flavor CHERRY Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 43376-322-30 1 in 1 CARTON 07/15/2020 1 30 mL in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 2 NDC: 43376-322-36 1 in 1 CARTON 07/15/2020 2 360 mL in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212102 07/15/2020 Labeler - UCB, Inc. (028526403)
Trademark Results [Fintepla]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 FINTEPLA 88284913 not registered Live/Pending |
ZOGENIX INTERNATIONAL LIMITED 2019-01-31 |
 FINTEPLA 87687367 not registered Dead/Abandoned |
ZOGENIX INTERNATIONAL LIMITED 2017-11-16 |
 FINTEPLA 86949118 5443859 Live/Registered |
Zogenix International Limited 2016-03-22 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.